
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Fluorométrie de tension-serre dans
Dans cet article
Résumé
Cet article décrit une amélioration de la Fluorométrie conventionnelle en tension-clamp (VCF) où les acides aminés non naturels fluorescents (fUAA) sont utilisés à la place des colorants maléimidés, pour sondage des réarrangements structurels dans les canaux ioniques. La procédure comprend l'injection d'ADN ovocytaire de Xenopus, la co-injection d'ARN / fUAA et les mesures simultanées de courant et de fluorescence.
Résumé
La Fluorométrie de la Tension-Clamp (VCF) a été la technique de choix pour étudier la structure et la fonction des protéines de la membrane électrogène, où les mesures en temps réel de la fluorescence et des courants rapportent simultanément sur les réarrangements locaux et la fonction globale, respectivement 1 . Alors que les techniques structurelles à haute résolution telles que la microscopie cryo-électronique ou la cristallographie aux rayons X fournissent des images statiques des protéines d'intérêt, VCF fournit des données structurelles dynamiques qui nous permettent de relier les réarrangements structurels (fluorescence) aux données fonctionnelles dynamiques (électrophysiologie). Jusqu'à récemment, la chimie réactif au thiol utilisée pour l'étiquetage fluorescent dirigé par le site des protéines a limité la portée de l'approche car toutes les cystéines accessibles, y compris les endogènes, seront étiquetées. Il était donc nécessaire de construire des protéines exemptes de cystéines endogènes. L'étiquetage était également limité aux sites accessibles à partir de l'extracellulairecôté. Cela a changé avec l' utilisation d'acides aminés non naturels fluorescents (fUAA) pour incorporer spécifiquement une petite sonde fluorescente en réponse à la suppression du codon d'arrêt en utilisant une paire ARNt et tRNA synthetase orthogonal 2 . L'amélioration du VCF nécessite seulement une procédure d'injection en deux étapes de l'injection d'ADN (paire d'ARNt / synthetase) suivie d'une co-injection d'ARN / fUAA. Maintenant, l'étiquetage des sites intracellulaires et enterrés est possible et l'utilisation de VCF a considérablement augmenté. La technique VCF devient ainsi attrayante pour l'étude d'une large gamme de protéines et, plus important encore, permet d'enquêter sur de nombreux mécanismes de régulation cytosolique.
Introduction
Plus de 200 acides aminés non naturels de diverses propriétés chimiques et physiques ont été incorporés génétiquement dans les protéines dans les cellules de E. coli , de levure et de mammifères 3 . L'acide aminé non naturel est incorporé en réponse à un codon d'arrêt spécifique par une paire d'ARNt / synthetase modifiée par voie orthogonale. L'approche génétique de la modification des protéines a fourni des informations précieuses sur la structure et la fonction des protéines. Ici, nous présentons un protocole d'utilisation de la Fluorométrie de tension-clamp (VCF) en combinaison avec une SAU fluorescente.
Dans VCF, l'observation simultanée de données fonctionnelles et de réarrangements structurels localisés autour de la sonde fluorescente (~ 5 Å) nous permet d'obtenir des informations dynamiques avec une résolution de milliseconde 1 . Les sondes fluorescentes modifient leur état de trempe lors d'un mouvement localisé de la protéine. Un mouvement de seulement 1-2 Å est suffisant pour conduire à des changements significatifs dans la fluorescenceIntensité 4 . Après l'identification du site d'intérêt dans la protéine cible, le site est muté par mutation ponctuelle. Classiquement, le résidu a été muté à une cystéine alors que maintenant, un codon d'arrêt ambré (TAG) est introduit pour l'incorporation de fUAA génétique. La protéine est ensuite transcrit in vitro .
Alors que d'autres systèmes d'expression ( p. Ex., Cellules de mammifères) peuvent être utilisés 5 , 6 , 7 , les ovocytes de Xenopus sont préférables pour les études de structure-fonction en raison de leur taille plus grande, conduisant à une manipulation plus facile et une plus grande intensité de fluorescence (plus de fluorophores) et, par conséquent, Rapport bruit-bruit. De plus, les ovocytes de Xenopus ont un faible effet de fond sur les protéines endogènes 2 , 8 et la pigmentation sombre sur les écussons des poteaux animaux contre la fluorescence de fond de tIl cytosol. Les ovocytes Xenopus sont éliminés chirurgicalement et l'ADN codant pour la paire orthogonale d'ARNt / tRNA-synthetase spécifique pour le fUAA est injecté dans le noyau des ovocytes. Après un temps d'incubation de 6 à 24 heures, l'ARN de la protéine est co-injecté avec le fUAA dans le cytosol des ovocytes, suivi d'une période d'incubation de 2 à 3 jours. Afin d'éviter tout dommage au fUAA (photoblanchage), les procédures incluant Anap doivent être effectuées sous la lumière rouge pour éviter l'excitation des fluorophores.
Les ovocytes sont étudiés sur une installation de tension-clamp d'ovocytes coupées, qui est montée sur un microscope de fluorescence vertical, et le courant électrique et les changements de fluorescence sont enregistrés simultanément 9 , 10 . Alternativement, on peut utiliser une pince de tension à deux électrodes 1 ou des configurations de bridage-bride 11 . La fluorescence est excitée par des longueurs d'onde appropriées avec un faible bruit RMS et Émission enregistrée à l'aide d'une photodiode liée à un amplificateur avec une amplification élevée.
Il existe plusieurs avantages de l'utilisation d'acides aminés non naturels fluorescents (fUAA) dans la fluorométrie tension-clamp. L'une est l'accès au côté cytosolique des protéines membranaires; De nombreux processus réglementaires sont situés ici ( p. Ex. Sites de liaison au Ca 2+ ou aux nucléotides, inactivation rapide et à l'état de fermeture des canaux ioniques à tension, ouverture des pores, couplage du module). Tous ces processus sont maintenant accessibles pour l'étiquetage fluorescent.
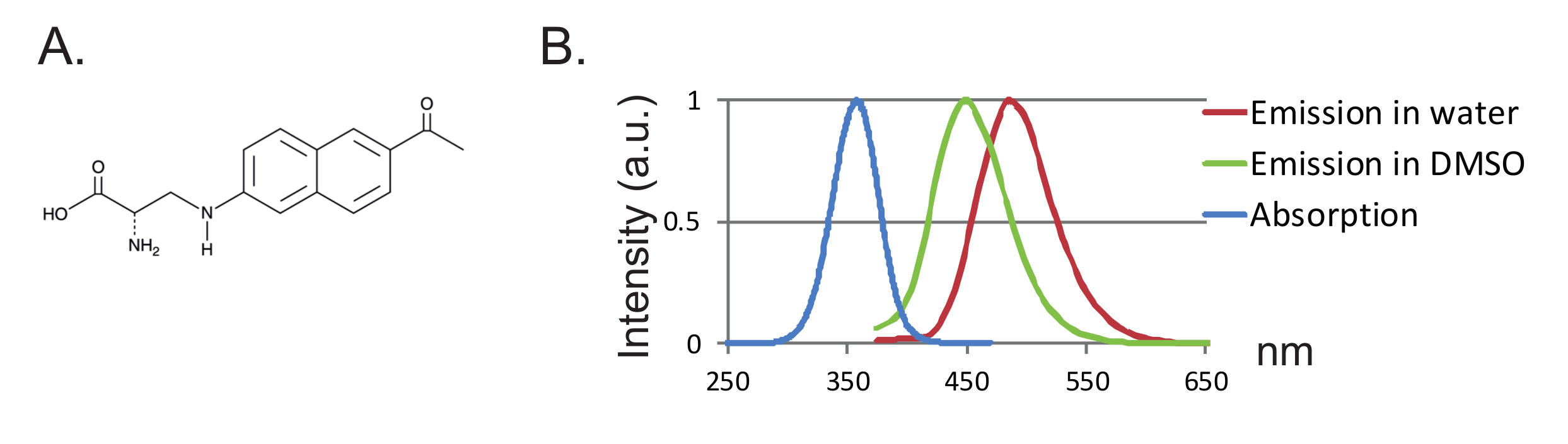
Un autre avantage est la petite taille de la sonde conduisant à moins de perturbation de la protéine. Jusqu'à présent, deux paires orthogonales d'ARNt / tRNA synthetase pour les FUAA ont été conçues 12 , 13 , où l'acide 3- (6-acétylnaphtalène-2-ylamino) -2-aminopropanoïque (Anap) est le seul fUAA qui a été utilisé dans les ovocytes Xenopus 2 ,"Xref"> 8. Anap est un fluorophore respectueux de l'environnement avec un poids moléculaire de 272,3 g / mol et n'est que légèrement plus grand que le tryptophane 12 ( figures 1A, 1B ). En raison de sa petite taille, moins d'effets stériques sont susceptibles d'être introduits par le fluorophore par rapport aux fluorophores classiques fixés via un lieur (typiquement plus de 500 g / mol). De plus, dans le cas d'Anap, le fluorophore est situé plus près du squelette de la protéine que ceux liés aux cystéines, et par conséquent, Anap sondage des réarrangements plus localisés. Enfin, l'élimination des cystéines endogènes dans le VCF conventionnel afin d'assurer l'étiquetage spécifique du site n'est plus une exigence dans l'UAA-VCF et donc (i) laisse les protéines dans (presque) leur état natif et (ii) permet l'application de VCF Pour étudier une gamme plus large de protéines dans laquelle la fonction peut être modifiée par la substitution de cysteine.

Figure 1 : Spectre Anap et Fluorescence. ( A ) Structure chimique d'Anap. ( B ) spectre d'absorption normalisé et spectres d'émission pour 1 nM Anap, démontrant la sensibilité de la fluorescence Anap à l'hydrophobicité du solvant. Les spectres d'émission ont été obtenus en excitant à 350 nm. Cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}
Un inconvénient de l'utilisation de SAU fluorescentes est qu'une population hétérogène de protéines peut résulter d'une lecture à distance de codon, d'une réinitiation translationnelle, de protéines tronquées C-terminales ou de diaphonie avec une aminoacylation endogène si la quantité d'ARNt aminoacylés est rare. Une telle expression de fuite doit toujours être vérifiée en l'absence du fUAA et de la paire tRNA / tRNA synthetase. Nous avons abordé la question de transLa réinitialisation lationnelle et comment la contourner pour les sites d'insertion N-terminaux précédemment 14 . Cependant, lorsque le fUAA, l'ARNt et l'ARNt synthetase sont présents en quantités saturées, il ne reste qu'une faible probabilité d'expression de fuite.
La principale différence de procédure entre fUAA-VCF et VCF conventionnel est l'injection et la manipulation des ovocytes; L'injection d'ADN codant pour l'ARNt et l'ARNt synthetase (pAnap) est suivie de l'introduction d'Anap, qui est soit co-injecté avec l'ARNm de protéine, soit ajouté à la solution d'incubation sous la forme d'un ester acétoxyméthyl (AM).
Access restricted. Please log in or start a trial to view this content.
Protocole
Les manipulations de grenouilles ont été effectuées conformément aux directives canadiennes et ont été approuvées par le comité d'éthique (CDEA, protocole n o 15-042) de l'Université de Montréal.
1. Préparation de l'ARNm pour l'incorporation de la FUAA
- Choisissez un site d'intérêt pour la protéine où les changements conformationnels devraient se produire. Sélectionnez un acide aminé dans cette région pour remplacer le fUAA.
REMARQUE: Le choix de la position est basé sur les réarrangements structurels attendus. Si une structure haute résolution existe et une hypothèse des mouvements attendus, l'anap devrait être placé de telle sorte que l'environnement chimique se modifiera; Cela pourrait être soit une modification de la constante diélectrique (environnement hydrophobe versus hydrophile) soit, plus probablement, tremper par un autre acide aminé. Les meilleurs extincteurs sont les tryptophans. Anap devrait être en contact avec l'extincteur dans un état (chevauchement du rayon Van-der-Waals) et libre dansL'autre. Si aucune structure ou modèle à haute résolution n'existe, il faudrait analyser la région d'intérêt. Dans les deux cas, il est conseillé de sélectionner plusieurs sites voisins pour augmenter la probabilité d'obtenir l'expression et le signal de fluorescence. Afin de minimiser les effets stériques pendant la maturation et / ou la fonction de la protéine, on peut choisir de remplacer les acides aminés grands et aromatiques (Phe, Trp, Tyr). Cependant, les auteurs ont constaté que la recherche d'une région d'intérêt pour l'insertion de la FUAA indépendamment de l'acide aminé substitué est plus productive. - Insérer un codon d'arrêt ambré (TAG) au site sélectionné en utilisant une mutagenèse dirigée 15 . Assurez-vous que la protéine d'intérêt ne se termine pas par un codon d'arrêt ambre (TAG). Si tel est le cas, muter à un autre (codage d'arrêt ocre ou opale). Amplifier, isoler et séquencer l'ADN. Obtenir l'ARNm protéique avec la transcription in vitro 16 et stocker l'ARNm à 20 ° C ou à 80 ° C.
- Obtenir chirurgicalement les ovocytes de stade V ou VI des grenouilles de Xenopus laevis et défolliculer avec la collagénase comme décrit précédemment 17 .
- Anesthésier les grenouilles avec un anesthésique approprié selon le protocole animal approuvé (ici: ester éthylique d'acide 3-aminobenzoïque). Quand ils ne répondent pas à une pincée douce à un bout de bout (réflexe de perte de retrait), ils sont convenablement anesthésiés pour une intervention chirurgicale.
- Enlever immédiatement les grenouilles de la solution anesthésique et rincer soigneusement sa peau à l'eau douce. Ce rinçage empêchera l'animal de tomber dans des niveaux plus profonds d'anesthésie en éliminant le produit chimique non absorbé de la surface de la peau.
- Retirer les ganglions ovaires d'un côté et ouvrir soigneusement les noeuds en utilisant deux pinces. Incuber et agiter les ovocytes dans la "solution standard d'ovocyte" (SOS) contenant 1% (p / v) de collagénase pendant 20 à 30 minutesÀ défolliculer. Laver trois fois avec une solution SOS.
- Choisir les ovocytes grands et sains individuellement et les incuber dans la solution de Barth complétée par des antibiotiques (100 U / mL de péniciline, 100 μg / mL de streptomycine, 10 mg / 100 ml de kanamycine) et 5% de sérum de cheval à 18 ° C pendant au moins 4 h avant l'injection .
REMARQUE: Après 2 à 4 chirurgies avec un délai de 4 mois entre les deux, Xenopus laevis est euthanasié par incubation prolongée (> 1 h) avec de l'ester éthylique d'acide 3-aminobenzoïque.
- Pour l'injection nucléaire d'ADN, prépare une pointe d'injection longue et mince pour pouvoir atteindre le noyau et éviter d'endommager l'ovocyte. Remplissez la pointe d'injection avec de l'huile et montez-la sur le dispositif de nanoinjector.
- Installez le nano-injecteur sous un microscope stéréo et utilisez des pinces pour briser la fin de la pointe. Ejectez l'huile jusqu'à ce qu'il n'y ait pas de bulles d'air piégées à l'intérieur de l'extrémité de la pointe.
- Placer 1 μL de 0,1 μg / &# 181; L pAnap dans de l'eau libre de nuclease contenant du NaOH (1% de NaOH 1 N) sur un parafil sous un stéréoscope et remplir le bout d'injection avec l'ADN.
- Transférer 40 ovocytes dans un plat d'injection recouvert de maille contenant de la solution de Barth complétée par des antibiotiques.
REMARQUE: Pour fabriquer un plat d'injection recouvert de maille, découpez une pièce de taille appropriée de maille de nylon de 800 μm pour remplir une boîte de Petri en polystyrène. Ajouter le chloroforme au centre, puis placez le maillage sur le dessus. Tenez le maillage à plat jusqu'à ce que les jeux de plastique. - Au fur et à mesure que le noyau d'ovocytes est situé dans le pôle animal (sombre), vissez la pointe d'injection au centre du pôle animal et empalez-le de sorte que la pointe atteigne près du centre de l'hémisphère animal (ou 2-3x la profondeur par rapport à l'injection d'ARN ). Injecter 9.2 nL de pAnap dans le noyau de chaque ovocyte. La pointe mince et le petit volume d'injection peuvent entraîner une injection irrégulière ou un embout bloqué. Vérifiez parfois si l'injection fonctionne par injection dans l'air.
REMARQUE: si l'ADN est correctement injecté dans le noyau est incertain. Attendez-vous donc que 10 à 40% des ovocytes n'expriment pas la paire d'ARNt / synthetase. Voir la discussion pour une élaboration plus approfondie. - Incuber les ovocytes dans 2 mL de solution de Barth complétée par des antibiotiques et 5% de sérum de cheval (HS) à 18 ° C pendant 6-24 h pour permettre une expression robuste d'ARNt spécifiques d'Anap et d'ARNt-synthetases.
NOTE: Le temps d'incubation de l'ADN peut durer plusieurs jours avant l'injection d'ARN, mais cela n'augmente pas l'expression. - Préparez le nano-injecteur pour l'injection d'ARN (identique à l'étape 2.2, mais la pointe de l'injection n'a pas besoin d'être aussi mince que pour l'injection d'ADN). Travailler uniquement sous la lumière rouge à partir de ce point pour empêcher le photoblanchage de l'Anap.
- Mélanger 1 μL d'Anap 1 mM avec 1 μL d'ARNm de 1-2 μg / μL directement sur un parafilm et remplir le bout d'injection avec la solution mélangée. Impelle juste en dessous de la membrane dans le végétal(Lumineux) et injecter 46 nL dans chaque ovocyte injecté par pAnap.
NOTE: La concentration d'ARNm requise dépend de la protéine d'intérêt. - Incuber les ovocytes protégés de la lumière dans une boîte ou enveloppés dans du papier d'aluminium, dans la solution de Barth complète les antibiotiques et 5% de sérum de cheval à 18 ° C pendant 2-3 jours. Échangez avec la solution fraîche de Barth tous les jours et retirez les ovocytes morts pour éviter toute contamination.
- Installez l'équipement de coupe-tension d'ovocytes coupé comme décrit précédemment 18 .
- Montez le système d'enregistrement d'électrophysiologie sur un microscope de fluorescence vertical en installant la chambre d'enregistrement sur un curseur qui permet de le déplacer entre le stéréoscope standard pour placer l'ovocyte et le microscope pour effectuer les mesures de fluorescence ( Figure 2c ).
NOTE: La géométrie de la chambre pour les ovocytes coupés n'est pas appropriée pour utiliser le traLa lumière allumée pour l'illumination pendant la manipulation. Par conséquent, une lampe halogène "col à col de cygne" avec filtre rouge est utilisée pour éclairer latéralement depuis le haut. Le condensateur du microscope peut être retiré pour créer un espace pour abaisser le niveau de la chambre d'électrophysiologie. - Connectez un système de détection de photodiode au port de sortie C-mount du microscope à fluorescence ( Figure 2a ). Connectez la lecture de photocourents à un deuxième canal d'entrée dans le processeur de signal numérique (DSP, convertisseur analogique / numérique - numérique / analogique).
- Utilisez une lampe halogène de 100 W et 12 V comme source lumineuse pour l'excitation de fluorescence.
REMARQUE: En variante, les brûleurs Hg peuvent être utilisés, mais doivent être réduits en intensité pour empêcher un photoblanchage trop rapide pendant les enregistrements. L'éclairage LED n'est recommandé que si les LED respectives affichent une intensité significative dans la plage d'excitation ( par exemple, ~ 350 nm pour Anap). La plupart des LED blanches n'atteignent pas loin le spectre UV. - InsUn obturateur électriquement contrôlé entre la source d'éclairage d'excitation et le microscope et connectez son contrôle (typiquement l'impulsion TTL) à une sortie numérique du DSP. Autour de l'impulsion TTL dans le logiciel d'enregistrement (voir la documentation du fabricant), de sorte que l'obturateur s'ouvre ~ 100 ms avant le début de l'enregistrement. De cette façon, toute vibration pendant le processus d'ouverture n'interfère pas avec l'enregistrement. Le temps dépend de la vitesse et des vibrations du volet. Fin de l'impulsion 5 ms avant la fin de l'enregistrement, comme illustré à la Figure 4 . De cette façon, la valeur de la fluorescence totale est également enregistrée.
- Insérez un cube de filtre approprié (filtre d'excitation, miroir dichroïque et filtre à émission) dans la tourelle de filtre-cube. Pour Anap, utilisez Ex: bande passante 377/50 nm, passe longue dichroïque de 409 nm et Em: bande passante 470/40 nm.
- Suivez les étapes de préparation pour la tension-clamp de l'ovocyte coupée comme décrit précédemment et visualisé 18 (préparation du pont d'agar, montage de l'oocyte, perméabilisation de la saponine). Cependant, travaillez sous la lumière rouge en tout temps pour éviter de blanchir le fluorophore avant les enregistrements. Lorsque vous placez l'ovocyte, assurez-vous que le pôle animal est tourné vers le haut. La pigmentation sous la membrane du pôle animal protège contre l'autofluorescence provenant du cytosol et réduit donc la fluorescence du fond.
- Faites glisser la chambre au microscope et concentrez-vous à l'aide d'un objectif 4X.
- Impulser l'ovocyte avec l'électrode V1 de détection de tension (3 M KCl), passer à l'objectif 40X d'immersion dans l'eau (NA 0,8 - 0,9). Concentrez-vous sur le poteau animal tourné vers le haut.
- Éteignez le feu rouge. Sélectionnez le bon cube de filtre en tournant la tourelle du cube de filtre et le port de sortie optique connecté à la photodiode. Allumez l'halogèneEn lampe à la plus haute intensité et basculer brièvement le déclencheur pendant 2 à 5 s pour lire l'intensité de fluorescence de fond provenant de l'ovocyte. Avec la configuration décrite, la valeur devrait être d'environ 50-200 pA pour Anap.
- Allumez la pince, basculez le commutateur de bain / protecteur sur actif et ajustez le potentiel de la membrane (V1 - V2) au potentiel de commande en tournant le bouton sur la trajectoire I.
- Sélectionnez le potentiel de maintien, le protocole d'étape, le nombre et la longueur des impulsions, etc. dans le logiciel d'enregistrement. Enregistrer les courants dépendants de la tension et les intensités de fluorescence Anap.
- Pour surveiller simultanément deux endroits dans la même protéine, muter un acide aminé extracellulaire et accessible en cysteine et éliminer d'autres cystéines pour assurer un étiquetage spécifique avec la thiol-chimie.
- Effectuez l'étape 2.1-2.5.
- Avant les enregistrements VCF, incuber des ovocytes dans du TMR-maléimide 5 μM dans une solution d'étiquetage pendant 15 min (ou autre colorantAvec des spectres non superposés par rapport à Anap).
- Lavez les ovocytes avec une solution d'étiquetage trois fois pour éliminer l'excès de colorant.
- Effectuez l'étape 4.1-4.6.
- Insérez un cube filtrant approprié pour le TMR (filtre d'excitation, miroir dichroïque et filtre à émission) dans la tourelle de filtre-cube. Passez au cube de filtre TMR en tournant la tourelle filtrante.
- Lisez la fluorescence d'arrière-plan pour TMR comme décrit pour Anap à l'étape 4.4.
REMARQUE: L'étiquetage avec la thiol-chimie entraîne une fluorescence de fond élevée due à un marquage non spécifique dans la membrane. Par conséquent, la fluorescence de fond TMR peut saturer l'amplificateur (> 2 000 pA). Dans ce cas, ne diminuez pas l'intensité de la lumière, mais simplement soustrayez la fluorescence d'arrière-plan en ajoutant un courant de décalage à la photodiode. Dans les systèmes disponibles dans le commerce, utilisez la fonction "échantillonnage et maintien" sur le système de détection. Notez la valeur de fluorescence d'arrière-plan (en utilisant un filtre de densité neutre 10X) dans un laboratoire jOurnal, car cette valeur ne sera pas enregistrée (saturation). - Enregistrez les courants dépendant de la tension et les intensités de fluorescence TMR simultanément comme à l'étape 4.6.
2. Préparation et injection d'ovocytes

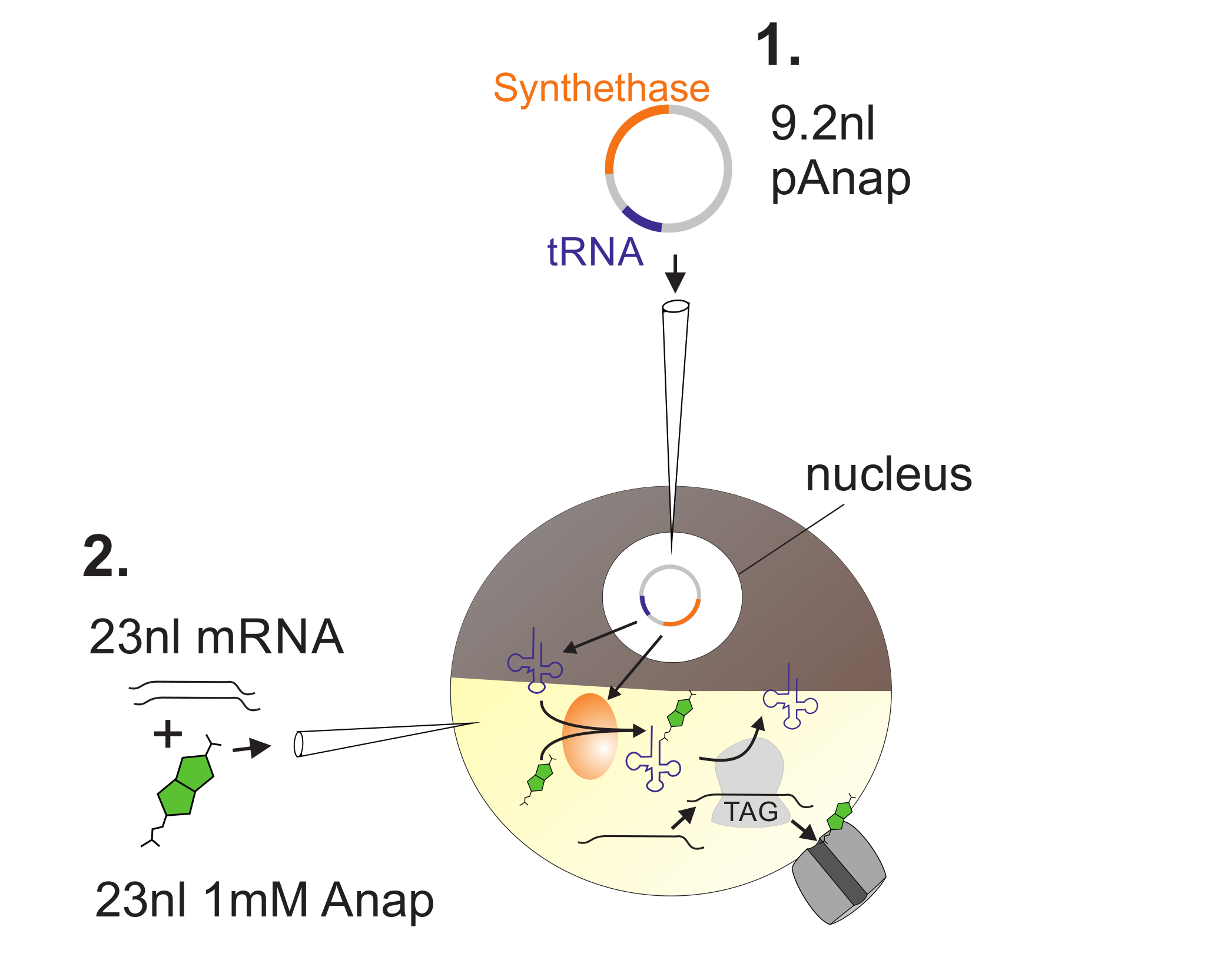
Figure 2 : Illustration de l'injection d'ADN et d'ARN dans les ovocytes Xenopus pour Anap Incorporation.
Tout d'abord, pAnap est injecté dans le noyau de l'ovocyte Xenopus ( 1 ). Après 6-24 h, Anap et l'ARN du canal sont co-injectés dans le pôle végétal ( 2 ). Anap sera orthogonalement aminoacylé avec l'ARNt portant un anti-codon anti-ambre, par l'aminoacyl-ARNt synthétase qui est codée par pAnap. De cette façon, les ARNt Anap-aminoacylés sont reconnus par le ribosome au codon d'arrêt ambré inséré dans la canalisationL ARN, entraînant la suppression du codon d'arrêt et l'insertion d'Anap. Cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}
3. Configuration VCF

Figure 3 < / Strong> : configuration VCF. ( A ) Vue latérale de la configuration VCF montrant le trajet lumineux à l'intérieur du microscope. Le cube de filtre contient un filtre d'excitation, un miroir dichroïque et un filtre à émission. ( B ) Les dimensions choisies de la chambre d'ovocytes sont de 3,4 cm pour le rayon de la chambre supérieure (1), 5,5 cm pour la longueur de la chambre inférieure (2), 1,4 cm pour la largeur de la chambre inférieure (3) et 1,7 cm pour la largeur de la chambre moyenne (4). ( C ) Vue de face de l'installation VCF. Le premier œil à gauche est destiné à monter l'ovocyte dans la chambre de serrage à tension coupée et à la perméabilisation. Ensuite, la chambre est glissée sous le microscope au deuxième œil à droite. Ici, l'électrode V1 est insérée dans l'ovocyte en utilisant l'objectif 4X, et la fluorescence est enregistrée à l'aide de l'objectif 40X d'immersion dans l'eau. Cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}
5. VCF à deux couleurs
Access restricted. Please log in or start a trial to view this content.
Résultats
La figure 4 montre un exemple d'enregistrements VCF obtenus à partir d'un exitoire exprimant des canaux Shaker avec une inactivation rapide enlevée (IR), L382stop-W434F en présence de pAnap et Anap. La mutation W434F bloque les courants ioniques de potassium, ce qui permet de mesurer les déplacements transitoires de la charge de déclenchement (courants de déclenchement). Les enregistrements simultanés des courants de déclenchement (traces supérieures) et...
Access restricted. Please log in or start a trial to view this content.
Discussion
L'aminoacylation in vivo des ARNt qui sont transcrites en continu avec l'ARNt-synthetase permet d'obtenir des niveaux d'expression élevés pour les mesures de fluorescence. Pour une incorporation fUAA efficace, il est essentiel que pAnap soit correctement injecté dans le noyau. En raison de l'incertitude quant à l'emplacement exact du noyau, 10 à 40% des injections d'ADN devraient échouer, ce qui entraîne des ovocytes non exprimant (ou exprimant des fuites). Par conséquent, il...
Access restricted. Please log in or start a trial to view this content.
Déclarations de divulgation
Les auteurs n'ont rien à dévoiler.
Remerciements
PAnap était un cadeau génial du Dr Peter Schultz (Scripps Research Institute). Ce travail a été financé par les Instituts du Canada pour subventions de recherche en santé MOP-102689 et MOP-136894 (à RB) et la Fondation canadienne pour l'innovation 950-225005.
Access restricted. Please log in or start a trial to view this content.
matériels
| Name | Company | Catalog Number | Comments |
| Solutions | |||
| Barth's solution | |||
| NaCl | Sigma-Aldrich | S7653 | 90 mM |
| KCl | Fisher Scientific | BP366-500 | 3 mM |
| MgSO4 | Sigma-Aldrich | M-9397 | 0.82 mM |
| CaCl2 | Sigma-Aldrich | C-7902 | 0.41 mM |
| Ca(NO3)2 | Sigma-Aldrich | C-1396 | 0.33 mM |
| HEPES | Sigma-Aldrich | H4034 | 5 mM |
| NaOH hydrate | BDH | BDH7225-4 | pH 7.6 |
| Penicilin | Invitrogen | 15140122 | 100 U/mL |
| Streptomycin | Invitrogen | 15140122 | 100 µg/mL |
| Kanamycin | Invitrogen | 15160054 | 10 mg/100mL |
| Horse Serum (HS) | Invitrogen | 16050122 | 5% |
| SOS Standard Oocyte Solution | |||
| NaCl | Sigma-Aldrich | 746398 | 102 mM |
| KCl | Sigma-Aldrich | 746436 | 3 mM |
| MgCl2 | Sigma-Aldrich | M9272 | 1 mM |
| HEPES | Sigma-Aldrich | H4034 | 5 mM |
| External recording solution | |||
| N-methyl-D-glucamine (NMDG) | Alfa Aesar | L14282 | 115 mM |
| HEPES | Sigma-Aldrich | H4034 | 10 mM |
| Calcium hydroxide | Sigma-Aldrich | 239232 | 2 mM |
| MES hydrate | Sigma-Aldrich | 258105 | pH 7.2 |
| Internal recording solution | |||
| N-methyl-D-glucamine (NMDG) | Alfa Aesar | L14282 | 115 mM |
| HEPES | Sigma-Aldrich | H4034 | 10 mM |
| Ethylenediamine Tetraacetic Acid (EDTA) | Fisher Scientific | E478-500 | 2 mM |
| MES hydrate | Sigma-Aldrich | 258105 | pH 7.2 |
| Labeling solution | |||
| KOH | Fisher Scientific | P250-1 | 115 mM |
| HEPES | Sigma-Aldrich | H4034 | 10 mM |
| Calcium hydroxide | Sigma-Aldrich | 239232 | 2 mM |
| MES hydrate | Sigma-Aldrich | 258105 | pH 7.2 |
| TMR stock solution | |||
| Tetramethylrhodamine-5-maleimide (TMR) | Molcular Probes by Life Technologies | T6027 | 5 mM in DMSO |
| Anap stock solution | |||
| Anap | ABZENA (TCRS) | Custom synthesis TCRS-170 | 1 mM in nuclease-free water and 1% NaOH 1 N |
| Name | Company | Catalog Number | Comments |
| Material/Equipment | |||
| pAnap | Addgene | 48696 | |
| High Performance Oocyte Clamp | Dagan Corporation | CA-1B | |
| Gpatch Acquisition software | Department of Anesthesiology, University of California, Los Angeles | ||
| Analysis software | Department of Anesthesiology, University of California, Los Angeles | ||
| Recording Chamber | Custom machined | ||
| Photo diode detection system | Dagan Corporation | PhotoMax-200/PIN | |
| Electrical shutter driver | UNIBLITZ | VCM-D1 |
Références
- Mannuzzu, L. M., Moronne, M. M., Isacoff, E. Y. Direct physical measure of conformational rearrangement underlying potassium channel gating. Science. 271 (5246), 213-216 (1996).
- Kalstrup, T., Blunck, R. Dynamics of internal pore opening in K(V) channels probed by a fluorescent unnatural amino acid. Proc Natl Acad Sci U S A. 110 (20), 8272-8277 (2013).
- Xiao, H., Schultz, P. G. At the Interface of Chemical and Biological Synthesis: An Expanded Genetic Code. Cold Spring Harb Perspect Biol. 8 (9), (2016).
- Blunck, R. Chapter 9. Handbook of Ion Channels. , CRC Press. 113-133 (2015).
- Chatterjee, A., Guo, J., Lee, H. S., Schultz, P. G. A genetically encoded fluorescent probe in mammalian cells. J Am Chem Soc. 135 (34), 12540-12543 (2013).
- DeBerg, H. A., Brzovic, P. S., Flynn, G. E., Zagotta, W. N., Stoll, S. Structure and Energetics of Allosteric Regulation of HCN2 Ion Channels by Cyclic Nucleotides. J Biol Chem. 291 (1), 371-381 (2016).
- Shen, B., et al. Genetically encoding unnatural amino acids in neural stem cells and optically reporting voltage-sensitive domain changes in differentiated neurons. Stem Cells. 29 (8), 1231-1240 (2011).
- Aman, T. K., Gordon, S. E., Zagotta, W. N. Regulation of CNGA1 Channel Gating by Interactions with the Membrane. J Biol Chem. 291 (19), 9939-9947 (2016).
- Haddad, G. A., Blunck, R. Mode shift of the voltage sensors in Shaker K+ channels is caused by energetic coupling to the pore domain. J Gen Physiol. 137 (5), 455-472 (2011).
- Batulan, Z., Haddad, G. A., Blunck, R. An intersubunit interaction between S4-S5 linker and S6 is responsible for the slow off-gating component in Shaker K+ channels. J Biol Chem. 285 (18), 14005-14019 (2010).
- Kusch, J., et al. How subunits cooperate in cAMP-induced activation of homotetrameric HCN2 channels. Nat Chem Biol. 8 (2), 162-169 (2012).
- Lee, H. S., Guo, J., Lemke, E. A., Dimla, R. D., Schultz, P. G. Genetic incorporation of a small, environmentally sensitive, fluorescent probe into proteins in Saccharomyces cerevisiae. J Am Chem Soc. 131 (36), 12921-12923 (2009).
- Summerer, D., et al. A genetically encoded fluorescent amino acid. Proc Natl Acad Sci U S A. 103 (26), 9785-9789 (2006).
- Kalstrup, T., Blunck, R. Reinitiation at non-canonical start codons leads to leak expression when incorporating unnatural amino acids. Sci Rep. 5, 11866(2015).
- Liu, H., Naismith, J. H. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 8, 91(2008).
- Beckert, B., Masquida, B. Synthesis of RNA by in vitro transcription. Methods Mol Biol. 703, 29-41 (2011).
- Goldin, A. L. Maintenance of Xenopus laevis and oocyte injection. Methods Enzymol. 207, 266-279 (1992).
- Rudokas, M. W., Varga, Z., Schubert, A. R., Asaro, A. B., Silva, J. R. The Xenopus oocyte cut-open vaseline gap voltage-clamp technique with fluorometry. J Vis Exp. (85), (2014).
- Zhao, J., Blunck, R. The isolated voltage sensing domain of the Shaker potassium channel forms a voltage-gated cation channel. Elife. 5, (2016).
- Posson, D. J., Ge, P., Miller, C., Bezanilla, F., Selvin, P. R. Small vertical movement of a K+ channel voltage sensor measured with luminescence energy transfer. Nature. 436 (7052), 848-851 (2005).
- Chanda, B., Asamoah, O. K., Blunck, R., Roux, B., Bezanilla, F. Gating charge displacement in voltage-gated ion channels involves limited transmembrane movement. Nature. 436 (7052), 852-856 (2005).
- Taraska, J. W., Puljung, M. C., Zagotta, W. N. Short-distance probes for protein backbone structure based on energy transfer between bimane and transition metal ions. Proc Natl Acad Sci U S A. 106 (38), 16227-16232 (2009).
- Baker, B. J., et al. Genetically encoded fluorescent sensors of membrane potential. Brain Cell Biol. 36 (1-4), 53-67 (2008).
- Miranda, P., et al. State-dependent FRET reports calcium- and voltage-dependent gating-ring motions in BK channels. Proc Natl Acad Sci U S A. , (2013).
- Sisido, M., Ninomiya, K., Ohtsuki, T., Hohsaka, T. Four-base codon/anticodon strategy and non-enzymatic aminoacylation for protein engineering with non-natural amino acids. Methods. 36 (3), 270-278 (2005).
- Hohsaka, T., Ashizuka, Y., Murakami, H., Sisido, M. Five-base codons for incorporation of nonnatural amino acids into proteins. Nucleic Acids Res. 29 (17), 3646-3651 (2001).
Access restricted. Please log in or start a trial to view this content.
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.