
Method Article
Generation der genomweiten Chromatin Konformation Capture Bibliotheken aus dicht inszenierten Anfang Drosophila Embryos
In diesem Artikel
Zusammenfassung
Diese Arbeit beschreibt ein Protokoll für die Erzeugung von hoher Auflösung in Situ Hi-C-Bibliotheken von dicht vor Gastrulation Drosophila Melanogaster inszeniert Embryonen.
Zusammenfassung
Die dreidimensionale Architektur des Chromatins zu untersuchen, bietet wertvolle Einblicke in die Mechanismen der Genregulation. Hier beschreiben wir ein Protokoll zur Durchführung der Chromatin Konformation Capture Technik in Situ Hi-C in Drosophila Melanogaster Embryo Populationen in Szene gesetzt. Das Ergebnis ist eine Sequenzierung-Bibliothek, die die Zuordnung aller Chromatin Interaktionen ermöglicht, die im Zellkern in einem einzigen Experiment auftreten. Embryo Sortierung erfolgt manuell mit einem fluoreszierenden Stereo-Mikroskop und eine transgene Fliegenschnur mit einem nuklearen Marker. Mit dieser Technik, Embryo Populationen aus jedem Zyklus nuclear Division und mit definierten Zellzyklus-Status erhalten Sie mit sehr hoher Reinheit. Das Protokoll kann auch an ältere Embryonen über Gastrulation sortieren angepasst werden. Sortierte Embryonen dienen als Eingänge für in Situ Hi-C. Alle Experimente, einschließlich Bibliothek Vorbereitung, Sequenzierung können in fünf Tagen abgeschlossen werden. Das Protokoll hat niedrige input-Anforderungen und arbeitet zuverlässig mit 20 Blastoderm Embryonen als input-Material. Das Endergebnis ist eine Sequenzierung-Bibliothek für die Sequenzierung der nächsten Generation. Nach der Sequenzierung, können die Daten zu genomweite Chromatin Interaktion Karten verarbeitet werden, die analysiert werden können, mit einem breiten Spektrum der verfügbaren Tools, Informationen über topologisch zuordnen Domänenstruktur (TAD), Chromatin Schleifen und Chromatin zu gewinnen Fächer bei Drosophila -Entwicklung.
Einleitung
Chromatin Konformation erfassen (3C) hat eine außerordentlich nützliche Methode, um die Topologie des Chromatins in der Kern-1Studie entwickelt. Die Variante 3C Hi-C ermöglicht Messung der Kontakt Frequenzen aller Chromatin-Interaktionen, die im Zellkern in einem einzigen Experiment2auftreten. Anwendung von Hi-C spielte eine wichtige Rolle bei der Entdeckung und Charakterisierung von vielen grundlegenden Prinzipien des Chromatin Organisation, z. B. TADs, Fächern und Schlaufen3,4,5.
Studium der Chromatin-Architektur im Rahmen der entwicklungspolitischen Übergänge und Zelldifferenzierung werden zunehmend verwendet, um die Mechanismen der Genregulation bei diesen Prozessen6,7,8zu entwirren, 9. Eines der Modellorganismen von großem Interesse ist Drosophila Melanogaster, deren Entwicklung und Genom gut charakterisiert sind. Jedoch führte nur wenige Studien, die untersuchen, Chromatin Architektur in Drosophila außerhalb von in-vitro- Gewebekultur Einstellungen wurden10,11. In Embryonen waren 16 – 18 h Post Befruchtung, TADs und Fächer erinnert an ähnliche Strukturen bei Säugetieren identifiziert10, welche wirft die Frage, welche Rolle sie in der Genregulation bei Drosophila Embryos spielen Entwicklung. Vor allem in den frühen Stadien der Entwicklung, vor der Gastrulation sind solche Studien technisch anspruchsvoll. Vor der Gastrulation unterziehen Drosophila Embryos 13 synchrone nuklearen Unternehmensbereiche, die in einem rasanten Tempo von 8 bis 60 min pro Zyklus12,13fortfahren. Darüber hinaus das Fehlen von visuellen Funktionen, die verschiedenen Phasen zu unterscheiden erschweren, dicht inszenierten Embryo Material in ausreichender Menge zu erhalten.
Um ein Protokoll zu entwickeln, das Chromatin Architekturstudium in der frühen Entwicklung von Drosophila mit nuklearen Kreislaufs Auflösung ermöglicht, wir haben zwei bestehende Techniken kombiniert: in Situ Hi-C, wodurch die Generation mit hoher Auflösung ganze Genom Kontakt Karten5und Embryo-Inszenierung mit einer transgenen Drosophila -Linie mit dem Ausdruck eines eGFP-PCNA Transgen13,14. Das Transgen lokalisiert in den Zellkern während der Interphase und verteilt im gesamten syncytial Blastoderm während der Mitose. Mit dieser Eigenschaft ist es möglich, verschiedene Phasen durch ihre nuklearen Dichte und mitotischen Embryonen durch die Streuung der GFP-Signal leicht zu unterscheiden.
Diese Techniken ermöglichen die dreidimensionale Struktur des Chromatins in hoher Auflösung von nur 20 Drosophila Embryos zu studieren. Dieses Protokoll enthält die Anweisungen für die Ernte und Sortierung Drosophila Embryos Populationen von Embryonen aus einem einzelnen Atom Abteilung Zyklus zu erhalten. Es beschreibt die weitere Verwendung der gewonnenen Embryonen durchzuführen in Situ Hi-C. Das Endergebnis ist ein Nukleotid-Bibliothek für Sequenzierung auf Maschinen der nächsten Generation Sequenzierung geeignet. Der daraus resultierende Sequenzierung liest können dann zu detaillierten Chromatin Interaktion Karten für das gesamte Genom von Drosophila verarbeitet werden.
Protokoll
1. Drosophila Embryos Sammlung
Hinweis: Eine gleichwertige Embryo-Sammlung kann durchgeführt werden, wie in einer früheren Publikation15dargestellt.
- Junge eGFP-PCNA fliegen übertragen (< 1 Woche alt) in ei Sammlung Käfige mit yeasted Sammlung Platten16 (1 % Ethanol, Essigsäure 1 % und 4 % Agar).
- Verschieben Sie Sammlung Käfige in Inkubator eingestellt bei 25 ° C. Inkubation für 1 – 2 Tage vor der Eizellentnahme verbessert Ei Ertrag erheblich. Wechselteller Sammlung zweimal am Tag.
- Entfernen Sie Platten mit Embryonen aus der Sammelkammer in Intervallen von 30 – 60 min.. Kleinere Intervalle führen zu weniger Embryonen, aber engere Verteilung der Entwicklungsstadien. Sammeln von mehreren Käfigen parallel also, die im Idealfall > 200 Eier werden alle 30 – 60 min gelegt.
- Speichern Sie die Platten bei 25 ° C, bis die Embryonen das gewünschte Alter erreichen. Für Blastoderm Embryonen (nukleare Zyklus 14) ca. 2 h inkubieren.
- Fügen Sie nach 2 h Inkubation Leitungswasser aus einer Spritzflasche Sammlung Platte so dass die gesamte Oberfläche mit Wasser bedeckt ist. Aussetzen von Embryonen und Hefe, mit einer weichen Bürste.
- Gießen Sie resuspendierte Embryonen aus der Sammlung Platte in ein Embryo fangkorb (kommerzielle Zelle Siebe mit 100 µm Porengröße oder hausgemachte Körbe17 gut funktionieren), Hinzufügen von zusätzlichen Leitungswasser aus einer Spritzflasche, bei Bedarf. Kombinieren Sie in diesem Stadium Embryonen von alle Platten, die parallel gesammelt wurden. Die gepoolte Probe repräsentiert einen einzelnen Batch.
- Waschen von Embryonen gut durch spülen den Korb mit Leitungswasser aus einer Spritzflasche für 30 s bis alle Hefe Rückstände abgespült werden.
- Dechorionate Embryonen indem man den Fangkorb zu einer 2,5 % Natriumhypochlorit-Lösung in Wasser. Leichte Erregung durch Umschwenken erleichtert die Entfernung von der Chorion. Weiter bis Embryonen ausreichend hydrophob sind, so dass sie an der Oberfläche der Lösung, Schwimmen wenn der Korb ist herausgehoben und wieder getaucht was ~1.75–2 min dauern sollte.
Achtung: Natriumhypochlorit ist ätzend. Tragen Sie geeignetsten persönliche Schutzausrüstung. Lösungen mit < 10 % Natriumhypochlorit in der Regel in der Spüle entsorgt werden, stellen Sie sicher, dass die Regelungen des Gastinstituts. - Entfernen Sie den Korb aus der Projektmappe und spülen Sie gründlich mit Leitungswasser aus einer Spritzflasche aus, bis der Bleichmittel Geruch nicht mehr spürbar ist.
(2) Embryo Fixierung
Hinweis: Optimale Fixation Bedingungen, vor allem die Konzentration an Waschmittel, Formaldehyd und die Dauer der Fixierung, empirisch ermittelte die Bühne der Embryonen passen müssen. Für Bühnen rund um den syncytial Blastoderm eine Endkonzentration von 0,5 % Triton x-100 und 1,8 % Formaldehyd in der wässrigen Phase gut funktionieren. Für die späteren Stadien Embryo Stadium 9 kann weitere Optimierung dieser Parameter erforderlich sein. Alle Lösungen, die während der Fixierung und Sortierung verwendet sollte Protease-Inhibitoren enthalten.
- Invertieren Sie fangkorb zu und legen Sie es über eine 15 mL konische Zentrifugation Röhre. Embryonen aus dem Korb in das Rohr mit einer Pasteurpipette Verzicht auf PBS-T spülen (PBS, 0,5 % Triton x-100).
- Lassen Sie die Embryonen an der Unterseite zu begleichen und passen Sie das Gesamtvolumen auf 2 mL mit PBS-T.
- 6 mL Heptan und 100 µL 37 % Formaldehyd in Wasser hinzufügen.
Achtung: Heptan und Formaldehyd sind giftig beim Einatmen oder nach Hautkontakt kontaktieren. Tragen geeigneten persönlichen Schutzausrüstung und arbeiten unter einem Abzug. Abfall, Heptan oder Formaldehyd enthalten muss separat das Gastinstitut vorschriftsmäßig entsorgt werden. - Starten Sie nach der Zugabe der Formaldehyd einen 15 min Timer und schütteln Sie kräftig das Rohr nach oben und unten für 1 min von hand. Die wässrige und organische Phase wird in Form einer Shampoo-ähnliche Konsistenz kombinieren.
- Regen Sie auf einem rotatorischen Mixer bis 10 min nach der Zugabe von Formaldehyd.
- Zentrifugieren Sie bei 500 X g für 1 min bei Raumtemperatur, Embryonen am unteren Ende der Röhre zu sammeln.
- Abzusaugen Sie die gesamte Shampoo-ähnliche Flüssigkeit und entsorgen Sie, kümmert sich nicht um alle Embryonen Aspirieren. Kleine Restmengen Shampoo-ähnliche überstand verursachen keine Probleme.
- 15 Minuten nach der Zugabe von Formaldehyd, Aufschwemmen der Embryonen in 5 mL PBS-T mit 125 mM Glycin das Formaldehyd zu stillen. Mischen Sie kräftig von oben und unten für 1 min schütteln.
- Bei 500 X g bei Raumtemperatur für 1 min zentrifugieren Sie und Aspirieren Sie überstand.
- Waschen von Embryonen durch resuspending ihnen in 5 mL eiskaltem PBS-T. Lassen Sie Embryonen Settle und aspirat alle überstand.
- Wiederholen Sie die Wäsche in Schritt 2.10 zwei weitere Male.
- Halten Sie die Embryonen auf Eis bis zu sortieren. In der Regel ist es eine gute Idee, 3 – 4 Chargen von Fly Embryonen, bevor Sie fortfahren zu sammeln, zu sortieren. Embryonen sollten jedoch am gleichen Tag sortiert werden. Längere Lagerung auf Eis oder im Kühlschrank führt zu veränderten Embryo Morphologie.
(3) Embryo sortieren
Hinweis: Sortierung kann auf Fluoreszenz Stereomikroskop ausgestattet mit einem GLP-Filter bei 60 – 80 X Vergrößerung erfolgen.
- Mit einer 1.000 µL Pipette, übertragen Sie einen Stapel von etwa 100 Embryonen zu, um ein kleines Glasgefäß geeignet für Sortierung, vorzugsweise von dunkler Farbe, und legen Sie ihn auf Eis.
-
Art-Embryonen durch Dichte und Zellzyklus Status als Atommacht (Abbildung 1) drücken Sie wünschenswert Embryonen in einem separaten Stapel mit einer Nadel oder Spritze Spitze.
- Entfernen Sie alle Embryonen mit verstreuten, nichtnukleare Verteilung von eGFP-PCNA (Abbildung 1E). Embryonen, die teilweise eine nichtnukleare GFP Signal zeigen sollte auch entfernt werden.
- Um die Sortierung zu erleichtern, montieren Sie eine Line-up von Referenz-Embryonen bei nuklearen Zyklus 12, 13 und 14 in jeder Charge mit den Bildern in Abbildung 1 als Leitfaden. Verwenden Sie diese Besetzung, um Embryonen von einem unbekannten Stadium mit einem Referenz-Embryonen anzupassen, um ihre Bühne zu bestimmen.
- Überprüfen Sie das Entwicklungsstadium für Referenz-Embryonen, Messen Sie die atomare Dichte imaging des Embryos und zählen die Anzahl der Nukleonen an der Oberfläche des Embryos auf einer Fläche von 2.500 µm2 mit imaging-Software, die Entfernungsinformationen bereitstellt.
Hinweis: Die erwartete Anzahl der Kerne für eine Fläche von 2.500 µm2 ist 12 bis 16 Kerne bei nuklearen Zyklus 12 und 20 bis 30 Kernen bei nuklearen Zyklus 1313.
- Sobald alle Embryonen in der entsprechenden Phase getrennt sind, fotografieren Sie die Embryonen für die Dokumentation und Qualitätskontrolle. Wenn das Stereo-Mikroskop nicht selbst mit einem Kameramodul ausgestattet ist, kann jedes Epifluoreszenz-Mikroskop mit GFP Filter verwendet werden.
- Pipette auf die gewünschte Embryonen mit einer 1.000 µL Pipette, Übertragung auf ein frisches Tubus und Platz auf dem Eis.
- Fortgesetzt, bis genügend Embryonen für das geplante Experiment sortiert werden. Für ältere Embryonen reichen als Stufe 9, in der Regel 20 Embryonen für eine in Situ Hi-C-Experiment. Bei nuklearen Zyklus 12 sind 80 Embryonen ein guter Ausgangspunkt. In früheren Zyklen sollte die Anzahl der Embryonen für jeden Zyklus etwa verdoppelt werden.
- Pool und geteilten Embryonen in 1,5 mL Röhrchen in einer Weise, dass eine Röhre genügend Embryonen für ein einzelnes in Situ Hi-C enthält experimentieren. Es ist ratsam, Rohre mit geringen DNA-Bindungseigenschaften zu verwenden, da das gleiche Rohr für das gesamte Protokoll verwendet werden wird und Adsorption von DNA kann zu erheblichen Verlusten bei niedrigen DNA-Konzentrationen führen.
- Röhren kurz bei 100 X g bei Raumtemperatur zu spinnen und überstand zu entfernen. Die Embryonen sollte so trocken wie möglich zum Einfrieren.
- Flash Einfrieren Embryonen durch Eintauchen der Rohre in flüssigem Stickstoff und Shop bei-80 ° C.
4. in Situ Hallo-C
- Lyse
- Legen Sie Rohre mit eingefrorenen Embryonen auf Eis.
- Aufschwemmen Sie Embryonen in 500 µL eiskalte Lyse-Puffer (10 mM Tris-Cl pH 8.0, 10 mM NaCl, 0,2 % IGEPAL CA-630, Protease-Inhibitoren, aufgelöst in Wasser). Dann warten Sie 1 min Embryonen auf der Unterseite des Rohres zu begleichen zu lassen.
- Grind Embryonen mit einem Metall Mikro Stößel, vorgekühlt auf Eis, soll das fest einen 1,5 mL Microcentrifuge Schlauch passen.
- Um zu vermeiden, die Embryonen zu agitieren, setzen Sie den Stößel langsam, bis es den Boden des Rohres berührt, nach unten drücken und dann Schleifen durch die Stößel zweimal in beide Richtungen drehen.
- Der Stößel sehr leicht anheben, wieder auf den Boden des Rohres schieben und Schleifen zu wiederholen.
- 4.1.3.2 10 Mal wiederholen, oder bis die Embryonen komplett lysiert werden. Die Lösung sollte homogen sein und keine große Reststücke von Embryonen bleiben sollte.
- Brüten Sie die homogenisierte Suspension für 15 min. Spin bei 1.000 X g, 4 ° C für 5 min auf Eis und verwerfen Sie überstand.
- Waschen Sie Pellets von resuspending in 500 µL eiskalte Lyse Puffer, pipettieren rauf und runter.
- Drehen Sie wieder wie in 4.1.4 und verwerfen Sie überstand.
- Aufschwemmen Sie gewaschenen Pellet in 100 µL 0,5 % Natrium-Dodecyl-Sulfat (SDS), pipettieren rauf und runter. Permeabilize Kerne durch Inkubation für 10 min bei 65 ° C in einem Heizblock. Stillen Sie SDS durch Zugabe von 50 µL 10 % Triton X-100 und 120 µL Wasser. Durch Streichen der Röhre mischen.
- Inkubation bei 37 ° C für 15 min. im Heizblock.
- Beschränkung Enzym Verdauung
- 25 µL 10-fach Restriktionsenzym Puffer und 20 U 5 U/µL MboI hinzufügen. Durch Streichen der Röhre mischen.
- Inkubation für 90 min bei 37 ° C im Heizblock unter leichte Erregung (750 u/min), um DNA zu verdauen.
- Fügen Sie weitere 20 U der MboI und weiter Inkubation für 90 min.
- Wärme zu inaktivieren MboI durch Inkubation bei 62 ° C für 20 Minuten.
- Überhang füllen
Hinweis: Ausfüllen des Überhangs mit biotinylierte dATP erlaubt die Auswahl der spezifischen aufgespaltenen Fragmente. Biotin-dATP an Ligatur Kreuzungen ist aus der Exonuclease Tätigkeit des T4 DNA-Polymerase (Abschnitt 4.6), geschützt, Biotin-dATP an unligated stumpfen enden effizient beseitigt ist. Der Pulldown mit Streptavidin beschichteten Perlen in Abschnitt 4.7 daher speziell bereichert für ligiertes, chimeric DNA-Fragmente.- Hinzufügen von 18 µL von 0,4 mM Biotin-14-dATP, 2,25 µL eines unveränderten dCTP, dGTP/dTTP-Mix (3,3 mM) und 8 µL 5 U/µL DNA-Polymerase I Klenow Fragment.
- Durch Streichen der Röhre mischen und Inkubation bei 37 ° C für 90 min im Heizblock.
- Ligatur
- Fügen Sie 657 µL Wasser, 120 µL 10 x T4 DNA Ligase Puffer, 100 µL 10 % Triton x-100, 6 µL 20 mg/mL Rinderserumalbumin (BSA) und Mix von flicking die Röhre. Schließlich fügen Sie 5 µL 5 U/µL T4 DNA-Ligase und Mischung von flicking die Röhre.
- Drehen Sie Rohr sanft (20 u/min) bei Raumtemperatur für 2 h.
- Fügen Sie eine zweite Tranche von 5 µL 5 U/µL T4 DNA-Ligase und drehen für 2 weitere h weiter.
- Spin-down Kerne bei 2.500 X g für 5 min und überstand verwerfen.
- DNA-Extraktion
- Aufschwemmen Sie Pellet in 500 µL Extraktionspuffer (50 mM Tris-Cl pH 8.0, 50 mM NaCl, 1 mM Ethylenediaminetetraacetic Säure (EDTA), 1 % SDS, aufgelöst in Wasser) und fügen Sie 20 µL 20 mg/mL Proteinase K. Mix durch Streichen der Röhre.
- Inkubation bei 55 ° C für 30 min, bei 1.000 u/min schütteln, um Protein zu verdauen.
- De-Crosslink 130 µL 5 M NaCl hinzugeben und über Nacht bei 68 ° C, schütteln bei 1.000 u/min inkubieren.
- Pipette Probe in eine neue 2 mL-Tube, bevorzugt mit niedrigen DNA-Bindungseigenschaften.
- Fügen Sie 0,1 X Mengen (63 µL) von 3 M Natrium Acetat pH 5,2 und 2 µL von 15 mg/mL GlycoBlue. Mischen Sie gut durch invertieren. Fügen Sie 1,6 X Mengen (1.008 µL) des reinen absoluten Ethanol und Mischung durch invertieren.
- Bei-80 ° C für 15 min. Zentrifugieren bei 20.000 X g bei 4 ° C für mindestens 30 min inkubieren. Das DNA-Pellet ist oft sehr klein, fast unsichtbar und kann nur durch die blaue Farbe des GlycoBlue beobachtet werden.
- Entfernen Sie überstand sehr vorsichtig ziehen die PIPETTENSPITZE in das Rohr entlang der gegenüberliegenden Wand wo das DNA-Pellet gelegen. Kleine verbleibende Tropfen sind oft leicht während dieser Schritt und die folgenden Waschungen entfernt, indem man sie aus den Rohren mit einer P10-Spitze statt pipettieren sie heraus.
- Waschen Sie Pellet durch Zugabe von 800 µL 70 % Ethanol. Mischen Sie durch invertieren und Zentrifugieren bei 20.000 X g bei Raumtemperatur für 5 min. dieses waschen mindestens einmal wiederholen.
- Entfernen Sie alle Spuren von Ethanol und lassen Sie das Rohr stehen bei geöffnetem Deckel für bis zu 5 min an der Luft trocknen. Sobald keine Flüssigkeit übrig ist, fügen Sie 50 µL 10 mM Tris-Cl pH 8.0. Immer wieder pipette die Lösung über die Fläche an der Wand des Rohres, wo das Pellet befand, um die DNA zu solubilisieren.
- Fügen Sie 1 µL von 20 mg/mL RNase A, Mix von flicking die Röhre und Inkubation bei 37 ° C für 15 min zu verdauen RNA. Die Probe kann nun über Nacht im Kühlschrank aufbewahrt oder auf unbestimmte Zeit bei-20 ° C eingefroren.
- Die Konzentration von DNA mit einem Fluoreszenzfarbstoff basierte Assay gemäß den Anweisungen des Herstellers zu überprüfen. Der Gesamtbetrag der DNA in der Probe soll mindestens 10 ng, sonst gibt es zu wenig Material für Verstärkung und Bibliothek Komplexität werden wahrscheinlich gering. In diesem Fall die Menge des Ausgangsmaterials war wahrscheinlich nicht ausreichend, oder Material war auf dem Weg vielleicht während Lyse und Niederschlag verloren.
- Biotin-Entfernung und DNA-Scheren
- Addieren Sie 12 µL 10 X T4-DNA-Polymerase-Puffer, 3 µL 1 mM dATP, 3 µL 1 mM dGTP und 46 µL Wasser. Durch Streichen der Röhre mischen. 5 µL 3 U/mL-T4-DNA-Polymerase, Mischen von flicking die Röhre und bei 20 ° C für 30 min inkubieren.
- Fügen Sie 3 µL 0,5 M EDTA, die Reaktion zu stoppen, und verwenden Sie Wasser ein Volumen von etwa 120 µL der Probe zu.
- Die DNA zu einer Größe von 200 – 400 bp mit einem Ultraschall-Gerät entsprechend den Anweisungen des Herstellers zu scheren. Mit den sonikator erwähnt in der Tabelle der Materialien, das folgende Programm ist angebracht: 2 Zyklen jeder der 50 s, 10 % Zoll, Stärke 5, 200 Zyklen/platzen.
- Biotin-pulldown
- Pipette 30 µL 10 mg/mL Streptavidin beschichteten magnetische Beads in einen neuen Schlauch, trennen Sie diese auf einem Magnetstativ und überstand verwerfen.
- Perlen in 1 x B & W Puffer (5 mM Tris-Cl pH 7.4, 0,5 mM EDTA, 1 M NaCl, aufgelöst in Wasser) + 0,1 % Triton x-100 und Mischung von Vortexen aufzuwirbeln. Ein Magnetstativ setzen Sie Rohr auf und 1 – 5 min. warten Sie, bis die Perlen, je nach Fabrikat und Modell getrennt sind.
- Aspirieren und überstand beim Schieben der Pipettenspitze an der Wand gegenüber befinden sich die Perlen zu verwerfen. Aufschwemmen Sie Perlen in 120 µL 2 x B & W-Puffer (10 mM Tris-Cl pH 7.4, 1 mM EDTA und 2 M NaCl). Durch Vortexen mischen.
- Sheared DNA zu einem neuen niedrigen DNA-Bindung Schlauch zu übertragen, und mischen mit 120 µL der Wulst Suspension in 2 X B & W Puffer durch aufschütteln. Drehen Sie Perlen mit der DNA-Probe bei 20 u/min für 15 min.
- Trennen Sie Perlen auf einem Magnetstativ zu und entsorgen Sie überstand.
- Aufschwemmen Perlen in 600 µL 1 x B & W + 0,1 % Triton x-100, und bei 55 ° C für 2 min schütteln bei 1.000 u/min inkubieren. Verwerfen Sie nach der Trennung überstand. Wiederholen Sie diese Wäsche einmal.
- Waschen Sie Perlen einmal mit 600 µL 10 mM Tris-Cl pH 8.0 und verwerfen Sie überstand nach Trennung zu.
- Perlen in 50 µL 10 mM Tris-Cl pH 8.0 aufzuwirbeln.
5. Sequenzierung Bibliothek Vorbereitung
Hinweis: Alle Bibliothek Schritte erfolgen mit Komponenten aus einer kommerziellen DNA-Bibliothek Vorbereitung kit (siehe Tabelle der Materialien). Jedoch können alternative Kits oder andere Reagenzien ersetzt werden. Niederschlag neigt, Formular in der Bibliothek-Vorbereitung-Agenten im Tiefkühllager. Es ist daher wichtig, um sicherzustellen, dass alle Niederschläge aufgelöst wird, vor der Verwendung der Reagenzien.
-
Ende Reparatur
- Übertragen Sie die Perle-Suspension in 50 µL 10 mM Tris-Cl pH 8.0 in eine neue PCR-Röhrchen.
- Fügen Sie 3 µL Ende Prep Enzym-Mix und 7 µL Ende Prep Reaktion Puffer. Mischen von pipettieren rauf und runter.
- Rohr auf einem Thermocycler übertragen und führen Sie das folgende Programm: 20 ° C für 30 min, 65 ° C für 30 min und Halt bei 4 ° C.
-
Adapter-Ligatur
- Fügen Sie 30 µL Ligation Master Mix, 2,5 µL 1,5 µM Sequenzierung Adapter (verdünnte bis 1,5 µM ab Lager lieferbar) und 1 µL Ligation Enhancer an der Wulst-Aufhängung. Mischen von pipettieren rauf und runter.
- Inkubation bei 20 ° C für 15 min in einem Thermocycler.
- 3 µL Enzym Benutzer hinzufügen. Mischen von pipettieren rauf und runter.
- Inkubation bei 37 ° C für 15 min in einem Thermocycler.
- Trennen Sie Perlen auf einem Magnetstativ zu und entfernen Sie überstand.
- Zum Waschen der Perlen, Aufschwemmen Perlen in 100 µL 1 x B & W Puffer + 0,1 % Triton x-100. Mix von aufschütteln und Übertragung auf einen neuen Microcentrifuge Schlauch. Trennen Sie Perlen auf einem Magnetstativ zu und entfernen Sie überstand.
- Wiederholen Sie dieses einmal mit 600 µL des gleichen Puffers waschen.
- Perlen in 600 µL 10 mM Tris-Cl pH 8.0, Mix von Vortexen, aufschwemmen und Perlen auf einen neuen Schlauch übertragen.
- Perlen auf einem Magnetstativ zu trennen, überstand verwerfen und Perlen in 50 µL 10 mM Tris-Cl pH 8.0 aufzuwirbeln.
-
PCR-Amplifikation
- Bereiten Sie zwei PCR-Röhrchen und in jedem, mischen Sie 25 µL der Polymerase Master Mix, 1,5 µL 10 µM nach vorne (nichtindizierte) PCR Primer und 1,5 µL 10 µM rückwärts (indizierte) PCR-Primer.
Hinweis: Nach vorne (nichtindizierte) PCR-Primer:
5'-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATC * T-3´.
Rückwärts (indizierte) PCR-Primer:
5'-CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC * T-3´. * zeigt Phosphorothioat Anleihen und Ns in den indizierten PCR-Primer. - Fügen Sie in jedem Röhrchen 22 µL Wulst Hemmung und Mischung hinzu, indem pipettieren rauf und runter.
- PCR mit dem folgenden Programm ausführen: 98 ° C für 1 min (98 ° C für 15 s, 65 ° C für 75 s, Rampen 1,5 ° C/s) 9 - 12 Mal wiederholt, 65 ° C für 5 min und Halt bei 4 ° c
Hinweis: Die Anzahl der Verstärkung Zyklen muss empirisch ermittelt werden. Allerdings haben wir festgestellt, dass Bibliotheken, die mehr als 12 Zyklen erforderlich im Allgemeinen von geringer Komplexität waren und nicht in qualitativ hochwertige Hi-C-Karten führen. Auf der anderen Seite wurden Bibliotheken, die weniger als 12 Zyklen erforderlich nicht negativ beeinflusst, durch die Verstärkung für eine volle 12 Zyklen. Daher ist es möglich, standardmäßig auf 12 Zyklen der Verstärkung. - Bündeln Sie die beiden PCR-Reaktionen in einem einzigen Microcentrifuge Schlauch, trennen Sie Perlen auf einem Magnetstativ zu und übertragen Sie den Überstand mit der Bibliothek zu einem neuen Schlauch.
- Bereiten Sie zwei PCR-Röhrchen und in jedem, mischen Sie 25 µL der Polymerase Master Mix, 1,5 µL 10 µM nach vorne (nichtindizierte) PCR Primer und 1,5 µL 10 µM rückwärts (indizierte) PCR-Primer.
-
Größenauswahl
- Bringen Sie Ampure XP Wulst Aussetzung auf Raumtemperatur und Mischung gut durch Schütteln.
- Volumen der gepoolten PCR-Reaktion auf genau 200 µL mit Wasser zu bringen. Während der PCR und die magnetische Trennung einige des ursprünglichen Volumens ist in der Regel verloren. Überprüfen Sie Volume zu, indem Sie die Pipette auf 200 µL und Aspirieren Sie das gesamte Volumen der Reaktion. Wenn Luft abgesaugt ist, Wasser mehr muss hinzugefügt werden. Übersteigt das Volumen 200 µL, die Lautstärke von Perlen in den Schritten 5.4.3 und 5.4.6 proportional hinzugefügt.
Hinweis: Die Bände in Klammern sind gültig, wenn das Gesamtvolumen der gepoolten PCR Reaktionen genau 200 µL ist. - 0,55 X Volumen (110 µL) Ampure XP Wulst Suspension und Mischen von pipettieren oben und unten mindestens 10 mal.
- In 5 min, separate Perlen auf einem Magnetstativ für 5 min bei Raumtemperatur inkubieren.
- Wechseln Sie überstand an einen neuen Schlauch Entsorgen Sie die Röhrchen mit den Perlen. Die Perlen haben DNA gebunden > 700 bp, die sequenziert werden zu groß ist.
- Hinzufügen des Überstands 0,2 X Volumen (40 µL, wodurch insgesamt 0,75 X Ampure Puffer in der Probe) Ampure XP Wulst Hemmung und Mix von oben und unten 10 mal pipettieren.
- In 5 min, separate Perlen auf einem Magnetstativ für 5 min bei Raumtemperatur inkubieren.
- Entsorgen Sie überstand die DNA enthält < 200 bp, inklusive kostenlose Primer, Grundierung Dimere und Fragmente zu klein sequenziert werden.
- Lassen Sie das Rohr auf die Magnetstativ. Waschen Sie Perlen, 700 µL Ethanol 80 % hinzufügen, kümmert sich um nicht zu stören das Wulst Pellet und inkubieren Sie für 30 s.
- Überstand verwerfen, dann nehmen Sie die u-Bahn aus der Magnetstativ und Perlen in 100 µL 10 mM Tris-Cl pH 8.0 Aufschwemmen. Mischen Sie, indem Sie nach oben und unten 10 mal pipettieren und bei Raumtemperatur für 1 min inkubieren.
- Fügen Sie 0,8 X Mengen (80 µL) Ampure XP Wulst Suspension. Mischen Sie, indem Sie nach oben und unten 10 mal pipettieren und in 5 min bei Raumtemperatur inkubieren. Diese zweite Runde der Untergrenze Größenauswahl sorgt dafür, dass die endgültige Bibliothek völlig frei von Primer und Grundierung Dimere ist.
- Trennen Sie Perlen auf einem Magnetstativ für 5 min zu und entsorgen Sie überstand.
- Waschen Sie die Wulst Pellet zweimal mit 700 µL Ethanol 80 % für 30 s jede, wobei das Rohr auf dem magnetischen Stand, wie oben beschrieben.
- Entfernen Sie mit dem Rohr noch auf die Magnetstativ alle Spuren von Ethanol. Es hilft, Tröpfchen von Ethanol aus dem Rohr mit einer Pipette P10 schieben. Verbleibende Ethanol für maximal 5 Minuten verdunsten zu lassen.
- Rohr aus der Magnetstativ und Perlen in 50 µL 10 mM Tris-Cl pH 8.0 aufzuwirbeln. Mischen Sie, indem Sie nach oben und unten 10 mal pipettieren.
- Inkubation bei Raumtemperatur für 5 min, dann separate Perlen auf einem Magnetstativ.
- Übertragen Sie überstand auf eine frische Rohr. Dies ist die letzte Hi-C-Bibliothek, bereit zu quantifizieren und sequenziert auf Maschinen der nächsten Generation Sequenzierung, gemäß den Anweisungen des Herstellers.
Ergebnisse
Sortiert Embryo Populationen an nuklearen Kreislaufs, 12, 13 und 14 (entspricht 01:30, 01:45 und 02:10 Stunden post-Düngung, bzw.12) und 3 – 4 h Post Düngung (hpf) wurden entsprechend den im Protokoll beschriebenen Verfahren. Von eGFP-PCNA Signal jeder Charge sortierte Embryo zu fotografieren, ist es möglich, die präzise Bühne und Zellzyklus Status jeder einzelnen Embryo zu dokumentieren, die in den nachgelagerten Experimenten verwendet wird. Beispielbilder von Embryonen aus sortierten Populationen sind in Abbildung 1 b-Egezeigt. Die Ausgabe des Protokolls in Situ Hi-C ist ein Nukleotid-Bibliothek bereit, auf Maschinen der nächsten Generation Sequenzierung sequenziert werden. Zu diesem Zweck ist eine endgültige Bibliothek Konzentration von mindestens 2 – 4 nM in der Regel erforderlich. Verwenden die empfohlenen Mengen von input-Material, ist diese Konzentration zuverlässig erreicht (Tabelle 1).
Die erwarteten Größenverteilung von DNA-Fragmenten nach Größenauswahl zwischen 300-600 bp, mit einem Maximum bei ca. 500 bp (Abbildung 2A(), je nach der genauen Scheren und Größe Selektionsparameter ist. Für die Sequenzierung empfehlen wir gepaart Ende liest mindestens 75 bp-Länge, die Anzahl der unkartierbar Einschränkung Fragmente des Genoms zu minimieren. Hochauflösende Karten mit 1 – 2 kb bin Größe erhalten Sie bei 400 Millionen mal gelesen. Wir empfehlen, mehrere biologische Wiederholungen in einer geringeren Tiefe von 150 Millionen Sequenzierung jeden, anstatt eine einzelne Replikation in sehr großer Tiefe Sequenzierung liest. Dies ermöglicht die Beurteilung der biologischen Variation und führt zu einer geringeren Anzahl von ausrangierten Lesevorgänge durch PCR Vervielfältigung. Für die grafische Darstellung können die Wiederholungen kombiniert werden. Es wird empfohlen vor der Festlegung auf eine Probe bei großer Tiefe Sequenzierung, auszuführen, Proben mit flachen Sequenzierung (ein paar Millionen gelesen je Probe) Basisbibliothek Qualitätsparameter wie in Abbildung 2 bzu bestimmen.
Analyse von Hi-C Daten erfordert erhebliche computational Ressourcen und Bioinformatik-Expertise. Als eine grobe Übersicht der gekoppelten liest das Referenz-Genom unabhängig zugeordnet sind, die daraus resultierende Ausrichtungen sind für Qualität und Ausrichtung gefiltert, dann eine Matrix der Kontakte auf einer gegebenen Auflösung oder Fragment platzebene erzeugt werden kann aus der gefilterten Ausrichtungen. Die Kontakt-Matrix ist die Grundlage für alle weiteren nachgeschalteten Analyse TADs, Loops und Fächer zu erkunden. Für die erste Analyse der Sequenzierung lautet, gibt es mehrere Bioinformatik Rohrleitungen zu ermöglichen, die Verarbeitung von Roh liest in Kontakt Matrizen ohne viel spezialisierte Bioinformatik wissen18,19, 20,21,22,23. Wie weitere Analyse durchgeführt wird, hängt die genaue biologische Frage unter Studie und erfordern große Erfahrung in Programmierung und scripting in R oder Python. Verschiedene Tools und Algorithmen TADs nennen sind jedoch verfügbar5,24,25,26,27,28, sowie Software zur Analyse und erkunden Sie Hi-C-Daten im Web-Browser und als eigenständige Desktop-Anwendungen29,30,31,32.
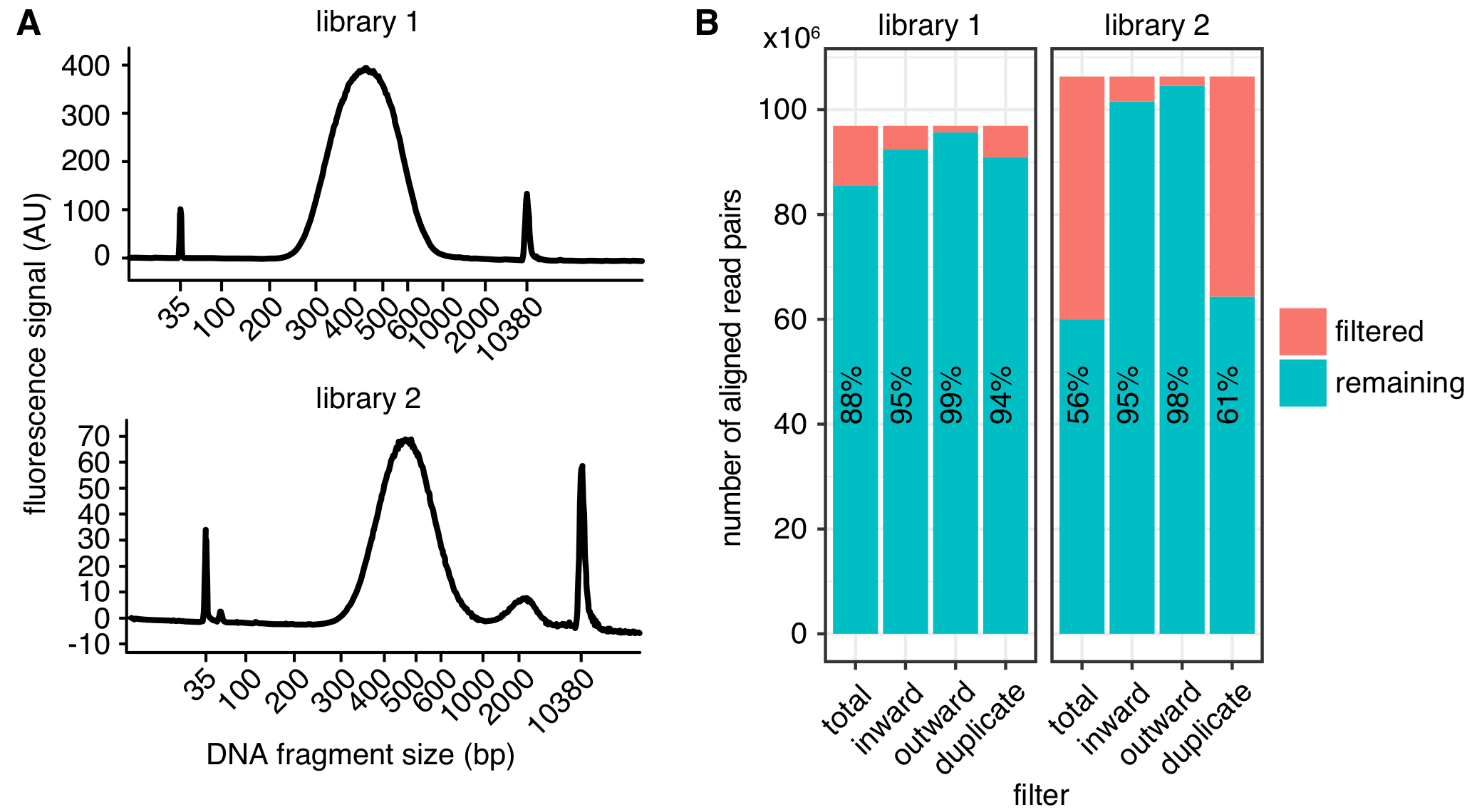
Nach der Bearbeitung kann die Qualität der Bibliothek mit verschiedenen Metriken (Abb. 2 b) ermittelt werden. Erstens sollte die Rate der PCR Duplikate, die die Anzahl der sequenzierten lesen Sie Paare aus dem gleichen ursprünglichen Molekül ist, so niedrig wie möglich, die Menge des vergeudeten Sequenz lautet begrenzen. Aber auch Bibliotheken mit > 40 % PCR Vervielfältigung kann in qualitativ hochwertige Kontakt Karten verarbeitet werden, wenn die Duplikate gefiltert werden. Zweitens sollte die Rate des gefilterten lautet aufgrund ihrer Ausrichtung, wie beschrieben in4, konsequent weniger als 10 % des ausgerichteten lesen Sie Paare sein.
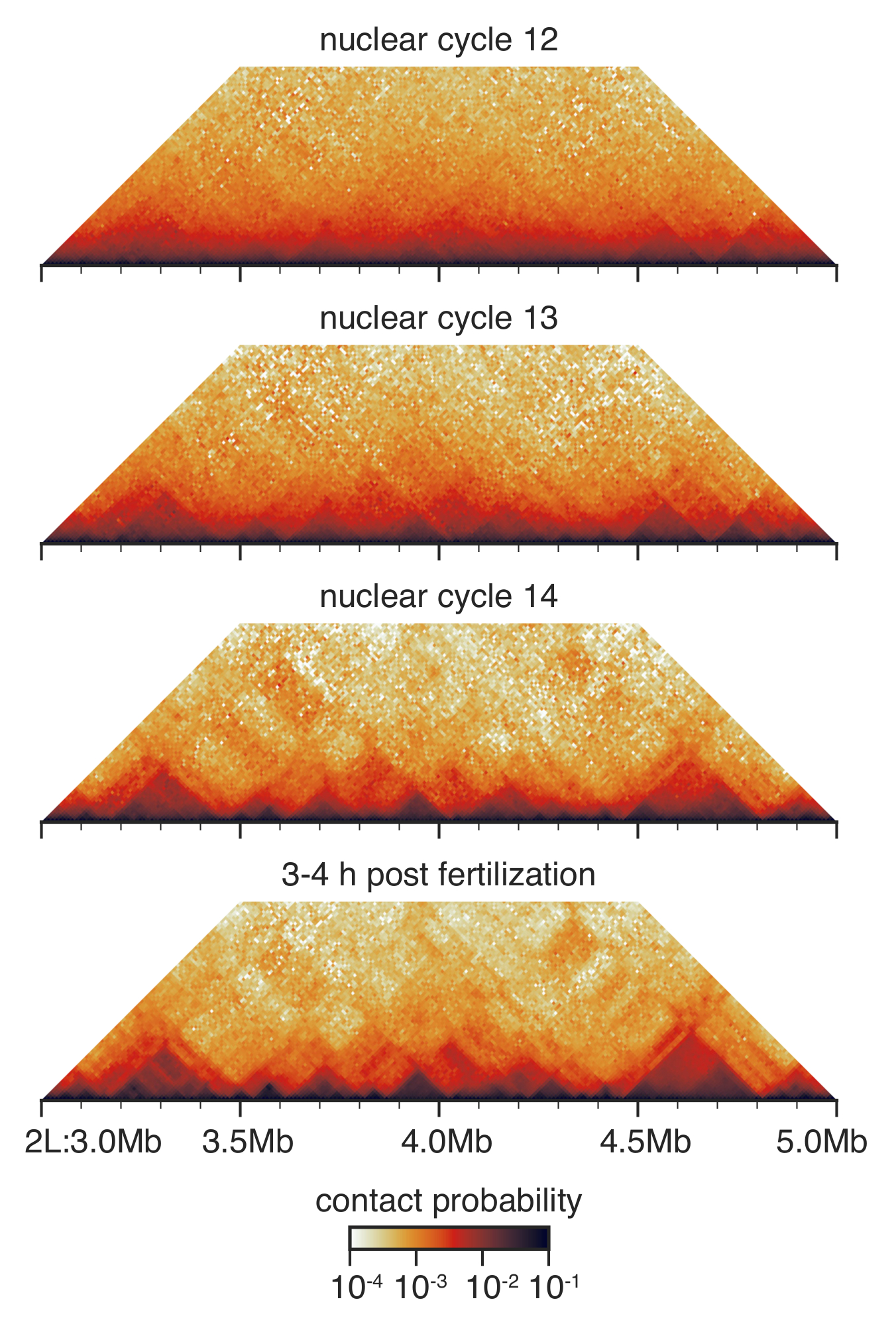
Während der Pre-gastrular Entwicklung von Drosophila zwischen nuklearen Kreislaufs 12 und 14 ist die nukleare Architektur drastisch umgebaut33 (Abbildung 3). Bei nuklearen Zyklus 12 Paar TADs werden erkannt und die Gesamtverteilung der Kontakte ist sehr glatt, ohne viele erkennbaren Eigenschaften. Dies ist dramatisch bei nuklearen Zyklus verändert, 13 und 14, wenn TADs zunehmend Prominente und unspezifischen weitreichende Kontakte sind erschöpft.

Abbildung 1: Repräsentative Bilder von eGFP-PCNA Embryonen bei der Sortierung. (A) eGFP-PCNA Signal aus einer unsortierten Bevölkerung von Embryonen nach 60 min. Sammlung und 2 h Inkubation bei 25 ° C (B-E) Beispiele von Embryonen aus sortierten Populationen an nuklearen Zyklus 12 (B), nukleare 13 (C), 14 (nuklearen Kreislaufs Zyklus ( D), und von Embryonen in der synchronen Mitose (E). Skalieren von Balken = 200 µm. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 2: Beispiele für in Situ Hi-C Bibliothek Qualitätsmetriken. (A) Bioanalyzer Spuren zeigen die Verteilung der DNA-Fragment Größen aus einer erfolgreichen Hi-C-Bibliothek (Library 1, oben) und aus einer Bibliothek mit einem Spitzenwert von Fragmenten, die sind zu groß für die Sequenzierung (Bibliothek 2, unten). Bibliothek 2 wurde erfolgreich sequenziert, aber auch größere Mengen an unerwünschten DNA-Fragmente zu verringerten Sequenzierung Renditen führen können. (B) Filterung Statistiken von zwei Hi-C-Bibliotheken: angezeigt ist die Anzahl der ausgerichteten lesen Sie Paare, die von der weiteren Analyse durch Lesen Sie Ausrichtung und Abstand (nach innen, nach außen)4 oder PCR Vervielfältigung (doppelte) ausgeschlossen werden. In jeder Bar sind die Anzahl der Lese-, vorbei an den Filter (verbleibende) und Versagen (gefiltert) aufgetragen. Der Anteil der liest den Filter passieren wird zusätzlich als Text angezeigt. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 3: Hi-C Interaktion Karten aus inszenierten Embryonen. Hallo-C Interaktion Karten sind Makulatur bei 10 kb Auflösung und ausgewogen wie vor33beschrieben. Dargestellt ist eine Region auf dem Chromosom 2 L. Klicken Sie bitte hier, um eine größere Version dieser Figur.
{kind=link}
| Bibliothek | Bühne | Anzahl der Embryonen | Menge DNA vor Scheren (ng) | PCR-Zyklen | Endgültige Bibliothek Konzentration (nM) |
| 1 | nukleare Zyklus 12 | 71 | 46 | 12 | 28.2 |
| 2 | nukleare Zyklus 12 | 46 | 40 | 12 | 22.2 |
| 3 | nukleare Zyklus 12 | 60 | 13 | 13 | 12.3 |
| 4 | nukleare Zyklus 13 | 36 | 39 | 12 | 22.2 |
| 5 | nukleare Zyklus 13 | 35 | 10 | 12 | 5.0 |
| 6 | nukleare Zyklus 13 | 48 | 18 | 12 | 8.7 |
| 7 | nukleare Zyklus 14 | 33 | 30 | 12 | 39,8 |
| 8 | nukleare Zyklus 14 | 24 | 36 | 12 | 20.4 |
| 9 | nukleare Zyklus 14 | 14 | 8 | 12 | 4.2 |
| 10 | 3-4 hpf | 17 | 30 | 12 | 24,0 |
| 11 | 3-4 hpf | 18 | 42 | 11 | 19.1 |
| 12 | 3-4 hpf | 22 | 63 | 11 | 48,4 |
Tabelle 1: Liste der repräsentativen Sequenzierung bibliotheksstatistik. Für jede Bibliothek in der Liste, die Anzahl der Embryonen, die für seine Generation, die Höhe der Gesamt-DNA vor Biotin Pulldown und Scheren, gemessen von Qubit, dienten Zyklen die Anzahl der PCR zur Verstärkung und die Endkonzentration der Sequenzierung Bibliothek verwendet nach Reinigung und Größenauswahl angegeben sind.
Diskussion
Die hier vorgestellten Protokoll ist sehr effektiv bei der Generierung von qualitativ hochwertige Karten der Chromatin-Architektur in den frühen Drosophila Embryos. Im Vergleich zu einer früheren Protokoll34, verwendet beschriebene Ansatz hier eine auf dem neuesten Stand in Situ Hi-C Verfahren5, was schnellere Verarbeitung, höheren Auflösung und weniger Reagenz Verwendung. Das gesamte Verfahren, einschließlich der in Situ -Hi-C-Protokoll wird voraussichtlich auf den unterschiedlichsten Bühnen und experimentellen Systemen neben Drosophilaarbeiten. Da das Protokoll geringem input Bedarf hat, könnte es auch auf isolierten Zellpopulationen verwendet werden. In Drosophilawenn einige Parameter mithilfe des Protokolls für Embryonen außerhalb des Bereichs hier beschrieben, insbesondere die Fixierung des Materials, müssen möglicherweise angepasst werden. Da ältere Embryonen eine hochgradig undurchlässige Kutikula entwickeln, können die Erhöhung der Konzentration von Formaldehyd und verlängert die Fixierung angebracht sein. Für die Sammlung von Embryonen in Phasen als nukleare Zyklus 14, Inkubationszeiten von Embryonen bei 25 ° C im Schritt 1.4 wie folgt angepasst werden müssen: nukleare Zyklus 12, 70 min; nukleare Zyklus 13, 90 min; 3 – 4 hpf, 03:30 Uhr.
Während 13 Spaltung Divisionen (Stufe 1-4) verdoppelt sich die Kerne Dichte etwa mit jeder Abteilung. Die Kerne können leicht durch ihre hellen GFP Fluoreszenz identifiziert werden. Während der Mitose eGFP-PCNA befindet sich nicht im Zellkern, und sein Signal über den Embryo verstreut ist. Diese Eigenschaft macht identifizierende Embryonen, die eine synchrone Spaltung Division möglich durchmachen. Für das Studium Chromatin Konformation, sind diese mitotischen Embryonen in der Regel nicht erwünscht, da die mitotische Organisation des Chromatins drastisch anders als die Interphase Organisation35. Es ist möglich, das Protokoll um speziell Embryonen durchläuft eine synchrone mitotische Teilung wählen anzupassen. In diesem Fall nur Embryonen mit verstreuten, nichtnukleare Verteilung von eGFP-PCNA aufbewahrt werden, und alle anderen Embryonen verworfen werden sollte. Da die atomare Dichte bestimmt werden kann, müssen alternative Methoden, um Embryonen durch ihre Morphologie in der übertragenen Lichtmikroskopie angesehen eingesetzt werden. Pol-Zellen und Kerne an die Peripherie der Embryo hindeuten, dass der Embryo mindestens nuklearen Kreislaufs 9, abgeschlossen wurde während sichtbare Cellularization an der Peripherie nuklearen Zyklus 1412anzeigt.
Hallo-C Experimente können erfolgreich mit einer großen Auswahl von Restriktionsenzymen5durchgeführt werden. Aktuelle Ansätze verwenden in der Regel Enzyme, die entweder eine 4-Base-Sequenz, wie MboI oder ein 6-Base Anerkennung-Website, z. B. HindIII zu erkennen. Der Vorteil von 4-Base Fräser über 6-Base Fräser ist, dass sie höhere mögliche Lösung angesichts genügend Sequenzierung Tiefe und eine gleichmäßigere Abdeckung von Restriktionsschnittstellen über das Genom. Es gibt keine klarer Vorteil bei der Wahl einer 4-Sockel-Fräser über weitere5,23,36,37. Die zwei am häufigsten verwendeten Enzyme, MboI und DpnII, erkennen beide die gleichen GATC-Anerkennung-Website. DpnII ist unempfindlicher gegen CpG Methylierung, kein Anliegen in Drosophila. Die hier vorgestellten Protokoll kann auch als ein Restriktionsenzym DpnII verwenden erfolgreich abgeschlossen werden. In Abschnitt 4.2. Restriktionsenzym und Puffer haben gemäß den Empfehlungen des Herstellers für DpnII Kompatibilität angepasst werden.
Weicht die Fragmentgröße der Sequenzierung Bibliothek deutlich aus dem Bereich in Abbildung 2Agezeigt, kann Clusterbildung während der Sequenzierung werden weniger effizient oder komplett ausfallen. In diesem Fall die Größenverteilung nach der Schur sollte überprüft werden und Scheren Parameter entsprechend angepasst. Gipfel in der Verteilung der DNA-Fragmente von sehr kleinen (< 100 bp) oder sehr groß (> 1.000 bp) Größen deutet auf Probleme mit Größenauswahl, z. B. tragen über Perlen oder überstand, die verworfen werden sollen. Diese Bibliotheken mit kleinen Peaks bei diesen unerwünschten Größen, z. B. eine abgebildet, sind noch sequenziert oft erfolgreich mit nur einem geringfügigen Rückgang clustering Effizienz.
Hohe Raten von PCR Doppelarbeit sollte vermieden werden, denn dies drastisch die Anzahl der nutzbaren Sequenz liest reduziert. Die Rate der PCR Duplikate steht in direktem Zusammenhang mit der Menge an input-Material. Mit mehr Input mindert daher in der Regel Probleme mit PCR Vervielfältigung.
Höhere Zahlen liest, gefiltert durch Lesen Sie Orientierung ()( )Abbildung 2 bunzureichende Verdauung, die das Ergebnis der Verwendung zu wenig Enzym, zuviel input-Material oder unvollständige Homogenisierung der Embryonen werden kann.
Offenlegungen
Die Autoren haben nichts preisgeben.
Danksagungen
Diese Forschung wurde von der Max-Planck-Gesellschaft finanziert. C.B.H. wurde durch ein Stipendium von der International Max Planck Research School – molekulare Biomedizin unterstützt. Wir danken Shelby Blythe und Eric Wieschaus für die freundliche Bereitstellung eGFP-PCNA Drosophila Melanogaster Linie.
Materialien
| Name | Company | Catalog Number | Comments |
| Biotin-14-dATP | Life Technologies | 19524016 | |
| MboI | New England Biolabs | R0147L | |
| DNA Polymerase I Klenow Fragment | New England Biolabs | M0210L | |
| T4 DNA Ligase | Thermo Fisher | EL0012 | T4 DNA Ligase Buffer included |
| T4 DNA Polymerase | New England Biolabs | M0203L | |
| Proteinase K | AppliChem | A4392 | |
| GlycoBlue | Life Technologies | AM9516 | |
| Complete Ultra EDTA-free protease inhibitors | Roche | 5892791001 | |
| NEBNext Multiplex Oligos for Illumina (Index Primers Set 1) | New England Biolabs | E7335 | Sequencing Adaptor, Forward (unindexed) PCR primer and Reverse (indexed) PCR primer and USER enzyme used in the Library preparation section are components of this kit |
| NEBNext Ultra II DNA Library Prep Kit | New England Biolabs | E7645 | End Prep Enzyme Mix, End Prep Reaction Buffer, Ligation Enhancer, Ligation Master Mix and Polymerase Master Mix used in the Library preparation section are components of this kit |
| Covaris S2 AFA System | Covaris | ||
| DNA LoBind Tubes, 1.5 mL | Eppendorf | 0030108051 | |
| Falcon cell strainer 100 µm | Corning | 352360 | Embryo collection baskets |
| 37% formaldehyde | VWR | 437536C | |
| Heptane | AppliChem | 122062.1612 | |
| M165 FC fluorescent stereo microscope | Leica | ||
| M165 FC DFC camera | Leica | ||
| Metal micro pestle | Carl Roth | P985.1 | Used to lyse embryos in step 4.1.4 |
| RNase A | AppliChem | A3832,0050 | |
| Dynabeads MyOne Streptavidin C1 | Life Technologies | 65002 | Streptavidin coated magnetic beads |
| Ampure XP beads | Beckman Coulter | A63881 | |
| Qubit 3.0 Fluorometer | Thermo Fisher Scientific | Q33216 | |
| Qubit assay tubes | Thermo Fisher Scientific | Q32856 | |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Q32854 | |
| Phosphate buffered saline (PBS) | Sigma-Aldrich | P4417 | |
| eGFP-PCNA flies | Gift from S. Blythe and E. Wieschaus | ||
| Sodium hypochlorite 13% | Thermo Fisher | AC219255000 | |
| Triton X-100 | AppliChem | A4975 | |
| Tris buffer pH 8.0 (1 M) for molecular biology | AppliChem | A4577 | |
| NaCl | AppliChem | A2942 | |
| IGEPAL CA-630 | Sigma-Aldrich | I8896 | |
| 1.5 mL microcentrifuge tubes | Greiner Bio-One | 616201 | |
| SDS for molecular biology | AppliChem | A2263 | |
| 10x CutSmart buffer | New England Biolabs | B7204S | Restriction enzyme buffer |
| PCR Nucleotide Mix | Sigma-Aldrich | 11814362001 | Unmodified dCTP, dGTP, dTTP |
| BSA, Molecular Biology Grade | New England Biolabs | B9000S | |
| EDTA 0.5 M solution for molecular biology | AppliChem | A4892 | |
| Sodium acetate 3 M pH 5.2 | Sigma-Aldrich | S7899 | |
| DynaMag-2 Magnet | Life Technologies | 12321D | Magnetic stand |
| Intelli-Mixer RM-2L | Omnilab | 5729802 | Rotator |
| ThermoMixer F1.5 | Eppendorf | 5384000012 | Mixer |
| Small Embryo Collection Cages | Flystuff.com | 59-100 | Egg collection cage |
| Centrifuge 5424 R | Eppendorf | 5404000413 | |
| C1000 Touch Thermal Cycler | Bio-Rad | 1851148 | |
| PCR tube strips | Greiner Bio-One | 673275 | |
| NEBuffer 2.1 | New England Biolabs | B7202S | T4 DNA Polymerase buffer |
Referenzen
- Bonev, B., Cavalli, G. Organization and function of the 3D genome. Nat Rev Genet. 17 (11), 661-678 (2016).
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326 (5950), 289-293 (2009).
- Dixon, J. R., et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 485 (7398), 376-380 (2012).
- Jin, F., et al. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. , (2013).
- Rao, S. S. P., et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell. 159 (7), 1665-1680 (2014).
- Darbellay, F., Duboule, D. Topological Domains, Metagenes, and the Emergence of Pleiotropic Regulations at Hox Loci. Current topics in developmental biology. 116, 299-314 (2016).
- Beagan, J. A., et al. Local Genome Topology Can Exhibit an Incompletely Rewired 3D-Folding State during Somatic Cell Reprogramming. Cell stem cell. 18 (5), 611-624 (2016).
- Andrey, G., et al. Characterization of hundreds of regulatory landscapes in developing limbs reveals two regimes of chromatin folding. Genome Res. 27 (2), 223-233 (2017).
- Krijger, P. H. L., de Laat, W. Regulation of disease-associated gene expression in the 3D genome. Nature Reviews. Molecular Cell Biology. 17 (12), 771-782 (2016).
- Sexton, T., et al. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 148 (3), 458-472 (2012).
- Ghavi-Helm, Y., et al. Enhancer loops appear stable during development and are associated with paused polymerase. Nature. 512 (7512), 96-100 (2014).
- Foe, V. E., Alberts, B. M. Studies of nuclear and cytoplasmic behaviour during the five mitotic cycles that precede gastrulation in Drosophila embryogenesis. J Cell Sci. 61, 31-70 (1983).
- Blythe, S. A., Wieschaus, E. F. Zygotic Genome Activation Triggers the DNA Replication Checkpoint at the Midblastula Transition. Cell. 160 (6), 1169-1181 (2015).
- Blythe, S. A., Wieschaus, E. F. Establishment and maintenance of heritable chromatin structure during early Drosophila embryogenesis. eLife. 5, e20148(2016).
- JoVE Science Education Database. Embryo and Larva Harvesting and Preparation. Biology I: yeast, Drosophila and C. elegans. Drosophila melanogaster. , JoVE, Cambridge, MA. (2017).
- Sicaeros, B., O'Dowd, D. K. Preparation of Neuronal Cultures from Midgastrula Stage Drosophila Embryos. Journal of Visualized Experiments. (5), (2007).
- Shermoen, A. W. Preparation of Baskets for Drosophila Egg Collections, Treatments, and Incubations. Cold Spring Harbor Protocols. (10), (2008).
- Ay, F., Noble, W. S. Analysis methods for studying the 3D architecture of the genome. Genome biology. 16 (1), 183(2015).
- Lazaris, C., Kelly, S., Ntziachristos, P., Aifantis, I., Tsirigos, A. HiC-bench: comprehensive and reproducible Hi-C data analysis designed for parameter exploration and benchmarking. BMC Genomics. 18 (1), (2017).
- Servant, N., et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biology. 16 (1), (2015).
- Durand, N. C., et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell systems. 3 (1), 95-98 (2016).
- Lajoie, B. R., Dekker, J., Kaplan, N. The Hitchhiker's guide to Hi-C analysis: Practical guidelines. Methods. 72, 65-75 (2015).
- Schmitt, A. D., Hu, M., Ren, B. Genome-wide mapping and analysis of chromosome architecture. Nature Reviews. Molecular Cell Biology. 17 (12), 743-755 (2016).
- Shin, H., et al. TopDom: an efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Res. 44 (7), e70(2016).
- Kruse, K., Hug, C. B., Hernández-Rodríguez, B., Vaquerizas, J. M. TADtool: visual parameter identification for TAD-calling algorithms. Bioinformatics. 32 (20), 3190-3192 (2016).
- Lévy-Leduc, C., Delattre, M., Mary-Huard, T., Robin, S. Two-dimensional segmentation for analyzing Hi-C data. Bioinformatics. 30 (17), Oxford, England. i386-i392 (2014).
- Filippova, D., Patro, R., Duggal, G., Kingsford, C. Identification of alternative topological domains in chromatin. Algorithms for molecular biology: AMB. 9 (1), 14(2014).
- Crane, E., et al. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature. 523 (7559), 240-244 (2015).
- Durand, N. C., et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell systems. 3 (1), 99-101 (2016).
- Zhou, X., et al. Exploring long-range genome interactions using the WashU Epigenome Browser. Nature Methods. 10 (5), 375-376 (2013).
- Ramírez, F., et al. High-resolution TADs reveal DNA sequences underlying genome organization in flies. bioRxiv. , 115063(2017).
- Kerpedjiev, P., et al. HiGlass: Web-based Visual Comparison And Exploration Of Genome Interaction Maps. bioRxiv. , 121889(2017).
- Hug, C. B., Grimaldi, A. G., Kruse, K., Vaquerizas, J. M. Chromatin Architecture Emerges during Zygotic Genome Activation Independent of Transcription. Cell. 169 (2), (2017).
- Berkum, N. L., et al. Hi-C: a method to study the three-dimensional architecture of genomes. Journal of Visualized Experiments: JoVE. (39), (2010).
- Naumova, N., et al. Organization of the mitotic chromosome. Science. 342 (6161), 948-953 (2013).
- Denker, A., de Laat, W. The second decade of 3C technologies: detailed insights into nuclear organization. Genes & development. 30 (12), 1357-1382 (2016).
- Belaghzal, H., Dekker, J., Gibcus, J. H. Hi-C 2.0: An optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods (San Diego, Calif). 123, 56-65 (2017).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten