
Method Article
Generazione di Genome-wide cromatina conformazione cattura librerie da strettamente in fasi precoce embrioni della drosofila
In questo articolo
Riepilogo
Questo lavoro descrive un protocollo per la generazione di alta risoluzione in situ Hi-C librerie da strettamente in scena pre-gastrulazione Drosophila melanogaster embrioni.
Abstract
Studiando l'architettura tridimensionale della cromatina offre preziose informazioni sui meccanismi di regolazione genica. Qui, descriviamo un protocollo per l'esecuzione la cromatina conformazione acquisizione tecnica in situ Hi-C su organizzato popolazioni di embrione di Drosophila melanogaster . Il risultato è una libreria di sequenziamento che permette la mappatura di tutte le interazioni di cromatina che si verificano nel nucleo in un singolo esperimento. L'ordinamento dell'embrione è fatto manualmente utilizzando un microscopio a fluorescenza stereo e una linea di volo transgenica contenenti un marcatore nucleare. Utilizzando questa tecnica, le popolazioni di embrione da ogni ciclo di divisione nucleare e con lo stato definito ciclo cellulare, può essere ottenuto con purezza molto elevata. Il protocollo può anche essere adattato per ordinare più vecchi embrioni di là di gastrulazione. Ordinato gli embrioni vengono utilizzati come input per in situ Hi-C. Tutti gli esperimenti, tra cui il sequenziamento preparazione libreria, possono essere completati in cinque giorni. Il protocollo ha bassi requisiti di ingresso e funziona in modo affidabile utilizzando 20 embrioni in fase blastoderm come materiale in entrata. Il risultato finale è una libreria di sequenziamento per il sequenziamento di nuova generazione. Dopo l'ordinamento, i dati possono essere trasformati in mappe di interazione della cromatina genoma che possono essere analizzate utilizzando una vasta gamma di strumenti disponibili per ottenere informazioni sull'associazione topologicamente struttura di dominio (TAD), cicli di cromatina e cromatina compartimenti durante lo sviluppo della drosofila .
Introduzione
Acquisizione di conformazione cromatinica (3C) è emerso come un metodo estremamente utile per studiare la topologia della cromatina nel nucleo1. La variante 3C Hi-C permette di misurare le frequenze di contatto di tutte le interazioni di cromatina che si verificano nel nucleo in un singolo esperimento2. Applicazione di Hi-C ha giocato un ruolo importante nella scoperta e caratterizzazione di molti principi fondamentali dell'organizzazione della cromatina, ad esempio TADs, comparti e cicli3,4,5.
Studi di architettura della cromatina nel contesto delle transizioni inerente allo sviluppo e differenziazione delle cellule sono sempre più utilizzati per svelare i meccanismi di regolazione genica durante questi processi6,7,8, 9. Uno degli organismi modello di grande interesse è la Drosophila melanogaster, cui sviluppo e genoma sono ben caratterizzati. Tuttavia, pochi studi che indagano l'architettura della cromatina in Drosophila di fuori della coltura in vitro del tessuto le impostazioni sono state condotti10,11. Negli embrioni fertilizzazione post 16 – 18 h, TADs e scomparti che ricorda di strutture simili a mammiferi erano identificati10, che solleva la questione di quale ruolo giocano nella regolazione genica durante l'embrione di Drosophila sviluppo. Soprattutto nelle prime fasi di sviluppo, prima della gastrulazione, tali studi sono tecnicamente impegnativi. Prima di gastrulazione, embrioni della drosofila subiscono 13 divisioni nucleari sincroni che procedono ad un ritmo estremamente rapido di 8 – 60 min per ciclo12,13. Oltre a questo, la mancanza di funzionalità visive a distinguere le diverse fasi rendono difficile ottenere materiale strettamente in fasi embrione in quantità sufficienti.
Al fine di sviluppare un protocollo che permette di studiare architettura della cromatina nello sviluppo iniziale di Drosophila a ciclo nucleare ad alta risoluzione, abbiamo combinato due tecniche esistenti: in situ Hi-C, che consente la generazione di alta risoluzione tutta genoma contatto mappe5e messa in scena di embrione tramite riga di Drosophila transgenica che esprimono un eGFP-PCNA transgene13,14. Questo transgene si localizza nel nucleo durante l'interfase e si disperde in tutto il blastoderm sinciziale durante la mitosi. Utilizzare questa proprietà, è possibile facilmente distinguere diverse fasi dalla loro densità nucleare e mitotici embrioni mediante la dispersione del segnale GFP.
Insieme, queste tecniche consentono di studiare la struttura tridimensionale della cromatina in alta risoluzione da poco più di 20 embrioni della drosofila . Questo protocollo include le istruzioni per la raccolta e cernita embrioni della drosofila per ottenere popolazioni di embrioni da un ciclo di singola divisione nucleare. Inoltre descrive come gli embrioni ottenuti vengono utilizzati per eseguire in situ Hi-C. Il risultato finale è una libreria di nucleotidi adatta per il sequenziamento su macchine di prossima generazione sequenziamento. Le letture di sequenziamento risultante possono quindi essere elaborate in cromatina dettagliata interazione mappe che coprono l'intero genoma di Drosophila .
Protocollo
1. raccolta di embrioni di drosophila
Nota: Una collezione di embrione equivalente può essere eseguita come indicato in una precedente pubblicazione15.
- Trasferimento giovane eGFP-PCNA mosche (< 1 settimana di età) in gabbie collezione uovo con yeasted collezione piastre16 (1% di etanolo, acido acetico 1% e 4% agar).
- Spostare gabbie insieme in un incubatore a 25 ° C. Incubazione per 1 – 2 giorni prima del prelievo degli ovociti migliora significativamente la resa di uovo. Cambiare collezione piatti due volte al giorno.
- Rimuovere le piastre contenenti embrioni dal vano di raccolta in intervalli di 30 – 60 min. Intervalli più piccoli comportare un minor numero di embrioni, ma più stretta distribuzione delle fasi di sviluppo. Raccogliere da gabbie multiple in parallelo così che idealmente > 200 uova ogni 30-60 min.
- Conservare le piastre a 25 ° C fino a quando gli embrioni raggiungono l'età desiderata. Per embrioni in fase blastoderm (ciclo nucleare 14), incubare per circa 2 h.
- Dopo 2 h di incubazione, aggiungere acqua di rubinetto da una spruzzetta per la piastra di raccolta in modo che l'intera superficie è ricoperta d'acqua. Sospendere gli embrioni e lievito utilizzando una spazzola morbida.
- Versare sedimento embrioni dalla piastra di raccolta in un cestello di raccolta di embrioni (filtri cella commerciale con porosità di 100 µm o cesti fatti in casa17 funzionano bene), l'aggiunta di acqua di rubinetto supplementare da una spruzzetta, se necessario. In questa fase, combinare gli embrioni da tutti i piatti che sono stati raccolti in parallelo. Il campione rappresenta un singolo batch.
- Lavare gli embrioni ben risciacquando il cestello con acqua di rubinetto da una spruzzetta per 30 s fino a quando tutti i residui di lievito viene lavato via.
- Dechorionate embrioni inserendo il cestello di raccolta in una soluzione di ipoclorito di sodio 2,5% in acqua. Luce agitazione agitando facilita la rimozione del corion. Continuare fino a quando gli embrioni sono sufficientemente idrofobi che galleggiano sulla superficie della soluzione quando il cestello è sollevato e sommersa di nuovo, che dovrebbe prendere ~1.75–2 min.
Attenzione: ipoclorito di sodio è corrosivo. Indossare adeguati dispositivi di protezione personale. Soluzioni contenenti < 10% di ipoclorito di sodio può solitamente essere smaltito nel lavandino, assicurati di controllare le regolazioni dell'Istituto ospitante. - Togliete il cestello dalla soluzione e sciacquare abbondantemente con acqua di rubinetto da una spruzzetta, fino a quando l'odore della candeggina non è più evidente.
2. embrione fissazione
Nota: Condizioni di fissaggio ottimale, soprattutto la concentrazione del detergente, formaldeide e la durata della fissazione, devono essere empiricamente determinato a montare il palco degli embrioni. Per le fasi intorno il blastoderm sinciziale, una concentrazione finale di 0,5% Triton X-100 e 1,8% formaldeide nella fase acquosa funzionano bene. Per fasi successive oltre stadio embrionale 9, ulteriore ottimizzazione di questi parametri può essere necessario. Tutte le soluzioni utilizzate durante la fissazione e l'ordinamento dovrebbero contenere inibitori della proteasi.
- Capovolgere il cestello di raccolta e posto sopra un tubo di centrifugazione conico da 15 mL. Lavare gli embrioni dal cesto nel tubo usando una pipetta di Pasteur erogazione PBS-T (PBS, 0,5% Triton X-100).
- Lasciate che gli embrioni depositano sul fondo e regolare il volume totale da 2 mL con PBS-T.
- Aggiungere 6 mL di eptano e 100 µ l di formaldeide al 37% in acqua.
Attenzione: Eptano e formaldeide sono tossici per inalazione o contatto pelle. Indossare adeguati dispositivi di protezione personale e lavorare in una cappa aspirante. Rifiuti contenenti eptano o formaldeide deve essere smaltito separatamente secondo le normative dell'Istituto ospitante. - Dopo l'aggiunta della formaldeide, avviare un timer 15 min e agitare vigorosamente il tubo su e giù per 1 min a mano. La fase acquosa e organica si combina alla consistenza forma un shampoo-like.
- Agitare su un mixer rotatorio fino a 10 min dopo l'aggiunta di formaldeide.
- Centrifugare a 500 g per 1 min a temperatura ambiente per raccogliere embrioni nella parte inferiore del tubo.
- Aspirare il liquido di shampoo-come intero e scartarla, avendo cura di non per aspirare eventuali embrioni. Piccole quantità residue del surnatante shampoo-come non causano problemi.
- 15 min dopo l'aggiunta di formaldeide, risospendere gli embrioni in 5 mL di PBS-T con glicina di 125 mM per placare la formaldeide. Mescolare vigorosamente agitando su e giù per 1 min.
- Centrifugare a 500 g a temperatura ambiente per 1 min e aspirare il surnatante.
- Lavare gli embrioni da loro risospendere in 5 mL di PBS ghiacciato-T. Lasciate che gli embrioni settle e aspirato tutto surnatante.
- Ripetere il lavaggio nel passaggio 2.10 altre due volte.
- Conservare gli embrioni su ghiaccio fino l'ordinamento. Di solito, è una buona idea per raccogliere 3 – 4 lotti di volare embrioni prima di procedere all'ordinamento. Tuttavia, gli embrioni devono essere ordinati nello stesso giorno. Stoccaggio prolungato su ghiaccio o in frigo conduce alla morfologia dell'embrione alterato.
3. embrione di ordinamento
Nota: L'ordinamento può essere fatto su qualsiasi stereo microscopio a fluorescenza equipaggiato con un filtro GFP 60 – 80 ingrandimenti.
- Utilizzando una pipetta di 1.000 µ l, trasferisce un lotto di circa 100 embrioni un piccolo recipiente di vetro adatto per l'ordinamento, preferibilmente di colore scuro e posizionarlo sul ghiaccio.
-
Embrioni di ordinamento di stato nucleare densità e ciclo cellulare (Figura 1) spingendo gli embrioni desiderabili in una pila separata utilizzando una punta dell'ago o una siringa.
- Rimuovere tutti gli embrioni con distribuzione disperso, non-nucleari di eGFP-PCNA (Figura 1E). Inoltre, gli embrioni che parzialmente mostrano un segnale GFP non nucleare dovrebbero essere rimosso.
- Per facilitare l'ordinamento, è possibile assemblare una line-up di embrioni di riferimento al ciclo nucleare 12, 13 e 14 in ogni batch utilizzando le immagini nella Figura 1 come guida. Utilizzare questa line-up per abbinare gli embrioni di una fase sconosciuto con uno degli embrioni di riferimento al fine di determinare la loro fase.
- Per verificare lo stadio di sviluppo per gli embrioni di riferimento, è possibile misurare la densità nucleare dall'embrione di imaging e contando il numero dei nuclei sulla superficie dell'embrione in un'area di 2.500 µm2 utilizzando software di imaging che fornisce informazioni sulla distanza.
Nota: Il numero previsto dei nuclei per un'area di 2.500 µm2 è 12 a 16 nuclei al ciclo nucleare 12 e 20-30 nuclei nucleare ciclo 1313.
- Una volta che tutti gli embrioni nello stadio appropriato sono separati, scattare foto degli embrioni per la documentazione e il controllo di qualità. Se il microscopio stereo non è di per sé dotato di un modulo di fotocamera, può essere utilizzato qualsiasi microscopio a epifluorescenza con filtri GFP.
- Pipettare fino gli embrioni desiderati utilizzando una pipetta di 1.000 µ l, trasferimento in una nuova provetta e posto sul ghiaccio.
- Continuare fino a quando non abbastanza embrioni vengono ordinati per l'esperimento pianificato. Per gli embrioni più anziani che tappa 9, generalmente 20 embrioni sono sufficienti per uno in situ esperimento Hi-C. Al ciclo nucleare 12, 80 embrioni sono un buon punto di partenza. Nei cicli precedenti, il numero di embrioni deve essere raddoppiato circa per ogni ciclo.
- Piscina e split embrioni in provette da 1,5 mL in modo tale che una provetta contiene abbastanza embrioni per un singolo in situ Hi-C sperimentare. Si consiglia di utilizzare tubi con basse caratteristiche di legame del DNA, dal momento che il tubo stesso verrà utilizzato per l'intero protocollo e adsorbimento del DNA può portare a perdite significative a basse concentrazioni di DNA.
- Gira tubi brevemente a 100 x g a temperatura ambiente e rimuovere il surnatante. Gli embrioni dovrebbero essere più asciutti possibile per il congelamento.
- Flash congelare gli embrioni immergendo i tubi in azoto liquido e conservare a-80 ° C.
4. in Situ Hi-C
- Lisi
- Inserire le provette con embrioni congelati sul ghiaccio.
- Risospendere embrioni in 500 µ l di tampone di lisi ghiacciata (10 mM Tris-Cl a pH 8.0, 10 millimetri di NaCl, 0,2% IGEPAL CA-630, inibitori della proteasi; disciolto in acqua). Quindi attendere 1 min per lasciare gli embrioni a stabilirsi nella parte inferiore del tubo.
- Embrioni di macinare con un pestello micro metal, pre-raffreddato su ghiaccio, che è progettato per adattarsi saldamente una microcentrifuga da 1,5 mL.
- Per evitare gli embrioni di agitazione, inserire il pestello lentamente fino a toccare il fondo del tubo, spingere verso il basso e poi macinare ruotando il pestello due volte in entrambe le direzioni.
- Sollevare leggermente il pestello, spingere verso il basso del tubo nuovamente e ripetere la rettifica.
- 4.1.3.2 ripetere per 10 volte, o fino a quando gli embrioni vengono lisati completamente. La soluzione deve essere omogenea, e nessun residui grossi pezzi di embrioni devono rimanere.
- Incubare la sospensione omogeneizzata in ghiaccio per 15 min. Spin a 1.000 x g, 4 ° C per 5 min ed eliminare il surnatante.
- Lavare pallina di risospensione in 500 µ l di tampone Lisi ghiacciata, su e giù il pipettaggio.
- Girare di nuovo come in 4.1.4 e gettare il surnatante.
- Risospendere il pellet lavato in 100 µ l di 0,5% sodio dodecil solfato (SDS), su e giù il pipettaggio. Permeabilize nuclei incubando per 10 min a 65 ° C in un blocco di riscaldamento. Placare la SDS aggiungendo 50 µ l di 10% Triton X-100 e 120 µ l di acqua. Mescolare, spostando il tubo.
- Incubare a 37 ° C per 15 min in blocco di calore.
- Digestione degli enzimi di restrizione
- Aggiungere 25 µ l di tampone di enzima di restrizione 10x e 20 U di 5 U / µ l MboI. Mescolare, spostando il tubo.
- Digerire DNA incubando per 90 min a 37 ° C in blocco di calore sotto leggera agitazione (750 giri/min).
- Aggiungere un altro 20 U di MboI e continuare a incubazione per 90 min.
- Calore-inattivare MboI incubando a 62 ° C per 20 min.
- Sporgenza Fill-in
Nota: Compilando la sporgenza con biotinilati dATP consente la selezione di specifici dei frammenti. Biotina-dATP alle giunzioni di legatura è protetta dall'attività esonucleasi di T4 DNA polimerasi (vedere paragrafo 4.6), mentre la biotina-dATP alle estremità smussate deossiHb viene rimosso in modo efficiente. Il pulldown con perline rivestite con streptavidina nella sezione 4.7 pertanto specificamente arricchisce per chimerici, dei frammenti di DNA.- Aggiungere 18 µ l di 0,4 mM biotina-14-dATP, 2,25 µ l di un mix di dCTP/dGTP/dTTP non modificato (3,3 mM) e 8 µ l di 5 U / µ l DNA polimerasi I frammento di Klenow.
- Mescolare, spostando il tubo e incubare a 37 ° C per 90 min in blocco di calore.
- Legatura
- Aggiungere 657 µ l di acqua, 120 µ l di 10 x T4 DNA ligasi Buffer, 100 µ l di 10% Triton X-100, 6 µ l di 20 mg/mL albumina di siero bovino (BSA) e mescolare, spostando il tubo. Infine aggiungere 5 µ l di mix e 5 U / µ l T4 DNA ligasi, spostando il tubo.
- Tubo ruotare delicatamente (20 giri/min) a temperatura ambiente per 2 h.
- Aggiungere una seconda rata di 5 µ l di 5 U / µ l T4 DNA ligasi e continuare a ruotare per 2 h più.
- Rotazione verso il basso i nuclei a 2.500 x g per 5 min ed eliminare il surnatante.
- Estrazione del DNA
- Risospendere il pellet in 500 µ l di tampone di estrazione (50 mM Tris-Cl a pH 8.0, 50 mM NaCl, 1 mM acido etilendiamminotetraacetico (EDTA), 1% SDS; disciolto in acqua) e aggiungere 20 µ l di proteinasi K. Mix di 20 mg/mL, spostando il tubo.
- Digerire le proteine incubando a 55 ° C per 30 minuti, agitando a 1.000 giri/min.
- A de-reticolare, aggiungere 130 µ l di 5 M di NaCl e incubare per una notte a 68 ° C, agitando a 1.000 giri/min.
- Campione di pipetta in una nuova provetta 2 mL, preferenzialmente con basse caratteristiche di legame del DNA.
- Aggiungere 0,1 x volumi (63 µ l) di 3M sodio acetato pH 5.2 e 2 µ l di 15 mg/mL GlycoBlue. Mescolare bene capovolgendo. Aggiungere 1,6 x volumi (1.008 µ l) di etanolo assoluto puro e mescolare capovolgendo.
- Incubare a-80 ° C per 15 min. centrifuga a 20.000 x g a 4 ° C per almeno 30 min. Il pellet di DNA è spesso molto piccola, quasi invisibile e può essere individuato solo a causa del colore blu di GlycoBlue.
- Rimuovere il surnatante con molta attenzione, muovendo il puntale della pipetta nel tubo lungo la parete opposta da dove si trova il pellet di DNA. Piccole goccioline restanti sono spesso facilmente rimosso durante questo passaggio e i seguenti lavaggi, spingendoli fuori i tubi utilizzando una punta di P10 piuttosto che di pipettaggio li fuori.
- Lavare la pallina con l'aggiunta di 800 µ l di etanolo al 70%. Miscelare accuratamente capovolgendo e centrifugare a 20.000 x g a temperatura ambiente per 5 min, ripetere questo lavaggio almeno una volta.
- Rimuovere tutte le tracce di etanolo e lasciare il tubo con il coperchio aperto per fino a 5 min a asciugare all'aria. Una volta che nessun liquido è restante, aggiungere 50 µ l di 10 mM Tris-Cl a pH 8.0. Pipettare ripetutamente la soluzione sopra la zona sulla parete del tubo dove era situato il pellet per solubilizzare il DNA.
- Aggiungere 1 µ l di 20 mg/mL RNasi A, mix, spostando il tubo e incubare a 37 ° C per 15 min da digerire RNA. Il campione ora può essere conservato in frigorifero durante la notte o congelato a-20 ° C a tempo indeterminato.
- Controllare la concentrazione di DNA usando un'analisi di tintura fluorescente basata secondo le istruzioni del produttore. La quantità totale di DNA nel campione dovrebbe essere almeno 10 ng, altrimenti troppo poco materiale è disponibile per l'amplificazione e complessità biblioteca sarà probabilmente basso. Quando questo accade, la quantità di materiale di partenza probabilmente non era sufficiente, o materiale è stato perso lungo la strada, forse durante la lisi e la precipitazione.
- Rimozione di biotina e del DNA di taglio
- Aggiungere insieme 12 µ l di tampone di T4 DNA polimerasi x 10, 3 µ l di 1 mM dATP, 3 µ l di 1 mM dGTP e 46 µ l di acqua. Mescolare, spostando il tubo. Aggiungere 5 µ l di 3 U/mL T4 DNA polimerasi, mescolare, spostando il tubo e incubare a 20 ° C per 30 min.
- 3 µ l di EDTA 0.5 M per arrestare la reazione e usare l'acqua per portare il campione ad un volume di circa 120 µ l.
- Taglio del DNA a una dimensione di 200 – 400 bp utilizzando un dispositivo di sonicazione secondo le istruzioni del produttore. Utilizzando il sonicatore menzionato nella Tabella materiali, il seguente programma è appropriato: 2 cicli ciascuno di 50 s, 10% dazio, intensità 5, 200 cicli/scatti in sequenza.
- Biotina pulldown
- Pipettare 30 µ l di 10 mg/mL streptavidina biglie magnetiche in un nuovo tubo, separarli su un supporto magnetico e gettare il surnatante.
- Risospendere perline in 1x tampone B & W (5 mM Tris-Cl a pH 7.4, 0,5 mM EDTA, NaCl di 1m; disciolto in acqua) + 0,1% Triton X-100 e mescolare nel Vortex. Mettete il tubo su un supporto magnetico e attendere per 1 – 5 min che le perle sono separate, a seconda della marca e modello.
- Aspirare e scartare il surnatante facendo scorrere la punta della pipetta lungo la parete opposta di dove si trovano le perle. Risospendere perline in 120 µ l di tampone B & W (10 mM Tris-Cl pH 7.4, 1 mM EDTA e 2 M NaCl): 2x. Mescolare nel Vortex.
- Trasferire DNA tranciato un nuovo tubo di associazione DNA basso e mescolare con 120 µ l della sospensione della perla nel buffer 2 X B & W nel Vortex. Ruotare perline con il campione di DNA a 20 rpm per 15 min.
- Perle su un supporto magnetico di separare ed eliminare il surnatante.
- Risospendere le perline a 600 µ l di 1x B & W + 0,1% Triton X-100 e incubare a 55 ° C per 2 min, agitazione a 1.000 giri/min. Dopo la separazione, gettare il surnatante. Ripetere questo lavaggio una volta.
- Lavare le perle di una volta con 600 µ l di 10 mM Tris-Cl a pH 8.0 e scartare il supernatante dopo la separazione.
- Risospendere perline in 50 µ l di 10 mM Tris-Cl a pH 8.0.
5. sequenziamento libreria preparazione
Nota: Tutti i passaggi di libreria vengono eseguiti utilizzando componenti da una preparazione commerciale della libreria DNA kit (Vedi Tabella materiali). Tuttavia, possono essere sostituite Kit alternativo o altri reagenti. Precipitazione tende a formare negli agenti di preparazione di libreria durante la conservazione nel congelatore. È quindi importante assicurarsi che tutte le precipitazioni sono dissolto prima di utilizzare i reagenti.
-
Riparazione di fine
- Trasferire la sospensione di perlina in 50 µ l di 10 mM Tris-Cl a pH 8.0 in una nuova provetta PCR.
- Aggiungere 3 µ l di fine preparazione enzima Mix e 7 µ l di tampone di reazione della preparazione della fine. Mescolare pipettando su e giù.
- Tubo di trasferimento di un termociclatore ed eseguire il seguente programma: 20 ° C per 30 min, 65 ° C per 30 min e tenere a 4 ° C.
-
Legatura di adattatore
- Aggiungere 30 µ l di legatura Master Mix, 2,5 µ l di 1,5 µM Sequencing adattatore (diluire a 1,5 µM da stock) e 1 µ l di legatura Enhancer per la sospensione della perla. Mescolare pipettando su e giù.
- Incubare a 20 ° C per 15 min in un termociclatore.
- 3 µ l di enzima di utente. Mescolare pipettando su e giù.
- Incubare a 37 ° C per 15 min in un termociclatore.
- Separare le perle su un supporto magnetico e rimuovere il surnatante.
- Per lavare perline, risospendere perline in 100 µ l di 1x buffer B & W + 0,1% Triton X-100. Mescolare nel vortex e trasferimento ad un nuovo tubo del microcentrifuge. Separare le perle su un supporto magnetico e rimuovere il surnatante.
- Ripetere questo lavaggio una volta utilizzando 600 µ l di buffer stesso.
- Risospendere perline in 600 µ l di 10 mM Tris-Cl a pH 8.0, mix nel vortex e perline di trasferimento ad un nuovo tubo.
- Separare le perle su un supporto magnetico, eliminare il surnatante e risospendere perline in 50 µ l di 10 mM Tris-Cl a pH 8.0.
-
Amplificazione di PCR
- Preparare due provette per PCR e in ognuno, mescolare 25 µ l di polimerasi Master Mix, 1,5 µ l di 10 µM in avanti (non indicizzati) PCR primer e 1,5 µ l di 10 µM Reverse primer PCR (indicizzata).
Nota: Forward (non indicizzati) PCR primer:
5'-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATC * T-3 ´.
Inversa (indicizzata) primer PCR:
5'-CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC * T-3 ´. * indica fosforotioato obbligazioni e Ns in indicizzata primer PCR. - In ogni provetta, aggiungere 22 µ l di sospensione tallone e mescolare pipettando su e giù.
- Eseguire PCR usando il seguente programma: 98 ° C per 1 min, (98 ° C per 15 s, 65 ° C per 75 s, rampa 1.5 ° C/s) ripetuto 9 - 12 volte, 65 ° C per 5 min e tenere a 4 ° C.
Nota: Il numero di cicli di amplificazione deve essere determinata empiricamente. Tuttavia, abbiamo trovato che le librerie che ha richiesto più di 12 cicli erano generalmente di bassa complessità e non hanno provocato Hi-C mappe di alta qualità. D'altra parte, le librerie che ha richiesto meno di 12 cicli non sono state colpite negativamente da amplificare per un pieno 12 cicli. Di conseguenza, è possibile per 12 cicli di amplificazione per impostazione predefinita. - Piscina le due reazioni di PCR in un tubo del microcentrifuge singolo, separare perline su un supporto magnetico e trasferire il supernatante contenente la libreria in una nuova provetta.
- Preparare due provette per PCR e in ognuno, mescolare 25 µ l di polimerasi Master Mix, 1,5 µ l di 10 µM in avanti (non indicizzati) PCR primer e 1,5 µ l di 10 µM Reverse primer PCR (indicizzata).
-
Selezione dimensione
- Portare Ampure XP sospensione tallone a temperatura ambiente e mescolare bene agitando.
- Portare il volume della reazione PCR in pool a esattamente 200 µ l con acqua. Durante la PCR e la separazione magnetica, alcuno del volume originale è solitamente perso. Verificare il volume impostando la pipetta a 200 µ l e aspirare l'intero volume della reazione. Se l'aria è aspirata, più l'acqua deve essere aggiunto. Se il volume supera 200 µ l, regolare il volume di perline aggiunto nei passaggi 5.4.3 e 5.4.6 proporzionalmente.
Nota: I volumi tra parentesi sono validi se il volume totale delle reazioni PCR pool è esattamente 200 µ l. - Aggiungere volumi x 0,55 (110 µ l) di sospensione tallone Ampure XP e Miscelare pipettando su e giù almeno 10 volte.
- Incubare a temperatura ambiente per 5 min, perline separate su un supporto magnetico per 5 min.
- Spostare surnatante in un nuovo tubo. Gettare la provetta contenente le perline. Le perle sono associati DNA > 700 bp, che è troppo grande per essere sequenziata.
- Al supernatante, aggiungere 0,2 x volumi (40 µ l, risultante in un totale di 0,75 x Ampure tampone del campione) di sospensione tallone Ampure XP e mescolare pipettando su e giù per 10 volte.
- Incubare a temperatura ambiente per 5 min, perline separate su un supporto magnetico per 5 min.
- Eliminare il surnatante che contiene DNA < 200 bp, che comprende primer gratuito, dimeri dell'iniettore e frammenti troppo piccoli da sequenziare.
- Lasciare il tubo sul supporto magnetico. Per lavare perline, aggiungere 700 µ l di etanolo di 80%, facendo attenzione a non disturbare il pellet perlina e incubare per 30 s.
- Eliminare il surnatante, poi prendere la metropolitana fuori il supporto magnetico e risospendere perline in 100 µ l di 10 mM Tris-Cl a pH 8.0. Mescolare pipettando su e giù per 10 volte e incubare a temperatura ambiente per 1 min.
- Aggiungere 0,8 x volumi (80 µ l) di sospensione tallone Ampure XP. Mescolare pipettando su e giù per 10 volte e incubare a temperatura ambiente per 5 min. Questo secondo turno di selezione dimensione limite inferiore assicura che la libreria finale è completamente priva di primer e dimeri dell'iniettore.
- Separare le perle su un supporto magnetico per 5 min ed eliminare il surnatante.
- Lavare la pallina di perle due volte con 700 µ l di etanolo di 80% per 30 s ciascuno, lasciando il tubo sul supporto magnetico, come sopra.
- Con il tubo ancora su supporto magnetico, rimuovere tutte le tracce di etanolo. Aiuta a spingere le goccioline di etanolo dal tubo usando una pipetta P10. Evaporare etanolo residuo per un massimo di 5 min.
- Prendere il tubo fuori il supporto magnetico e risospendere perline in 50 µ l di 10 mM Tris-Cl a pH 8.0. Mescolare pipettando su e giù per 10 volte.
- Incubare a temperatura ambiente per 5 minuti, poi separate perline su un supporto magnetico.
- Trasferire il surnatante in una nuova provetta. Questa è la libreria di Hi-C finale, pronta per essere quantificato e sequenziati sulla prossime generazione di macchine di sequenziamento, secondo le istruzioni del produttore.
Risultati
Ordinato di popolazioni di embrione al ciclo nucleare 12, 13 e 14 (corrispondenti a 01:30, 01:45 e 02:10 ore post fecondazione, rispettivamente12) e fecondazione post di 3 – 4 h (hpf) sono stati ottenuti secondo le procedure descritte nel protocollo. Scattando foto del segnale eGFP-PCNA di ciascun lotto di embrione ordinato, è possibile documentare il preciso stato di fase e ciclo cellulare di ogni singolo embrione che viene utilizzato negli esperimenti a valle. Immagini di esempio degli embrioni da popolazioni ordinati sono mostrate in Figura 1B-E. L'output del protocollo in situ Hi-C è una libreria di nucleotide pronta ad essere sequenziata sulla prossime generazione di macchine di sequenziamento. Per questo scopo, una concentrazione di raccolta finale di almeno 2 – 4 nM è richiesta solitamente. Utilizzando le quantità raccomandate di materiale in entrata, questa concentrazione è attendibilmente realizzato (tabella 1).
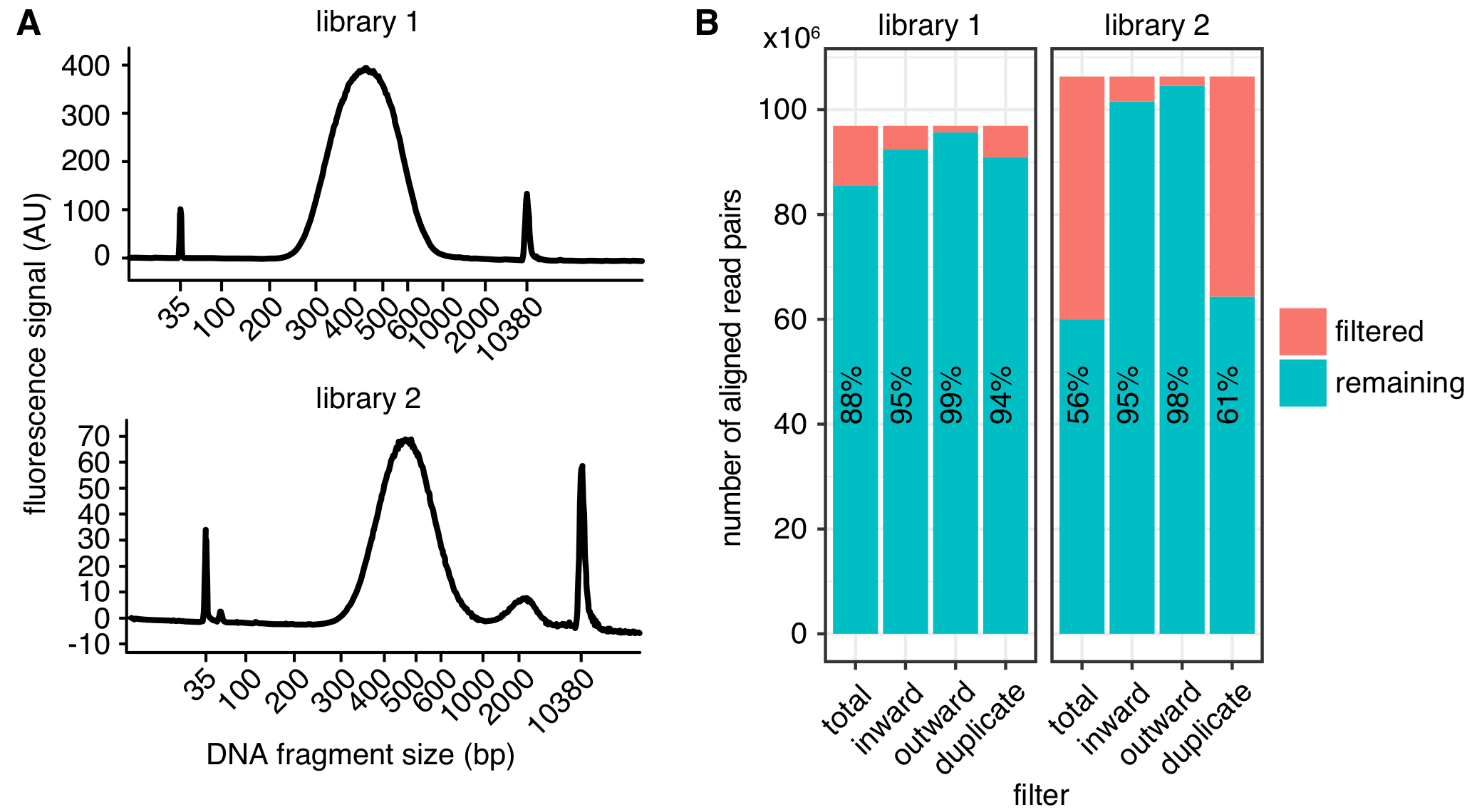
La distribuzione di dimensione prevista dei frammenti del DNA dopo la selezione della dimensione sono tra 300 – 600 bp, con un massimo a circa 500 bp (Figura 2A), a seconda la tosatura esatta e parametri di selezione dimensione. Per una sequenza, si consiglia di fine accoppiato letture di almeno 75 lunghezza bp per ridurre al minimo il numero di frammenti di restrizione unmappable nel genoma. Mappe ad alta risoluzione con 1 – 2 kb bin dimensione possono essere ottenute da 400 milioni di letture. Si consiglia di sequenziamento più replicati biologici ad una profondità inferiore di 150 milioni legge ogni, invece di sequenziamento di una singola replica a profondità molto elevate. Questo permette di valutare la variazione biologica e conduce a un minor numero di letture scartati a causa della duplicazione di PCR. Per la rappresentazione visiva, le repliche possono essere combinati. Prima di impegnarsi in un campione ad alta profondità di sequenziamento, si consiglia di eseguire i campioni mediante sequenziamento superficiale (qualche milione letture per campione) per determinare i parametri di qualità di libreria di base come in Figura 2B.
Analisi dei dati di Hi-C richiede significativa esperienza di bioinformatica e risorse computazionale. Come un quadro di massima, le letture accoppiate vengono mappate in modo indipendente per il genoma di riferimento, i tracciati risultanti sono filtrati per la qualità e l'orientamento, quindi una matrice di contatti a livello di risoluzione o un frammento di una determinata collocazione può essere generata dal filtrato allineamenti. La matrice di contatto è la base di tutto più a valle analisi esplorare TADs, cicli e scomparti. Per l'analisi iniziale del sequenziamento si legge, sono disponibili diverse condutture di bioinformatica che ne consentono l'elaborazione di crude letture in matrici di contatto senza molto specializzati bioinformatica conoscenza18,19, 2021,,22,23. Come ulteriore analisi è trasportata fuori dipende in larga misura esatta domanda biologica sotto studio e potrebbero richiedere significativa esperienza nella programmazione e di scripting in R o Python. Tuttavia, diversi strumenti e algoritmi di chiamare TADs sono disponibili5,24,25,26,27,28, nonché software per analizzare e Esplora Hi-C dati nel browser web e come applicazioni desktop autonome29,30,31,32.
Una volta elaborata, la qualità della biblioteca può essere determinata utilizzando diversi parametri (Figura 2B). In primo luogo, il tasso di duplicati PCR, che è il numero di coppie di lettura sequenziate derivanti dalla stessa molecola originale, dovrebbe essere più basso possibile limitare la quantità di letture di sequenza sprecato. Tuttavia, anche le librerie con > 40% duplicazione di PCR possa essere trasformati in contattare mappe di alta qualità se i duplicati vengono filtrati. In secondo luogo, il tasso di letture filtrate a causa del loro orientamento, come descritto in4, deve costantemente essere inferiore al 10% delle coppie lettura allineate.
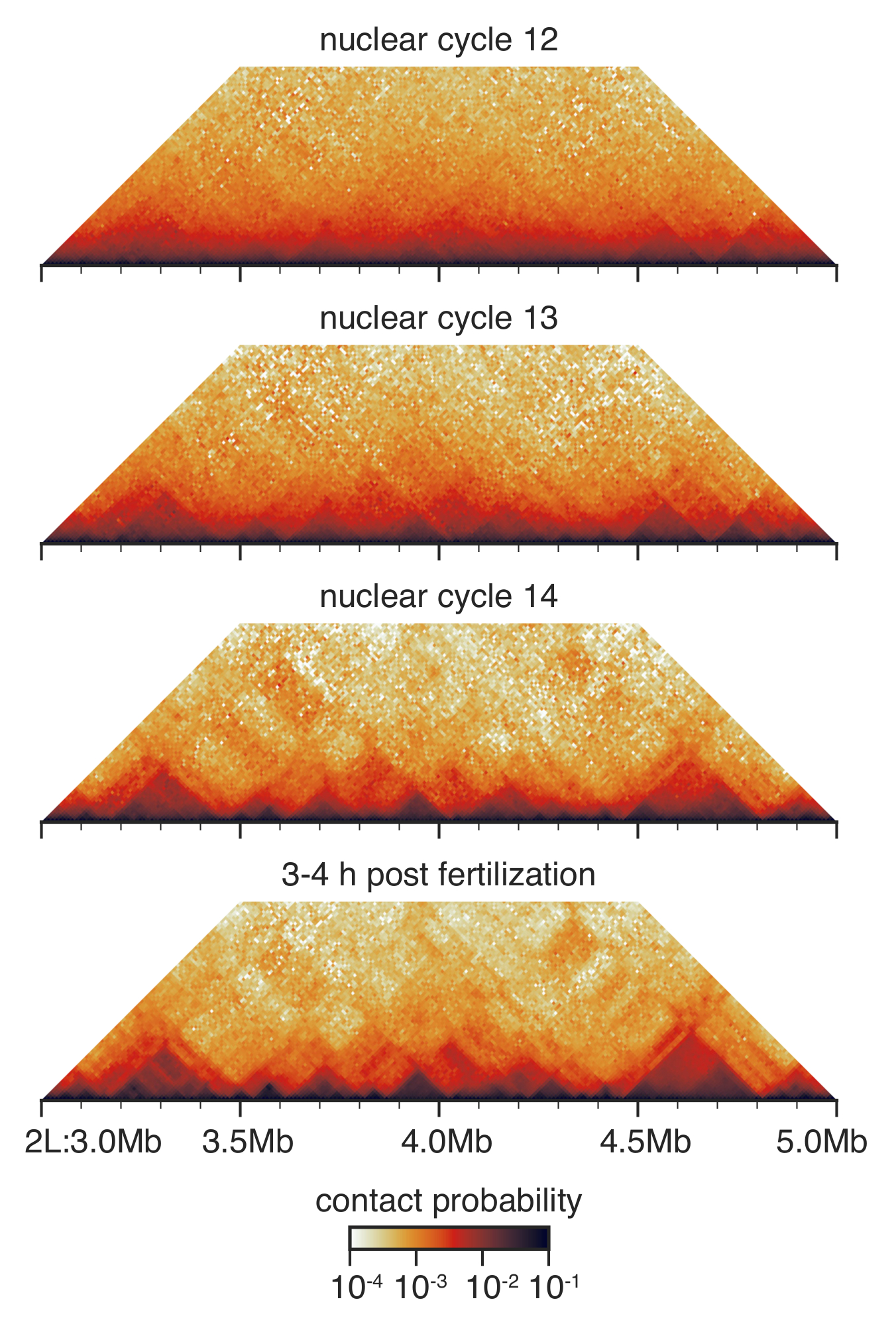
Durante lo sviluppo pre-gastrular di Drosophila tra ciclo nucleare 12 e 14, l'architettura nucleare è drasticamente ristrutturato33 (Figura 3). Al ciclo nucleare 12, pochi TADs vengono rilevati e la distribuzione complessiva dei contatti è molto liscia, senza molte caratteristiche distinguibili. Ciò è stata cambiata al ciclo nucleare 13 e 14, quando TADs sono sempre più prominenti e aspecifici contatti a lungo raggio sono esaurite.

Figura 1: immagini rappresentative degli embrioni di eGFP-PCNA durante l'ordinamento. Segnale di eGFP-PCNA (A) da una popolazione indifferenziato di embrioni dopo 60 min raccolta e 2 ore di incubazione a 25 ° C (B-E) esempi di embrioni da popolazioni ordinati al ciclo nucleare 12 (B), nucleare ciclo 13 (C), 14 (ciclo nucleare D) e da embrioni in fase di mitosi sincrone (E). Scala bar = 200 µm. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: Esempi di metriche di qualità in situ Hi-C biblioteca. (A) Bioanalyzer tracce mostrano la distribuzione delle dimensioni del frammento del DNA da un successo Hi-C library (libreria 1, alto) e da una libreria che consente di visualizzare un picco di frammenti che sono troppo grandi per il sequenziamento (Biblioteca 2, in basso). Libreria 2 è stato sequenziato con successo, ma anche grandi quantità di frammenti di DNA indesiderati possono condurre ai rendimenti di sequenziamento in diminuzione. (B) filtro statistiche delle due librerie di Hi-C: sono visualizzati il numero di coppie di lettura allineate che sono esclusi da ulteriori analisi a causa di leggi orientamento e distanza (verso l'interno, verso l'esterno)4 o duplicazione di PCR (duplicato). In ogni battuta, vengono stampato il numero di letture passando il filtro (rimanenti) e in mancanza di (filtrato). La percentuale di letture passando il filtro viene inoltre visualizzata come testo. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: mappe da embrioni in fasi: interazione Hi-C. Hi-C interazione mappe sono cestinate a risoluzione di 10 kb e bilanciate come descritto prima33. Mostrato è una regione sul cromosoma 2 L. per favore clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
| Biblioteca | Fase | Numero di embrioni | Quantità del DNA prima tosatura (ng) | Cicli di PCR | Concentrazione finale biblioteca (nM) |
| 1 | nucleare ciclo 12 | 71 | 46 | 12 | 28.2 |
| 2 | nucleare ciclo 12 | 46 | 40 | 12 | 22,2 |
| 3 | nucleare ciclo 12 | 60 | 13 | 13 | 12.3 |
| 4 | nucleare ciclo 13 | 36 | 39 | 12 | 22,2 |
| 5 | nucleare ciclo 13 | 35 | 10 | 12 | 5.0 |
| 6 | nucleare ciclo 13 | 48 | 18 | 12 | 8.7 |
| 7 | ciclo nucleare 14 | 33 | 30 | 12 | 39,8 |
| 8 | ciclo nucleare 14 | 24 | 36 | 12 | 20,4 |
| 9 | ciclo nucleare 14 | 14 | 8 | 12 | 4.2 |
| 10 | 3-4 hpf | 17 | 30 | 12 | 24.0 |
| 11 | 3-4 hpf | 18 | 42 | 11 | 19,1 |
| 12 | 3-4 hpf | 22 | 63 | 11 | 48,4 |
Tabella 1: elenco delle statistiche delle biblioteche di sequenziamento rappresentante. Per ogni libreria nell'elenco, il numero di embrioni che sono stati utilizzati per la sua generazione, la quantità di DNA totale prima di biotina pulldown e tosatura misurata da Qubit, numero di PCR di cicli utilizzati per l'amplificazione e la concentrazione finale della libreria di sequenziamento Dopo la selezione di purificazione e le dimensioni sono indicate.
Discussione
Il protocollo presentato qui è molto efficace nella generazione di mappe di alta qualità dell'architettura della cromatina in primi embrioni della drosofila . Rispetto ad un precedente protocollo34, l'approccio descritto qui utilizza un sono aggiornati in situ Hi-C procedura5, con conseguente elaborazione più veloce, maggiore risoluzione e meno uso di reagente. La procedura complessiva, compreso il protocollo di Hi-C in situ è previsto di lavorare su una vasta gamma di sistemi sperimentali oltre Drosophilae fasi. Poiché il protocollo ha un requisito di ingresso basso, potrebbe essere utilizzato anche su popolazioni di cellule isolate. In Drosophila, quando utilizza il protocollo per gli embrioni nell'intervallo descritto qui, alcuni parametri, in particolare la fissazione del materiale, potrebbe essere necessario essere regolata. Poiché più vecchi embrioni si sviluppano una cuticola altamente impermeabile, aumentare la concentrazione di formaldeide e prolungare la fissazione può essere appropriati. Per la raccolta degli embrioni nelle fasi diverse nucleare ciclo 14, i tempi di incubazione degli embrioni a 25 ° C nel passaggio 1.4 devono essere regolato come segue: ciclo nucleare 12, 70 min; ciclo nucleare 13, 90 min; 3 – 4 hpf, 03:30 h.
Durante le divisioni di 13 clivaggio (fase 1-4), la densità di nuclei raddoppia ad ogni divisione. I nuclei possono essere identificati facilmente da loro brillante fluorescenza di GFP. Durante la mitosi, eGFP-PCNA non si trova nel nucleo, e il suo segnale è dispersa in tutto l'embrione. Questa caratteristica rende l'identificazione degli embrioni che stanno subendo una divisione di clivaggio sincrono possibile. Per studiare la conformazione della cromatina, questi embrioni mitotici non sono solitamente desiderabili, poiché l'organizzazione mitotica della cromatina è drasticamente diverso da quello di organizzazione l'interfase35. È possibile adattare il protocollo specificamente selezionare embrioni sottoposti a una divisione mitotica sincrona. In questo caso, dovrebbero essere tenuti solo gli embrioni con distribuzione disperso, non-nucleari di eGFP-PCNA, e tutti gli altri embrioni devono essere eliminati. Poiché la densità nucleare non può essere determinata, metodi alternativi di embrioni in fase di loro morfologia hanno visualizzata in microscopia chiara trasmessa devono essere impiegati. Presenza di cellule del palo e nuclei alla periferia dell'embrione indicano che l'embrione ha completato il ciclo nucleare almeno 9, mentre indica cellularization visibili alla periferia nucleare ciclo 1412.
Hi-C esperimenti possono essere effettuati con successo usando una vasta selezione di enzimi di restrizione5. Approcci attuali utilizzano in genere gli enzimi che riconoscono una sequenza di 4-base, ad esempio MboI o un sito di riconoscimento 6-base, ad esempio HindIII. Il vantaggio di 4-base frese su frese 6-base è che essi offrono maggiore risoluzione potenziale, dato abbastanza profondità di sequenziamento e una copertura più uniforme dei siti di restrizione in tutto il genoma. Non c'è nessun chiaro vantaggio nella scelta di una lama 4-base sopra un altro5,23,36,37. I due enzimi più comunemente usati, MboI e DpnII, entrambe riconoscono il sito di riconoscimento stesso GATC. DpnII è meno sensibile alla metilazione di CpG, che è di alcuna preoccupazione in drosofila. Il protocollo presentato qui può anche essere completato con successo utilizzando DpnII come un enzima di restrizione. Nella sezione 4.2. enzima di restrizione e buffer devono essere regolati per la compatibilità DpnII, secondo le raccomandazioni del produttore.
Se la dimensione del frammento della biblioteca sequenziamento si discosta significativamente dalla gamma mostrata nella Figura 2A, formazione del cluster durante la sequenza può essere meno efficiente o fallire completamente. In questo caso, la distribuzione delle dimensioni dopo la tosatura deve essere controllata e parametri di taglio regolata di conseguenza. Picchi nella distribuzione di frammenti di DNA di piccolissime (< 100 bp) o molto grandi (> 1.000 bp) dimensioni indica problemi con selezione di dimensioni, ad esempio trasportare sopra di perline o surnatante che dovrebbero essere scartati. Spesso queste librerie con piccoli picchi a queste dimensioni indesiderabili, come quello nella foto, è ancora sequenziati con successo con solo una diminuzione secondaria in clustering di efficienza.
Alti tassi di duplicazione di PCR dovrebbero essere evitati perché questo riduce drasticamente il numero di sequenza utilizzabile letture. Il tasso di PCR duplicati è direttamente correlato alla quantità di materiale in ingresso. Pertanto utilizzando ulteriori input di solito allevia i problemi con duplicazione di PCR.
Un numero maggiore di letture filtrati a causa di leggere orientamento (Figura 2B) indica digestione insufficiente, che può essere il risultato dell'utilizzo di enzima troppo poco, troppo input materiale o incompleta omogeneizzazione degli embrioni.
Divulgazioni
Gli autori non hanno nulla a rivelare.
Riconoscimenti
Questa ricerca è stata finanziata dalla società Max Planck. C.B.H. è stato sostenuto da una borsa di studio della scuola di ricerca internazionale Max Planck – biomedicina molecolare. Ringraziamo Shelby Blythe ed Eric Wieschaus per gentilmente fornire la linea di melanogaster della drosofila eGFP-PCNA.
Materiali
| Name | Company | Catalog Number | Comments |
| Biotin-14-dATP | Life Technologies | 19524016 | |
| MboI | New England Biolabs | R0147L | |
| DNA Polymerase I Klenow Fragment | New England Biolabs | M0210L | |
| T4 DNA Ligase | Thermo Fisher | EL0012 | T4 DNA Ligase Buffer included |
| T4 DNA Polymerase | New England Biolabs | M0203L | |
| Proteinase K | AppliChem | A4392 | |
| GlycoBlue | Life Technologies | AM9516 | |
| Complete Ultra EDTA-free protease inhibitors | Roche | 5892791001 | |
| NEBNext Multiplex Oligos for Illumina (Index Primers Set 1) | New England Biolabs | E7335 | Sequencing Adaptor, Forward (unindexed) PCR primer and Reverse (indexed) PCR primer and USER enzyme used in the Library preparation section are components of this kit |
| NEBNext Ultra II DNA Library Prep Kit | New England Biolabs | E7645 | End Prep Enzyme Mix, End Prep Reaction Buffer, Ligation Enhancer, Ligation Master Mix and Polymerase Master Mix used in the Library preparation section are components of this kit |
| Covaris S2 AFA System | Covaris | ||
| DNA LoBind Tubes, 1.5 mL | Eppendorf | 0030108051 | |
| Falcon cell strainer 100 µm | Corning | 352360 | Embryo collection baskets |
| 37% formaldehyde | VWR | 437536C | |
| Heptane | AppliChem | 122062.1612 | |
| M165 FC fluorescent stereo microscope | Leica | ||
| M165 FC DFC camera | Leica | ||
| Metal micro pestle | Carl Roth | P985.1 | Used to lyse embryos in step 4.1.4 |
| RNase A | AppliChem | A3832,0050 | |
| Dynabeads MyOne Streptavidin C1 | Life Technologies | 65002 | Streptavidin coated magnetic beads |
| Ampure XP beads | Beckman Coulter | A63881 | |
| Qubit 3.0 Fluorometer | Thermo Fisher Scientific | Q33216 | |
| Qubit assay tubes | Thermo Fisher Scientific | Q32856 | |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Q32854 | |
| Phosphate buffered saline (PBS) | Sigma-Aldrich | P4417 | |
| eGFP-PCNA flies | Gift from S. Blythe and E. Wieschaus | ||
| Sodium hypochlorite 13% | Thermo Fisher | AC219255000 | |
| Triton X-100 | AppliChem | A4975 | |
| Tris buffer pH 8.0 (1 M) for molecular biology | AppliChem | A4577 | |
| NaCl | AppliChem | A2942 | |
| IGEPAL CA-630 | Sigma-Aldrich | I8896 | |
| 1.5 mL microcentrifuge tubes | Greiner Bio-One | 616201 | |
| SDS for molecular biology | AppliChem | A2263 | |
| 10x CutSmart buffer | New England Biolabs | B7204S | Restriction enzyme buffer |
| PCR Nucleotide Mix | Sigma-Aldrich | 11814362001 | Unmodified dCTP, dGTP, dTTP |
| BSA, Molecular Biology Grade | New England Biolabs | B9000S | |
| EDTA 0.5 M solution for molecular biology | AppliChem | A4892 | |
| Sodium acetate 3 M pH 5.2 | Sigma-Aldrich | S7899 | |
| DynaMag-2 Magnet | Life Technologies | 12321D | Magnetic stand |
| Intelli-Mixer RM-2L | Omnilab | 5729802 | Rotator |
| ThermoMixer F1.5 | Eppendorf | 5384000012 | Mixer |
| Small Embryo Collection Cages | Flystuff.com | 59-100 | Egg collection cage |
| Centrifuge 5424 R | Eppendorf | 5404000413 | |
| C1000 Touch Thermal Cycler | Bio-Rad | 1851148 | |
| PCR tube strips | Greiner Bio-One | 673275 | |
| NEBuffer 2.1 | New England Biolabs | B7202S | T4 DNA Polymerase buffer |
Riferimenti
- Bonev, B., Cavalli, G. Organization and function of the 3D genome. Nat Rev Genet. 17 (11), 661-678 (2016).
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326 (5950), 289-293 (2009).
- Dixon, J. R., et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 485 (7398), 376-380 (2012).
- Jin, F., et al. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. , (2013).
- Rao, S. S. P., et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell. 159 (7), 1665-1680 (2014).
- Darbellay, F., Duboule, D. Topological Domains, Metagenes, and the Emergence of Pleiotropic Regulations at Hox Loci. Current topics in developmental biology. 116, 299-314 (2016).
- Beagan, J. A., et al. Local Genome Topology Can Exhibit an Incompletely Rewired 3D-Folding State during Somatic Cell Reprogramming. Cell stem cell. 18 (5), 611-624 (2016).
- Andrey, G., et al. Characterization of hundreds of regulatory landscapes in developing limbs reveals two regimes of chromatin folding. Genome Res. 27 (2), 223-233 (2017).
- Krijger, P. H. L., de Laat, W. Regulation of disease-associated gene expression in the 3D genome. Nature Reviews. Molecular Cell Biology. 17 (12), 771-782 (2016).
- Sexton, T., et al. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 148 (3), 458-472 (2012).
- Ghavi-Helm, Y., et al. Enhancer loops appear stable during development and are associated with paused polymerase. Nature. 512 (7512), 96-100 (2014).
- Foe, V. E., Alberts, B. M. Studies of nuclear and cytoplasmic behaviour during the five mitotic cycles that precede gastrulation in Drosophila embryogenesis. J Cell Sci. 61, 31-70 (1983).
- Blythe, S. A., Wieschaus, E. F. Zygotic Genome Activation Triggers the DNA Replication Checkpoint at the Midblastula Transition. Cell. 160 (6), 1169-1181 (2015).
- Blythe, S. A., Wieschaus, E. F. Establishment and maintenance of heritable chromatin structure during early Drosophila embryogenesis. eLife. 5, e20148(2016).
- JoVE Science Education Database. Embryo and Larva Harvesting and Preparation. Biology I: yeast, Drosophila and C. elegans. Drosophila melanogaster. , JoVE, Cambridge, MA. (2017).
- Sicaeros, B., O'Dowd, D. K. Preparation of Neuronal Cultures from Midgastrula Stage Drosophila Embryos. Journal of Visualized Experiments. (5), (2007).
- Shermoen, A. W. Preparation of Baskets for Drosophila Egg Collections, Treatments, and Incubations. Cold Spring Harbor Protocols. (10), (2008).
- Ay, F., Noble, W. S. Analysis methods for studying the 3D architecture of the genome. Genome biology. 16 (1), 183(2015).
- Lazaris, C., Kelly, S., Ntziachristos, P., Aifantis, I., Tsirigos, A. HiC-bench: comprehensive and reproducible Hi-C data analysis designed for parameter exploration and benchmarking. BMC Genomics. 18 (1), (2017).
- Servant, N., et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biology. 16 (1), (2015).
- Durand, N. C., et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell systems. 3 (1), 95-98 (2016).
- Lajoie, B. R., Dekker, J., Kaplan, N. The Hitchhiker's guide to Hi-C analysis: Practical guidelines. Methods. 72, 65-75 (2015).
- Schmitt, A. D., Hu, M., Ren, B. Genome-wide mapping and analysis of chromosome architecture. Nature Reviews. Molecular Cell Biology. 17 (12), 743-755 (2016).
- Shin, H., et al. TopDom: an efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Res. 44 (7), e70(2016).
- Kruse, K., Hug, C. B., Hernández-Rodríguez, B., Vaquerizas, J. M. TADtool: visual parameter identification for TAD-calling algorithms. Bioinformatics. 32 (20), 3190-3192 (2016).
- Lévy-Leduc, C., Delattre, M., Mary-Huard, T., Robin, S. Two-dimensional segmentation for analyzing Hi-C data. Bioinformatics. 30 (17), Oxford, England. i386-i392 (2014).
- Filippova, D., Patro, R., Duggal, G., Kingsford, C. Identification of alternative topological domains in chromatin. Algorithms for molecular biology: AMB. 9 (1), 14(2014).
- Crane, E., et al. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature. 523 (7559), 240-244 (2015).
- Durand, N. C., et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell systems. 3 (1), 99-101 (2016).
- Zhou, X., et al. Exploring long-range genome interactions using the WashU Epigenome Browser. Nature Methods. 10 (5), 375-376 (2013).
- Ramírez, F., et al. High-resolution TADs reveal DNA sequences underlying genome organization in flies. bioRxiv. , 115063(2017).
- Kerpedjiev, P., et al. HiGlass: Web-based Visual Comparison And Exploration Of Genome Interaction Maps. bioRxiv. , 121889(2017).
- Hug, C. B., Grimaldi, A. G., Kruse, K., Vaquerizas, J. M. Chromatin Architecture Emerges during Zygotic Genome Activation Independent of Transcription. Cell. 169 (2), (2017).
- Berkum, N. L., et al. Hi-C: a method to study the three-dimensional architecture of genomes. Journal of Visualized Experiments: JoVE. (39), (2010).
- Naumova, N., et al. Organization of the mitotic chromosome. Science. 342 (6161), 948-953 (2013).
- Denker, A., de Laat, W. The second decade of 3C technologies: detailed insights into nuclear organization. Genes & development. 30 (12), 1357-1382 (2016).
- Belaghzal, H., Dekker, J., Gibcus, J. H. Hi-C 2.0: An optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods (San Diego, Calif). 123, 56-65 (2017).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati