
Method Article
Geração de todo o genoma da cromatina conformação captura bibliotecas de firmemente encenado embriões adiantados da drosófila
Neste Artigo
Resumo
Este trabalho descreve um protocolo para a geração de alta resolução em situ Hi-C bibliotecas de encenado firmemente pre-gastrulação Drosophila melanogaster embriões.
Resumo
Investigando a arquitetura tridimensional da cromatina oferece inestimável insight sobre os mecanismos de regulação gênica. Aqui, descrevemos um protocolo para a realização da cromatina conformação captura técnica em situ Hi-C na encenado populações do embrião de Drosophila melanogaster . O resultado é uma biblioteca de sequenciamento que permite o mapeamento de todas as interações de cromatina que ocorrem no núcleo em uma única experiência. Classificação do embrião é feito manualmente, usando um microscópio estéreo fluorescente e uma linha de mosca transgénica contendo um marcador nuclear. Usar esta técnica, as populações de embrião de cada ciclo de divisão nuclear e com o status do ciclo celular definido, pode ser obtido com altíssima pureza. O protocolo também pode ser adaptado para classificar os embriões mais velhos além da gastrulação. Classificada de embriões usados como entradas em situ Hi-C. Todos os experimentos, incluindo sequenciamento preparação da biblioteca, podem ser concluídos em cinco dias. O protocolo tem requisitos de entrada baixos e funciona usando confiantemente 20 embriões de estágio de Drosophila como material de entrada. O resultado final é uma biblioteca de sequenciamento para sequenciamento de próxima geração. Após o sequenciamento, os dados podem ser processados em mapas de interação de todo o genoma da cromatina que podem ser analisados usando uma ampla gama de ferramentas disponíveis para obter informação sobre topologicamente associando a estrutura de domínio (TAD), loops de cromatina e cromatina compartimentos durante o desenvolvimento da drosófila .
Introdução
Captura de conformação da cromatina (3c) surgiu como um método extremamente útil para o estudo da topologia da cromatina no núcleo1. A variante 3C Hi-C permite medir as frequências de contato de todas as interações de cromatina que ocorrem no núcleo em um único experimento2. Aplicação do Hi-C tem desempenhado um papel importante na descoberta e caracterização de muitos princípios fundamentais de organização da cromatina, como TADs, compartimentos e loops3,4,5.
Estudos de arquitetura de cromatina no contexto das transições do desenvolvimento e diferenciação celular são cada vez mais usados para desvendar os mecanismos de regulação gênica durante estes processos6,7,8, 9. Um dos organismos modelo de grande interesse é a Drosophila melanogaster, cujo desenvolvimento e genoma são bem caracterizadas. No entanto, alguns estudos que investigam a arquitetura da cromatina em Drosophila fora em vitro tecido cultura configurações foram realizadas10,11. Em embriões, fertilização post de 16 – 18 h, Tad e compartimentos reminiscentes de estruturas semelhantes em mamíferos foram identificados10, que levanta a questão de qual o papel que eles estão jogando no Regulamento do gene durante o embrião da drosófila desenvolvimento. Especialmente nas fases iniciais de desenvolvimento, antes da gastrulação, tais estudos são tecnicamente desafiadoras. Antes de gastrulação, embriões da drosófila passam por 13 divisões nucleares síncronas que produziam a um ritmo extremamente rápido de 8 – 60 min por ciclo de12,13. Além disso, a falta de recursos visuais para distinguir as diferentes fases tornam difícil obter material de embrião firmemente encenado em quantidades suficientes.
A fim de desenvolver um protocolo que permite estudar arquitetura da cromatina no desenvolvimento adiantado de Drosophila em resolução do ciclo nuclear, nós combinamos duas técnicas existentes: em situ Hi-C, que permite a geração de alta resolução toda genoma contato mapas5e encenação de embrião usando uma linha de Drosophila transgênica expressando uma eGFP-PCNA transgene13,14. Este transgene localiza-se no núcleo durante a interfase e dispersa por toda a Drosophila sincicial durante a mitose. Usando essa propriedade, é possível facilmente distinguir diferentes fases por sua densidade nuclear e mitóticos embriões pela dispersão do sinal de GFP.
Juntas, estas técnicas permitem estudar a estrutura tridimensional da cromatina em alta resolução de até 20 embriões da drosófila . Este protocolo inclui as instruções para colheita e classificação de embriões da drosófila para obter populações de embriões de um ciclo de divisão nuclear. Mais descreve como os embriões obtidos são usados para executar em situ Hi-C. O resultado final é uma biblioteca de nucleotídeo apropriada para sequenciamento em máquinas de sequenciamento na próxima geração. As leituras de sequenciamento resultante então podem ser processadas em cromatina detalhadas interação mapas cobrindo todo o genoma de Drosophila .
Protocolo
1. coleta de embrião de drosophila
Nota: Uma colheita de embriões equivalente pode ser executada, como mostrado em um anterior publicação15.
- Transferir o jovem eGFP-PCNA moscas (< 1 semana de idade) em gaiolas de coleção ovo com coleção levedada placas16 (1% de etanol, ácido acético a 1% e 4% de ágar).
- Gaiolas de coleção de movimento para uma incubadora, situado a 25 ° C. Incubação de 1-2 dias antes da recolha de ovos significativamente melhora o rendimento do ovo. Mudar as placas da coleção duas vezes por dia.
- Remova placas contendo embriões da câmara de recolha em intervalos de 30 a 60 min. Intervalos menores resultam em menos embriões, mas distribuição mais apertado dos estádios de desenvolvimento. Coletar de várias gaiolas em paralelo então que idealmente > 200 ovos cada 30-60 min.
- Armazene as placas a 25 ° C até os embriões atinjam a idade desejada. Para a fase de Drosophila embriões (ciclo nuclear 14), incubar durante aproximadamente 2 h.
- Após 2 h de incubação, adicione água da torneira de uma garrafa de água para a colecta para que toda a superfície é coberta com água. Suspender os embriões e uma escova macia do fermento.
- Despeje embriões ressuspensão de coleta em uma cesta de coleção do embrião (filtros de célula comercial com tamanho de poro de 100 µm ou cestas caseiros17 funcionam bem), adicionar água da torneira adicional de uma garrafa de água, se necessário. Nesta fase, combine embriões de todas as placas que foram coletados em paralelo. As amostras representa um único lote.
- Lave os embriões enxaguando bem o cesto com água da torneira de uma garrafa de água por 30 s até que todos os resíduos do fermento é lavada.
- Dechorionate embriões, colocando o cesto de coleta em uma solução de hipoclorito de sódio 2,5% em água. Leve agitação e agitando facilita a remoção do cório. Continue até que os embriões são suficientemente hidrofóbicos para que flutuam na superfície da solução quando a cesta é levantada para fora e submersa novamente, que deverá ter ~1.75–2 min.
Atenção: o hipoclorito de sódio é corrosiva. Use equipamento de protecção adequado. Soluções que contenham < hipoclorito de sódio 10% geralmente pode ser Descartado na pia, certifique-se de verificar as normas do Instituto de acolhimento. - Retire o cesto da solução e enxaguar com água da torneira de uma garrafa de água, até que o cheiro de água sanitária não é mais perceptível.
2. o embrião fixação
Nota: Condições de fixação ideal, principalmente a concentração de detergente, formol e a duração da fixação, precisa ser determinada empiricamente caber o palco dos embriões. Para estágios em torno da Drosophila sincicial, uma concentração final de 0.5% Triton X-100 e 1,8% formaldeído na fase aquosa funcionar bem. Para fases posteriores, além da fase de embrião 9, mais otimização desses parâmetros pode ser necessária. Todas as soluções utilizadas durante a fixação e classificação devem conter inibidores de protease.
- Inverter a cesta de coleção e coloque sobre um tubo de centrifugação cônico de 15 mL. Lave os embriões da cesta no tubo com uma pipeta Pasteur dispensação PBS-T (PBS, 0,5% Triton X-100).
- Deixe os embriões se estabelecer no fundo e ajustar o volume total de 2 mL com PBS-T.
- Adicione 6 mL de heptano e 100 µ l de 37% de formaldeído em água.
Cuidado: Heptano e formaldeído são tóxicos quando inalados ou contato após a pele. Usar equipamento de protecção adequado e em uma coifa. Resíduos contendo heptano ou formol tem que ser eliminados separadamente de acordo com regulamentos do Instituto anfitrião. - Após a adição do formaldeído, iniciar um timer de 15 min e agite vigorosamente o tubo de cima e para baixo por 1 min com a mão. A fase aquosa e orgânica combinará a consistência de formulário como um xampu.
- Agite em um misturador rotatórios até 10 min após a adição de formaldeído.
- Centrifugar a 500 x g por 1 min à temperatura ambiente para coletar embriões na parte inferior do tubo.
- Aspirar o líquido de xampu, como todo e descartá-lo, tomando cuidado para não aspirar qualquer embriões. Pequenas quantidades restantes de xampu, como sobrenadante não causam problemas.
- 15 min após a adição de formaldeído, resuspenda os embriões em 5 mL de PBS-T com glicina 125mm para saciar o formaldeído. Misture vigorosamente, agitando para cima e para baixo por 1 min.
- Centrifugar a 500 x g em temperatura ambiente por 1 min e aspire o sobrenadante.
- Lavagem de embriões por resuspending-los em 5 mL de PBS gelado-T. Deixe para resolver os embriões e Aspire todo o sobrenadante.
- Repeti a lavagem na etapa 2.10 mais duas vezes.
- Manter os embriões no gelo até a classificação. Geralmente, é uma boa ideia para coletar 3 – 4 lotes de embriões voar antes de prosseguir para classificação. No entanto, os embriões devem ser enviados no mesmo dia. Armazenamento prolongado no gelo ou na geladeira leva a morfologia alterada do embrião.
3. embrião classificação
Nota: Classificação pode ser feito em qualquer microscópio estéreo fluorescente, equipado com um filtro GFP na ampliação de 60 – 80 X.
- Utilizando uma pipeta de 1.000 µ l, transferir um lote de cerca de 100 embriões para um recipiente de vidro pequeno apropriado para triagem, de preferência de uma cor escura e colocá-lo no gelo.
-
Embriões de classificação pela densidade e ciclo celular arsenal nuclear (Figura 1), empurrando os embriões desejáveis em uma pilha separada usando uma ponta de agulha ou seringa.
- Remova todos os embriões com distribuição dispersa, não-nucleares de eGFP-PCNA (Figura 1E). Também, os embriões que parcialmente mostram um sinal não-nucleares de GFP devem ser removidos.
- Para ajudar na triagem, monte um line-up de embriões de referência no ciclo nuclear 12, 13 e 14 em cada lote usando as imagens na Figura 1 como um guia. Use esta formação para coincidir com os embriões de um estágio desconhecido com um dos embriões da referência a fim de determinar o seu estágio.
- Para verificar o grau de desenvolvimento de embriões de referência, medir a densidade nuclear pela imagem latente do embrião e contando o número de núcleos na superfície do embrião em uma área de 2.500 µm2 usando o software de imagem que fornece informações de distância.
Nota: O número esperado de núcleos para uma área de 2.500 µm2 é de 12 a 16 núcleos no ciclo nuclear 12 e 20 a 30 núcleos no ciclo nuclear 1313.
- Uma vez que todos os embriões no estágio apropriado são separados, tire fotos dos embriões para documentação e controle de qualidade. Se o microscópio estéreo não é equipado com um módulo de câmera em si, pode ser utilizado qualquer microscópio de epifluorescência com filtros GFP.
- Pipetar para os embriões desejados usando uma pipeta de 1.000 µ l, transferência para um novo tubo e lugar no gelo.
- Continue até bastante embriões são classificados para o experimento planejado. Para embriões mais velhos do que a etapa 9, geralmente, 20 embriões são suficientes para um em situ experiência Hi-C. No ciclo nuclear 12, 80 embriões são um bom ponto de partida. Em ciclos anteriores, o número de embriões aproximadamente deve ser duplicado para cada ciclo.
- Piscina e divisão de embriões em tubos de 1,5 mL de tal forma que um tubo contém suficiente embriões para um único em situ Hi-C experimentam. É aconselhável a utilização de tubos com características de ligação de DNA baixas, desde que o mesmo tubo será usado para o protocolo inteiro e adsorção de DNA pode levar a perdas significativas em baixas concentrações de DNA.
- Girar os tubos brevemente no 100 x g , à temperatura ambiente e remover o sobrenadante. Os embriões devem ser tão secos quanto possível para o congelamento.
- Flash congelar embriões submergindo os tubos em nitrogênio líquido e loja a-80 ° C.
4. in Situ Oi-C
- Lise
- Colocar os tubos com embriões congelados no gelo.
- Resuspenda embriões em 500 µ l de tampão de Lise gelada (10 mM Tris-Cl pH 8.0, 10 mM de NaCl, 0,2% IGEPAL CA-630, inibidores da protease; dissolvido em água). Então espere 1 min para deixa embriões-se na parte inferior do tubo.
- Embriões de moagem usando um pilão de metal micro, pré-refrigerado no gelo, que é projetado para caber firmemente um tubo de microcentrifugadora de 1,5 mL.
- Para evitar que os embriões de agitação, insira o pilão lentamente até que toque o fundo do tubo, empurre para baixo e depois triturá girando o pilão duas vezes em ambas as direções.
- Levantar o pilão muito ligeiramente, empurre para o fundo do tubo novamente e repita a moagem.
- Repita 4.1.3.2 10 vezes, ou até que os embriões lysed completamente. A solução deve ser homogênea, e não residuais pedaços grandes de embriões devem permanecer.
- Incube a suspensão homogeneizada no gelo por 15 min. Spin a 1.000 x g, 4 ° C por 5 min e descartar o sobrenadante.
- Lave o pellet por resuspending em 500 µ l gelada do lysis, pipetagem para cima e para baixo.
- Gire novamente como em 4.1.4 e descartar o sobrenadante.
- Resuspenda o pellet lavado em 100 µ l de 0,5% sulfato dodecyl de sódio (SDS), pipetagem para cima e para baixo. Permeabilize núcleos incubando durante 10 minutos a 65 ° C, em um bloco de aquecimento. Saciar SDS pela adição de 50 µ l de água de X-100 e 120 µ l Triton 10%. Misture por agredir o tubo.
- Incube a 37 ° C por 15 min no bloco do calor.
- Digestão da enzima de restrição
- Adicione 25 µ l de 10x buffer de enzima de restrição e 20 U de 5 U / µ l de MboI. Misture por agredir o tubo.
- Digerir o ADN incubando por 90 min a 37 ° C, no bloco de calor sob agitação ligeira (750 rpm).
- Adicione mais 20 U de MboI e continue a incubação por 90 min.
- Calor-inativar MboI incubando a 62 ° C por 20 min.
- Preenchimento de saliência
Nota: Preencher a saliência com biotinilado dATP permite a seleção de fragmentos específicos ligados. Biotina-dATP nos cruzamentos de ligadura é protegido contra a atividade do exonuclease de T4 DNA Polymerase (seção 4.6), Considerando que a biotina-dATP na unligated extremidades sem corte é eficientemente removido. O pulldown usando grânulos streptavidin-revestido na seção 4.7, portanto, especificamente enriquece de fragmentos de DNA quiméricoes, ligados.- Adicione 18 µ l de 0,4 mM biotina-14-dATP, 2.25 µ l de uma mistura sem modificações do dCTP/dGTP/dTTP 3,3 mM (cada) e 8 µ l de 5 U / µ l DNA polimerase I Klenow fragmento.
- Misture por agredir o tubo e incubar a 37 ° C por 90 min no bloco do calor.
- Ligadura
- Adicionar 657 µ l de água, 120 µ l de 10 x T4 DNA Ligase Buffer, 100 µ l de 10% Triton X-100, 6 µ l de 20 mg/mL albumina de soro bovino (BSA) e misture por agredir o tubo. Finalmente, adicione 5 µ l de 5 U / µ l T4 DNA Ligase e misture por agredir o tubo.
- Gire tubo suavemente (20 rpm) em temperatura ambiente por 2 h.
- Adicione uma segunda parcela de 5 µ l de 5 U / µ l T4 DNA Ligase e continue girando para 2 h mais.
- Spin para baixo núcleos a 2.500 x g por 5 min e descartar o sobrenadante.
- Extração de DNA
- Resuspenda o pellet em 500 µ l de solução tampão (pH 50 mM Tris-Cl 8.0, 50mm NaCl, 1mm de ácido etilenodiaminotetracético (EDTA), 1% SDS; dissolvido em água) e adicionar 20 µ l de proteinase de 20 mg/mL K. Mix por agredir o tubo.
- Digerir a proteína por incubar a 55 ° C por 30 min, agitando a 1.000 rpm.
- Para de-crosslink, adicione 130 µ l de 5 M NaCl e incubar durante uma noite em 68 ° C, agitando a 1.000 rpm.
- Amostra da pipeta em um novo tubo de 2 mL, preferencialmente com baixas características de ligação de DNA.
- Adicione 0,1 x volumes (63 µ l) de 3M de sódio acetato pH 5.2 e 2 µ l de 15 mg/mL GlycoBlue. Misture bem por inversão. Adicione volumes de 1,6 x (1.008 µ l) de etanol absoluto puro e misture por inversão.
- Incube a-80 ° C durante 15 min. centrifugar a 20.000 x g a 4 ° C, durante pelo menos 30 min. A pelota do ADN é muitas vezes muito pequena, quase invisível e só pode ser manchada devido a cor azul de GlycoBlue.
- Remova o sobrenadante cuidadosamente, movendo a ponta da pipeta no tubo ao longo da parede oposta, de onde se encontra a pelota do ADN. Pequenas gotículas restantes são muitas vezes facilmente removidas durante esta etapa e as lavagens seguintes, empurrando-os sem os tubos usando uma ponta P10 em vez de pipetagem-los para fora.
- Lave o pellet adicionando 800 µ l de etanol 70%. Misture por inversão e centrifugar a 20.000 x g em temperatura ambiente por 5 min. repetir esta lavagem pelo menos uma vez.
- Remover todos os vestígios de álcool etílico e deixar o tubo de pé com a tampa aberta para até 5 min. secar ao ar livre. Uma vez que nenhum líquido é restantes, adicione 50 µ l de pH 10 mM Tris-Cl 8.0. Pipete repetidamente a solução sobre a área na parede do tubo onde localizava-se a pelota para solubilizar o DNA.
- Adicione 1 µ l de 20 mg/mL RNase A, mix por agredir o tubo e incubar a 37 ° C por 15 min a síntese do RNA. Agora, a amostra pode ser armazenada na geladeira durante a noite ou congelada a-20 ° C indefinidamente.
- Verifica a concentração de DNA usando um tintura fluorescente com base de ensaio de acordo com as instruções do fabricante. A quantidade total de DNA da amostra deve ser de pelo menos 10 ng, caso contrário muito pouco material está disponível para a amplificação e complexidade de biblioteca provavelmente será baixa. Quando isso acontece, a quantidade de material começar provavelmente não foi suficiente, ou material foi perdido ao longo do caminho, talvez durante a Lise e precipitação.
- Remoção de biotina e DNA shearing
- Adicione juntos 12 µ l de tampão de T4 DNA Polymerase x 10, 3 µ l de 1 mM dATP, 3 µ l de 1 mM dGTP e 46 µ l de água. Misture por agredir o tubo. Adicionar 5 µ l de 3 U/mL T4 DNA Polymerase, misture por agredir o tubo e incubar a 20 ° C por 30 min.
- Adicione 3 µ l de EDTA 0,5 M para parar a reação e usar a água para levar a amostra para um volume de aproximadamente 120 µ l.
- Cisalhamento do DNA de um tamanho de 200-400 bp usando um dispositivo sonication, de acordo com as instruções do fabricante. Usando o sonicador mencionado na Tabela de materiais, o programa a seguir é apropriado: 2 ciclos de cada um dos 50 s, 10% de imposto, intensidade 5, 200 ciclos/explosão.
- Biotina pulldown
- Pipetar 30 µ l de 10 mg/mL cobertas com estreptavidina grânulos magnético para um novo tubo, separá-los num suporte magnético e descartar o sobrenadante.
- Resuspenda grânulos em 1x tampão B & W (5 mM Tris-Cl pH 7,4, EDTA de 0,5 mM, 1 M NaCl; dissolvido em água) + 0,1% Triton X-100 e mistura vortexing. Coloque o tubo num suporte magnético e aguarde 1 – 5 min, até que os grânulos são separados, dependendo da marca e modelo.
- Aspirar e desprezar o sobrenadante deslizar a ponta da pipeta ao longo da parede em frente de onde se encontram os grânulos. Resuspenda grânulos em 120 µ l de 2 x B & W tampão (pH 10 mM Tris-Cl 7.4, 1 mM EDTA e 2 M NaCl). Misture-se pela utilização do vortex.
- Transferência de DNA cortado para um novo tubo de ligação de DNA baixo e misture com 120 µ l da suspensão do grânulo em 2 X B & W buffer vortexing. Gire a contas com a amostra de DNA a 20 rpm por 15 min.
- Separar os grânulos num suporte magnético e descartar o sobrenadante.
- Ressuspender pérolas em 600 µ l de 1 x B & W + 0,1% Triton X-100 e incubar a 55 ° C por 2 min, agitando a 1.000 rpm. Após a separação, desprezar o sobrenadante. Repeti esta lavagem uma vez.
- Lave os grânulos de uma vez com 600 µ l de pH 10 mM Tris-Cl 8.0 e descartar o sobrenadante após a separação.
- Resuspenda grânulos em 50 µ l de pH 10 mM Tris-Cl 8.0.
5. sequenciamento biblioteca preparação
Nota: Todas as etapas de biblioteca são feitas usando componentes de uma preparação de biblioteca de DNA comercial kit (veja a Tabela de materiais). No entanto, kits alternativos ou outros reagentes podem ser substituídos. Precipitação tende a se formar em agentes de preparação da biblioteca durante o armazenamento do congelador. Portanto, é importante se certificar de que todos os precipitação é dissolvida antes de utilizar os reagentes.
-
Reparação de fim
- Transferi a suspensão do grânulo em 50 µ l de pH de 10 mM Tris-Cl 8.0 para um novo tubo PCR.
- Adicione 3 µ l do Mix de enzima preparação final e 7 µ l de tampão de reação de preparação final. Misture pipetando para cima e para baixo.
- Tubo de transferência para um termociclador e executar o programa a seguir: 20 ° C por 30 min, 65 ° C por 30 min e espera a 4 ° C.
-
Ligadura de adaptador
- Adicionar 30 µ l de ligadura Master Mix, 2,5 µ l de 1,5 µM sequenciamento adaptador (diluir a 1,5 µM do estoque) e 1 µ l de ligadura Enhancer para a suspensão do grânulo. Misture pipetando para cima e para baixo.
- Incube a 20 ° C por 15 min em um termociclador.
- Adicione 3 µ l da enzima do usuário. Misture pipetando para cima e para baixo.
- Incube a 37 ° C por 15 min em um termociclador.
- Separar os grânulos num suporte magnético e remover o sobrenadante.
- Para lavar os grânulos, resuspenda grânulos em 100 µ l de 1x tampão B & W + 0,1% Triton X-100. Mix por num Vortex e transferência para um novo tubo de microcentrifugadora. Separar os grânulos num suporte magnético e remover o sobrenadante.
- Repeti esta lavagem uma vez usando 600 µ l de buffer mesmo.
- Resuspenda grânulos em 600 µ l de pH 10 mM Tris-Cl 8.0, mix por utilização do Vortex e grânulos de transferência para um novo tubo.
- Separar os grânulos num suporte magnético, descartar o sobrenadante e ressuspender grânulos em 50 µ l de pH de 10 mM Tris-Cl 8.0.
-
Amplificação por PCR
- Prepare dois tubos PCR e em cada um, misture 25 µ l de Polymerase Master Mix, 1,5 µ l de 10 µM para a frente (não indexadas) PCR primer e 1,5 µ l de 10 µM reverso primer PCR (indexado).
Nota: Para a frente (não indexadas) da primeira demão do PCR:
5'-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATC * T-3´.
Cartilha PCR reversa (indexada):
5'-CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC * T-3´. * indica phosphorothioate títulos e Ns em primer PCR indexado. - Em cada tubo, adicione 22 µ l da suspensão do grânulo e mistura pipetando para cima e para baixo.
- Executar o PCR usando o programa a seguir: 98 ° C por 1 min, (98 ° C por 15 s, 65 ° C para 75 s, subida de 1,5 ° C/s) repetido 9 - 12 vezes, 65 ° C por 5 min e espera a 4 ° C.
Nota: O número de ciclos de amplificação tem que ser determinado empiricamente. No entanto, descobrimos que as bibliotecas que necessárias mais de 12 ciclos foram, em geral, de baixa complexidade e não resultou em alta qualidade que Hi-C mapas. Por outro lado, as bibliotecas que necessárias menos de 12 ciclos não foram afetadas negativamente amplificando para um total de 12 ciclos. Portanto, é possível para o padrão de 12 ciclos de amplificação. - Juntar as duas reações de PCR em um tubo único microcentrifuga, separar contas num suporte magnético e transferir o sobrenadante contendo a biblioteca para um novo tubo.
- Prepare dois tubos PCR e em cada um, misture 25 µ l de Polymerase Master Mix, 1,5 µ l de 10 µM para a frente (não indexadas) PCR primer e 1,5 µ l de 10 µM reverso primer PCR (indexado).
-
Seleção de tamanho
- Trazer Ampure XP suspensão do grânulo à temperatura ambiente e misture bem por agitação.
- Trazer o volume da reação de PCR em pool para exatamente 200 µ l com água. Durante a PCR e a separação magnética, alguns do volume original é normalmente perdido. Verifique se o volume, definindo a pipeta a 200 µ l e Aspire todo o volume da reação. Se o ar é aspirado, mais água precisa ser adicionado. Se o volume for superior a 200 µ l, ajuste o volume dos grânulos adicionado nas etapas 5.4.3 e 5.4.6 proporcionalmente.
Nota: Os volumes entre parênteses são válidos se o volume total das reações de PCR em pool é exatamente 200 µ l. - Adicione 0,55 x volumes (110 µ l) de suspensão de grânulo Ampure XP e misture pipetando para cima e para baixo pelo menos 10 vezes.
- Incube a temperatura ambiente por 5 min, grânulos separados em um carrinho magnético por 5 min.
- Mova o sobrenadante para um tubo novo. Descarte o tubo contendo os grânulos. Os grânulos têm ligado DNA > 700 bp, que é muito grande para ser sequenciado.
- Para o sobrenadante, adicione 0,2 x volumes (40 µ l, resultando em um total de 0,75 x Ampure buffer na amostra) de suspensão de grânulo Ampure XP e misture pipetando acima e para baixo 10 vezes.
- Incube a temperatura ambiente por 5 min, grânulos separados em um carrinho magnético por 5 min.
- Descartar o sobrenadante, que contém ADN < 200 bp, que inclui primers livre, dímero da primeira demão e fragmentos muito pequenos para ser sequenciado.
- Deixe o tubo sobre o suporte magnético. Lave os grânulos, adicionar 700 µ l de etanol 80%, tendo o cuidado para não perturbar a pelota do grânulo e incube por 30 s.
- Descartar o sobrenadante, então pegue o tubo fora o suporte magnético e Resuspenda grânulos em 100 µ l de pH 10 mM Tris-Cl 8.0. Misture pipetando acima e para baixo 10 vezes e incubar a temperatura ambiente por 1 min.
- Adicione 0,8 x volumes (80 µ l) de suspensão de grânulo Ampure XP. Misture pipetando acima e para baixo 10 vezes e incubar a temperatura ambiente por 5 min. Esta segunda rodada de seleção de tamanho de limite inferior garante que a biblioteca final é completamente livre de primers e dímeros da primeira demão.
- Separar os grânulos num suporte magnético por 5 min e descartar o sobrenadante.
- Lave o pellet de grânulo duas vezes com 700 µ l de etanol 80% por 30 s cada um, deixando o tubo no carrinho magnético, como acima.
- Com o tubo ainda no stand magnético, remova todos os vestígios de álcool etílico. Ajuda a empurrar as gotas de etanol fora do tubo usando uma pipeta de P10. Deixe o etanol residual evaporar por um período máximo de 5 min.
- Pegue o tubo fora o suporte magnético e Resuspenda grânulos em 50 µ l de pH 10 mM Tris-Cl 8.0. Misture pipetando acima e para baixo 10 vezes.
- Incube a temperatura ambiente por 5 min e, em seguida, grânulos separados num suporte magnético.
- Transferi o sobrenadante para um tubo fresco. Esta é a biblioteca de Hi-C final, pronta para ser quantificada e sequenciados em máquinas de sequenciamento de geração seguinte, de acordo com as instruções do fabricante.
Resultados
Classificados de populações de embrião no ciclo nuclear, 12, 13 e 14 (correspondente a 01:30, 01:45 e 02:10 horas pós fertilização, respectivamente12) e fertilização de post de 3-4 h (hpf) foram obtidos de acordo com os procedimentos descritos no protocolo. Tirando fotos do sinal de cada lote de embriões classificados eGFP-PCNA, é possível documentar o status de palco e ciclo celular preciso de cada embrião único que é usado em experimentos a jusante. Fotos de exemplo de embriões de populações classificadas são mostradas na figura 1B-E. A saída do protocolo em situ Hi-C é uma biblioteca de nucleotídeo pronta a ser sequenciado em máquinas de sequenciamento na próxima geração. Para este efeito, uma concentração final de biblioteca de pelo menos 2-4 nM é geralmente necessária. Usando as quantidades recomendadas de material de entrada, esta concentração é confiável alcançado (tabela 1).
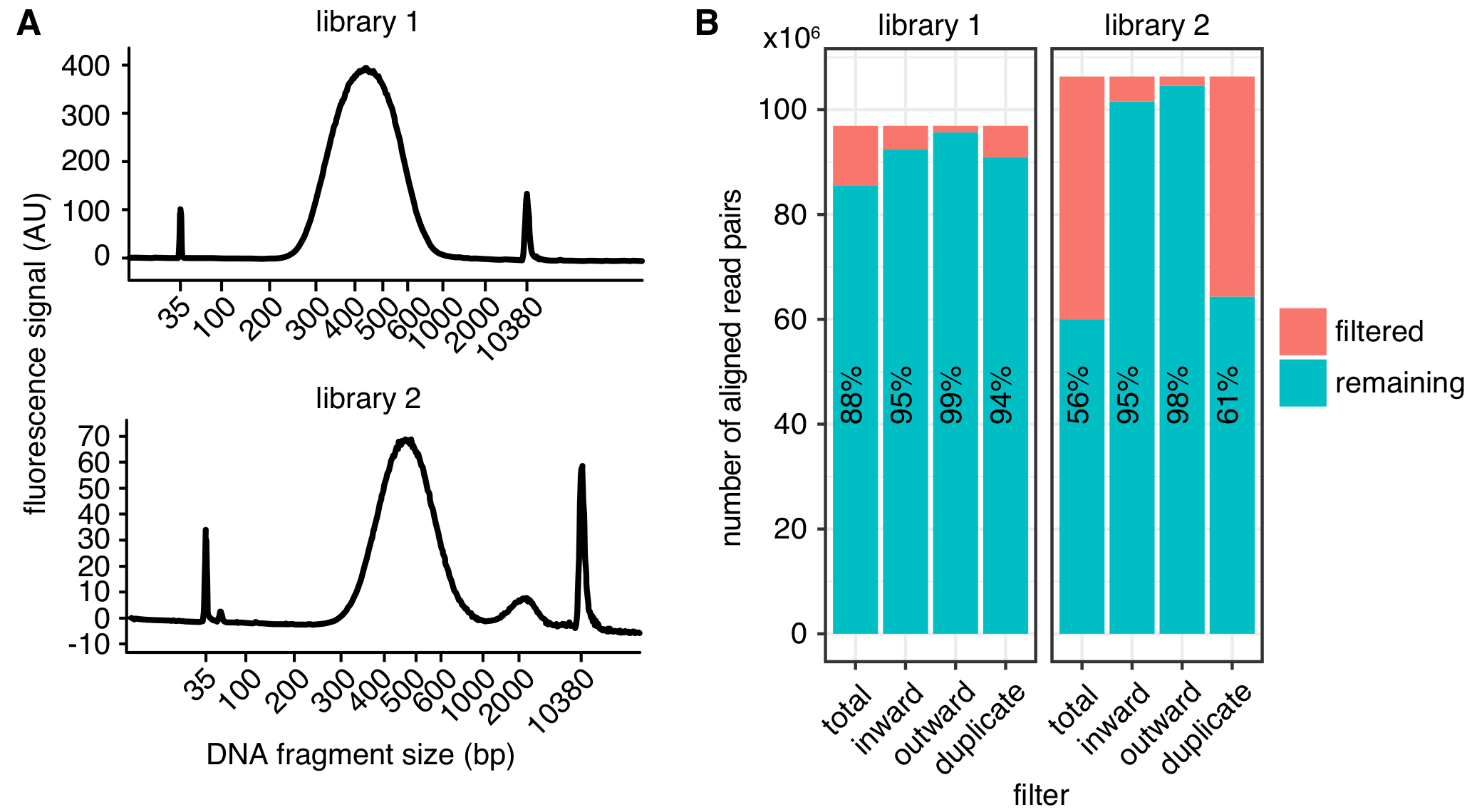
A distribuição de tamanho esperado de fragmentos de DNA após seleção de tamanho entre 300 – 600 bp, com um máximo em cerca de 500 bp (Figura 2A), dependendo do corte exato e parâmetros de seleção de tamanho. Para sequenciamento, recomendamos emparelhado-final leituras pelo menos 75 comprimento de bp para minimizar o número de fragmentos de restrição não mapeável no genoma. Mapas de alta resolução com tamanho 1-2 kb podem ser obtidos leituras 400 milhões. Recomendamos que várias repetições biológicas a uma menor profundidade de 150 milhões de sequenciamento lê cada um, em vez de um único replicar a profundidade muito elevada de sequenciamento. Isto permite a avaliação da variação biológica e leva a um menor número de leituras descartados devido a duplicação de PCR. Para uma representação visual, as réplicas podem ser combinadas. Antes de cometer a uma amostra de alta profundidade de sequenciamento, recomendamos executar amostras usando sequenciamento superficial (alguns milhões de leituras por exemplo) para determinar os parâmetros de qualidade da biblioteca básica como na Figura 2B.
Análise de dados Hi-C requer significativa experiência computacional de recursos e bioinformática. Como uma visão geral de áspera, o lê emparelhado é mapeado independentemente para o genoma de referência, os alinhamentos resultantes são filtrados para qualidade e orientação, em seguida, uma matriz de contatos em um nível de resolução ou fragmento de dado bin pode ser gerado de filtrada alinhamentos. A matriz de contato é a base para a análise de tudo mais a jusante explorando TADs, loops e compartimentos. Para a análise inicial do sequenciamento diz, várias condutas de Bioinformática estão disponíveis que permitem processamento de crus leituras em matrizes contatos sem muita bioinformática especializado conhecimento18,19, 20,21,22,23. Como é realizada uma análise mais aprofundada depende em grande parte a pergunta exata biológica em estudo e podem exigir significativa experiência em programação e scripting em R ou Python. No entanto, várias ferramentas e algoritmos de chamar TADs estão disponíveis5,24,25,26,,27,28, bem como software para analisar e Explore dados Hi-C no navegador da web e como stand-alone aplicações desktop29,30,31,32.
Uma vez processadas, a qualidade da biblioteca pode ser determinada usando métricas diferentes (Figura 2B). Primeiro, a taxa de duplicatas PCR, que é o número de pares de leitura sequenciadas decorrentes da mesma molécula original, deve ser tão baixa quanto possível limitar a quantidade de leituras de sequência desperdiçado. No entanto, mesmo as bibliotecas com > 40% duplicação de PCR pode ser processada em contatos mapas de alta qualidade se as duplicatas são filtradas. Em segundo lugar, a taxa de leituras filtradas devido à sua orientação, conforme descrito em4, deve consistentemente ser inferior a 10% dos pares leitura alinhados.
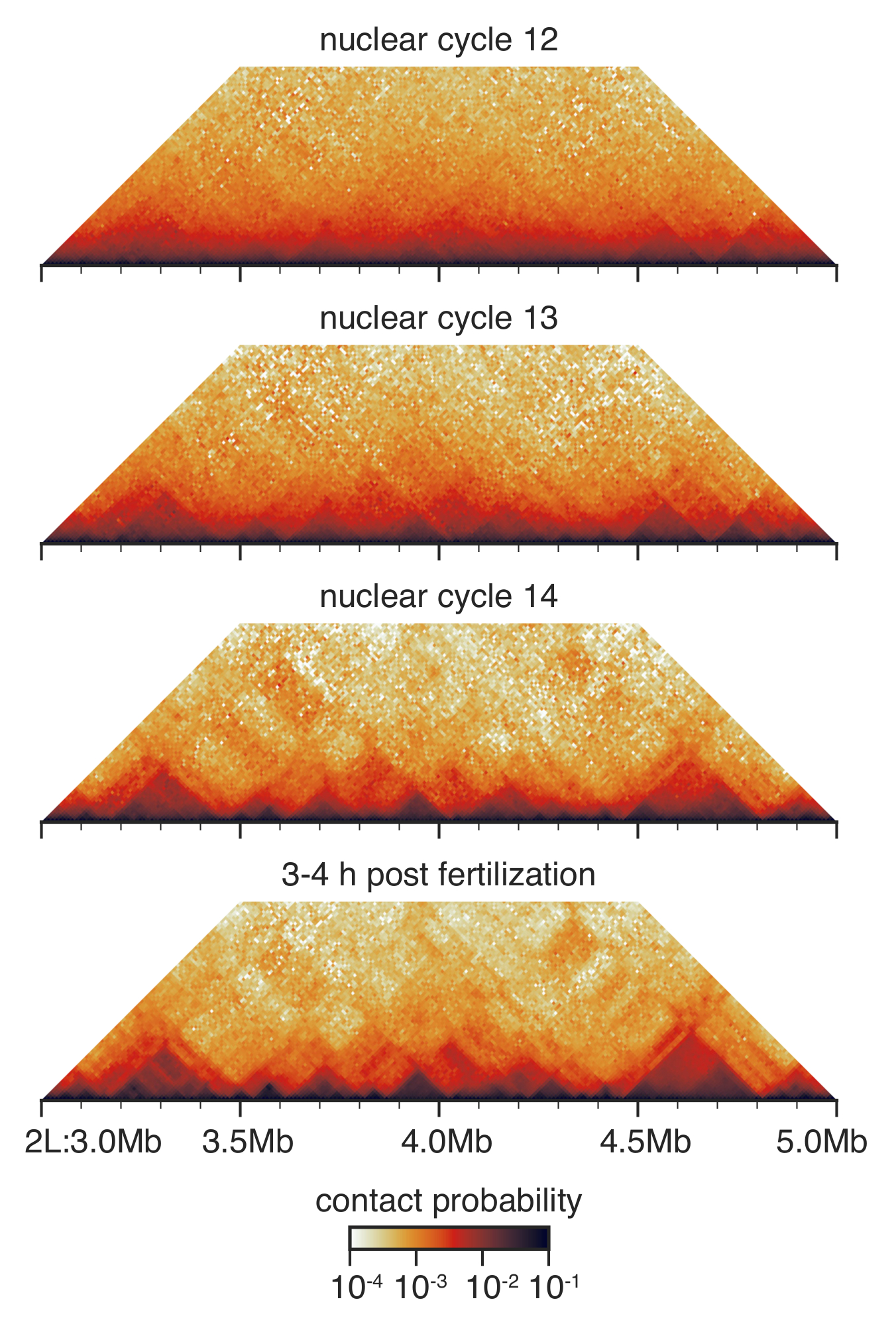
Durante o desenvolvimento pré-gastrular de Drosophila entre ciclo nuclear 12 e 14, a arquitetura nuclear é drasticamente remodelada33 (Figura 3). No ciclo nuclear 12, TADs poucos são detectados, e a distribuição geral de contatos é muito lisa e sem muitas características perceptíveis. Isto é dramaticamente alterado no ciclo nuclear 13 e 14, quando TADs são cada vez mais proeminentes e inespecíficos contatos de longo alcance estão esgotados.

Figura 1: fotos representativas dos embriões eGFP-PCNA durante a classificação. Sinal de PCNA-eGFP (A) de uma população não seleccionada de embriões após 60 min coleção e incubação de 2 h a 25 ° C (B-E) exemplos de embriões de populações classificadas no ciclo nuclear 12 (B), nuclear ciclo 13 (C), 14 (ciclo nuclear D) e de embriões submetidos a mitose síncrona (E). Escala de barras = 200 µm. clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: Exemplos em situ Hi-C biblioteca de métricas de qualidade. Traços Bioanalyzer (A) mostrando a distribuição de tamanhos de fragmento de DNA de uma bem sucedida Hi-C library (biblioteca de 1, superior) e de uma biblioteca que exibe um pico de fragmentos que são demasiado grandes para sequenciamento (biblioteca 2, inferior). Biblioteca 2 com êxito foi sequenciada, mas ainda maiores quantidades de fragmentos de DNA indesejadas podem levar à diminuição da sequenciação de rendimentos. (B) filtragem estatísticas de duas bibliotecas de Hi-C: exibido é o número de pares de leitura alinhados que são excluídos da análise adicional devido à leitura orientação e distância (interno, externo)4 ou duplicação de PCR (duplicada). Em cada bar, o número de leituras, passando o filtro (restante) e falhando (filtrado) é plotado. A percentagem de leituras passando o filtro além disso é mostrada como texto. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: interação C mapas a partir de embriões encenados. Interação C mapas são guardados em resolução de 10 kb e equilibrados como descrito antes33. Mostrada é uma região no cromossomo 2 L. , por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
| Biblioteca | Estágio | Número de embriões | Quantidade de DNA antes de cisalhamento (ng) | Ciclos PCR | Concentração final de biblioteca (nM) |
| 1 | ciclo nuclear 12 | 71 | 46 | 12 | 28,2 |
| 2 | ciclo nuclear 12 | 46 | 40 | 12 | 22.2 |
| 3 | ciclo nuclear 12 | 60 | 13 | 13 | 12.3 |
| 4 | ciclo nuclear 13 | 36 | 39 | 12 | 22.2 |
| 5 | ciclo nuclear 13 | 35 | 10 | 12 | 5.0 |
| 6 | ciclo nuclear 13 | 48 | 18 | 12 | 8.7 |
| 7 | ciclo nuclear 14 | 33 | 30 | 12 | 39,8 |
| 8 | ciclo nuclear 14 | 24 | 36 | 12 | 20,4 |
| 9 | ciclo nuclear 14 | 14 | 8 | 12 | 4.2 |
| 10 | 3-4 hpf | 17 | 30 | 12 | 24.0 |
| 11 | 3-4 hpf | 18 | 42 | 11 | 19.1 |
| 12 | 3-4 hpf | 22 | 63 | 11 | 48,4 |
Tabela 1: lista de estatísticas de biblioteca de sequenciamento representativa. Para cada biblioteca na lista, o número de embriões que foram usados para sua geração, a quantidade de DNA total antes de biotina pulldown e distorção medido pelo Qubit, o número de PCR ciclos utilizados para a amplificação e a concentração final da biblioteca de sequenciamento após a seleção de purificação e tamanho são indicados.
Discussão
O protocolo aqui apresentado é muito eficaz na geração de mapas de alta qualidade da arquitetura da cromatina em embriões adiantados da drosófila . Comparado a um anterior protocolo34, a abordagem descrita aqui usa um atualizado em situ Hi-C procedimento5, resultando em processamento mais rápido, maior resolução e menos uso de reagente. O procedimento geral, incluindo o protocolo em situ Hi-C é esperado para trabalhar em uma grande variedade de fases e sistemas experimentais, além de Drosophila. Desde que o protocolo tem um requisito de entrada baixo, também poderia ser usado em populações de células isoladas. Em Drosophila, quando usando o protocolo de embriões fora do intervalo descrito aqui, alguns parâmetros, nomeadamente a fixação do material, talvez precise ser ajustado. Desde que os embriões mais velhos desenvolvem uma cutícula altamente impermeável, aumentando a concentração de formaldeído e prolongar a fixação podem ser apropriados. Para coleta de embriões em estágios diferentes do ciclo nuclear 14, os tempos de incubação de embriões a 25 ° C na etapa 1.4 precisam ser ajustado da seguinte maneira: ciclo nuclear 12, 70 min; ciclo nuclear 13, 90 min; 3 – 4 hpf, 03:30 h.
Durante as divisões de 13 clivagem (fase 1-4), a densidade de núcleos mais ou menos duplas com cada divisão. Os núcleos podem ser facilmente identificados por sua fluorescência de GFP. Durante a mitose, eGFP-PCNA não está localizado no núcleo, e seu sinal é dispersa em todo o embrião. Esta característica torna a identificação de embriões que estão passando por uma divisão de clivagem síncrono possível. Para estudar a conformação da cromatina, estes embriões mitóticas geralmente não são desejáveis, desde que a organização mitótica da cromatina é drasticamente diferente do que a organização de interfase35. É possível adaptar o protocolo para selecionar especificamente embriões submetidos a uma divisão mitótica síncrona. Neste caso, somente embriões com distribuição dispersa, não-nucleares de eGFP-PCNA devem ser mantidos, e todos os outros embriões devem ser descartados. Desde que a densidade nuclear não pode ser determinada, devem ser empregados métodos alternativos para o estágio de embriões por sua morfologia visualizados em microscopia de luz transmitida. Presença de células do polo e núcleos na periferia de embrião indicam que o embrião completou o ciclo nuclear pelo menos 9, Considerando que celularização visível na periferia indica ciclo nuclear 1412.
Oi-C experiências podem ser executadas com êxito usando uma grande variedade de enzimas de restrição5. As abordagens atuais normalmente usam enzimas que reconhecem ou uma sequência de 4-base, tais como MboI, ou um site de reconhecimento 6-base, tais como HindIII. A vantagem de 4-base cortadores sobre cortadores 6-base é que eles oferecem maior resolução potencial, dada a profundidade suficiente sequenciamento e uma cobertura mais uniforme dos sítios de restrição em todo o genoma. Não há nenhuma vantagem clara na hora de escolher um cortador 4-base sobre outro5,23,36,37. As duas enzimas mais comumente usadas, MboI e DpnII, ambos reconhecem o mesmo site de reconhecimento do GATC. DpnII é menos sensível a metilação de CpG, que é de nenhum interesse em Drosophila. O protocolo apresentado aqui pode também ser concluído com êxito usando DpnII como uma enzima de restrição. Na seção 4.2. enzima de restrição e reserva tem que ser ajustado para compatibilidade de DpnII, de acordo com as recomendações do fabricante.
Se o tamanho do fragmento da biblioteca de sequenciamento diverge significativamente o intervalo, mostrado na Figura 2A, formação de cluster durante o sequenciamento pode ser menos eficiente ou falhar completamente. Neste caso, a distribuição de tamanho após a tosquia deve ser verificada e parâmetros de corte ajustada em conformidade. Picos na distribuição de fragmentos de DNA do muito pequeno (< 100 bp) ou muito grandes (> 1.000 bp) tamanhos indica problemas com a seleção de tamanho, tais como transportar ao longo de grânulos ou sobrenadante que deveriam ser descartados. Muitas vezes essas bibliotecas com pequenos picos a esses tamanhos indesejáveis, tais como o retratado, é ainda sequenciados com sucesso com apenas uma menor diminuição na eficiência de clustering.
Altas taxas de duplicação de PCR devem ser evitadas porque isso reduz drasticamente o número de leituras de sequência utilizável. A taxa de PCR duplicatas está diretamente relacionada à quantidade de material de entrada. Usar mais entrada, portanto, geralmente alivia problemas com duplicação de PCR.
Os números mais altos de leituras filtradas devido à orientação de leitura (,Figura 2B) indicam insuficiente digestão, que pode ser o resultado de usar pouca enzima, material muito entrada ou homogeneização incompleta dos embriões.
Divulgações
Os autores não têm nada para divulgar.
Agradecimentos
Esta pesquisa foi financiada pela sociedade Max Planck. C.B.H foi apoiado por uma bolsa de estudos da escola International Max Planck Research – Biomedicina Molecular. Agradecemos Shelby Blythe e Eric Wieschaus gentilmente fornecendo linha eGFP-PCNA Drosophila melanogaster.
Materiais
| Name | Company | Catalog Number | Comments |
| Biotin-14-dATP | Life Technologies | 19524016 | |
| MboI | New England Biolabs | R0147L | |
| DNA Polymerase I Klenow Fragment | New England Biolabs | M0210L | |
| T4 DNA Ligase | Thermo Fisher | EL0012 | T4 DNA Ligase Buffer included |
| T4 DNA Polymerase | New England Biolabs | M0203L | |
| Proteinase K | AppliChem | A4392 | |
| GlycoBlue | Life Technologies | AM9516 | |
| Complete Ultra EDTA-free protease inhibitors | Roche | 5892791001 | |
| NEBNext Multiplex Oligos for Illumina (Index Primers Set 1) | New England Biolabs | E7335 | Sequencing Adaptor, Forward (unindexed) PCR primer and Reverse (indexed) PCR primer and USER enzyme used in the Library preparation section are components of this kit |
| NEBNext Ultra II DNA Library Prep Kit | New England Biolabs | E7645 | End Prep Enzyme Mix, End Prep Reaction Buffer, Ligation Enhancer, Ligation Master Mix and Polymerase Master Mix used in the Library preparation section are components of this kit |
| Covaris S2 AFA System | Covaris | ||
| DNA LoBind Tubes, 1.5 mL | Eppendorf | 0030108051 | |
| Falcon cell strainer 100 µm | Corning | 352360 | Embryo collection baskets |
| 37% formaldehyde | VWR | 437536C | |
| Heptane | AppliChem | 122062.1612 | |
| M165 FC fluorescent stereo microscope | Leica | ||
| M165 FC DFC camera | Leica | ||
| Metal micro pestle | Carl Roth | P985.1 | Used to lyse embryos in step 4.1.4 |
| RNase A | AppliChem | A3832,0050 | |
| Dynabeads MyOne Streptavidin C1 | Life Technologies | 65002 | Streptavidin coated magnetic beads |
| Ampure XP beads | Beckman Coulter | A63881 | |
| Qubit 3.0 Fluorometer | Thermo Fisher Scientific | Q33216 | |
| Qubit assay tubes | Thermo Fisher Scientific | Q32856 | |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Q32854 | |
| Phosphate buffered saline (PBS) | Sigma-Aldrich | P4417 | |
| eGFP-PCNA flies | Gift from S. Blythe and E. Wieschaus | ||
| Sodium hypochlorite 13% | Thermo Fisher | AC219255000 | |
| Triton X-100 | AppliChem | A4975 | |
| Tris buffer pH 8.0 (1 M) for molecular biology | AppliChem | A4577 | |
| NaCl | AppliChem | A2942 | |
| IGEPAL CA-630 | Sigma-Aldrich | I8896 | |
| 1.5 mL microcentrifuge tubes | Greiner Bio-One | 616201 | |
| SDS for molecular biology | AppliChem | A2263 | |
| 10x CutSmart buffer | New England Biolabs | B7204S | Restriction enzyme buffer |
| PCR Nucleotide Mix | Sigma-Aldrich | 11814362001 | Unmodified dCTP, dGTP, dTTP |
| BSA, Molecular Biology Grade | New England Biolabs | B9000S | |
| EDTA 0.5 M solution for molecular biology | AppliChem | A4892 | |
| Sodium acetate 3 M pH 5.2 | Sigma-Aldrich | S7899 | |
| DynaMag-2 Magnet | Life Technologies | 12321D | Magnetic stand |
| Intelli-Mixer RM-2L | Omnilab | 5729802 | Rotator |
| ThermoMixer F1.5 | Eppendorf | 5384000012 | Mixer |
| Small Embryo Collection Cages | Flystuff.com | 59-100 | Egg collection cage |
| Centrifuge 5424 R | Eppendorf | 5404000413 | |
| C1000 Touch Thermal Cycler | Bio-Rad | 1851148 | |
| PCR tube strips | Greiner Bio-One | 673275 | |
| NEBuffer 2.1 | New England Biolabs | B7202S | T4 DNA Polymerase buffer |
Referências
- Bonev, B., Cavalli, G. Organization and function of the 3D genome. Nat Rev Genet. 17 (11), 661-678 (2016).
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326 (5950), 289-293 (2009).
- Dixon, J. R., et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 485 (7398), 376-380 (2012).
- Jin, F., et al. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. , (2013).
- Rao, S. S. P., et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell. 159 (7), 1665-1680 (2014).
- Darbellay, F., Duboule, D. Topological Domains, Metagenes, and the Emergence of Pleiotropic Regulations at Hox Loci. Current topics in developmental biology. 116, 299-314 (2016).
- Beagan, J. A., et al. Local Genome Topology Can Exhibit an Incompletely Rewired 3D-Folding State during Somatic Cell Reprogramming. Cell stem cell. 18 (5), 611-624 (2016).
- Andrey, G., et al. Characterization of hundreds of regulatory landscapes in developing limbs reveals two regimes of chromatin folding. Genome Res. 27 (2), 223-233 (2017).
- Krijger, P. H. L., de Laat, W. Regulation of disease-associated gene expression in the 3D genome. Nature Reviews. Molecular Cell Biology. 17 (12), 771-782 (2016).
- Sexton, T., et al. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 148 (3), 458-472 (2012).
- Ghavi-Helm, Y., et al. Enhancer loops appear stable during development and are associated with paused polymerase. Nature. 512 (7512), 96-100 (2014).
- Foe, V. E., Alberts, B. M. Studies of nuclear and cytoplasmic behaviour during the five mitotic cycles that precede gastrulation in Drosophila embryogenesis. J Cell Sci. 61, 31-70 (1983).
- Blythe, S. A., Wieschaus, E. F. Zygotic Genome Activation Triggers the DNA Replication Checkpoint at the Midblastula Transition. Cell. 160 (6), 1169-1181 (2015).
- Blythe, S. A., Wieschaus, E. F. Establishment and maintenance of heritable chromatin structure during early Drosophila embryogenesis. eLife. 5, e20148(2016).
- JoVE Science Education Database. Embryo and Larva Harvesting and Preparation. Biology I: yeast, Drosophila and C. elegans. Drosophila melanogaster. , JoVE, Cambridge, MA. (2017).
- Sicaeros, B., O'Dowd, D. K. Preparation of Neuronal Cultures from Midgastrula Stage Drosophila Embryos. Journal of Visualized Experiments. (5), (2007).
- Shermoen, A. W. Preparation of Baskets for Drosophila Egg Collections, Treatments, and Incubations. Cold Spring Harbor Protocols. (10), (2008).
- Ay, F., Noble, W. S. Analysis methods for studying the 3D architecture of the genome. Genome biology. 16 (1), 183(2015).
- Lazaris, C., Kelly, S., Ntziachristos, P., Aifantis, I., Tsirigos, A. HiC-bench: comprehensive and reproducible Hi-C data analysis designed for parameter exploration and benchmarking. BMC Genomics. 18 (1), (2017).
- Servant, N., et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biology. 16 (1), (2015).
- Durand, N. C., et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell systems. 3 (1), 95-98 (2016).
- Lajoie, B. R., Dekker, J., Kaplan, N. The Hitchhiker's guide to Hi-C analysis: Practical guidelines. Methods. 72, 65-75 (2015).
- Schmitt, A. D., Hu, M., Ren, B. Genome-wide mapping and analysis of chromosome architecture. Nature Reviews. Molecular Cell Biology. 17 (12), 743-755 (2016).
- Shin, H., et al. TopDom: an efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Res. 44 (7), e70(2016).
- Kruse, K., Hug, C. B., Hernández-Rodríguez, B., Vaquerizas, J. M. TADtool: visual parameter identification for TAD-calling algorithms. Bioinformatics. 32 (20), 3190-3192 (2016).
- Lévy-Leduc, C., Delattre, M., Mary-Huard, T., Robin, S. Two-dimensional segmentation for analyzing Hi-C data. Bioinformatics. 30 (17), Oxford, England. i386-i392 (2014).
- Filippova, D., Patro, R., Duggal, G., Kingsford, C. Identification of alternative topological domains in chromatin. Algorithms for molecular biology: AMB. 9 (1), 14(2014).
- Crane, E., et al. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature. 523 (7559), 240-244 (2015).
- Durand, N. C., et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell systems. 3 (1), 99-101 (2016).
- Zhou, X., et al. Exploring long-range genome interactions using the WashU Epigenome Browser. Nature Methods. 10 (5), 375-376 (2013).
- Ramírez, F., et al. High-resolution TADs reveal DNA sequences underlying genome organization in flies. bioRxiv. , 115063(2017).
- Kerpedjiev, P., et al. HiGlass: Web-based Visual Comparison And Exploration Of Genome Interaction Maps. bioRxiv. , 121889(2017).
- Hug, C. B., Grimaldi, A. G., Kruse, K., Vaquerizas, J. M. Chromatin Architecture Emerges during Zygotic Genome Activation Independent of Transcription. Cell. 169 (2), (2017).
- Berkum, N. L., et al. Hi-C: a method to study the three-dimensional architecture of genomes. Journal of Visualized Experiments: JoVE. (39), (2010).
- Naumova, N., et al. Organization of the mitotic chromosome. Science. 342 (6161), 948-953 (2013).
- Denker, A., de Laat, W. The second decade of 3C technologies: detailed insights into nuclear organization. Genes & development. 30 (12), 1357-1382 (2016).
- Belaghzal, H., Dekker, J., Gibcus, J. H. Hi-C 2.0: An optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods (San Diego, Calif). 123, 56-65 (2017).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados