
Method Article
Direkte Kraftmessungen der subzellulären Mechanik im Einschluss mit optischen Pinzetten
In diesem Artikel
Zusammenfassung
Hier stellen wir ein Protokoll vor, um die intrazellulären mechanischen Eigenschaften isolierter embryonaler Zebrafischzellen im dreidimensionalen Einschluss mit direkter Kraftmessung durch eine optische Falle zu untersuchen.
Zusammenfassung
Während der Entwicklung eines mehrzelligen Organismus teilt sich eine einzelne befruchtete Zelle und führt zu mehreren Geweben mit unterschiedlichen Funktionen. Die Gewebemorphogenese geht Hand in Hand mit molekularen und strukturellen Veränderungen auf Einzelzellebene, die zu Variationen subzellulärer mechanischer Eigenschaften führen. Infolgedessen widerstehen selbst innerhalb derselben Zelle verschiedene Organellen und Kompartimente unterschiedlich mechanischen Belastungen; und Mechanotransduktionswege können ihre mechanischen Eigenschaften aktiv regulieren. Die Fähigkeit einer Zelle, sich an die Mikroumgebung der Gewebenische anzupassen, ist daher zum Teil auf die Fähigkeit zurückzuführen, mechanische Belastungen zu erkennen und darauf zu reagieren. Wir haben kürzlich ein neues Mechanosensationsparadigma vorgeschlagen, bei dem die Kernverformung und -positionierung es einer Zelle ermöglicht, die physikalische 3D-Umgebung zu messen und der Zelle ein Gefühl der Propriozeption zu verleihen, um Veränderungen in der Zellform zu entschlüsseln. In diesem Artikel beschreiben wir eine neue Methode zur Messung der Kräfte und Materialeigenschaften, die den Zellkern in lebenden Zellen formen, beispielhaft für adhärente Zellen und mechanisch begrenzte Zellen. Die Messungen können nicht-invasiv mit optischen Fallen im Inneren von Zellen durchgeführt werden, und die Kräfte sind durch kalibrierfreie Detektion des Lichtimpulses direkt zugänglich. Dies ermöglicht die Messung der Mechanik des Zellkerns unabhängig von Zelloberflächenverformungen und die Dissektion exterozeptiver und interozeptiver Mechanotransduktionswege. Wichtig ist, dass das Trapping-Experiment mit optischer Mikroskopie kombiniert werden kann, um die zelluläre Reaktion und subzelluläre Dynamik unter Verwendung von Fluoreszenzbildgebung des Zytoskeletts, der Calciumionen oder der Kernmorphologie zu untersuchen. Die vorgestellte Methode ist einfach anzuwenden, kompatibel mit kommerziellen Lösungen für Kraftmessungen und kann leicht erweitert werden, um die Mechanik anderer subzellulärer Kompartimente, z. B. Mitochondrien, Stressfasern und Endosomen, zu untersuchen.
Einleitung
Die Gewebemorphogenese ist ein komplexer Prozess, bei dem biochemische Signale und physikalische Kräfte räumlich zeitlich koordiniert werden. Im sich entwickelnden Embryo diktieren Gradienten biochemischer Signalfaktoren die Schicksalsspezifikation und gewährleisten eine korrekte Gewebestrukturierung1,2. Gleichzeitig spielen intrinsische und extrinsische Kräfte eine Rolle beim Aufbau der Architektur des Embryos3,4. Der Einfluss der Zellkortexmechanik in diesem Zusammenhang wurde ausführlich untersucht5,6. Die enge Verbindung zwischen mechano-chemischen Prozessen während der Morphogenese beruht auf den Eigenschaften einzelner Zellen, mechanische Kräfte in ihrer Gewebemikroumgebung zu erfassen und darauf zu reagieren. Zellen entschlüsseln dabei mechanische Signale durch das Vorhandensein kraftempfindlicher subzellulärer und molekularer Elemente, die mechanische Informationen in spezifische Signalwege umwandeln, die das Zellverhalten, das Zellschicksal und die Zellmechanik steuern.
Ein Kennzeichen von Entwicklungsprozessen ist, dass sich Zellen als Gruppen organisieren, um mehrzellige Strukturen aufzubauen. Daher ordnen sich einzelne Zellen selten neu an und bewegen sich allein, sondern sind in einem engen Soziotop assoziiert, in dem sie kollektives Verhalten wie suprazelluläre Migration7, (Un-)Jamming-Übergänge8,9 oder Blastozystenverdichtung10 zeigen. Mechanische Kräfte, die innerhalb und zwischen Zellen erzeugt werden, dienen als wichtige Hinweise, um die kollektive Zelldynamik zu bestimmen7,11. Aber selbst wenn sich Zellen alleine bewegen, wie z.B. Vorläuferzellen, die sich zwischen Gewebeblättern oder engen Gewebenischen quetschen, erfahren sie beim Navigieren in einer dreidimensionalen Umgebung umfangreiche anisotrope mechanische Kräfte. Diese mechanischen Belastungen der Zellen haben tiefgreifende Auswirkungen auf das Zellverhalten12,13. Es wurden mehrere Mechanismen untersucht, die auf dem Kern als Hauptelement der Mechanotransduktion zusammenlaufen14,15, als passives oder aktives mechanisches Element während der Migration innerhalb einer dichten 3D-Gewebeumgebung15,16.
Wir haben kürzlich einen Mechanismus vorgeschlagen, der Zellen ausrüstet, um Formverformungen zu messen, wobei der Kern als elastischer intrazellulärer Mechano-Gauge verwendet wird12. Der Kern, der die größte Organelle in einer Zelle ist, erfährt große Verformungen, wenn Zellen polarisieren, wandern oder ihre Form unter mechanischer Dehnung, Enge oder osmotischem Stress ändern16,17,18,19. Wir fanden heraus, dass die Kernhüllendehnung zusammen mit der intrazellulären Positionierung des Kerns den Zellen Informationen über das Ausmaß und die Art der Zelldeformation (wie Zellkompression versus Zellschwellung) liefert. Die Dehnung des Zellkerns ist mit einer Entfaltung der inneren Kernmembran (INM) verbunden, die die kalziumabhängige cPLA2-Lipase-Aktivität (zytosolische Phospholipase A2) am INM fördert, gefolgt von der Freisetzung von Arachidonsäure (AA) und einer schnellen Aktivierung von Myosin II am Zellkortex. Dies führt zu einer erhöhten Zellkontraktilität und amöboiden Zellmigration oberhalb einer Schwelle der kortikalen Kontraktilität6. Die mechanosensitive Reaktion auf Zelldeformation tritt in weniger als einer Minute auf und ist bei Derentriegelung reversibel, was darauf hindeutet, dass der Kern als Dehnungsmessstreifen für die zelluläre Propriozeption fungiert, die das adaptive Zellverhalten unter mechanischen Stressbedingungen reguliert. Es wird gezeigt, dass dieser mechanosensitive Signalweg in Vorläuferstammzellen aus Zebrafischembryonen sowohl in pluripotenten als auch in liniengebundenen Zellen aktiv ist12 und in verschiedenen Spezies und Zelllinien konserviert ist20.
Zusätzlich zu den kernigen Eigenschaften als Zell-Mechanosensor werden Kernarchitektur und -mechanik während der Entwicklung und als Reaktion auf die Zellschicksalspezifikation intrinsisch reguliert21, wodurch die zelluläre Mechano-Empfindlichkeit abgestimmt wird22,23. Die Folge könnte eine Änderung der nuklearen Compliance sein, die morphologische Veränderungen und Übergänge von einem prämigratorischen zu einem migrationären Zustand und umgekehrt ermöglicht8.
Es wurden mehrere Techniken zur Messung der Zellkernmechanik angewandt, wie z. B. Rasterkraftmikroskopie24,25, Mikropipettenaspiration26,27, mikrofluidische Technologie28 und Mikronadeln29. Viele dieser Techniken sind jedoch invasiv in dem Sinne, dass die gesamte Zelle deformiert werden muss, was die Messung der mechanischen Eigenschaften und kraftabhängigen Reaktionen des Kerns selbst einschränkt. Um die gleichzeitige Verformung der Zelloberfläche und ihres mechanosensitiven Zellkortex30 zu umgehen, wurden isolierte Kerne in verschiedenen Kontexten untersucht31,32. Es kann jedoch nicht ausgeschlossen werden, dass die nukleare Isolation mit einer Veränderung der mechanischen Kerneigenschaften und ihrer Regulation verbunden ist (Referenz24 und eigene unveröffentlichte Beobachtungen).
Optische Pinzetten (OTs) sind eine vielseitige Technologie, die eine Fülle von Experimenten in der Zellmekanalobiologie ermöglicht hat und maßgeblich zu unserem Verständnis beigetragen hat, wie molekulare Maschinen chemische in mechanische Energie umwandeln33,34. Optische Pinzetten verwenden einen eng fokussierten Laserstrahl, um optische Kräfte auf dielektrische Partikel auszuüben, deren Brechungsindex höher ist als der des umgebenden Mediums33. Solche Kräfte können in der Größenordnung von Hunderten von Pico-Newton liegen und zu einer effektiven Eingrenzung des Teilchens innerhalb des Laserfallenfokus führen, was eine Manipulation des gefangenen Teilchens in drei Dimensionen ermöglicht. Die Verwendung von Licht hat einen wichtigen Vorteil, da die Messung nicht-invasiv in lebenden Zellen durchgeführt werden kann. Optische Manipulationen beschränken sich weiter auf den Fallenfokus des Laserstrahls. Daher kann die Manipulation ohne Stimulation der umgebenden Zellmembranen durchgeführt werden und stört nicht den Aktinkortex oder mechanosensitive Prozesse an der Plasmamembran, wie z.B. die kraftabhängige Aktivierung von Ionenkanälen.
Die Schwierigkeit des optischen Pinzettenansatzes besteht darin, die auf die Mikrosphäre ausgeübten Kräfte mit klassischen Ansätzen genau zu bestimmen, die auf der kalibrierten indirekten Kraft basierend auf dem Äquipartitionstheorem oder der Verwendung definierter Stokes-Widerstandskräfte zur Messung einer laserleistungsabhängigen Fluchtkraft beruhen35. Während diese Methoden in einem In-vitro-Experiment einfach umzusetzen sind, können sie in der Regel nicht in eine zelluläre Umgebung übersetzt werden. Es wurden mehrere Strategien in das Feld eingeführt, die auf einer direkten Kraftkalibrierung beruhen, die aus den ersten Prinzipien der Impulserhaltung abgeleitet wurde36,37. Im Gegensatz zu anderen Kraftspektroskopie-Ansätzen werden Kraftmessungen aus einem lokalen Austausch des Lichtimpulses mit dem beliebig geformten gefangenen Teilchen abgeleitet38,39. In unserem Versuchsaufbau werden Änderungen des Lichtimpulses, die durch optische Kräfte entstehen, direkt gemessen, ohne dass eine In-situ-Trap-Kalibrierung erforderlich ist40,41,42,43. So werden die Messungen in einer viskosen Umgebung wie dem Inneren der Zelle oder sogar innerhalb eines Gewebes möglich und Kräfte können leicht bis auf die pN-Ebene quantifiziert werden.
In diesem Protokoll beschreiben wir einen Assay zur mechanischen Manipulation intrazellulärer Organellen oder Strukturen und zur quantitativen Beurteilung ihrer mechanischen Eigenschaften durch einen optischen Pinzettenaufbau. Dieser Aufbau ist in ein rotierendes Scheibenfluoreszenzmikroskop integriert, das eine parallele Abbildung des zellulären Verhaltens oder der intrazellulären Dynamik ermöglicht. Der Assay ermöglicht die Charakterisierung der mechanischen Eigenschaften bestimmter zellulärer Kompartimente, wie z.B. des Zellkerns, und untersucht gleichzeitig die mögliche Mechanoresponse und Aktivierung molekularer Signalwege als Folge der Verformung selbst. Darüber hinaus ermöglicht das optische Einfangen von injizierten Mikrokügelchen in Zellen eine Erhöhung der Eindringkraft dank eines deutlich höheren Brechungsindex der Polystyrolperle (n = 1,59) im Vergleich zum intrinsischen Brechungskontrast44 des Kerns (n ~ 1,35) gegenüber Zytoplasma (n ~ 1,38). Die vorgestellte Strategie kann leicht an die Untersuchung anderer intrazellulärer Strukturen und Organellen sowie an andere Ansätze angepasst werden, die aktive Mikrorheologie, die Verwendung mehrerer optischer Fallen zur gleichzeitigen Untersuchung der gleichen / unterschiedlichen subzellulären Strukturen und Messungen zur Zellmechanobiologie im lebenden Embryo umfassen.
Protokoll
Alle verwendeten Protokolle wurden vom Institutional Animal Care and Use Ethic Committee (PRBB-IACUEC) genehmigt und gemäß den nationalen und europäischen Vorschriften umgesetzt. Alle Experimente wurden nach den Prinzipien der 3R durchgeführt. Zebrafische (Danio rerio) wurden wie zuvor beschrieben gepflegt.
1. Präparation isolierter primär embryonaler Vorläuferstammzellen von Zebrafischen
- Zubereitung von Mikropipetten und Agarose
HINWEIS: Ein vollständiges Mikroinjektionsprotokoll für Zebrafischembryonen finden Sie unter Referenz45.- Ziehen Sie mit einem Mikropipettenzieher eine 1,0 mm große Glaskapillare, um zwei Nadeln45 zu erhalten. Bewahren Sie die unbenutzten Nadeln in einer 150 mm Petrischale auf, die an einem Spielteigkissen befestigt ist, oder in einem Innen-Außen-Laborbandring, um die dünne Spitze vor Beschädigungen während des Transports zu schützen.
- 1% hochreine Agarose in E3 (5 mM NaCl, 0,17 mM KCl, 0,33 mM CaCl2, 0,33 mM MgSO4) in einer Standard-Küchen-/Labormikrowelle für 10 s schmelzen. Erhitzen Sie die Mischung wiederholt für kurze Zeiträume (einige Sekunden), bis die Agarose schmilzt.
- Wenn die Agarose vollständig geschmolzen ist, lassen Sie sie kurz abkühlen und gießen Sie sie dann in eine 10 cm lange Petrischale. Fügen Sie langsam die dreieckige Mikroinjektionsform (siehe Materialtabelle) auf der Oberseite der Agarose hinzu, um das Auftreten von Blasen zu vermeiden. Drücken Sie die Form nicht und stellen Sie sicher, dass sie auf der Agaroseoberfläche bleibt.
- Wenn die Agarose vollständig erstarrt ist, entfernen Sie die dreieckige Form sehr langsam, indem Sie eine sanfte Kraft ausüben, um Brüche in der Agarose zu vermeiden. Die Platte kann kopfüber bei 4 °C für 2-4 Wochen gelagert werden.
- 30 min vor der Mikroinjektion den Teller aus dem Kühlschrank nehmen und E3 vorgewärmt auf 28 °C geben, damit er sich bei Raumtemperatur stabilisiert.
- Zubereitung von Injektionsmischungen
- Zur Herstellung der Injektionsmischung werden 1 μm Mikrokügelchen (Polystyrol, nicht fluoreszierend) im Verhältnis 1:5 in RNase-freiem Wasser verdünnt.
- mRNA für die vorübergehende Expression von fluoreszierenden Markern oder die Expression rekombinanter Genkonstrukte und/oder die Co-Injektion von Morpholino in der gewünschten Konzentration vorbereiten.
HINWEIS: Eine typische Injektionsmischung für die Co-Injektion von Mikrokügelchen zusammen mit 100 pg mRNA pro Embryo, um beispielsweise den Zellkern mit H2A-mCherry zu markieren, ist: 1 μL Perlen + 1 μL mRNA (Stammkonzentration beträgt 1 μg/μL) + 2,5 μL RNA-freies Wasser + 0,5 μL Phenolrot (Stammlösung 0,5%, Phenolrot ist nicht obligatorisch; es wird für eine bessere Visualisierung des injizierten Tropfens verwendet, aber die nicht markierte Injektion Tropfen ist auch für einen erfahrenen Experimentator sichtbar). Die RNA-Injektion kann auch nützlich sein, um injizierte Embryonen auszuwählen. Fluoreszierende Mikrokügelchen können injiziert werden, anstatt nicht fluoreszierend, um sie zu visualisieren.
- Beladung und Kalibrierung von Mikroinjektionsnadeln
- Schalten Sie den Mikroinjektor mit der Option Time-Gated ein . Diese Einstellung ist sehr wichtig, um das Injektionsvolumen richtig zu kalibrieren. Stellen Sie die Gating-Zeit auf ca. 500 ms ein.
- Laden Sie 3 μL der Injektionsmischung mit einer Mikroladerpipette in die Nadel.
- Führen Sie die Nadel in den Mikromanipulator ein und verschließen Sie sie fest. Überprüfen Sie, ob sich der Mikromanipulator in einer guten Position befindet und genügend Freiheit hat, sich auf der Injektionsplatte in x-y-Richtung zu bewegen.
- Messen Sie die Tropfengröße mit einem Mikrometer-Objektträger (5 mm/100 Divisionen) mit einem Tropfen Mineralöl auf top45 und schleudern Sie einen Tropfen der Injektionsmischung direkt in das Mineralöl.
- Schneiden Sie die Nadel mit einer scharfen Pinzette in einem steilen Winkel zu, um eine scharfe spitze Spitze zu erzeugen. Stellen Sie die Tropfengröße auf 0,1 mm ein, was 0,5 nL Spritzgussmaterial entspricht.
HINWEIS: Wenn durch das Schneiden der Nadel dieses Volumen überschritten wird, wird empfohlen, den Kalibriervorgang mit einer neuen Nadel zu wiederholen. Die Gating-Zeit des Mikroinjektors kann leicht an das Tropfenvolumen angepasst werden; kurze Gating-Zeiten entsprechen jedoch einem großen Nadeldurchmesser, der die Embryonen möglicherweise schädigt.
- Mikroinjektion von Zebrafischembryonen im Einzelzellstadium
- Sammeln Sie Zebrafischembryonen kurz nach der Befruchtung zur Mikroinjektion der Perlenmischung direkt in den Embryo im Einzelzellstadium (Zygote), bevor die erste Zellteilung erfolgt.
HINWEIS: Dies gewährleistet eine ordnungsgemäße Verteilung der Mikrosphären und eine ausreichend hohe Ausbeute an isolierten Blastomeren mit mindestens einer Mikrosphäre pro Zelle in späteren Entwicklungsstadien, in denen Experimente durchgeführt werden (Blastula-Gastrula-Stadium). Einrückungsexperimente können immer noch durchgeführt werden, wenn sich zwei Kugeln innerhalb der Zelle befinden, aber Zellen, die keine Kügelchen haben, sollten ausgeschlossen werden (obwohl eine Einrückung ohne Kugeln möglich ist). AB-Wildtyp-Stämme wurden in diesem Protokoll verwendet, aber jeder andere Stamm, z. B. TL, kann verwendet werden. - Einzellige Embryonen (Zygote) in eine vorgewärmte dreieckige 1% ige Agaroseform geben, wie in Abbildung 1A gezeigt, mit einer Pasteurpipette aus Kunststoff.
- Entfernen Sie zusätzliches Medium mit derselben Pipette, um zu vermeiden, dass die Embryonen herumschweben. Schieben Sie die Embryonen vorsichtig mit einem Pinsel in die dreieckige Form. Halten Sie etwas Abstand zwischen den Embryonen, um die korrekte Orientierung zu erleichtern (Abbildung 1B).
- Richten Sie die Embryonen vorsichtig mit einem Pinsel aus, so dass die Embryonen seitlich ausgerichtet sind, wobei die eine Zelle der Zygote deutlich sichtbar ist, wie in Abbildung 1B dargestellt. Eine ideale Orientierung für die Mikroinjektion wird erreicht, wenn die eine Zelle des Embryos der Nadelrichtung (Injektion über den Tierpol des Embryos) oder umgekehrt der Dotterzelle (Injektion über den pflanzenden Pol des Embryos) zugewandt ist, wie in Abbildung 1C gezeigt.
- Halten Sie die Schüssel mit einer Hand und positionieren Sie mit der anderen Hand die Nadelspitze mit dem Mikromanipulator-Controller. Senken Sie die Nadelspitze in Richtung der Embryonen.
- Durchbohren Sie das Chorion und betreten Sie den einzelligen Embryo mit der Nadel, während Sie den Eingriff durch das Stereomikroskop überwachen. Stellen Sie sicher, dass die Nadel richtig platziert wird und nach dem Injizieren die richtige Position des injizierten Tropfens ist, wie in Abbildung 1C dargestellt.
- Wiederholen Sie dies für alle Embryonen: Bewegen Sie die Nadel nach oben, schieben Sie die Schale mit den Embryonen, bis der nächste Embryo zentriert ist, senken Sie die Nadel und injizieren Sie sie.
- Sobald der gesamte Satz embryonen injiziert ist, entfernen Sie die Embryonen aus der Agaroseform / Petrischale, indem Sie einige E3 spülen und sie mit einer Pasteur-Pipette aus Kunststoff in eine neue Petrischale geben. Es wird empfohlen, genügend Medien auf die Injektionsplatte zu legen, um ein Austrocknen der Embryonen während des Mikroinjektionsverfahrens zu vermeiden.
- Wiederholen Sie den Vorgang, bis die gewünschte Anzahl von Embryonen injiziert wird. Die Embryonen müssen sich in einem Zellstadium befinden, um eine maximale und homogene Ausbreitung der Kügelchen zu gewährleisten.
HINWEIS: Dieses Verfahren ist für frühe Blastula-Embryonen optimiert und muss wahrscheinlich optimiert werden, wenn verschiedene Entwicklungsstadien untersucht werden sollen. - Legen Sie die injizierten Embryonen für ca. 4 h oder bis zum gewünschten Stadium (Abbildung 1D) in einen Inkubator bei 28-31 °C, bevor Sie mit dem Protokoll für die primäre Zellkultur fortfahren.
HINWEIS: Lassen Sie die Embryonen optional über das Blastulastadium (oder den gewünschten Messzeitpunkt) hinaus entwickeln, um das Überleben zu sichern und Toxizitätsartefakte auszuschließen. In den Larvenstadien montieren Sie anästhesierte Larven mit Tricain in 0,75% Agarose und bilden die Verteilung der Mikrosphären in verschiedenen Geweben ab. Um eine Stammlösung herzustellen, mischen Sie: 400 mg Tricainpulver in 97,9 ml destilliertem Wasser, etwa 2,1 ml 1 M TRIS-Base (pH 9) und stellen Sie es auf pH 7 ein. Diese Lösung kann bei 4 °C gelagert werden. Um Tricain als Anästhetikum zu verwenden, verdünnen Sie 4,2 ml Stammlösung in 100 ml Eimedium (oder gewünschtem Medium); in diesem Fall wurde E3 verwendet. Weitere Informationen finden Sie unter reference46 .
- Sammeln Sie Zebrafischembryonen kurz nach der Befruchtung zur Mikroinjektion der Perlenmischung direkt in den Embryo im Einzelzellstadium (Zygote), bevor die erste Zellteilung erfolgt.
2. Einzelzellpräparation und Färbung
- Die Kugelstadiumsembryonen (4 hpf, Stunden nach der Befruchtung) mit einer Pasteurpipette aus Kunststoff in eine Glasschale geben. Wählen Sie die Embryonen aus, die positiv für das Signal der injizierten Perlen sind und die das fluoreszierende Protein im Falle einer mRNA-Injektion exprimieren. Einige Embryonen können eine hohe Perlenhäufung aufweisen und ausgeschlossen werden.
- Dekreionieren Sie die Embryonen manuell mit einer Pinzette. Transfer von ca. 10-15 Embryonen in 1,5 ml Reaktionsbehälter mit einer Pasteurpipette aus Glas.
HINWEIS: Wenn die Embryonen dehorioniert sind, haften sie am Kunststoff, und die Verwendung von Glaswaren ist erforderlich. Alternativ zur Glasplatte kann eine Kunststoff-Petrischale mit einer dünnen Schicht aus 1% Agarose verwendet werden. Die manuelle Dekationierung sollte der enzymatischen Pronasebehandlung vorgezogen werden, um proteolytische Schäden an Zelloberflächenproteinen und mögliche Veränderungen der mechanischen Zell- und Gewebeeigenschaften zu verhindern und längere Erholungszeiten zu verhindern47.
- Dekreionieren Sie die Embryonen manuell mit einer Pinzette. Transfer von ca. 10-15 Embryonen in 1,5 ml Reaktionsbehälter mit einer Pasteurpipette aus Glas.
- Entfernen Sie die E3-Medien und geben Sie 500 μL vorgewärmtes CO2-unabhängiges Gewebekulturmedium (DMEM-F12; mit L-Glutamin und 15 mM HEPES, ohne Natriumbicarbonat und Phenolrot, ergänzt mit 10 Einheiten Penicillin und 10 mg/L Streptomycin).
HINWEIS: Verwenden Sie keine CO2-abhängigen Medien, es sei denn, es wird ein Mikroskop-Inkubator verwendet. Die Verwendung von z.B. RPMI unter karbonatgepufferten Bedingungen verursacht Veränderungen des pH-Wertes des Mediums und kann das Überleben der Zellen beeinträchtigen. Ein weiterer wichtiger Aspekt ist die Vermeidung von Kulturmedien, die Serum enthalten. Serum kann Lysophosphatidsäure (LPA) enthalten, einen starken Aktivator des Rho/ROCK-Signalwegs, der in der Lage ist, die zelluläre Kontraktilität und Motilität in Stammzellen des Vorläufers zu kontrollieren6. Die Osmolarität des Mediums sollte bei 300 mOsm gehalten werden, um osmotische Herausforderungen zu vermeiden, die die Kernmorphologie oder -mechanik beeinträchtigen könnten12. - Dissoziieren Sie Zellen manuell, indem Sie die Röhre vorsichtig schütteln. Stellen Sie sicher, dass der Inhalt der Röhre trüb wird und keine großen Brocken für das Auge sichtbar sind. Vermeiden Sie die Bildung von Blasen, um die Beschädigung und den Verlust von Zellen zu minimieren.
- Zentrifuge bei 200 x g für 3 min. Das Pellet muss deutlich sichtbar sein.
- Entfernen Sie den Überstand und führen Sie einen der unten beschriebenen Schritte aus.
- Wenn keine Färbung erforderlich ist, fügen Sie 500 μL DMEM hinzu. Vorsichtig resuspendieren Sie mit einer 200 μL Pipette, indem Sie einen Flüssigkeitsstrahl auf das Pellet richten. Üben Sie keine übermäßige Scherkraft auf die Zellen aus. Schaumbildung weist auf eine Schädigung der Zellen hin.
- Für die Markierung des Zellkerns mit DNA-Farbstoffen wie Hoechst mischen Sie 0,5 μL DNA-Hoechst (Stamm 2 mg/ml) in 1.000 μL DMEM, um 1 μg/ml Endkonzentration zu erhalten. 500 μL dieser Färbelösung in die Zellen geben und vorsichtig resuspendieren. 7 min im Dunkeln inkubieren.
- Um die Zellen mit einem fluoreszierenden chemischen Calciumindikator Calbryte-520 zu färben, fügen Sie Calbryte-520 zu einer Konzentration von 5 μM in DMEM hinzu. 20 min im Dunkeln inkubieren.
HINWEIS: Die in den Schritten 2.5.2 und 2.5.3 angegebenen Protokolle wurden für diese spezifischen Produkte optimiert. Andere Färbungen können mit den vom Hersteller angegebenen Protokollen durchgeführt werden.
- Zentrifugieren Sie erneut mit den gleichen Einstellungen wie in Schritt 2.4; Entfernen Sie den Überstand und resuspenieren Sie die Zellen vorsichtig (um die Bildung von Clustern zu vermeiden) in 50 μL DMEM für Proben in Suspension oder 20 μL DMEM für Zellen im Eingeschlossenen.
3. Herstellung von optischen Fangkammern mit Polydimethylsiloxan (PDMS)-Abstand
HINWEIS: Optische Kraftmessungen auf der Grundlage der Lichtimpulserkennung erfordern die Erfassung des gesamten Lichts, das aus den optischen Fallen austritt40. Für die Robustheit des invarianten Kalibrierfaktors α (pN/V) muss die Lichtverteilung an der hinteren Brennebene (BFP) des optischen Kraftsensors eine genaue Übereinstimmung mit dem Photonenimpuls aufweisen. Dieser bestimmt den Abstand von der Oberfläche der Sammellinse zur Fangebene auf ca. 2 mm, was der maximalen Höhe der optischen Fangkammern entspricht.
- PDMS Spin-Beschichtung von #1.5 Glasbodenschalen.
HINWEIS: Das folgende Rezept wird für ca. 40 Gerichte zur Verfügung gestellt. Die resultierende Mikrokammer wird unterschiedliche Höhen haben, je nachdem, ob Experimente an suspendierten oder eingeschlossenen Zellen durchgeführt werden sollen (Abbildung 1D).- Mischen Sie 9 mL des Basispolymers PDMS und 1 mL PDMS-Härter in einem 50 mL konischen Rohr. Mischen Sie die beiden Produkte aktiv, um eine ordnungsgemäße Verteilung des Härters zu gewährleisten.
- Entgasen Sie die Mischung, um Blasen mit einer Vakuumpumpe zu vermeiden. Führen Sie das konische Rohr in eine Vakuumflasche ein und evakuieren Sie die Kammer. Warten Sie, bis keine Blasen mehr in der Mischung vorhanden sind.
HINWEIS: Öffnen Sie das Vakuum langsam, um Schaumbildung und Verschütten des PDMS aus dem Falkenrohr zu verhindern. - Legen Sie die Glasbodenschale auf das Spin-Coater-Futter (Abbildung 2A). Seien Sie vorsichtig, um nicht zu kratzen, Fingerabdrücke zu nehmen oder das Gericht schmutzig zu machen. Schützen Sie die Spin-Coater-Box mit Aluminiumfolie vor PDMS-Leckagen.
- Für OT-Kammern für Experimente an Zellen in Suspension etwa 250 μL PDMS-Mischung in der Mitte der bodenigen Schale hinzufügen und bei 750 U / min für 1 min drehen. Die Höhe der PDMS-Schicht wird ca. 50 μm betragen48.
- Für OT-Kammern für Experimente an begrenzten Zellen fügen Sie einen kleinen Tropfen PDMS (ca. 50 μL) hinzu und drehen Sie ihn mit 4.000 U / min für 5 min. Die Höhe der PDMS-Schicht beträgt ca. 10 μm. Ein detailliertes Protokoll zum Abrufen unterschiedlicher PDMS-Dicken finden Sie unter Referenz48.
- Härten Sie die PDMS-beschichteten Glasbodenschalen bei 70 °C für 1 h aus.
- Schneiden Sie mit einem Skalpell ein 1 x 1 cm großes Quadrat auf die PDMS-Schicht und ziehen Sie es mit einer Pinzette ab (Abbildung 2C). Im Falle von eingeschlossenen Zellen pdMS-Ablagerungen mit Isopropanol waschen.
- Kammerbeschichtung für Experimente mit leicht befestigten Zellen in Suspension
- Fügen Sie 100 μL Concanavalin A (ConA) bei 0,5 mg / ml hinzu, um die gesamte Oberfläche des quadratischen Hohlraums zu bedecken, und lassen Sie es 30 minuten lang inkubieren.
HINWEIS: ConA ist ein Lektin, das an Zelloberflächenzucker bindet und einzelne Zellen auf die Deckglasoberfläche koppelt. - Entfernen Sie den ConA-Tropfen und spülen Sie die Oberfläche vorsichtig mit DMEM-Medium ab, ohne die mit ConA behandelte Oberfläche zu zerkratzen.
- 30 μL der zuvor vorbereiteten Probe (Schritt 2.6) in den Brunnen geben und vorsichtig resuspendieren, um Zellcluster zu entfernen.
- Schließen Sie den Hohlraum, indem Sie vorsichtig ein 22 x 22 mm # 1.5-Deckglas auf die PDMS-Felgen legen (vermeiden Sie es, es abrupt fallen zu lassen, verwenden Sie nach Möglichkeit eine Pinzette, Abbildung 2B, C).
HINWEIS: Jede Deckglasdicke würde für die obere Glasabdeckung funktionieren (die Sammellinse hat einen Arbeitsabstand von 2 mm).
- Fügen Sie 100 μL Concanavalin A (ConA) bei 0,5 mg / ml hinzu, um die gesamte Oberfläche des quadratischen Hohlraums zu bedecken, und lassen Sie es 30 minuten lang inkubieren.
- Kammervorbereitung für Experimente mit Zellen im Confinement
- Geben Sie einen 10-μL-Tropfen lösungshaltiger Zellen (Schritt 2.6) in den quadratischen Hohlraum (Abbildung 2B).
- Die Probe wird sehr vorsichtig mit einem 22 x 22 mm großen Deckglas versehen, so dass sich der Tropfen im gesamten Bereich ausbreitet und keine Blasen beobachtet werden. Auch hier ist es praktisch, eine Pinzette zu verwenden, wie in Abbildung 2C gezeigt, um zu verhindern, dass das Deckglas abrupt herunterfällt.
4. Alternative Optionen für OT-Kammerabstand
HINWEIS: Diese Schritte können befolgt werden, wenn keine Mikrofabrik oder kein Spin coater verfügbar ist.
- Kammervorbereitung für Experimente mit Zellen in Suspension
HINWEIS: Falls kein Spin Coater verfügbar ist, kann ein Abstandhalter mit normalem, doppelseitigem Klebeband (ca. 100 μm Höhe) hergestellt werden.- Schneiden Sie ein Stück doppelseitiges Klebeband mit einem ca. 10 cm x 10 cm großen quadratischen Loch in der Mitte (gleiche Abmessungen wie in PDMS, Abbildung 2B).
- Entfernen Sie eine der Schutzschichten des Klebebandes, indem Sie es abziehen, und legen Sie die unbedeckte Seite des Klebebands in die Mitte einer Glasbodenschale Nr. 1,5 H. Drücken Sie vorsichtig, um die gesamte Oberfläche auf dem Glas haften zu lassen, während Luftblasen vermieden werden, und entfernen Sie dann die verbleibende Schutzschicht des Bandes, indem Sie es abziehen.
- Folgen Sie den Anweisungen in Schritt 3.2.
- Kammervorbereitung für Experimente mit Zellen im Confinement
HINWEIS: Um Zellen präzise einzuschließen, können monodisperse Mikropartikel mit einem bekannten Durchmesser als Abstandshalter zwischen den beiden Deckgläsern verwendet werden.- 10 μm Polystyrolperlen zu suspendierten Zellen in einer Konzentration von 104 Kügelchen/μL geben.
- Geben Sie einen 10 μL Tropfen Lösung, der Zellen und Perlen enthält, auf ein 22 x 60 mm großes Deckglas.
- Die Probe wird sehr vorsichtig mit einem weiteren 22 x 60 mm großen Deckglas versehen, so dass sich der Tropfen in der gesamten Fläche ausbreitet und keine Blasen beobachtet werden. Um das obere Deckglas vorsichtig zu positionieren (vermeiden Sie, dass es abrupt herunterfällt), ist es bequem, eine Pinzette zu verwenden.
- Da die Probe austrocknen kann, empfiehlt es sich, die Vorbereitung zügig durchzuführen.
5. Einrichten der optischen Falle für intrazelluläre Messungen
HINWEIS: Die folgenden Schritte sind für eine kommerzielle optische Pinzettenplattform optimiert, die aus einem optischen Mikromanipulationsmodul auf der Basis einer akusto-optischen Ablenkung (AOD) und einem optischen Kraftsensor auf der Grundlage einer direkten Erkennung von Lichtimpulsänderungen besteht (Abbildung 2, Referenz12,40,49). Details und optische Komponenten des Aufbaus finden Sie in Abbildung 2F. Um die kraftinduzierte Verformung während der optischen Pinzettenmanipulationen zu beobachten, wird ein Nipkow-Spinnscheiben-Konfokalmikroskop für die zweifarbige Fluoreszenzbildgebung in den linken Anschluss des inversen Mikroskops eingekoppelt. Ohne den Mangel an Allgemeingültigkeit kann dieses Protokoll mit jedem dynamischen OTs-System angewendet werden, das mit direkten Kraftmessungen auf der Grundlage der Lichtimpulserkennung ausgestattet ist. Detaillierte Schritt-für-Schritt-Verfahren stehen zur Verfügung, um selbstgebaute optische Gradientenfallen für In-vivo-Anwendungen zu konstruieren50. Diejenigen, die auf AOD-Modulation basieren, zeichnen sich durch eventuelle Experimente mit mehreren Fallen und schnellen Messungen aus51,52. In der Literatur existieren mehrere Protokolle zur Konstruktion eines lichtimpulsbasierten Instruments36,39,40,53, und jede andere Bildgebungsmodalität (differentieller Interferenzkontrast, Weitfeldfluoreszenz usw.) kann verwendet werden.
- Optische Pinzette Start-up
- Um die Ausgangsleistungsstabilität zu optimieren, schalten Sie den Laser mindestens 30 Minuten vor dem Experiment mit einer beträchtlich hohen Leistung (z. B. 3 W) ein.
- Schalten Sie das Elektronikmodul der optischen Mikromanipulations- und Kraftmesseinheiten ein.
HINWEIS: Wenden Sie alle Laserschutzmaßnahmen an und verwenden Sie nur Geräte, die vom institutionellen Vorstand genehmigt wurden. Verwenden Sie niemals die Okulare des optischen Mikroskops, wenn der Laser eingeschaltet ist. Verwenden Sie immer eine zugelassene IR-Schutzbrille (OD7 im Bereich von 950-1080 nm), blockieren Sie das IR-Laserlicht mit dem Verschluss im Epifluoreszenzport 2 und führen Sie die optische Trapping-Software erst aus, wenn die Ausrichtung des optischen Kraftsensors nach Schritt 5.3 abgeschlossen ist. Verwenden Sie im Allgemeinen keine stark reflektierende Probe, da die Rückreflexion den Laser beschädigen könnte. - Steuern Sie die Fallenleistung mit dem rotierenden HWP (Abbildung 2F) am Eingang des optischen Mikromanipulationsmoduls.
HINWEIS: Das kommerzielle optische Mikromanipulationsmodul, das in diesem Protokoll verwendet wird, enthält diese Funktion bereits. Integrieren Sie dieses Tool für selbstgebaute optische Fangsysteme zur Leistungsregelung, damit höhere und stabilere Laserleistungen verwendet werden können.
- Verwenden Sie eine leere Mikrokammer für die Kalibrierung
- Schneiden Sie ein 1 x 1 cm großes Quadrat auf ein doppelseitiges Klebeband und befestigen Sie es auf einem 1 mm dicken Objektträger.
- Fügen Sie Wasser in das Quadrat hinzu und schließen Sie es von oben mit einem #1.5-Deckglas (22 x 22 mm). Die Zugabe eines etwas höheren Wasservolumens, z. B. 30-40 μL, wird empfohlen, um Blasen in der abgedeckten Kammer zu vermeiden. Wischen Sie die Kalibrierkammer vorsichtig ab, falls Wasser aus ihr austritt.
- Ausrichtung des optischen Kraftsensors
- Geben Sie einen Wassertropfen auf das 60x/1.2 Wassertauchobjektiv. Stellen Sie die Kalibrierkammer auf die Bühne, wobei das Deckglas Nr. 1.5 dem Objektiv zugewandt ist. Konzentrieren Sie sich auf die untere Oberfläche, wo sich die Zellproben schließlich befinden werden.
- Fügen Sie einen Tropfen Tauchöl auf den oberen Glasträger hinzu, der die Probe bedeckt (Abbildung 2D). Senken Sie die Sammellinse der Kraftsensoreinheit vorsichtig ab, bis sie den Öltröpfchen berührt.
HINWEIS: Der Tröpfchen muss groß genug sein, damit er die gesamte Linse bedeckt, die das aus den Fallen austretende Laserlicht sammelt. Normalerweise reichen 200 μL aus, um die gesamte Oberfläche abzudecken und einen stabilen Tauchkontakt zu gewährleisten. Seien Sie konservativ und vermeiden Sie eine Überfüllung, da sie in die Probe eindringen könnte. - Sehen Sie sich gemäß dem Protokoll des Herstellers für die Ausrichtung des optischen Kraftsensors das Bild der Probenebene auf der Hilfskamera an, die zur Positionierung der OTs verwendet wird (AUX, Abbildung 2F). Senken Sie den optischen Kraftsensor sehr vorsichtig ab, bis der Feldstopp (FS, Abbildung 2F-G) auf der Probenebene konjugiert erscheint. Dies gewährleistet ordnungsgemäße direkte Kraftmessungen aus der probeninvarianten Detektion von Lichtimpulsänderungen40.
HINWEIS: Schließen Sie den FS so weit, dass sein Bild kleiner als das Sichtfeld (FOV) und damit sichtbar wird. Seien Sie besonders vorsichtig und drücken Sie die Sammellinse des optischen Kraftsensors nicht gegen die Probe. Die vertikale Position des optischen Kraftsensors kann alternativ aus der Analyse der Einschlusslichtverteilung am BFP für Lichtkegel mit definierter numerischer Apertur (NA) bestimmt werden. - Stellen Sie sicher, dass sich keine Luftblasen im Öltröpfchen befinden. diese können sich direkt auf die Kraftmessungen auswirken. Um nach Luftblasen zu suchen, setzen Sie die Bertrand-Linse ein (BL, Abbildung 2G) und beobachten Sie den Bildweg durch das Okular. Wenn Schmutz oder Luftblasen sichtbar sind oder mehr Öl benötigt wird (Abbildung S1A), reinigen Sie die Linse und die Kammer mit staubfreiem Linsengewebe und wiederholen Sie den Vorgang in den Schritten 5.3.2 und 5.3.3. Ein ungehinderter optischer Pfad ist in Abbildung S1B dargestellt.
- Mit den seitlichen Schrauben, die auf der Halterung des optischen Kraftsensors angebracht sind, zentrieren Sie das FS in das Sichtfeld. Öffnen Sie aus Gründen der Genauigkeit das FS so, dass es fast das auf der Hilfskamera sichtbare Sichtfeld ausfüllt (AUX, Abbildung 2F).
6. Optische Pinzettenoptimierung
HINWEIS: Die direkte Kraftmessung beruht ausschließlich auf der Änderung des Lichtimpulses, der sich aus der auf das eingeschlossene Partikel ausgeübten Kraft ergibt, und daher muss im Gegensatz zu indirekten Methoden die Fallensteifigkeit nicht vor jedem Experiment kalibriert werden. Die gerätespezifische Umrechnung von Durchbiegungs-/Kraftfaktor (α; pN/V, Referenz41) wird vom Hersteller kalibriert und ist somit versuchsinvariant. Da der Laserspot jedoch über eine Fläche von 70 μm x 70 μm manipuliert wird, sind die Schritte 6.2-6.5 entscheidend, um eine optimale Überfangung und Leistungsstabilität zu gewährleisten. Die folgenden Schritte werden in der Herstellersoftware mitgeliefert, so dass die OTs halbautomatisch über den Arbeitsbereich optimiert werden.
- Starten Sie die OTs-Software und die Erfassungssoftware für Kamera-AUX.
- Subtrahieren Sie die anfängliche Spannungsbasislinie, indem Sie im Untermenü Systemkalibrierung der optischen Pinzette-Treibersoftware auf den Schritt 1: Elektronik-Offset klicken.
- Um eine Trap-Leistungsabflachung im ot-Arbeitsbereich durchzuführen, stellen Sie die Trap-Leistung auf die Hälfte ihres Maximums ein, indem Sie das HWP entsprechend drehen. Ändern Sie die Fallenleistung nicht durch Ändern der Laserleistung, sondern mit dem rotierenden HWP (Abbildung 2F). Klicken Sie auf Schritt 2: Einschalten , um die automatisierte Routine für die Leistungsreduzierung von Trap zu starten.
HINWEIS: Dies ist ein kritischer Schritt, um die Variation der Fallenleistung im Arbeitsbereich des Betriebssystems auszugleichen (Abbildung S1D). Eine erfolgreiche Routine reduziert die Variation der Fallenleistung auf 2% im Arbeitsbereich des Betriebssystems und konvergiert nach 2 Minuten. - Um eine Kalibrierung der Trap-Position durchzuführen, entfernen Sie den IR-Filter, sodass das Licht des Lasers auf der Kamera sichtbar ist. Finden Sie den IR-Spot, indem Sie die Bildebene auf die Unterseite der Mikrokammer fokussieren. Erhalten Sie den kleinstmöglichen IR-Spot, indem Sie die Bildebene (Objektivposition) und den Histogrammkontrast in der AUX-Erfassungssoftware der Kamera einstellen. Reduzieren Sie bei Bedarf die Leistung der optischen Falle, indem Sie das HWP drehen (Abbildung 2F). Klicken Sie auf Schritt 3: Position, um die automatische Routine- oder Trap-Positionierungskalibrierung zu starten.
HINWEIS: Diese Routine ermöglicht die genaue Übereinstimmung der Positionskoordinaten des OT in Kamera-AUX mit den AOD-Lenkwinkeln. Eine erfolgreiche Routine generiert das Angle-to-Position-Mapping in wenigen Sekunden. - Anfängliche Momentumkompensation
HINWEIS: Die Bewegung der optischen Falle über die Probe verursacht Variationen in der Licht-Impuls-Verteilung am BFP (Abbildung S1E, F). Dies führt zu kraftunabhängigen Signaländerungen in Bezug auf die Laserposition über dem Arbeitsbereich, obwohl die Fallenleistung wie in Schritt 6.3 abgeflacht wurde. Die Folge ist eine Variation der Kraftbasislinie aufgrund der Position (unabhängig von einer tatsächlichen Kraft, die auf die optisch eingeschlossene Perle wirkt), die vor jedem Experiment korrigiert werden muss.- Legen Sie die Trap-Leistung fest, die in den Experimenten verwendet wird, indem Sie das HWP drehen (Abbildung 2F).
- Klicken Sie im Untermenü Extras auf die Option Globaler Offset. Dadurch wird der Offset-Cancel-Assistent der software für optische Pinzetten geöffnet, der die anfängliche Impulsbasis korrigiert.
- Klicken Sie auf Offset | Kompensieren Sie , um das Anfangsimpuls der Positionsvariante zu korrigieren.
HINWEIS: Wenn sich in den laufenden Wochen keine Änderung auf den optischen Pfad auswirkt, bleiben die Karten für die Leistungsreduzierung der Falle (Schritt 6.3) und die Position (Schritt 6.4) unveränderlich. Wir empfehlen daher, immer die gleiche Kombination von optischen Elementen (dichroitische Spiegel, Filter usw.) zu verwenden, die den Laserfallenpfad beeinflussen können, oder eine neue Trap-Leistungsreduzierungsroutine durchzuführen. In Bezug auf die anfängliche Impulskompensation (Schritt 6.5) bietet der Hersteller der OTs-Plattform eine On-the-Fly-Kalibrierung an, die für jede neue Fangleistung und experimentelle Sitzung geändert werden muss. Die Schritte 6.3 und 6.4 müssen auf dem in Schritt 5.2 beschriebenen leeren Kalibrierobjektträger ausgeführt werden. In einer Probe, die Zellen oder andere Objekte enthält, sollte Schritt 6.5 frei von Objekten durchgeführt werden, die die Lichtstreuung im Arbeitsbereich des Betriebssystems verändern können.
- Optional können Sie eine Mikrosphäre einfangen und die Falle mit einer bekannten Geschwindigkeit bewegen, während Sie das Kraftsignal aufzeichnen. Stellen Sie beispielsweise die Falle so ein, dass sie eine dreieckige Oszillation ausführt: Das aufgezeichnete Kraftsignal ist ein quadratisches Signal.
HINWEIS: Der Kraftwert sollte linear mit der Geschwindigkeit ansteigen, entsprechend der Schleppkraft, die auf die Perle wirkt. Dieser Test dient als Positivkontrolle dafür, dass Kraftmessungen korrekt durchgeführt werden38. Alternativ kann der optische Kraftsensor verwendet werden, um die optische Fangsteifigkeit κ [pN/μm] und den Positionskalibrierungsfaktor β [μm/V] aus der Leistungsspektralanalyse zu erhalten35. Bei korrekter Ausrichtung beträgt der vom Hersteller angegebene invariante Kalibrierfaktor α = κ·β [pN/V].- Initiieren Sie eine Echtzeit-Kraftmessung, indem Sie in der Herstellersoftware im Untermenü Maßnahmen auf Plot 1 klicken. Dies liefert eine Ablesung der aktuellen optischen Fangkraft und -leistung.
- Öffnen Sie den Dialog Oszillationsparameter aus dem Untermenü Extras . Legen Sie in den Auswahlringen Form und Typ eine Wellenform mit dreieckigem Raum fest. Stellen Sie als Beispiel eine Amplitude von 10 μm und eine Frequenz von 3 Hz ein. Dies führt zu einer viskosen Kraft von etwa 1 pN auf eine Mikroperle mit einem Durchmesser von 1 μm38.
- Klicken Sie im AUX-Fenster der Kamera mit der rechten Maustaste auf die Mikroperle und wählen Sie Oszillation starten. Die Kraftmessung wird zu einem quadratischen Kraftsignal mit Plateaus bei ±1 pN.
- Klicken Sie mit der rechten Maustaste auf die Mikroperle und wählen Sie Stop Oscillating.
7. Spinnscheiben-Konfokalmikroskopie
- Schalten Sie das konfokale Mikroskop- und Zubehörgerät mit rotierender Scheibe, die integrierten Laser-Engines und die Erfassungskameras ein.
- Starten Sie die Imaging-Software.
- Stellen Sie Bildgebungskanäle für die Hoechst-Färbung des Zellkerns und GFP für die Zellplasmamembran ein.
- Aktivieren Sie die 405 nm und 488 nm Anregungslaserlinien.
- Fügen Sie ein dichroitisches Multiband hinzu, um die Anregung an die Probe zu reflektieren, damit das emittierte Licht zu den Kameras gelangen kann.
- Teilen Sie die Fluoreszenzemission mit einem 500 nm langen, dichroitischen Spiegel.
- Verwenden Sie die Emissionsfilter DAPI/BFP (~445 nm) und GFP (~521 nm) vor den beiden Erfassungskameras. Siehe Abbildung 2F,G.
- Stellen Sie die Belichtungszeit für jeden Kanal auf 100 ms ein.
- Stellen Sie die Laseremission so ein, dass sie eine Leistung von 5 mW in der Probenebene erhält. Um die Leistung zu messen, verwenden Sie einen handelsüblichen Leistungsmesser.
- Legen Sie das Imaging-Protokoll fest. Um ein spektrales Durchbluten vom Hoechst-Kanal in den GFP-Kanal zu vermeiden, müssen die beiden Farbstoffe nacheinander abgebildet werden.
HINWEIS: Wenn eine Hardwaresynchronisation zwischen den AODs der optischen Falle und der Kameraerfassung besteht, stellen Sie sicher, dass die Triggerpolarität korrekt eingestellt ist. Wenden Sie sich im Zweifelsfall an Ihren Facility Manager oder Mikroskophersteller.
8. Durchführung der Kerneinrückungsexperimente
HINWEIS: Schalten Sie die optischen Fallen immer aus - sowohl mit der Software als auch mit dem Schließen des Verschlusses am Epifluoreszenzanschluss 2 -, wenn Sie das Kraftsensormodul anheben und die Probe wechseln. Wenn nicht, könnten schwere Schäden an optischen Elementen und dem Experimentator auftreten. Seien Sie vorsichtig mit dem seitlichen Abstand zwischen Linsenhalter und unterem Schalenrand, wenn Sie nach Zellen suchen, um zu vermeiden, dass die Linse in die Tisch-/Kulturschale stößt (Abbildung 2).
- Legen Sie die Probe in das Mikroskop und befolgen Sie Schritt 5.3 dieses Protokolls.
- Setzen Sie mit dem rotierenden HWP (Abbildung 2F) die Fallenleistung auf 200 mW als Ausgangswert, wenn die Steifigkeit des untersuchten Kerns oder der untersuchten intrazellulären Struktur nicht bekannt ist. Übersetzen Sie den Arbeitsbereich des OTs (mit dem Mikroskoptisch) an einen ort ohne Zellen, um die anfängliche Impulsbasislinie durch Schritt 6.5 zu kompensieren.
HINWEIS: Abhängig von der Steifigkeit der subzellulären Struktur sollte der Fallenleistungswert auf niedrigere oder höhere Werte eingestellt werden, um eine ähnliche Eindringtiefe zu erhalten. - Suchen Sie mit dem Software-Controller für Mikroskoptische durch Transmissionshellfeldmikroskopie nach einer Zelle mit einer oder zwei Perlen (Abbildung 3A).
- Definieren Sie eine Trap-Trajektorie.
- Öffnen Sie das Dialogfenster Leitkurve (Trajectory ) im Untermenü Werkzeuge (Tools ) und wählen Sie im Auswahlring Für Leitkurventyp (Trajectory Type) die Option Verschiebung (Displacement ).
- Schreiben Sie in das Numerische Blatt die Verschiebung und die Zeit jedes nachfolgenden Leitkurvenschritts. Hier sind zwei Beispiele.
- Programmieren Sie für ein Spannungsrelaxationsexperiment trapezförmige Lasten, wie in Abbildung 3B dargestellt. In Tabelle S1 wurden zwei trapezförmige Vertiefungen mit einem Verfahrweg von 5 μm aufgebracht; Geschwindigkeit von 5 μm/s; Wartezeit vor dem Rückzug: 10 s.
- Für ein sich wiederholendes Eindringexperiment mit konstanter Geschwindigkeit, um eine dreieckige Routine ohne Verweilzeit auf dem Kern zu erhalten, stellen Sie die Trajektorienamplitude, z. B. 5 μm, und die Zeit für den Schritt ein, z. B. 2 s für eine Geschwindigkeit von 2,5 μm/s. In Tabelle S2 wird dies achtmal bei gleicher Geschwindigkeit angewendet.
HINWEIS: Diese Werte müssen für jeden Zelltyp und jedes Experiment bestimmt werden, aber die folgenden Parameter einer trapezförmigen Routine erfassen die wichtigsten Dynamiken in dem hier vorgestellten Experiment. Die Wartezeit sollte ausreichen, damit der Kern nach dem Einrücken seine vollständige Spannungsentspannung zeigt
- Einfangen einer Mikrosphäre
- Stellen Sie die Bildebene mit dem Mikroskoptisch-Software-Controller leicht über der Perle ein.
- Aktivieren Sie Traps mit der OTs-Software und klicken Sie auf die Perle im AUX-Imaging-Fenster der Kamera (kalibriert nach Schritt 6.4). Ein erfolgreicher Einschluss der Perle durch die optische Falle reduziert die Bewegung der Perle stark.
- Klicken und ziehen Sie die Perle über das Zytoplasma und platzieren Sie sie in einem Abstand von ~ 2 μm von der Kernhülle (Abbildung 3A). Stellen Sie sicher, dass die Flugbahn so eingestellt ist, dass der Perleneinzug senkrecht zur Kernmembran verläuft.
- Wenn dies für Positionsmessungen der Perle relativ zur Falle erforderlich ist, scannen Sie optional die Falle über die Perle, um die Überfangsteifigkeit zu bestimmen, k [pN/μm]54, wodurch Δxbead = -F/k (siehe Diskussion). Das in diesem Protokoll verwendete optische Mikromanipulationsmodul verfügt über eine integrierte Routine für diesen Zweck.
- Öffnen Sie im Untermenü Extras den Dialog Partikelscan.
- Wählen Sie die Trap, die Sie scannen möchten, und Hochfrequenz als Scanmethode aus. Wählen Sie die Richtung (x oder y) der Einzugsleitkurve für die Perlenabtastmessung aus.
- Es erscheint ein Fenster mit der Messung der Fangsteifigkeit. Ziehen Sie im Diagramm die beiden Cursor, um den linearen Überfüllungsbereich auszuwählen, der F = -kx entspricht. Die lineare Anpassung an den ausgewählten Datenteil wird automatisch aktualisiert.
HINWEIS: Stellen Sie die Anfangsposition der Perle weit von der Zellmembran entfernt (~ 5 μm) ein, da Licht-Impuls-Ablenkungen an der Mittel-Zell-Grenzfläche die Angemessenheit von Kraftmessungen beeinflussen. Wenn sich der Kern zu nahe an der Zellmembran befindet, versuchen Sie, den Kern von der gegenüberliegenden Stelle einzudrücken. Verwerfen Sie die Zelle, wenn dies nicht möglich ist.
- Starten Sie die Bildaufnahme, indem Sie in der Imaging-Software auf die Aufnahmeschaltfläche klicken.
- Starten Sie das Speichern von Trap-Position und Force-Messdaten, indem Sie auf Data | Speichern Sie im Echtzeit-Force-Reading-Fenster (geöffnet wie in Schritt 6.6.1).
HINWEIS: Die optische Falle ist mit einem Triggereingang ausgestattet, der an den Timing-Ausgang der Kamera angeschlossen werden kann. So werden Bild- und Kraftdaten hardwaresynchronisiert und die Elektronik ist in der Lage, die Trap-Zyklen mit der Anzahl der Frames der Bilder während der Aufnahme abzubilden. - Starten Sie die zuvor geladene Leitkurve, indem Sie mit der rechten Maustaste auf die Perle klicken und Leitkurve starten auswählen.
- Warten Sie, bis die Flugbahn abgeschlossen ist und sich das System stabilisiert hat.
- Stoppen Sie das Speichern von Überfallkraftmessdaten. Ein Dialogfeld zum Speichern von Daten wird angezeigt.
HINWEIS: Um die Datenspeicherung zu optimieren, können Daten durch Auswahl des dezimierenden Parameters in diesem Dialogfeld (10, 100 oder 1000) dezimiert werden. - Stoppen Sie die Bildaufnahme und zeichnen Sie die Ergebnisse in der Nachbearbeitungssoftware der Wahl des Benutzers.
- Wenn die Mikrosphäre während der Routine verloren geht und der Kern nicht eingerückt werden kann (Abbildung S2), verwerfen Sie die Messung und erhöhen Sie die Leistung. Beachten Sie, dass Schritt 6.5 wiederholt werden muss. In unseren Händen werden mindestens 95% der Routinen erfolgreich abgeschlossen, ohne die Perle aus der Falle zu verlieren.
Ergebnisse
Mikroinjektion von Fangperlen:
Mikrosphären, die in den einzelligen Zebrafisch-Embryo injiziert wurden, verteilten sich während der Morphogenese über die gesamte Tierkappe. Für eine klarere Visualisierung wiederholten wir das Injektionsprotokoll mit rot fluoreszierenden Mikrokügelchen und nahmen volumetrische Bilder mit unserem konfokalen Mikroskop in verschiedenen Entwicklungsstadien auf. In Abbildung 4A-D werden injizierte Kügelchen im Zytoplasma von Vorläuferstammzellen in vivo bei 5 hfp visualisiert. Später erschienen Mikrosphären, die sich bei 24 hpf über den gesamten Embryo ausbreiteten (Abbildung 4E). Embryonen in beiden Stadien entwickelten sich normal und die Überlebensraten waren vergleichbar mit nicht injizierten oder pseudo-injizierten Kontrollembryonen (siehe Abbildung S3). Dies steht im Einklang mit anderen Studien, die über ein unbeirrtes Überleben von per perleninjizierten Zebrafischen bis zu 5 Tage nach der Befruchtung berichten55.
Unser Spinnscheiben-Konfokalmikroskop ist kompatibel mit der Mehrkanal-Fluoreszenzmikrokopie. In Abbildung 5A zeigen wir isolierte Stammzellen mit einer oder zwei Kügelchen im Zytoplasma. Mehrere fluoreszierende Markierungen können verwendet werden, um verschiedene Aspekte der Zelle zu untersuchen (Abbildung 5B). Die Kernmorphologie kann mit einem Hoechst-Farbstoff oder unter Verwendung einer H2A::mCherry-mRNA-Expression verfolgt werden, während die innere Kernmembran mit Lap2b-eGFP12 analysiert werden kann. Die Dynamik des Actomyosin-Kortex sowie der intrazelluläre Kalziumspiegel können mit einer my12.1::eGFP transgenen Linie56 bzw. Calbryte-520 Inkubation beobachtet werden. Das hier beschriebene Protokoll zielt darauf ab, die Zellkernmechanik von immobilisierten Wildtypzellen auf adhäsiven Substraten (später als Suspension bezeichnet) und im mechanischen Einschluss zu vergleichen. Isolierte Stammzellen, die in Mikrokammern von 10 μm Höhe eingeschlossen waren, zeigten eine teilweise Entfaltung der inneren Kernmembran (INM) und eine anschließende Zunahme der Actomyosin-Kontraktilität12. In Abbildung 5C sind eingeschlossene Zellen mit einer oder zwei Kügelchen im Zytoplasma dargestellt. Ein erfolgreicher Einschluss wird über abgeflachte, expandierte Zellen mit einem breiteren Querschnitt des Zellkerns sichtbar. Die Kernmembran wird in geschlossenen Zellen weiter entfaltet und sollte im Vergleich zu Zellen in Suspension geglättet erscheinen (Abbildung 5C).
Kraft-Zeit- und Kraft-Verformungsanalyse
Die Analyse der erhaltenen Ergebnisse hängt stark von der untersuchten Probe und der Fragestellung ab und kann daher hier nicht verallgemeinert werden. Eine gängige Methode zur Analyse der Eindringungsmessung besteht beispielsweise darin, einen Elastizitätsmodul zu extrahieren, indem ein modifiziertes Hertz-Modell an die Krafteinrückungsdaten angepasst wird57. Die Annahme für eine solche Behandlung muss jedoch sorgfältig beurteilt werden und ist möglicherweise nicht immer richtig begründet (z. B. ist die untersuchte Struktur isotrop, homogen, wobei die lineare Elastizität und die Vertiefungen kleiner als der Perlenradius sind). Wir betrachten hier daher nur modellunabhängige Messungen, die es erlauben, das mechanische Verhalten der untersuchten Struktur zwischen verschiedenen experimentellen Szenarien zu vergleichen.
Als Ausgangspunkt liefert die Messung der Steigung der Kraft-Verschiebungs-Kurve in einer bestimmten Eindringtiefe ein Maß für eine modellunabhängige strukturelle Steifigkeit58 des Kerns. Dieser Wert kann dann aus mehreren Proben gesammelt und zwischen unterschiedlichen experimentellen Einstellungen und Probenstörungen verglichen werden.
Eindringungsmessung
In den folgenden Zeilen konzentrieren wir uns auf die mechanische Reaktion des Zellkerns während der Zellverformung im Einschluss. Experimente in Schritt 8 dieses Protokolls führen typischerweise zu Kraftspitzen von bis zu 200 pN für Eindringtiefen von ca. 2-3 μm. Diese Werte können jedoch je nach Zelltyp und experimentellen Bedingungen sehr unterschiedlich sein, wobei weichere Kerne zu einer geringeren Kraft für einen bestimmten Einzug führen. Es wird dabei benötigt, um die Kernverformung zusammen mit der Kraft genau zu messen, um eine genaue mechanische Charakterisierung des Zellkerns zu ermöglichen. In diesem Abschnitt erhalten wir die Zellkernsteifigkeit aus repräsentativen Krafteindringmessungen.
In Abbildung 6 zeigen wir die Verformungen der distalen und proximalen Seite eines Kerns in einer suspendierten und eingeschlossenen Zelle. Ein reichhaltiges mechanisches Verhalten kann beobachtet werden. In einer typischen suspendierten Zelle auf einem Adhäsionssubstrat war der Kern durch die Perle stark eingerückt, aber auch bei sich wiederholenden Schiebeereignissen leicht verschoben. Wir maßen den Perleneinzug auf den Kern, indem wir die Kymographen analysierten, die aus der Fluoreszenzbildgebung von Hoechst-gefärbten Zellkernen gewonnen wurden. Kymographen wurden mit dem Multi Kymograph-Plugin von Fiji entlang der Einrückungsrichtung (Abbildung 6A,B) einfach berechnet und zur weiteren Verarbeitung in Matlab (Version 2021, Mathworks) importiert. Eine Schrittfunktion wurde an das rohe Intensitätsprofil angepasst, mit dem Ziel, die begrenzenden Kanten des Kerns entlang der Flugbahn der Eindringroutine zu verfolgen. Wie man sehen kann, enthält es genaue Informationen über die Kernformänderung (Abbildung 6 und Abbildung S2). Wir haben die folgende Doppel-Sigmoid-Kurve als analytische Version einer Schrittfunktion verwendet:
 (Gleichung 1)
(Gleichung 1)
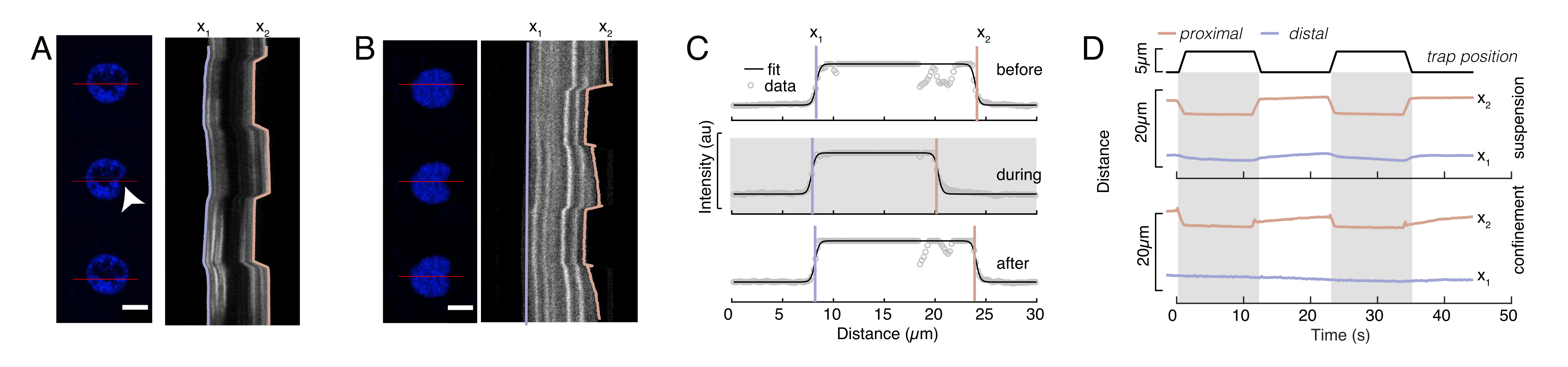
Dabei bezeichnen x1 und x2 die distalen und proximalen Ränder des Kerns, während A und B die maximalen und Hintergrundgrauwerte des blauen Kanals (Hoechst-Farbstoff) des Bildes sind (Abbildung 6B). Die Kantenbreite wurde berücksichtigt (e0 = 0,25 mm). Während die eingerückte, proximale Kernkante (x2) der Flugbahn folgte, die von der optischen Fallenroutine nach dem Mikrosphären-Kern-Kontakt angewendet wurde, zeigt die gegenüberliegende, distale Kante (x1) eine Relaxationsdynamik, wie sie für ein viskoelastisches Material wie das Zytoplasma erwartet wird (Abbildung 6D). Im Gegensatz dazu zeigen Kerne in Zellen, die in 10 μm hohen Mikrokammern eingeschlossen sind, kein solches Translokationsverhalten des Kerns beim Einrücken innerhalb der Zelle (Abbildung 6B,D). Ebenfalls in Abbildung 6D gezeigt, bleiben die hinteren Kanten der Kerne unverändert durch die von der proximalen Seite drückende Perle, höchstwahrscheinlich aufgrund stärkerer Kräfte, die sich aus der Zellkontraktilität und der Reibung ergeben, die gegen die Eindringkraft wirken. Um die korrekte Verformungstiefe zu erhalten, wurde die Verschiebung x1 vom eingerückten Maß x2 subtrahiert: Δx = x2 - x1 (siehe auch Abbildung 6D).
Datenanalyse erzwingen
Die Kraft, die die Kernverformung verursacht, wurde aus der Änderung des Lichtimpulses gemessen, die von der optisch eingeschlossenen Mikroperle ausging (Abbildung 7A). Die Kraft beim Anlegen trapezförmiger Trajektorien (Schritt 8.4.3, Abbildung 7B) nahm zunächst linear zu, bis sich die Falle nicht mehr bewegte, entspannte sich dann aber auf einen stationären Wert. Dieses Verhalten zeigte ein viskoelastisches Material mit Verlust- und Speichermodulen an. Direkt nach dem Einrückungsereignis erreichte die Kraft einen Spitzenwert, Fp, gefolgt von einer Spannungsrelaxation (Abbildung 7C):
 (Gleichung 2)
(Gleichung 2)
wobei F0 die gespeicherte Kraft für die elastische Komponente und f(t) eine dimensionslose Relaxationsfunktion ist. Wir haben dieses Verhalten auf drei Arten analysiert:
1. Betrachten eines linearen Standardkörpers mit einer exponentiellen Spannungsrelaxation, d.h. f(t) = e-t/τ, schematisch dargestellt in Abbildung 7C-Einschub.
2. Verwendung eines allgemeinen, doppelt exponentiellen Zerfalls:
F(t) = A + B1e-t/τ1 + B2e-t/τ2.
3. Verwendung eines Potenzgesetzes, gefolgt von einem exponentiellen Zerfall59:
f(t) = t-pe-t/τ, eingepasst in Abbildung 7C.
Während die Anpassung für Modell 1 einfach durchgeführt werden kann, empfehlen wir, die anfänglichen Vermutungen für (τ1, τ2) und (p, τ) für die Modelle 2 bzw. 3 zu schätzen. Dies kann jeweils durch Anpassen von Linien an die Daten in logarithmisch-versuslineen (Abbildung 7D, links) und logarithmisch-versus-logarithmischen (Abbildung 7D, rechts) Skalen erfolgen. Tabelle S3 fasst die Ergebnisse für das in Abbildung 7 analysierte Beispiel zusammen. Im folgenden Abschnitt betrachten wir die Kombination eines Potenzgesetzes und eines Exponentialgesetzes zur Charakterisierung der Zellkernmechanik.
Kraftverschiebungsverhältnis
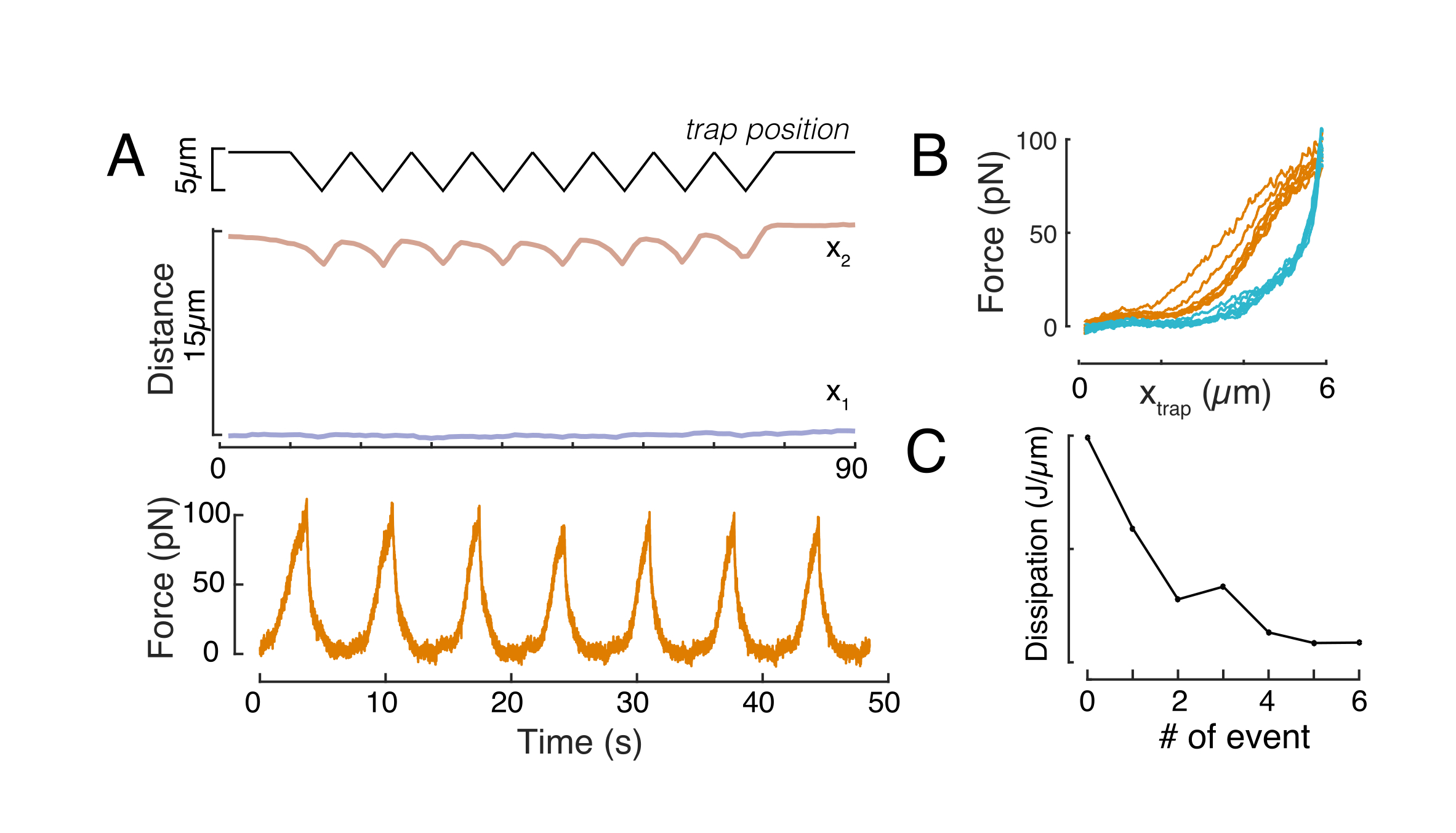
Ebenso kann der beschriebene Versuchsaufbau verwendet werden, um das Kraft-Verschiebungs-Verhältnis mehrerer Eindringereignisse zu erhalten. Durch die Durchführung von Dreiecksroutinen (Schritt 8.4.4, Abbildung 8A) ist es möglich, die Kraft auf die Verformung zu beziehen und eine Kraft-Einrückungskurve darzustellen. Ein beispielhaftes Ergebnis ist in Abbildung 8B dargestellt, in dem eine flache Grundlinie die Neigung sanft änderte, sobald die Perle mit dem Kern in Kontakt kam. Die Identifizierung des wahren Kontaktpunkts in den verrauschten Daten ist eine Herausforderung, und es muss darauf geachtet werden, ob der Kontaktbereich für elastische Modelle geeignet ist60. In diesem speziellen Experiment konnte auch gesehen werden, dass die nachfolgenden Vertiefungen zu Kurven mit tieferen Kontaktpunkten führen, die auf eine zu langsame Erholung der Kernform nach perlenretraktion und eine Änderung des hysteretischen Zyklus hinweisen, der durch die viskoelastischen Materialeigenschaften des Kerns definiert ist (Abbildung 8C). Daher sollte sich der Forscher bewusst sein, wenn dies geschieht, und dies in die analytische Pipeline integrieren oder die Anzahl der nachfolgenden Messungen so einschränken, dass dieser Effekt die Messung nicht verändert.
Kernmechanik in Zellen in Suspension und unter 10 μm Einschluss
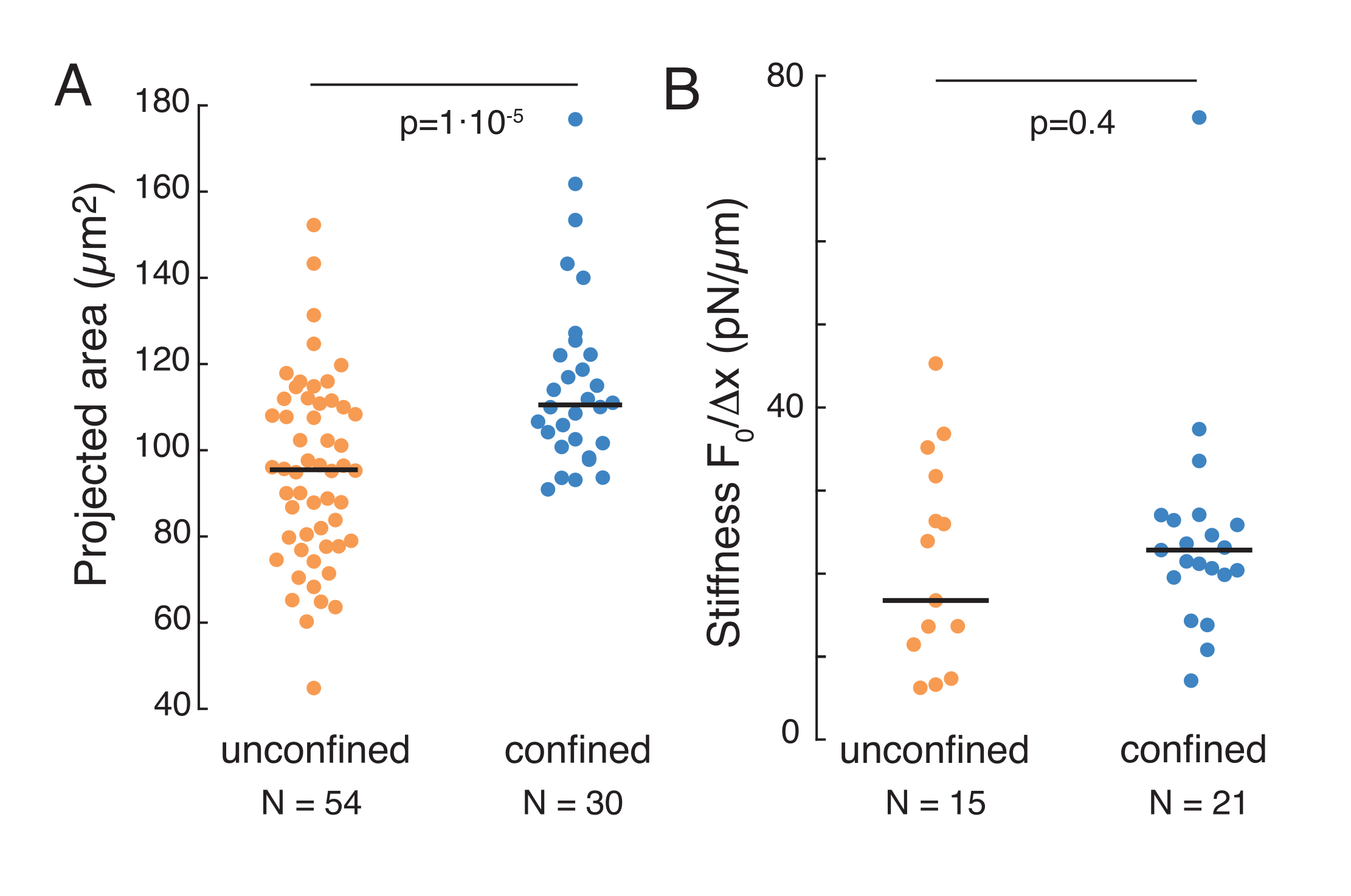
Der oben genannte Ansatz wurde verwendet, um die Dynamik der Kernspannungsrelaxation in suspendierten Zellen auf Adhäsionssubstraten und eingeschlossenen Zellen zu analysieren. Unsere Ergebnisse zeigen, dass der Einschluss zu einer Ausdehnung der projizierten Fläche führt (Abbildung 9A), aber zu einer unbedeutenden Änderung der Kernsteifigkeit (Abbildung 9B). Wir haben eine ähnliche Relaxation mit τ = 6,08 ± 1,1 s (nicht geschlossen) und τ = 4,00 ± 0,6 s (Einschluss) gemessen, was auf eine schnelle viskoelastische Dissipation hinweist, gefolgt von einem gespeicherten Kraftwert, der dem Elastizitätsmodul des Kerns entspricht. Um experimentelle Variationen zu berücksichtigen, die durch unterschiedliche Anfangsbedingungen in den Eindringroutinen erzeugt werden können, wurden die gemessenen gespeicherten Kräfte auf die Eindringtiefe normalisiert, wie  . Dieser Parameter berücksichtigt die Kernsteifigkeit und beschreibt die Kraft oder die Spannung, die für eine bestimmte Vertiefung erforderlich ist. Wir erhielten eine ähnliche Steifigkeit unter Eingeschlossenheit und in nicht eingeschlossenen Zellen: = 20,1 ± 12,6 pN/μm bzw . = 24,6 ± 13,6 pN/μm (mittlere ± Standardabweichung).
. Dieser Parameter berücksichtigt die Kernsteifigkeit und beschreibt die Kraft oder die Spannung, die für eine bestimmte Vertiefung erforderlich ist. Wir erhielten eine ähnliche Steifigkeit unter Eingeschlossenheit und in nicht eingeschlossenen Zellen: = 20,1 ± 12,6 pN/μm bzw . = 24,6 ± 13,6 pN/μm (mittlere ± Standardabweichung).

Abbildung 1: Mikroinjektion von Zebrafischembryonen im einzelligen Stadium (Zygote). (A) Injektionsplatte: Für die Injektion wird eine dreieckige Injektionsplatte verwendet. Die Platte besteht aus 1% hochreiner Agarose in E3 (Eimedium). Die Ober- und Seitenansicht wird auf der rechten Seite angezeigt. (B) Positionierung des Embryos: Richten Sie die Embryonen vorsichtig mit einem Pinsel aus und orientieren Sie sich so, dass die Einzelle deutlich sichtbar und mit der Nadel leicht zugänglich ist. Wir schlagen vor, die Embryonen mit der Zelle auf der gegenüberliegenden Seite der Nadel auszurichten, wie in der Skizze gezeigt. (C) Injektionsverfahren in den Embryo im Einzelzellstadium: Durchstechen des Chorions, das den Embryo und die Einzelzelle umgibt, mit der Nadel. Stellen Sie sicher, dass sich die Nadelspitze in der Zelle befindet, und lassen Sie den Druck zur Injektion los. (D) Die Embryonen werden bei 28-31 °C inkubiert, bis sie sich bis zum Blastula-Stadium (Kugel) (4 hpf) entwickeln. Führen Sie das Zellisolationsprotokoll und die Zellfärbung (Schritt 2) durch und bereiten Sie die optische Fangkammer mit isolierten Zellen in Suspension und/oder Einschluss in Kombination mit der entsprechenden Substratoberflächenbeschichtung vor (Schritt 3). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Vorbereitung der optischen Pinzettenapparatur. (A) Schichtung von PDMS mit definierter Höhe auf Glasbodenschalen. Der PDMS-Abfall verteilt sich aufgrund der Fliehkraft gleichmäßig. (B) Vorbereitung der Probenkammer aus der PDMS-Schicht. 1: Schneiden Sie ein Quadrat mit einem Skalpell, 2: beschichten Sie den inneren Brunnen mit Concanavalin A (ConA), Waschen und Samenzellen; 3: Decken Sie mit einem Glasschieber oder Abdeckschein ab, um den Brunnen zu versiegeln. (C) Bild des quadratischen Schneidens mit einem Skalpell und Entfernen des PDMS-Brunnens mit einer Pinzette. (D) Montage der Sammellinse des optischen Kraftsensors über der Fangkammer. Ein Tropfen Tauchöl dient als Immersionsmedium zwischen der Auffanglinse und der oberen Glasabdeckung. Schematisch nicht zu skalieren. Seien Sie vorsichtig, wenn Sie die Auffanglinse absenken, um die Glasabdeckung der Probenschale nicht zu berühren. (E) Bild der Krafterfassungseinheit in Kontakt mit der Probe. (F) Schematische Darstellung des Versuchsaufbaus. Das optische Mikromanipulationsmodul verwendet einen Dauerstrichlaserstrahl (5W, λ = 1064 nm) mit Leistungsregelung durch eine Halbwellenplatte (HWP) und einen Polarisationsstrahlteiler (BS). Nach der Modulation mit einem Paar AODs wird es an den oberen Epifluoreszenzport eines inversen Mikroskops gekoppelt. Der Laserstrahl wird dann von einem dichroitischen Kurzpassspiegel (IR-DM) mit 950 nm reflektiert, wodurch die Transmission der Fluoreszenzanregung und -emission ermöglicht wird. Der Fanglaser wird in die hintere Epifluoreszenzöffnung des Mikroskops (oberer Revolver) geführt. Die OTs werden auf der Brennebene eines Wasserimmersionsobjektivs (60x, NA = 1,2) erzeugt. Der optische Kraftsensor wird vom Mikroskoprevolver belichtet und fängt das aus den OTs austretende Laserlicht mit einer Ölimmersionslinse mit hohem NA ein. Gleichzeitig ermöglicht der Kraftsensor eine Hellfeldausleuchtung. Die konfokale Einheit mit rotierender Scheibe ist mit dem linken Port gekoppelt. Es ist mit zwei integrierten Laser-Engines (ILE) ausgestattet, die sieben Fluoreszenz-Anregungslaser und zwei hinterleuchtete sCMOS-Kameras steuern, was eine parallele Dual-Fluorophor-Bildgebung ermöglicht Abb: TI, Transilluminator; FS, Feldstopp; AOD, akustoptischer Deflektor; HWP, Halbwellenplatte; CAM, Kamera (G) Foto der optischen Fangvorrichtung. Roter Kreis kennzeichnet die Bertrand-Linse, die manuell in den Strahlengang geschaltet werden kann. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

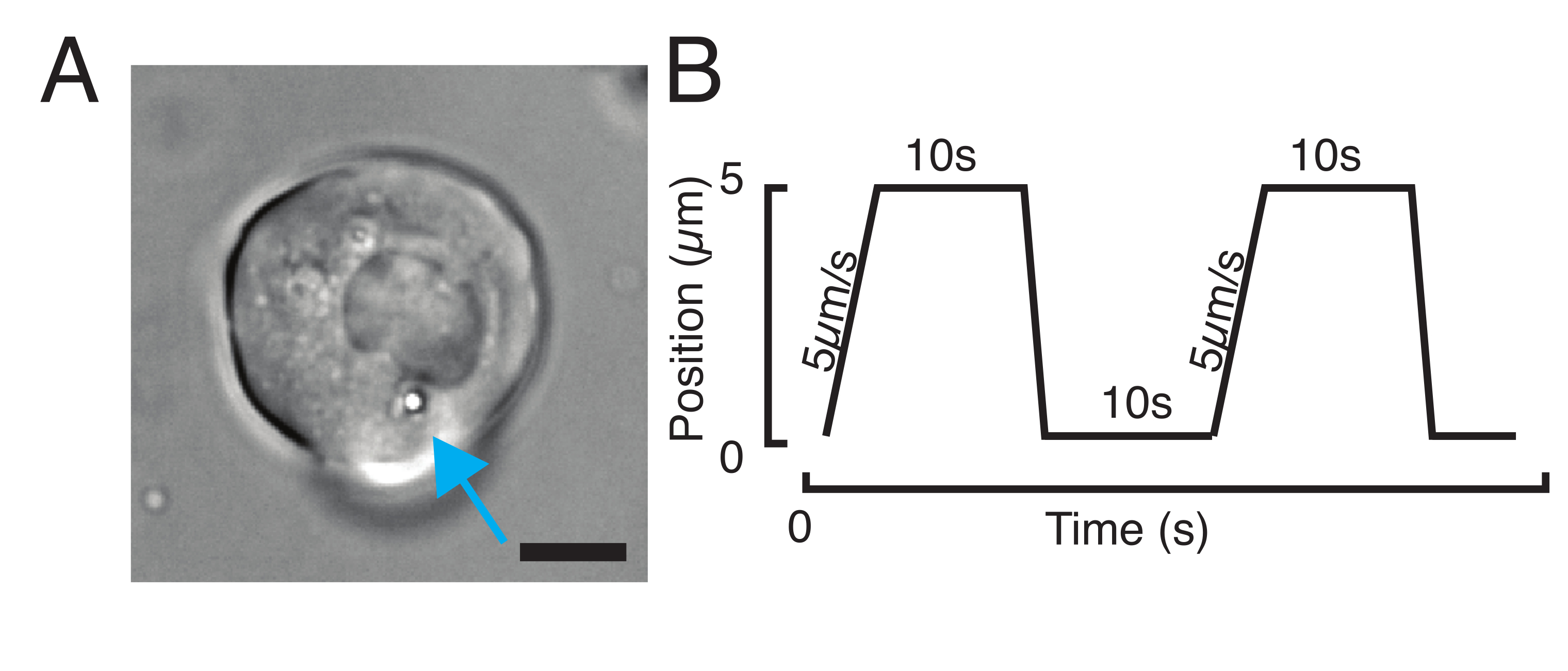
Abbildung 3: Auswahl der richtigen Proben und Parameter. (A) Repräsentatives Bild einer isolierten Zebrafisch-Vorläuferstammzelle mit einer einzigen Mikrosphäre, die nahe genug am Kern positioniert ist, um das Eindringexperiment durchzuführen. Maßstabsbalken = 10 μm. (B) Beispielhafte Trap-Trajektorie; Eindringtiefe 5 μm; Eindringgeschwindigkeit = 5 μm/s; Entspannungszeit 10 s. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}

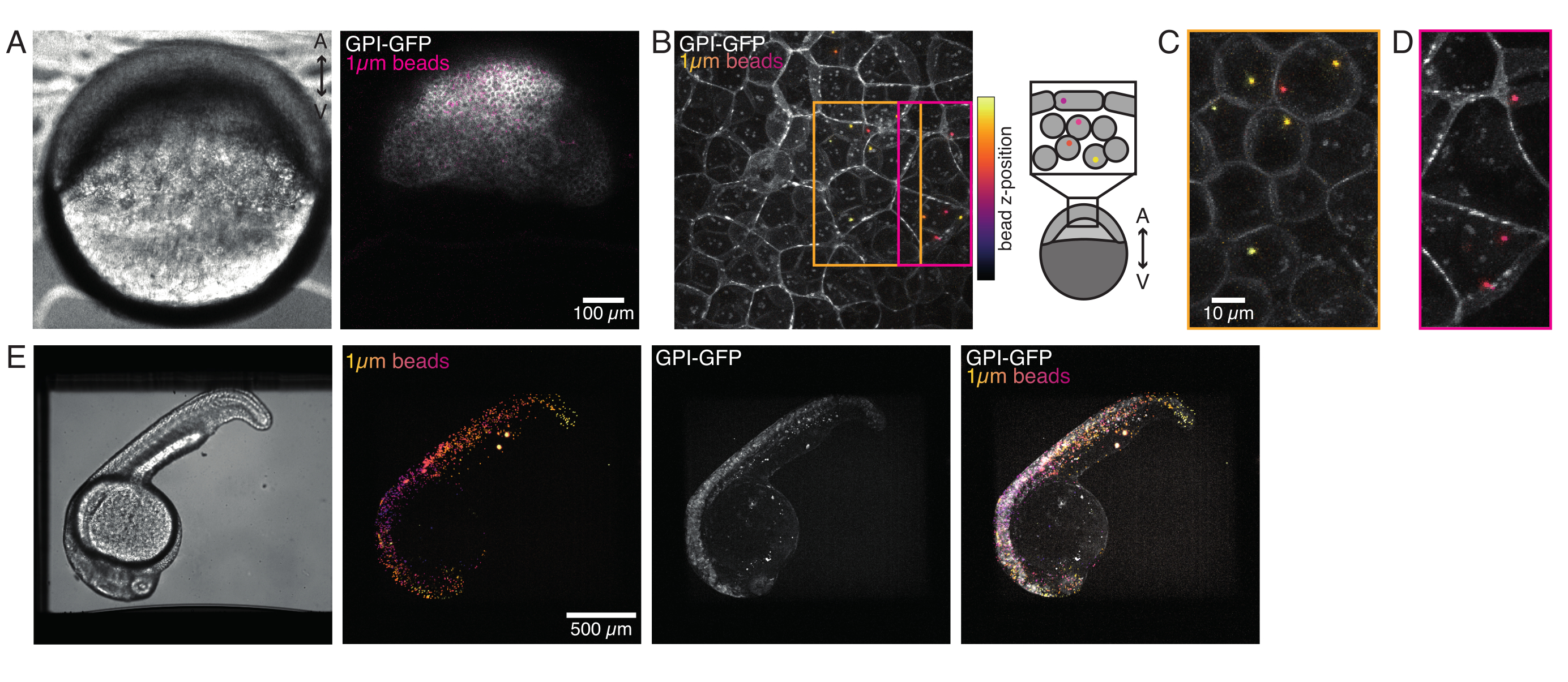
Abbildung 4: Mikroperlenlokalisation in Zebrafischembryonen während der Entwicklung. 0,5 nL von 1 μm roten Fluoreszenzperlen werden zusammen mit GPI-GFP mRNA (100 pg/Embryo, Plasmamembran) in WT-Embryonen injiziert, um Perlenlokalisationen sichtbar zu machen. (A-D) Verteilung der Mikrosphäre 5 h nach der Injektion in einen Embryo, der in 0,75% Agarose montiert ist. (A) Hellfeld- und Fluoreszenzbild. Die Kügelchen sind homogen über das embryonale Gewebe verteilt, wie in einer konfokalen Mikroaufnahme zu sehen ist. (B) Maximale Projektion des konfokalen Fluoreszenz-Z-Stacks. Die Perlen sind farblich von lila bis gelb entsprechend ihrer Z-Position im Bildstapel codiert. Lila/Magenta entspricht den meisten äußeren Kügelchen/Zellen (EVL; epitheliale Umhüllungsschicht; oder Vorläuferstammzellen, die sich in der Nähe der EVL-Oberfläche befinden), Gelb entspricht inneren Kügelchen (Vorläufer-Tiefenzellen), wie in der Skizze rechts gezeigt. (C) Schnitt und maximale Projektion eines Unterstapels von (B), der dem Bereich in der orangefarbenen Box entspricht: Ein großer Teil der tiefen Zellen enthält 1-2 Perlen. (D) Schnitt und maximale Projektion eines Unterstapels von (B), der der Magenta-Box entspricht: Einige EVL-Zellen enthalten 1-2 Perlen. (E) Hellfeldbild und maximale Projektion eines Z-Stapels eines 24 hpf Embryos, der in 0,75% Agarose montiert und mit Tricain betäubt wurde. Die Embryonen wurden 15 Minuten lang mit Tricain vorinkubiert. Von links nach rechts: Mikrosphären (1 μm Durchmesser), GPI-GFP und Bildüberlappung. Die Perlen verteilten sich über den gesamten Körper des Embryos. Scalebar-Dimension, die in jedem Bedienfeld angegeben ist. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 5: Isolierte Zebrafisch-Stammzellen mit unterschiedlicher Markierung. (A) Transmissionslichtmikroskopische Aufnahme von Suspensionszellen mit 1 (oben) oder 2 (unten) injizierten Kügelchen. Cyanfarbene Pfeile zeigen auf Perlen. (B) Fluoreszierende konfokale Bilder von Suspensionszellen mit unterschiedlichen Färbungen. Oben links: Lap2b-eGFP (innere Kernmembran, 80 pg/Embryo) und H2A-mCherry. Oben rechts: GPI-GFP (Plasmamembran, 100 pg/Embryo) und DNA-Hoechst (gefärbt wie in Abschnitt 2 beschrieben). Unten links: MyI12.1-eGFP (transgene Linie) und DNA-Hoechst. Unten rechts: Calbryte488 und DNA-Hoechst (gefärbt wie in Abschnitt 2 beschrieben). (C) Transmissionslichtmikroskopische Aufnahme von eingeschlossenen Zellen mit 1 (oben) oder 2 (unten) injizierten Kügelchen. Cyanfarbene Pfeile zeigen auf Perlen. Maßstabsbalken = 10 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 6: Abschätzung der Kernverformung aus rotierenden Scheibenfilmen. (A,B) Zeitraffer eines Eindringexperiments des Kerns in (A) einer suspendierten Zelle und (B) einer eingeschlossenen Zelle. Maßstabsleiste 10 μm. Repräsentative Schnappschüsse eines Hoechst-markierten Kerns werden 5 s vor, während und 5 s nach dem Einrücken mit einer optisch gefangenen Mikrosphäre (weiße Pfeilspitze) gezeigt. Kymographen entlang des Einrückungssegments (rote Linie, rechtes Feld). x1 und x2 sind die distalen und proximalen (nahe der Perle) Grenzen des Kerns während des Eindringexperiments, das aus der Anpassung des Intensitätsprofils an Gleichung 1 extrahiert wurde. (C) Intensitätsprofile entlang des Eindringsegments für drei verschiedene Rahmen (vor, während und nach dem Eindrücken) und angepasst an Gleichung 1, um die distalen, x1 und proximalen x2 Positionen der Kernkanten zu beurteilen. (D) Repräsentative Trajektorien von x1(t) in Blau und x2(t) in Bernstein während eines Eindringversuchs von suspendierten und eingeschlossenen Zellen (10 μm). Schattierte Bereiche zeigen die Vertiefung an, der Abstand zwischen x1 und x2 den Kerndurchmesser. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 7: Kraftsignalverarbeitung. (A) Schematische Darstellung einer optisch eingeschlossenen Mikrokugel, die den Zellkern beim Einrücken verformt. Kernmembran und optische Kräfte werden durch die schwarzen Pfeile angezeigt. Die Änderung des Strahlimpulses wird durch den grünen Pfeil Schmollmund angezeigt. (B) Trap-Trajektorie (oben) und Kraft (unten), die von der optisch gefangenen Mikrosphäre während eines wiederholten Kerneindringexperiments erfahren werden. (C) Zerfall der Kraftrelaxation nach dem Kraftpeak in der maximalen Eindringtiefe. Inset zeigt ein Schema eines linearen Standardkörpers, dessen Dynamik sich hier den phänomenologischen Beobachtungen annähert. (D) Links: Logarithmus der normalisierten Kraft versus Zeit. Die schattierten Bereiche geben den Datenteil an, der für den doppelten exponentiellen Zerfall (rote Linien) verwendet wird. Rechts: Logarithmus der normalisierten Kraft gegenüber dem Logarithmus der Zeit. Der schattierte Bereich gibt den Datenteil an, der zur Anpassung an das Potenzgesetz verwendet wird. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 8: Krafteindringroutine mit dreieckigen Fallenverschiebungen. (A) Repräsentative Trajektorie von x1(t) in blau und x2(t) in Bernstein während eines dreieckigen Eindringexperiments, das an einer Zelle in 10 μm Einschlusshöhe durchgeführt wurde. Oben: Trap-Position. Mitte: Kernformanalyse. Der Abstand zwischen x1 und x2 gibt den Kerndurchmesser an. Unten: Kraftsignal. (B) Force vs Trap-Position für acht aufeinanderfolgende Einrückungen. (C) Entwicklung der Dissipation des Kerns für jedes nachfolgende Eindringungsereignis, abgeleitet aus der Hysterese zwischen dem Annäherungs- und dem Rückzugsteil der f-d-Kurve. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 9. Kerneigenschaften von Zellen in Suspension (Klebefläche) und Einschluss aus trapezförmigen Routinen. (A) Projizierte Fläche des Zellkerns von Zellen in Suspension und unter 10 μm Einschluss. Schwarzer Balken stellt den Median dar. (B) Kernsteifigkeit von Zellen in Suspension und unter Eingeschlossenheit. Schwarzer Balken stellt den Median dar. P-Werte, die aus dem Kruskal-Wallis-Test mit MatLab abgeleitet wurden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Ergänzende Tabelle 1: Trapezförmige Trajektorie, definiert durch die optische Pinzetten-Software. Erste (zweite) Zeile ist der x (y) Abstand, den die Trap linear verschiebt. In der dritten Zeile wird die Dauer eines bestimmten Schritts in Sekunden festgelegt. Diese Trajektorie besteht aus sieben Punkten und entspricht dem Trapez, das in Abbildung 7B zweimal gegen den Kern belastet wird. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Ergänzende Tabelle 2: Dreieckige Trajektorie, die von der optischen Pinzettensoftware definiert wird. Analog zu Tabelle 2 besteht diese Trajektorie aus 16 Punkten, was acht Eindringereignissen in einer Tiefe von 5 μm und einer Geschwindigkeit von 2,5 μm/s entspricht .
Ergänzende Tabelle 3: Anpassungsparameter für die Daten in Abbildung 7. IG: Erste Vermutung. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Ergänzende Abbildung S1: Ausrichtung des optischen Kraftsensors und Impulsbasislinienkompensation. (A) Feldstopp, aufgenommen an der Hilfskamera (AUX, Abbildung 2) durch das Bertrand-Objektiv. Im Tauchöl erscheint eine Luftblase, die durch das Okular nicht sichtbar ist. (B) Sauberer optischer Pfad. Für eine genaue Ausrichtung öffnen Sie den Feldstopp und lassen Sie ihn mit dem NA = 1,2-Lichtkegel übereinstimmen. (C) Bild der Probenebene. Das rote Quadrat zeigt den OT-Arbeitsbereich an. Maßstabsbalken: 20 μm. (D) Trap-Leistung gemessen über das FOV, entlang weißer Doppelpfeile in C. In Rot die Variation der Trap-Leistung, wenn keine Korrektur angewendet wird. In Blau wurde die Trap-Leistung über das gesamte Sichtfeld korrigiert. (E) X-Komponente der Impulsbasislinie entlang desselben Bereichs. In rot, nicht korrigierte Spur. In Blau, Spur korrigiert für Fallenleistung. In grün, Trace korrigiert für Momentum Baseline mit Global Offset Compensation in der Software des Herstellers. (F) Wie in E für die Y-Komponente. Beachten Sie, dass im Normalbetrieb die schattierten Komponenten für mechanische und Kraftmessungen verwendet werden, z. B. x-Kraftkomponente während der Bewegung entlang der x-Koordinate und die y-Kraftkomponente während der Bewegung entlang der y-Achse. Nachdem alle Korrekturen implementiert sind, wird ein RMSD-Rauschen von <0,5 pN erhalten. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Abbildung S2: Eine fehlgeschlagene Routine aufgrund schwacher Fallen. (A) Kymograph, der eine Kerneinrückung aus einer fehlgeschlagenen Routine zeigt. Nur kurze, transiente Verformungen sind durch ein Entweichen der Perle aus der Falle sichtbar. Wichtig ist, dass sich der Trapping-Laser immer noch ohne Perle bewegt, um die vordefinierte Leitkurve (grüne gepunktete Linie) zu vervollständigen. Maßstabsleiste = 10 μm. (B) Oben: Trap-Position versus Zeit. Mitte: Kantenverfolgungsergebnis der eingerückten proximalen und distalen Kernkante. Beachten Sie, dass sich die distale Kante nicht ohne die Vertiefung bewegt, wie sie üblicherweise für abgeschlossene Routinen an isolierten Zellen auf Adhäsionssubstraten beobachtet wird. Unten: Kraft versus Zeit zeigt den Verlust der Mikrosphäre, der durch eine Verringerung des thermischen Rauschens und einen plötzlichen Abfall auf Null angezeigt wird. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Abbildung S3: Überleben der injizierten Embryonen. Embryonen, denen 1 μm Perlen und 100 pg/Embryo mRNA in den im Protokoll beschriebenen Konzentrationen injiziert wurden, wurden mit nicht injizierten Embryonen verglichen und zeigen 24 h nach der Befruchtung keine signifikanten Unterschiede. Mittelwert und Standardabweichung von drei unabhängigen Experimenten mit N > 21 Embryonen pro Bedingung für jedes Experiment. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Diskussion
In diesem Protokoll beschreiben wir eine einzigartige Methode, um die mechanischen Eigenschaften des Zellkerns in den lebenden Zellen zu untersuchen. Im Gegensatz zu anderen Techniken der Kraftspektroskopie ermöglichte uns das nicht-invasive optische Trapping, den Beitrag der Zellmembran und des Zytoskeletts von der Zellkernsteifigkeit zu entkoppeln. Wichtig ist, dass die optische Mikromanipulation mit der multimodalen Mikroskopie kompatibel ist, die es dem Experimentator ermöglicht, verschiedene Prozesse zu untersuchen, die an der zellkernden Mechanobiologie beteiligt sind. Als repräsentatives Ergebnis haben wir die DNA-Hoechst-Färbung verwendet, um die Kernverformung beim Einrücken durch Kräfte in der Größenordnung von mehreren hundert picoNewton zu messen.
Mögliche Anwendungen unserer Methode über die in diesem Protokoll beschriebenen Beispiele hinaus
Die Möglichkeit, quantitative mechanische Informationen aus Messungen in lebenden Zellen ohne äußere Störungen zu extrahieren, ermöglicht eine Fülle beispielloser Möglichkeiten, die gerade erst erforscht werden. Somit kann das vorgestellte Protokoll unserer optischen Mikromanipulationsplattform mit großer Vielseitigkeit auf komplexere Experimente ausgeweitet werden. Akusto-optische Deflektoren (AOD) können mehrere optische Fallen für synchrone Kraftmessungen über verschiedene Zellstandorte hinweg erzeugen sowie für die aktive Mikrorheologie in einem weiten Frequenzbereich51,61 verwendet werden. Wie bereits erwähnt, kann die Kraftreaktion beim Einrücken die maximale Fangkraft überwinden, was zu einem Entweichen der Perle aus der optischen Falle führt. In diesem Fall kann mit dem AOD eine Kraftrückmeldung konfiguriert werden, um die optische Kraft einzuklemmen. Alles in allem können mit dieser Plattform mehrere mikrorheologische Ansätze, wie die in diesem Protokoll beschriebene Spannungsrelaxation, aber auch aktive Mikrorheologie oder Kriechkonformität experimentell gewonnen und durch neuartige Softwarepakete gründlich analysiert werden61,62,63,64,65 . Darüber hinaus ist die Anwendung von Kräften nicht auf den Zellkern beschränkt, sondern könnte prinzipiell durchgeführt werden, um verschiedene intrazelluläre Strukturen und in komplexen Geweben zu messen, wie es sich gezeigt hat, um fließende rote Blutkörperchen in intakten Blutgefäßen einzufangen66,67 oder Chloroplasten und Mitochondrien einzufangen und zu verformen68 . Die Licht-Impuls-Kalibrierung ist unabhängig von der Form und Größe des eingeschlossenen Objekts und ermöglicht somit direkte Kraftmessungen an jeder Kraftsonde mit beliebiger Form38,39. Die Verwendung von injizierten Mikrosphären ermöglichte es uns, hohe Kräfte auf den Kern mit relativ geringer Laserleistung im Vergleich zur direkten Manipulation zellulärer Strukturen anzuwenden69,70,71. Bei einer ausreichend hohen Brechungsindexdifferenz ist jedoch keine extern angelegte Kraftsonde erforderlich und intrazelluläre Organellen können direkt ohne injizierte Kügelchen manipuliert werden (unveröffentlichte Beobachtungen und Referenz70).
Mögliche Modifikationen unserer Methode zur Erweiterung der Anwendungen
Je nach Experiment können unterschiedliche Größen von Mikrokügelchen injiziert werden, aber die relativen Kontrollen müssen durchgeführt werden. Zum Beispiel, um Zellen in späteren Stadien zu untersuchen, können kleinere Perlen injiziert werden. Dadurch wird die maximale Kraft reduziert, die von der optischen Falle ausgeübt werden kann (wie in Referenz55 gezeigt). Größere Perlen können injiziert werden, um höhere Kräfte auszuüben, aber diese können die Embryonalentwicklung je nach Größe oder Stadium des Interesses beeinflussen. In Experimenten, bei denen eine Mikroperleninjektion keine Option ist, können verschiedene Organellen, die Brechungsindizes aufweisen, die Unterschiede zu denen des Zytoplasmas aufweisen, immer noch optisch manipuliert werden, was zu optischen Kräften führt, die durch Lichtimpulsänderungen messbar sind42. Wie bereits erwähnt, wurden diese Methoden von Bambardekar et al. eingesetzt, um Zell-Zell-Verbindungen im Drosophila-Embryo zu verformen70. Ebenso hat der Zellkern einen niedrigeren Brechungsindex als der umgebende Medium44, was eine perlenfreie Einrückung (unveröffentlichte Beobachtungen und Referenz72) ermöglicht, wenn auch mit einer geringeren Fangstärke. So kann der Kern nicht leicht eingeschlossen werden und entkommt der Falle.
Der spinbeschichtete PDMS-Abstandhalter wird mit einer bequemen und schnellen Methode hergestellt, ist aber für Labore ohne Zugang zu einer Mikro-/Nanofabrik oder technischen Labors möglicherweise unerreichbar. Somit kann der Abstandhalter einfach aus Laborband oder Parafilm zusammengesetzt werden (Schritt 4). Das Protokoll kann auch angepasst werden, indem mikrofluidische Kanäle hergestellt werden, die die Lieferung einzelner Zellen in vordefinierte Messtöpfe oder in eine Kammer mit einer definierten Höhe automatisieren, um den Einschlusseffekt innerhalb derselben Probe abzuschätzen. Solche mikrofluidischen Vorrichtungen müssen jedoch so ausgelegt sein, dass sie in den Raum zwischen dem Mikroskopobjektiv und der Sammellinse des optischen Kraftsensors von etwa 2 mm passen (siehe Schritt 3). Beachten Sie, dass der optische Kraftsensor in der entsprechenden Höhe positioniert werden muss, damit keine optischen Aberrationen durch Defokussierung die Photonenimpulsmessung beeinflussen.
Andere Änderungen könnten den Wechsel von biologischen Reportern beinhalten. Wir fanden heraus, dass die Hoechst-Fluoreszenz spektral in den GFP-Kanal blutet und wir daher die Kombination mit mCherry-markiertem Histon als Kernmarker für die gleichzeitige Messung in zwei Fluoreszenzkanälen bevorzugen. Alternativ kann die Kernverformung leicht mit einem auf die innere Kernmembran ausgerichteten Etikett wie Lap2b-GFP verfolgt werden (Abbildung 2).
Die Eindringung auf den Zellkern lag in der Größenordnung von 2-3 Mikrometern, die wir durch Bildanalyse der beugungsbegrenzten Spinnscheiben-Konfokalmikroskopie genau messen konnten. Bei steiferen Kernen oder kleineren Kräften ist die Vertiefung mit diesem Ansatz kaum messbar. Die absolut kraftkalibrierte optische Pinzette kann aber auch für Positionsmessungen der gefangenen Perle in situ mittels BFP-Interferometrie mit Nanometergenauigkeit kalibriert werden51. Mit diesem Ansatz können das Spannungssignal und der optische Kraftsensor durch den Parameter β [nm/V] in die Position der eingeschlossenen Sonde übersetzt werden, während der invariante Parameter α [pN/V] durch die bereits erwähnte Licht-Impuls-Kalibrierung41 Kraftwerte liefert (Details siehe unten).
Fehlerbehebung
Wir fanden heraus, dass die folgenden Herausforderungen während des Experiments auftreten konnten:
Es bildet sich keine stabile Falle und die Mikrosphäre entweicht leicht
Jeder Schmutz auf dem Mikroskopobjektiv oder ein falsch ausgerichteter Korrekturkragen kann zum Versagen einer stabilen Falle führen. Wenn keine sofortige Lösung gefunden wird, messen Sie die Punktspreizfunktion der Objektivlinse. Wenn sich die interessierende Probe tief in einem optisch dichten Gewebe befindet, kann der Laserfokus schwere optische Aberrationen aufweisen, die zu instabilem Einfangen führen (dieser Effekt ist in isolierten Zellen normalerweise vernachlässigbar, wird aber in dickeren Geweben deutlicher). Bei hoher Steifigkeit könnte die Rückstellkraft des Kerns die Fluchtkraft der Falle übersteigen, so dass die Mikrosphäre verloren geht und die Eindringroutine versagt. Anfangs wird der Kernmembranrand proximal zur optischen Falle kaum eingerückt (Abbildung S2A). In diesem Fall wird der Trapping-Laser nicht mehr durch Kraft und Brownsche Bewegung beeinflusst, was zu einem Kraftabfall auf Null und einer Abnahme des Signalrauschens führt (Abbildung S2B). In diesem Fall kann die Laserleistung erhöht werden, um eine stärkere Falle zu haben, die Amplitude der trapezförmigen Flugbahn, die die Perle in den Kern drückt, kann reduziert werden, oder die Ausgangsposition der eingeschlossenen Mikroperle kann weiter vom Kern entfernt sein.
Die Zelle bewegt sich während der Stimulation
Wenn Zellen nicht ausreichend gebunden sind, bewegt die optische Gradientenfalle die Zellen während der intrazellulären Eindringroutine, so dass die Kräfte und die zugrunde liegende Mechanik des Kerns artefaktisch sind. Um eine Verschiebung der gesamten Zelle zu verhindern, empfehlen wir, die Konzentration von Zelladhäsionsmolekülen auf der Oberfläche, z.B. ConA, zu erhöhen.
Anfängliche Momentumkompensation
Wenn eine anfängliche Impulskompensationsroutine in der OTs-Plattform nicht verfügbar ist (Schritt 6.5), muss ein künstliches, kraftunabhängiges Basissignal korrigiert werden. Dies ist als Steigung auf der Kraftkurve sichtbar, auch wenn keine Wulst eingeklemmt ist (Abbildung S1E). Um die Korrektur durchzuführen, muss die gleiche Trajektorie ohne Perle außerhalb der Zelle an genau der gleichen Position durchgeführt werden. Verschieben Sie dazu die Zelle mithilfe des Stufensteuerelements von der Trap weg. Als Referenz ändert sich der Kraftversatz um 5 pN über das Sichtfeld bei 200 mW in unserem System; Daher wird es für kurze Trajektorien vernachlässigbar. Alternativ kann ein Piezo-Scan-Tisch verwendet werden, um die Zellen auf der Probe zu bewegen, wobei die Laserposition konstant bleibt.
Kritische Schritte des vorgestellten Protokolls
Mikrosphären sollten im rechten 1-Zell-Stadium injiziert werden, um eine maximale Verteilung über den Embryo zu gewährleisten. Perlen sollten nicht fluoreszierend sein, damit kein Licht in die für die Bildgebung verwendeten Fluoreszenzkanäle gelangt. So sind aufgrund ihrer Helligkeit (Anregung: 405 nm; Emission: 445 nm) sogar typische rot fluoreszierende Kügelchen deutlich sichtbar im Blaukanal, der zur Abbildung des Zellkerns nach der Hoechst-Färbung verwendet wird. Eine stabile Befestigung der Zelle am Substrat ist entscheidend, um eine laterale Verschiebung während der Eindringroutine zu verhindern. Bewegt sich die Zelle während der Routine, werden Kräfte unterschätzt. Sollte dies häufig vorkommen, optimieren Sie das Anhangsprotokoll. Für Gewebekulturzellen führen andere Zelladhäsionsproteine wie Fibronektin, Kollagen oder Poly-L-Lysin zu einer zufriedenstellenden Bindung (unveröffentlichte Beobachtungen). Während des Einschlusses sind die Zellen einer plötzlichen und starken mechanischen Belastung ausgesetzt. Dies kann zu Schäden an den Zellen führen und häufig trifft der Experimentator auf geplatzte Zellen, wenn der Vorgang nicht sorgfältig durchgeführt wird. Wenn die Einschlusshöhe zu klein ist, leiden alle Zellen unter Kernumhüllungsbruch oder irreversiblen Schäden. Um diese zu mildern, senken Sie den oberen Deckslip langsamer ab und/ oder vergrößern Sie den Abstand zwischen den Deckblättern.
Einschränkungen der Technik und Vorschläge, um sie zu überwinden
Eine klare Einschränkung der Technik ist das Eindringen des Laserlichts in tiefe Gewebeabschnitte, was zu Aberrationen und instabilem Einfangen führt. Somit hängt eine untere Grenze der Eindringtiefe von der Klarheit der Probe, der anzuwendenden Aberrationskorrektur73 und der eingesetzten Laserleistung ab. Es sollte berücksichtigt werden, dass eine höhere Laserleistung zu einer thermischen Anregung der Probe in der Nähe der Mikrosphäre führt. Die Erwärmung der Probe durch den Laserspot mit einer Wellenlänge von 1064 nm wird jedoch minimiert, um eine plausible hitzebedingte Belastung unserer biologischen Proben zu vermeiden74.
Eine weitere Einschränkung ist die maximale Kraft, die gemessen werden kann. Obwohl die direkte Lichtimpulsdetektion Kraftmessungen weit über das lineare Ansprechregime der optischen Falle40,41 hinaus ermöglicht, liegt die maximal aufgebrachte Kraft in der Größenordnung von einigen hundert picoNewton. Begrenzt wird dies durch die Laserleistung und die Folgeschadensschwelle von weichem biologischem Material sowie die Brechungsindexdifferenzen, die normalerweise nicht größer als 0,1 oder 0,344 sind. Mehrere Methoden wurden vorgeschlagen, um die Kraftnachweisgrenze zu erhöhen, z. B. mit strukturiertem Licht75, entspiegelten Mikrokugeln76, Partikeln mit hohem Brechungsindex77 oder hochdotierten Quantenpunkten78.
OTs können für Positionsmessungen im Nanometerbereich durch BFP-Interferometrie verwendet werden, so dass die Position der Perle innerhalb der Falle Δx = β Sx ist, wobei Sx das Spannungssignal des Sensors ist und β [μm/V] on-the-fly nach verschiedenen Protokollen kalibriert werden kann35,54. Für einen optischen Kraftsensor kann nachgewiesen werden, dass sich der Spannungs-Kraft-invariante Umrechnungsfaktor α [pN/V] direkt auf β und die Fallensteifigkeit k [pN/μm] bis α = kβ 37 bezieht. Ein Beispiel ist die Anwendung der hier vorgestellten experimentellen Routinen auf sehr steife Kerne, für die Kräfte bei vernünftigen Laserleistungen (200-500 mW) nicht ausreichen, um Eindringwerte zu induzieren, die groß genug sind. In diesem Fall muss die Perle mit dem Kern in Kontakt gebracht und die Fangsteifigkeit vor der Messung kalibriert werden (Schritt 8.6). Der Einzug d des Kerns als Funktion der Kraft kann indirekt bestimmt werden als:
d = xtrap - F/k
wobei xtrap die Trap-Position ist. Anders als der invariante Lichtimpulsfaktor α [pN/V] muss der Faktor β [μm/V] vor jedem Experiment kalibriert werden, da er von vielen lokalen Variablen abhängt, die die Überfangdynamik bestimmen, wie z. B. der Partikelgröße, der Größe des optischen Trap-Spots und der relativen Brechungsindizes.
Offenlegungen
Die Autoren haben nichts preiszugeben.
Danksagungen
MK würdigt die finanzielle Unterstützung des spanischen Ministeriums für Wirtschaft und Wettbewerbsfähigkeit durch den Plan Nacional (PGC2018-097882-A-I00), FEDER (EQC2018-005048-P), Severo Ochoa Programm für Exzellenzzentren in F&E (CEX2019-000910-S; RYC-2016-21062), von der Fundació Privada Cellex, der Fundació Mir-Puig und der Generalitat de Catalunya durch das CERCA- und Forschungsprogramm (2017 SGR 1012), zusätzlich zur Finanzierung durch ERC (MechanoSystems) und HFSP (CDA00023/2018). V.R. würdigt die Unterstützung des spanischen Ministeriums für Wissenschaft und Innovation für die EMBL-Partnerschaft, das Centro de Excelencia Severo Ochoa, den Plan Nacional von MINECO (BFU2017-86296-P, PID2020-117011GB-I00) und die Generalitat de Catalunya (CERCA). V.V. erkennt die Unterstützung des ICFOstepstone PhD-Programms an, das vom Forschungs- und Innovationsprogramm Horizon 2020 der Europäischen Union im Rahmen der Marie-Skłodowska-Curie-Finanzhilfevereinbarung 665884 finanziert wird. Wir danken Arnau Farré für die kritische Lektüre des Manuskripts; Maria Marsal für die Hilfe bei der Bildgebung und Montage des 24 hpf Embryos und; Senda Jiménez-Delgado für die Unterstützung bei Zebrafisch-Mikronjektionen.
Materialien
| Name | Company | Catalog Number | Comments |
| #1.5 22 mm cover glasses | Ted Pella | 260148 | |
| #1.5 22x60 mm Coverglasses | Ted Pella | 260152 | |
| #1.5H glass bottom dishes | Willco | GWST-5040 | |
| 10-um beads | Supelco | 72986 | |
| 1-mm glass capillaries | Harvard Apparatus | 30-0020 GC100F-15 | |
| 1-um polystyrene microbeads | Sigma | 89904 | |
| 1-um red-fluorescent beads | ThermoFisher | F8816 | |
| Agar | ThermoFisher | 16500500 | |
| Aqcuisition cameras sCMOS | Andor | Sona-4BV11-UNI | |
| Auxiliary camera (Figure 3, AUX) | Blackfly, FLIR | BFS-U3-200S6M-C | |
| Calbryte 520 | AAT Bioquest | 520 AM | |
| Centrifuge | Eppendorf | 5453000011 | |
| Concanavalin A | Sigma | C5275 | |
| DMEM | Sigma | D2906 | |
| DNA-Hoechst | ThermoFisher | 33342 | |
| Double scotch tape | Biesse Adesivi | ||
| E3 | 5 mM NaCl. 0.17 mM KCl. 0.33 mM CaCl2. 0.33 mM MgSO4 | ||
| Eclipse Ti2 | Nikon | ||
| Forceps | Fine Science Tools | 11252-20 | |
| GPI-GFP | |||
| H2A-mCh | |||
| Image acquisition software | Fusion-Andor | ||
| Immersion Oil | Cargille | Type B: 16484 | |
| IR protection googles | Thorlabs | LG1 | |
| Lap2b-eGFP | |||
| Micro loader pipette | Eppendorf | GELoader | |
| Microinjector | World Precision Instruments | SYS-PV820 | |
| MicroManager 2.0 | |||
| Micromiter slide | ID5243 GXMGRAT-5 5mm/100 divisions | ||
| Mineral oil | Sigma | M3616 | |
| Motorized stage | ASI | ||
| Needle puller | Sutter instrument Co. | Model P-97 | |
| Optical tweezers platform | Impetux Optics | Sensocell | |
| OTs software (LightAce) | Impetux Optics | ||
| PDMS | Sigma | Sylgard 184 | |
| PDMS Curing agent | Sigma | Sylgard 184 | |
| Post processing software (Matlab) | Mathworks | ||
| RNAse free water | Thermofisher | AM9937 | |
| Short-pass dichroic mirror (Figure 3, IR-F) | Semrock | FF01-950/SP-25 | |
| Spin-coater | Specialty Coating Systems | Spincoat G3P-8 | |
| Spinning-disk confocal microscope | Andor | DragonFly 502 | |
| Stereomicroscope | Leica M80 | ||
| Triangular microinjection mold | Adaptive Science Tools | TU1 | |
| Universal oven | Memmert | UNB 200 | |
| Water immersion objective | Nikon | MRD07602 |
Referenzen
- Chan, C. J., Heisenberg, C. P., Hiiragi, T. Coordination of Morphogenesis and Cell-Fate Specification in Development. Current Biology. 27 (18), 1024-1035 (2017).
- Heller, E., Fuchs, E. Tissue patterning and cellular mechanics. Journal of Cell Biology. 211 (2), 219-231 (2015).
- Heisenberg, C. P., Bellaïche, Y. Forces in tissue morphogenesis and patterning. Cell. 153 (5), 948-962 (2013).
- Petridou, N. I., Spiró, Z., Heisenberg, C. P. Multiscale force sensing in development. Nature Cell Biology. 19 (6), 581-588 (2017).
- Krieg, M., et al. Tensile forces govern germ-layer organization in zebrafish. Nature Cell Biology. 10 (4), 429-436 (2008).
- Ruprecht, V., et al. Cortical contractility triggers a stochastic switch to fast amoeboid cell motility. Cell. 160 (4), 673-685 (2015).
- Shellard, A., Mayor, R. Supracellular migration - Beyond collective cell migration. Journal of Cell Science. 132 (8), (2019).
- Mongera, A., et al. A fluid-to-solid jamming transition underlies vertebrate body axis elongation. Nature. 561 (7723), 401-405 (2018).
- Atia, L., et al. Geometric constraints during epithelial jamming. Nature Physics. 14 (6), 613-620 (2018).
- Turlier, H., Maître, J. -. L. Mechanics of tissue compaction. Seminars in Cell & Developmental Biology. 47-48, 110-117 (2015).
- Ladoux, B., Mège, R. M. Mechanobiology of collective cell behaviours. Nature Reviews Molecular Cell Biology. 18 (12), 743-757 (2017).
- Venturini, V., et al. The nucleus measures shape changes for cellular proprioception to control dynamic cell behavior. Science. 370 (6514), (2020).
- Charras, G., Sahai, E. Physical influences of the extracellular environment on cell migration. Nature Reviews Molecular Cell Biology. 15 (12), 813-824 (2014).
- Kirby, T. J., Lammerding, J. Emerging views of the nucleus as a cellular mechanosensor. Nature Cell Biology. 20 (4), 373-381 (2018).
- Lee, H. P., et al. The nuclear piston activates mechanosensitive ion channels to generate cell migration paths in confining microenvironments. Science Advances. 7 (2), (2021).
- Friedl, P., Wolf, K., Lammerding, J. Nuclear mechanics during cell migration. Current Opinion in Cell Biology. 23 (1), 55-64 (2011).
- Versaevel, M., Riaz, M., Grevesse, T., Gabriele, S. Cell confinement: Putting the squeeze on the nucleus. Soft Matter. 9 (29), 6665-6676 (2013).
- Zuela-Sopilniak, N., et al. Measuring nucleus mechanics within a living multicellular organism: Physical decoupling and attenuated recovery rate are physiological protective mechanisms of the cell nucleus under high mechanical load. Molecular Biology of the Cell. 31 (17), 1943-1950 (2020).
- Kim, D. H., Wirtz, D. Cytoskeletal tension induces the polarized architecture of the nucleus. Biomaterials. 48, 161-172 (2015).
- Lomakin, A. J., et al. The nucleus acts as a ruler tailoring cell responses to spatial constraints. Science. 370 (6514), (2020).
- Hampoelz, B., et al. Microtubule-induced nuclear envelope fluctuations control chromatin dynamics in Drosophila embryos. Development. 138 (16), 3377-3386 (2011).
- Heo, S. J., et al. Differentiation alters stem cell nuclear architecture, mechanics, and mechano-sensitivity. eLife. 5, 1-21 (2016).
- Cosgrove, B. D., et al. Nuclear envelope wrinkling predicts mesenchymal progenitor cell mechano-response in 2D and 3D microenvironments. Biomaterials. 270, 120662 (2021).
- Liu, H., et al. In situ mechanical characterization of the cell nucleus by atomic force microscopy. ACS Nano. 8 (4), 3821-3828 (2014).
- Hobson, C. M., et al. Correlating nuclear morphology and external force with combined atomic force microscopy and light sheet imaging separates roles of chromatin and lamin A/C in nuclear mechanics. Molecular Biology of the Cell. 31 (16), 1788-1801 (2020).
- Pajerowski, J. D., Dahl, K. N., Zhong, F. L., Sammak, P. J., Discher, D. E. Physical plasticity of the nucleus in stem cell differentiation. Proceedings of the National Academy of Sciences of the United States of America. 104 (40), 15619-15624 (2007).
- Rowat, A. C., Lammerding, J., Ipsen, J. H. Mechanical properties of the cell nucleus and the effect of emerin deficiency. Biophysical Journal. 91 (12), 4649-4664 (2006).
- Davidson, P. M., et al. High-throughput microfluidic micropipette aspiration device to probe time-scale dependent nuclear mechanics in intact cells. Lab on a Chip. 19 (21), 3652-3663 (2019).
- Lombardi, M., Zwerger, M., Lammerding, J. Biophysical assays to probe the mechanical properties of the interphase cell nucleus: Substrate strain application and microneedle manipulation. Journal of Visualized Experiments: JoVE. (55), (2011).
- Luo, T., Mohan, K., Iglesias, P. A., Robinson, D. N. Molecular mechanisms of cellular mechanosensing. Nature Materials. 12 (11), 1064-1071 (2013).
- Dahl, K. N., Engler, A. J., Pajerowski, J. D., Discher, D. E. Power-law rheology of isolated nuclei with deformation mapping of nuclear substructures. Biophysical Journal. 89 (4), 2855-2864 (2005).
- Guilluy, C., et al. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nature Cell Biology. 16 (4), 376-381 (2014).
- Bustamante, C. J., Wang, M. D. Optical tweezers in single-molecule biophysics. Nature Reviews Methods Primers. , 1-29 (2021).
- Svoboda, K., Block, S. M. Force and velocity measured for single kinesin molecules. Cell. 77 (5), 773-784 (1994).
- Berg-Sørensen, K., Flyvbjerg, H. Power spectrum analysis for optical tweezers. Review of Scientific Instruments. 75 (3), 594-612 (2004).
- Smith, S. B., Cui, Y., Bustamante, C. Optical-trap force transducer that operates by direct measurement of light momentum. Methods in Enzymology. 361 (1994), 134-162 (2003).
- Farré, A., Montes-Usategui, M. A force detection technique for single-beam optical traps based on direct measurement of light momentum changes. Optics Express. 18 (11), 11955 (2010).
- Català, F., Marsà, F., Montes-Usategui, M., Farré, A., Martín-Badosa, E. Extending calibration-free force measurements to optically-trapped rod-shaped samples. Scientific Reports. 7, 1-10 (2017).
- Bui, A. A. M., et al. Calibration of force detection for arbitrarily shaped particles in optical tweezers. Scientific Reports. 8 (1), 1-12 (2018).
- Farré, A., Marsà, F., Montes-Usategui, M. Beyond the hookean spring model: Direct measurement of optical forces through light momentum changes. Methods in Molecular Biology. 1486, (2017).
- Farré, A., Marsà, F., Montes-Usategui, M. Optimized back-focal-plane interferometry directly measures forces of optically trapped particles. Optics Express. 20 (11), 12270 (2012).
- Jun, Y., Tripathy, S. K., Narayanareddy, B. R. J., Mattson-Hoss, M. K., Gross, S. P. Calibration of optical tweezers for in vivo force measurements: How do different approaches compare. Biophysical Journal. 107 (6), 1474-1484 (2014).
- Mas, J., Farré, A., Sancho-Parramon, J., Martín-Badosa, E., Montes-Usategui, M. Force measurements with optical tweezers inside living cells. Optical Trapping and Optical Micromanipulation XI. 9164, (2014).
- Schürmann, M., Scholze, J., Müller, P., Guck, J., Chan, C. J. Cell nuclei have lower refractive index and mass density than cytoplasm. Journal of Biophotonics. 9 (10), 1068-1076 (2016).
- Rosen, J. N., Sweeney, M. F., Mably, J. D. Microinjection of zebrafish embryos to analyze gene function. Journal of Visualized Experiments: JoVE. (25), (2009).
- Westerfield, M. . The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish. (Danio rerio), 5th Edition. , (2007).
- Schubert, R., et al. Assay for characterizing the recovery of vertebrate cells for adhesion measurements by single-cell force spectroscopy. FEBS Letters. 588 (19), 3639-3648 (2014).
- Koschwanez, J. H., Carlson, R. H., Meldrum, D. R. Thin PDMS films using long spin times or tert-butyl alcohol as a solvent. PLoS One. 4 (2), 2-6 (2009).
- Das, R., et al. Mechanical stretch inhibition sensitizes proprioceptors to compressive stresses. bioRxiv. , (2021).
- Chardès, C., Clement, R., Blanc, O., Lenne, P. F. Probing cell mechanics with bead-free optical tweezers in the drosophila embryo. Journal of Visualized Experiments: JoVE. (141), (2018).
- Staunton, J. R., Blehm, B., Devine, A., Tanner, K. In situ calibration of position detection in an optical trap for active microrheology in viscous materials. Optics Express. 25 (3), 1746 (2017).
- Bola, R., Treptow, D., Marzoa, A., Montes-Usategui, M., Martin-Badosa, E. Acousto-holographic optical tweezers. Optics Letters. 45 (10), 2938-2941 (2020).
- Thalhammer, G., Obmascher, L., Ritsch-Marte, M. Direct measurement of axial optical forces. Optics Express. 23 (5), 6112 (2015).
- Vermeulen, K. C., et al. Calibrating bead displacements in optical tweezers using acousto-optic deflectors. Review of Scientific Instruments. 77 (1), 1-6 (2006).
- Dzementsei, A., Barooji, Y. F., Ober, E. A., Oddershede, L. B. Foregut organ progenitors and their niche display distinct viscoelastic properties in vivo during early morphogenesis stages. bioRxiv. , 1-35 (2021).
- Behrndt, M., et al. Forces driving epithelial spreading in zebrafish gastrulation. Science. 338 (6104), 257-260 (2012).
- Krieg, M., et al. Atomic force microscopy-based mechanobiology. Nature Reviews Physics. , (2018).
- A-Hassan, E., et al. Relative microelastic mapping of living cells by atomic force microscopy. Biophysical Journal. 74 (3), 1564-1578 (1998).
- Khalilgharibi, N., et al. Stress relaxation in epithelial monolayers is controlled by the actomyosin cortex. Nature Physics. 15, (2019).
- Crick, S. L., Yin, F. C. Assessing micromechanical properties of cells with atomic force microscopy: importance of the contact point. Biomechanics and Modeling in Mechanobiology. 6 (3), 199-210 (2007).
- Hurst, S., Vos, B. E., Betz, T. Intracellular softening and fluidification reveals a mechanical switch of cytoskeletal material contributions during division. bioRxiv. , 425761 (2021).
- Kaplan, J. L., Bonfanti, A., Kabla, A. RHEOS.jl - A Julia package for rheology data analysis. arXiv. 4, 1-5 (2020).
- Bonfanti, A., Kaplan, J. L., Charras, G., Kabla, A. Fractional viscoelastic models for power-law materials. Soft Matter. 16 (26), 6002-6020 (2020).
- Rivas-Barbosa, R., Escobedo-Sánchez, M. A., Tassieri, M., Laurati, M. i-Rheo: determining the linear viscoelastic moduli of colloidal dispersions from step-stress measurements. Physical Chemistry Chemical Physics: PCCP. 22 (7), 3839-3848 (2020).
- Tassieri, M., et al. i-Rheo: Measuring the materials' linear viscoelastic properties "in a step". Journal of Rheology. 60 (4), 649-660 (2016).
- Zhong, M. C., Wei, X. B., Zhou, J. H., Wang, Z. Q., Li, Y. M. Trapping red blood cells in living animals using optical tweezers. Nature Communications. 4, 1767-1768 (2013).
- Harlepp, S., Thalmann, F., Follain, G., Goetz, J. G. Hemodynamic forces can be accurately measured in vivo with optical tweezers. Molecular Biology of the Cell. 28 (23), 3252-3260 (2017).
- Bayoudh, S., Mehta, M., Rubinsztein-Dunlop, H., Heckenberg, N. R., Critchley, C. Micromanipulation of chloroplasts using optical tweezers. Journal of Microscopy. 203 (2), 214-222 (2001).
- Favre-Bulle, I. A., Stilgoe, A. B., Rubinsztein-Dunlop, H., Scott, E. K. Optical trapping of otoliths drives vestibular behaviours in larval zebrafish. Nature Communications. 8 (1), 630 (2017).
- Bambardekar, K., Clément, R., Blanc, O., Chardès, C., Lenne, P. F. Direct laser manipulation reveals the mechanics of cell contacts in vivo. Proceedings of the National Academy of Sciences of the United States of America. 112 (5), 1416-1421 (2015).
- Ferro, V., Chuai, M., McGloin, D., Weijer, C. J. Measurement of junctional tension in epithelial cells at the onset of primitive streak formation in the chick embryo via non-destructive optical manipulation. Development (Cambridge). 147 (3), (2020).
- Hörner, F., et al. Holographic optical tweezers-based in vivo manipulations in zebrafish embryos. Journal of Biophotonics. 10 (11), 1492-1501 (2017).
- Zhong, M. -. C., Wang, Z. -. Q., Li, Y. -. M. Aberration compensation for optical trapping of cells within living mice. Applied Optics. 56 (7), 1972 (2017).
- Català, F., Marsà, F., Montes-Usategui, M., Farré, A., Martín-Badosa, E. Influence of experimental parameters on the laser heating of an optical trap. Scientific Reports. 7 (1), 1-9 (2017).
- Taylor, M. A., Waleed, M., Stilgoe, A. B., Rubinsztein-Dunlop, H., Bowen, W. P. Enhanced optical trapping via structured scattering. Nature Photonics. 9 (10), 669-673 (2015).
- Bormuth, V., et al. Optical trapping of coated microspheres. Optics Express. 16 (18), 13831-13844 (2008).
- Sudhakar, S., et al. Germanium nanospheres for ultraresolution picotensiometry of kinesin motors. Science. 371 (6530), (2021).
- Shan, X., et al. Optical tweezers beyond refractive index mismatch using highly doped upconversion nanoparticles. Nature Nanotechnology. 16 (5), 531-537 (2021).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten