
Method Article
Misure di forza diretta della meccanica subcellulare nel confinamento mediante pinzette ottiche
In questo articolo
Riepilogo
Qui, presentiamo un protocollo per studiare le proprietà meccaniche intracellulari di cellule embrionali isolate di zebrafish in confinamento tridimensionale con misurazione della forza diretta mediante una trappola ottica.
Abstract
Durante lo sviluppo di un organismo multicellulare, una singola cellula fecondata si divide e dà origine a più tessuti con funzioni diverse. La morfogenesi tissutale va di pari passo con i cambiamenti molecolari e strutturali a livello di singola cellula che si traducono in variazioni delle proprietà meccaniche subcellulari. Di conseguenza, anche all'interno della stessa cellula, organelli e compartimenti diversi resistono in modo diverso alle sollecitazioni meccaniche; e le vie di meccanotrasduzione possono regolare attivamente le loro proprietà meccaniche. La capacità di una cellula di adattarsi al microambiente della nicchia tissutale è quindi in parte dovuta alla capacità di percepire e rispondere alle sollecitazioni meccaniche. Recentemente abbiamo proposto un nuovo paradigma di meccanosensazione in cui la deformazione e il posizionamento nucleare consentono a una cellula di misurare l'ambiente fisico 3D e dotano la cellula di un senso di propriocezione per decodificare i cambiamenti nella forma della cellula. In questo articolo, descriviamo un nuovo metodo per misurare le forze e le proprietà del materiale che modellano il nucleo cellulare all'interno delle cellule viventi, esemplificato su cellule aderenti e cellule confinate meccanicamente. Le misurazioni possono essere eseguite in modo non invasivo con trappole ottiche all'interno delle cellule e le forze sono direttamente accessibili attraverso il rilevamento senza calibrazione del momento luminoso. Ciò consente di misurare la meccanica del nucleo indipendentemente dalle deformazioni della superficie cellulare e di consentire la dissezione delle vie di meccanotrasduzione esterocettiva e interocettiva. È importante sottolineare che l'esperimento di trapping può essere combinato con la microscopia ottica per studiare la risposta cellulare e le dinamiche subcellulari utilizzando l'imaging a fluorescenza del citoscheletro, degli ioni calcio o della morfologia nucleare. Il metodo presentato è semplice da applicare, compatibile con soluzioni commerciali per le misurazioni della forza e può essere facilmente esteso per studiare la meccanica di altri compartimenti subcellulari, ad esempio mitocondri, fibre di stress ed endosomi.
Introduzione
La morfogenesi tissutale è un processo complesso in cui i segnali biochimici e le forze fisiche sono coordinati spaziotemporalmente. Nell'embrione in via di sviluppo, i gradienti dei fattori di segnalazione biochimica dettano le specifiche del destino e assicurano un corretto pattern tissutale1,2. Allo stesso tempo, le forze intrinseche ed estrinseche svolgono un ruolo nella costruzione dell'architettura dell'embrione3,4. L'influenza della meccanica della corteccia cellulare in questo contesto è stata ampiamente studiata5,6. La stretta interconnessione tra i processi meccano-chimici durante la morfogenesi si basa sulle proprietà delle singole cellule per rilevare e rispondere alle forze meccaniche nel loro microambiente tissutale. Le cellule, quindi, decodificano i segnali meccanici attraverso la presenza di elementi subcellulari e molecolari sensibili alla forza che trasducono informazioni meccaniche in specifiche vie di segnalazione che controllano il comportamento cellulare, il destino cellulare e la meccanica cellulare.
Un segno distintivo dei processi di sviluppo è che le cellule si organizzano come gruppi per costruire strutture multicellulari. Come tali, le singole cellule raramente riorganizzano e si muovono da sole, ma sono associate in un sociotopo stretto in cui mostrano un comportamento collettivo come la migrazione sopracellulare7, le transizioni (un)jamming8,9 o la compattazione della blastocisti10. Le forze meccaniche generate all'interno e tra le cellule servono come spunti importanti per istruire la dinamica cellulare collettiva7,11. Ma anche quando le cellule si muovono da sole, come le cellule progenitrici che si fanno strada tra fogli di tessuto o strette nicchie di tessuto, sperimentano estese forze meccaniche anisotropiche quando navigano in un ambiente tridimensionale. Queste sollecitazioni meccaniche sulle cellule hanno profonde conseguenze sul comportamento cellulare12,13. Sono stati studiati diversi meccanismi che convergono sul nucleo come un importante elemento di meccanotrasduzione14,15, come elemento meccanico passivo o attivo durante la migrazione all'interno di un ambiente di tessuto 3D denso15,16.
Recentemente abbiamo proposto un meccanismo che equipaggia le cellule per misurare le deformazioni della forma utilizzando il nucleo come meccanometro intracellulare elastico12. Il nucleo, essendo il più grande organello in una cellula, subisce grandi deformazioni quando le cellule si polarizzano, migrano o cambiano forma sotto allungamento meccanico, confinamento o stress osmotico16,17,18,19. Abbiamo scoperto che l'allungamento dell'involucro nucleare insieme al posizionamento intracellulare del nucleo fornisce alle cellule informazioni sulla grandezza e il tipo di deformazione cellulare (come la compressione cellulare rispetto al gonfiore cellulare). Lo stiramento del nucleo è associato a uno sviluppo della membrana nucleare interna (INM), che promuove l'attività della lipasi cPLA2 calcio-dipendente (fosfolipasi citosolica A2) all'INM seguita dal rilascio di acido arachidonico (AA) e dalla rapida attivazione della miosina II nella corteccia cellulare. Ciò porta ad un aumento della contrattilità cellulare e alla migrazione delle cellule ameboidi al di sopra di una soglia di contrattilità corticale6. La risposta meccanosensibile alla deformazione cellulare si verifica in meno di un minuto ed è reversibile al momento del rilascio del confinamento, suggerendo che il nucleo agisce come un estensimetro per la propriocezione cellulare che regola il comportamento cellulare adattativo in condizioni di stress meccanico. Questa via meccanosensibile ha dimostrato di essere attiva nelle cellule staminali progenitrici derivate da embrioni di zebrafish, sia in cellule pluripotenti che impegnate nel lignaggio12 ed è conservata in diverse specie e linee cellulari20.
Oltre alle proprietà nucleari come cellula-meccanosensore, l'architettura e la meccanica nucleare sono intrinsecamente regolate durante lo sviluppo e in risposta alla specifica del destino cellulare21, quindi sintonizzando la meccano-sensibilità cellulare22,23. La conseguenza potrebbe essere un cambiamento nella conformità nucleare che consenta cambiamenti morfologici e transizioni da uno stato premigratorio a uno migratorio e viceversa8.
Sono state applicate diverse tecniche per misurare la meccanica del nucleo cellulare, come la microscopia a forza atomica24,25, l'aspirazione a micropipetta26,27, la tecnologia microfluidica28 e i microaghi29. Tuttavia, molte di queste tecniche sono invasive nel senso che l'intera cellula deve essere deformata, limitando la misurazione delle caratteristiche meccaniche e delle risposte forza-dipendenti del nucleo stesso. Per aggirare la deformazione simultanea della superficie cellulare e della sua corteccia cellulare meccanosensibile30, sono stati studiati nuclei isolati in vari contesti31,32. Tuttavia, non si può escludere che l'isolamento nucleare sia associato a un cambiamento delle proprietà dei nuclei meccanici e della loro regolazione (riferimento24 e proprie osservazioni non pubblicate).
Le pinzette ottiche (OT) sono una tecnologia versatile che ha permesso una pletora di esperimenti in meccanobiologia cellulare e sono stati determinanti nella nostra comprensione di come le macchine molecolari convertono la chimica in energia meccanica33,34. Le pinzette ottiche utilizzano un raggio laser strettamente focalizzato per esercitare forze ottiche sulle particelle dielettriche che hanno un indice di rifrazione superiore al mezzo circostante33. Tali forze possono essere dell'ordine di centinaia di pico-Newton e risultare in un efficace confinamento della particella all'interno della messa a fuoco della trappola laser, consentendo la manipolazione della particella intrappolata in tre dimensioni. L'uso della luce ha un vantaggio importante in quanto la misurazione può essere eseguita in modo non invasivo all'interno di cellule viventi. Le manipolazioni ottiche sono ulteriormente limitate alla messa a fuoco della trappola del raggio laser. Quindi, la manipolazione può essere eseguita senza stimolare le membrane cellulari circostanti e non perturba la corteccia di actina o i processi meccanosensibili sulla membrana plasmatica, come l'attivazione forza-dipendente dei canali ionici.
La difficoltà dell'approccio della pinzetta ottica è quella di determinare con precisione le forze applicate alla microsfera utilizzando approcci classici che si basano sulla calibrazione della forza indiretta basata sul teorema di equipartizione o sull'uso di forze di Resistenza di Stokes definite per misurare una forza di fuga dipendente dalla potenza laser35. Mentre questi metodi sono semplici da implementare in un esperimento in vitro, di solito non possono essere tradotti in un ambiente cellulare. Sono state introdotte sul campo diverse strategie che si basano su una calibrazione diretta della forza, derivata dai primi principi di conservazione della quantità di moto36,37. A differenza di altri approcci di spettroscopia di forza, le misurazioni della forza sono dedotte da uno scambio locale di momento luminoso con la particella intrappolata di forma arbitraria38,39. Nel nostro set-up sperimentale, i cambiamenti nel momento della luce derivanti dalle forze ottiche vengono misurati direttamente senza la necessità di calibrazione della trappola in situ40,41,42,43. Pertanto, le misurazioni diventano possibili in un ambiente viscoso come l'interno della cellula o anche all'interno di un tessuto, e le forze possono essere facilmente quantificate fino al livello pN.
In questo protocollo, descriviamo un test per manipolare meccanicamente organelli o strutture intracellulari e valutare quantitativamente le loro proprietà meccaniche mediante una pinzetta ottica. Questa configurazione è integrata in un microscopio fluorescente a disco rotante che consente l'imaging parallelo del comportamento cellulare o della dinamica intracellulare. Il saggio consente la caratterizzazione delle proprietà meccaniche di specifici compartimenti cellulari, come il nucleo, studiando contemporaneamente la possibile meccanoresponsazione e attivazione delle vie di segnalazione molecolare a seguito della deformazione stessa. Inoltre, l'intrappolamento ottico delle microsfere iniettate all'interno delle cellule consente un aumento della forza di indentazione grazie ad un indice di rifrazione notevolmente più elevato del tallone di polistirene (n = 1,59) rispetto al contrasto rifrattivo intrinseco44 del nucleo (n ~ 1,35) rispetto al citoplasma (n ~ 1,38). La strategia presentata può essere facilmente adattata allo studio di altre strutture intracellulari e organelli, nonché ad altri approcci che coinvolgono la microreologia attiva, l'uso di più trappole ottiche per sondare contemporaneamente le stesse / diverse strutture subcellulari e misurazioni mirate alla meccanobiologia cellulare nell'embrione vivo.
Protocollo
Tutti i protocolli utilizzati sono stati approvati dall'Institutional Animal Care and Use Ethic Committee (PRBB-IACUEC) e implementati secondo le normative nazionali ed europee. Tutti gli esperimenti sono stati condotti in conformità con i principi delle 3R. I pesci zebra (Danio rerio) sono stati mantenuti come descritto in precedenza.
1. Preparazione di cellule staminali progenitrici embrionali embrionali primarie isolate
- Preparazione di micropipette e agarosio
NOTA: per un protocollo completo di microiniezione di embrioni di zebrafish, vedere riferimento45.- Con un estrattore di micropipette, tirare un capillare di vetro da 1,0 mm per ottenere due aghi45. Conservare gli aghi inutilizzati in una capsula di Petri da 150 mm attaccata a un cuscino di pasta da gioco o in un anello di nastro da laboratorio interno-esterno per proteggere la punta sottile da danni durante il trasporto.
- Fondere l'agarosio ultrapuro all'1% in E3 (5 mM NaCl, 0,17 mM KCl, 0,33 mM CaCl2, 0,33 mM MgSO4) in una cucina standard / forno a microonde da laboratorio per 10 s. Riscaldare ripetutamente la miscela per brevi periodi di tempo (pochi secondi) fino a quando l'agarosio si scioglie.
- Quando l'agarosio è completamente sciolto, lasciarlo raffreddare brevemente, quindi versarlo in una capsula di Petri di 10 cm. Aggiungere lentamente lo stampo triangolare per microiniezione (vedi Tabella dei materiali) sulla parte superiore dell'agarosio evitando la comparsa di bolle. Non spingere lo stampo, assicurandosi che rimanga sulla superficie dell'agarosio.
- Quando l'agarosio si solidifica completamente, rimuovere lo stampo triangolare molto lentamente esercitando una forza delicata per evitare eventuali rotture nell'agarosio. La piastra può essere conservata a testa in giù a 4 °C per 2-4 settimane.
- 30 minuti prima della microiniezione, estrarre la piastra dal frigorifero e aggiungere E3 preriscaldato a 28 °C per farlo stabilizzare a temperatura ambiente.
- Preparazione della miscela di iniezione
- Per preparare la miscela di iniezione, diluire microsfere da 1 μm (polistirene, non fluorescente) in rapporto 1:5 in acqua priva di RNasi.
- Preparare l'mRNA per l'espressione transitoria di marcatori fluorescenti o l'espressione di costrutti genici ricombinanti e/o la co-iniezione di morfolino alla concentrazione desiderata.
NOTA: Una tipica miscela di iniezione per la co-iniezione di microsfere insieme a 100 pg di mRNA per embrione da etichettare, ad esempio, il nucleo con H2A-mCherry è: 1 μL di perline + 1 μL di mRNA (la concentrazione di stock è 1 μg/μL) + 2,5 μL di acqua priva di RNA + 0,5 μL di rosso fenolo (soluzione madre 0,5%, il rosso fenolo non è obbligatorio; viene utilizzato per una migliore visualizzazione della goccia iniettata ma l'iniezione non marcata drop è visibile anche per uno sperimentatore esperto). L'iniezione di RNA può anche essere utile per selezionare embrioni iniettati. Le microsfere fluorescenti possono essere iniettate, invece che non fluorescenti, per visualizzarle.
- Caricamento e calibrazione dell'ago per microiniezione
- Accendere il microiniettore utilizzando l'opzione Time-Gated . Questa impostazione è molto importante per calibrare correttamente il volume di iniezione. Impostare il tempo di gating a circa 500 ms.
- Caricare 3 μL della miscela di iniezione nell'ago utilizzando una pipetta micro-caricatore.
- Inserire l'ago nel micromanipolatore e sigillare ermeticamente. Controllare se il micromanipolatore è in una buona posizione e ha abbastanza libertà per muoversi in direzione x-y sulla piastra di iniezione.
- Misurare la dimensione della goccia utilizzando un vetrino micrometrico (divisioni 5 mm/100) con una goccia di olio minerale in cima45 ed espellendo una goccia della miscela di iniezione direttamente nell'olio minerale.
- Ritaglia l'ago con una pinza affilata ad un angolo ripido per generare una punta appuntita affilata. Regolare la dimensione della goccia a 0,1 mm, corrispondente a 0,5 nL di materiale iniettato.
NOTA: Se tagliando l'ago si supera questo volume, si consiglia di ripetere la procedura di calibrazione con un nuovo ago. Il tempo di gating del microiniettore può essere leggermente regolato per adattarsi al volume della goccia; tuttavia, brevi tempi di gating corrispondono a un grande diametro dell'ago, che potenzialmente danneggia gli embrioni.
- Microiniezione di embrioni di zebrafish allo stadio di una cellula
- Raccogliere embrioni di zebrafish poco dopo la fecondazione per la microiniezione della miscela di perline direttamente nell'embrione allo stadio unicellulare (zigote) prima che si verifichi la prima divisione cellulare.
NOTA: Ciò garantisce una corretta distribuzione delle microsfere e una resa sufficientemente elevata di blastomeri isolati con almeno una microsfera per cellula nelle fasi successive dello sviluppo in cui vengono eseguiti esperimenti (stadio blastula-gastrula). Gli esperimenti di indentazione possono ancora essere eseguiti se ci sono due sfere all'interno della cellula, ma le cellule che non hanno perline dovrebbero essere escluse (anche se è possibile la rientranza senza sfere). In questo protocollo sono stati utilizzati ceppi wildtype AB, ma è possibile utilizzare qualsiasi altro ceppo, ad esempio TL. - Posizionare embrioni allo stadio unicellulare (zigote) in uno stampo di agarosio all'1% di forma triangolare preriscaldata, come mostrato nella Figura 1A, utilizzando una pipetta pasteur di plastica.
- Rimuovere il mezzo extra con la stessa pipetta per evitare che gli embrioni galleggino intorno. Spingere delicatamente gli embrioni nello stampo triangolare tramite un pennello. Mantenere un po' di spazio tra gli embrioni per facilitare il corretto orientamento (Figura 1B).
- Allineare delicatamente gli embrioni con un pennello in modo che gli embrioni siano orientati lateralmente, con l'unica cellula dello zigote chiaramente visibile, come mostrato nella Figura 1B. Un orientamento ideale per la microiniezione si raggiunge quando l'unica cellula dell'embrione è rivolta verso la direzione dell'ago ( iniezione attraverso il polo animale dell'embrione) o in modo opposto di fronte alla cellula del tuorlo ( iniezione attraverso il polo vegetale dell'embrione), come mostrato nella Figura 1C.
- Tenere il piatto con una mano e usare l'altra mano per posizionare la punta dell'ago utilizzando il controller del micromanipolatore. Abbassare la punta dell'ago verso gli embrioni.
- Perforare il corion ed entrare nell'embrione unicellulare con l'ago mentre si monitora la procedura attraverso lo stereomicroscopio. Garantire il corretto posizionamento dell'ago e, dopo l'iniezione, la corretta posizione della goccia iniettata come mostrato nella Figura 1C.
- Ripeti per tutti gli embrioni: sposta l'ago verso l'alto, fai scorrere il piatto con gli embrioni fino a quando l'embrione successivo è centrato, abbassa l'ago e iniettalo.
- Una volta iniettato l'intero set di embrioni, rimuovere gli embrioni dalla muffa di agarosio / capsula di Petri lavando un po 'di E3 e metterli in una nuova capsula di Petri usando una pipetta di plastica Pasteur. Si raccomanda di posizionare mezzi sufficienti sulla piastra di iniezione per evitare l'essiccazione degli embrioni durante la procedura di microiniezione.
- Ripetere la procedura fino a quando non viene iniettato il numero desiderato di embrioni. Gli embrioni devono essere in uno stadio cellulare per garantire la diffusione massima e omogenea delle perline.
NOTA: Questa procedura è ottimizzata per gli embrioni precoci di blastula e probabilmente deve essere ottimizzata se si devono studiare diversi stadi di sviluppo. - Posizionare gli embrioni iniettati all'interno di un incubatore a 28-31 °C per circa 4 ore o fino allo stadio desiderato (Figura 1D) prima di procedere con il protocollo per la coltura cellulare primaria.
NOTA: Facoltativamente, lasciare che gli embrioni si sviluppino oltre lo stadio di blastula (o il punto temporale di misurazione desiderato) per garantire la sopravvivenza ed escludere artefatti tossici. Negli stadi larvali, montare larve anestetizzate con tricaina in agarosio allo 0,75% e visualizzare la distribuzione delle microsfere in vari tessuti. Per ottenere una soluzione madre, mescolare: 400 mg di polvere di tricaina in 97,9 mL di acqua distillata, circa 2,1 mL di 1 M TRIS-base (pH 9) e regolare a pH 7. Questa soluzione può essere conservata a 4 °C. Per utilizzare la tricaina come anestetico, diluire 4,2 mL di soluzione madre in 100 mL di terreno dell'uovo (o mezzo desiderato); in questo caso, è stato utilizzato l'E3. Consultare reference46 per i dettagli.
- Raccogliere embrioni di zebrafish poco dopo la fecondazione per la microiniezione della miscela di perline direttamente nell'embrione allo stadio unicellulare (zigote) prima che si verifichi la prima divisione cellulare.
2. Preparazione e colorazione unicellulare
- Posizionare gli embrioni dello stadio della sfera (4 hpf, ore dopo la fecondazione) in un piatto di vetro usando una pipetta pasteur di plastica. Selezionare gli embrioni che sono positivi per il segnale delle perle iniettate e che esprimono la proteina fluorescente in caso di iniezione di mRNA. Alcuni embrioni potrebbero mostrare un elevato raggruppamento di perline e possono essere esclusi.
- Decorionare manualmente gli embrioni usando una pinza. Trasferire circa 10-15 embrioni in contenitori di reazione da 1,5 ml utilizzando una pipetta pasteur di vetro.
NOTA: Quando gli embrioni sono deconizionati, si attaccano alla plastica ed è richiesto l'uso di oggetti in vetro. In alternativa alla lastra di vetro, è possibile utilizzare una piastra di Petri in plastica con uno strato sottile di agarosio all'1%. La decorionazione manuale deve essere preferita al trattamento enzimatico con pronasi per prevenire danni proteolitici alle proteine della superficie cellulare e potenziali cambiamenti nelle proprietà meccaniche delle cellule e dei tessuti, prevenendo tempi di recupero prolungati47.
- Decorionare manualmente gli embrioni usando una pinza. Trasferire circa 10-15 embrioni in contenitori di reazione da 1,5 ml utilizzando una pipetta pasteur di vetro.
- Rimuovere il mezzo E3 e aggiungere 500 μL di terreno di coltura tissutale preriscaldato co2-indipendente (DMEM-F12; con L-glutammina e 15 mM HEPES, senza bicarbonato di sodio e rosso fenolo integrato con 10 unità di penicillina e 10 mg/ L di streptomicina).
NOTA: non utilizzare mezzi dipendenti dalla CO2 a meno che non si utilizzi un incubatore per microscopio. L'uso di, ad esempio, RPMI in condizioni tamponate con carbonato causa cambiamenti nel pH del mezzo e può influenzare la sopravvivenza cellulare. Un altro aspetto chiave è quello di evitare i terreni di coltura che contengono siero. Il siero può contenere acido lisofosfatidico (LPA), un potente attivatore della via Rho/ROCK, in grado di controllare la contrattilità cellulare e la motilità nelle cellule staminali progenitrici6. L'osmolarità del mezzo dovrebbe essere mantenuta a 300 mOsm per evitare sfide osmotiche che potrebbero interferire con la morfologia o la meccanica nucleare12. - Dissociare manualmente le cellule scuotendo delicatamente il tubo. Assicurarsi che il contenuto del tubo diventi torbido senza grossi pezzi visibili dall'occhio. Evitare la formazione di bolle per ridurre al minimo il danno e la perdita di cellule.
- Centrifuga a 200 x g per 3 min. Il pellet deve essere chiaramente visibile.
- Rimuovere il surnatante e seguire uno dei passaggi descritti di seguito.
- Se non è necessaria alcuna colorazione, aggiungere 500 μL di DMEM. Risospendare delicatamente con una pipetta da 200 μL puntando un getto liquido sul pellet. Non esercitare un'eccessiva forza di taglio sulle cellule. La formazione di schiuma indica danni alle cellule.
- Per etichettare il nucleo con coloranti a DNA come Hoechst, mescolare 0,5 μL di DNA-Hoechst (stock 2 mg/mL) in 1.000 μL di DMEM per ottenere 1 μg/mL di concentrazione finale. Aggiungere 500 μL di questa soluzione colorante alle cellule e risospensare delicatamente. Incubare per 7 minuti al buio.
- Per macchiare le cellule con un indicatore chimico fluorescente di calcio Calbryte-520, aggiungere Calbryte-520 a una concentrazione di 5 μM in DMEM. Incubare per 20 minuti al buio.
NOTA: I protocolli indicati nei passaggi 2.5.2 e 2.5.3 sono stati ottimizzati per questi prodotti specifici. Altre colorazioni possono essere eseguite utilizzando i protocolli indicati dal produttore.
- Centrifugare di nuovo utilizzando le stesse impostazioni del passaggio 2.4; rimuovere il surnatante e risospescere delicatamente le cellule (per evitare la formazione di cluster) in 50 μL di DMEM per i campioni in sospensione o 20 μL di DMEM per le cellule in confinamento.
3. Preparazione di camere di intrappolamento ottico utilizzando la spaziatura tra polidimetilsilossano (PDMS)
NOTA: le misurazioni della forza ottica basate sul rilevamento del momento luminoso richiedono la cattura di tutta la luce che emerge dalle trappole ottiche40. Affinché la robustezza del fattore di calibrazione invariante α (pN/V), la distribuzione della luce sul piano focale posteriore (BFP) del sensore di forza ottica deve avere una corrispondenza accurata con il momento fotonico. Questo determina la distanza dalla superficie della lente di raccolta al piano di intrappolamento a circa 2 mm, che è l'altezza massima delle camere di intrappolamento ottico.
- Rivestimento PDMS di piatti con fondo di vetro #1.5.
NOTA: La seguente ricetta è prevista per circa 40 piatti. La microcamera risultante avrà altezze diverse a seconda che gli esperimenti debbano essere condotti su cellule sospese o confinate (Figura 1D).- Miscelare 9 mL del polimero di base PDMS e 1 mL di agente polimerizzante PDMS in un tubo conico da 50 mL. Mescolare attivamente i due prodotti per garantire una corretta distribuzione dell'agente polimerizzante.
- Degassare la miscela per evitare bolle utilizzando una pompa per vuoto. Introdurre il tubo conico in una bottiglia sottovuoto ed evacuare la camera. Attendere che non siano presenti bolle nella miscela.
NOTA: Aprire lentamente il vuoto per evitare schiuma e fuoriuscite del PDMS dal tubo del falco. - Posizionare il piatto con fondo di vetro sul mandrino dello spin-coater (Figura 2A). Sii gentile per non graffiare, prendere le impronte digitali o sporcare il piatto. Proteggere la scatola di spin-coater da perdite PDMS con un foglio di alluminio.
- Per le camere OT per esperimenti su celle in sospensione, aggiungere circa 250 μL di miscela PDMS al centro del piatto inferiore e ruotarla a 750 giri / min per 1 minuto. L'altezza dello strato PDMS sarà di circa 50 μm48.
- Per le camere OT per esperimenti su celle confinate, aggiungere una piccola goccia di PDMS (circa 50 μL) e ruotarla a 4.000 giri / min per 5 minuti. L'altezza dello strato PDMS sarà di circa 10 μm. Per un protocollo dettagliato su come ottenere diversi spessori PDMS, vedere reference48.
- Polimerizzare le piastre con fondo di vetro rivestite PDMS a 70 °C per 1 ora.
- Tagliare un quadrato di 1 x 1 cm sullo strato PDMS con un bisturi e staccarlo con una pinzetta (Figura 2C). Nel caso di celle confinate, lavare i detriti PDMS con isopropanolo.
- Rivestimento della camera per esperimenti con celle leggermente attaccate in sospensione
- Aggiungere 100 μL di Concanavalina A (ConA) a 0,5 mg/mL per coprire l'intera superficie della cavità quadrata e lasciarla incubare per 30 min.
NOTA: ConA è una lectina che si lega agli zuccheri della superficie cellulare e accoppia le singole cellule sulla superficie del vetro di copertura. - Rimuovere la goccia di ConA e risciacquare accuratamente la superficie con il mezzo DMEM senza graffiare la superficie trattata conA.
- Aggiungere 30 μL del campione precedentemente preparato (fase 2.6) nel pozzetto e risospesci delicatamente per eliminare eventuali cluster cellulari.
- Chiudere la cavità posizionando delicatamente un vetro di copertura #1.5 da 22 x 22 mm sopra i cerchi PDMS (evitare di lasciarlo cadere bruscamente, utilizzare una pinza se possibile, Figura 2B,C).
NOTA: qualsiasi spessore di copertura funzionerebbe per la copertura superiore in vetro (la lente di raccolta ha una distanza di lavoro di 2 mm).
- Aggiungere 100 μL di Concanavalina A (ConA) a 0,5 mg/mL per coprire l'intera superficie della cavità quadrata e lasciarla incubare per 30 min.
- Preparazione della camera per esperimenti con cellule in confinamento
- Introdurre una goccia di 10 μL di soluzione contenente cellule (fase 2.6) nella cavità quadrata (Figura 2B).
- Molto delicatamente, inserire il campione con un vetro di copertura 22 x 22 mm in modo tale che la goccia si diffonda in tutta l'area e non si osservino bolle. Ancora una volta, è conveniente usare una pinza, come mostrato nella Figura 2C, per evitare che il vetro di copertura cada bruscamente.
4. Opzioni alternative per la spaziatura della camera OT
NOTA: questi passaggi possono essere seguiti se non è disponibile un'officina di microfabbricazione o una centrifuga.
- Preparazione della camera per esperimenti con cellule in sospensione
NOTA: Nel caso in cui non sia disponibile uno spin coater, è possibile realizzare un distanziatore utilizzando un normale scotch biadesivo (circa 100 μm di altezza).- Tagliare un pezzo di scotch biadesivo con un foro quadrato di circa 10 cm x 10 cm al centro (stesse dimensioni del PDMS, Figura 2B).
- Rimuovere uno degli strati protettivi del nastro staccandolo e posizionare il lato scoperto del nastro al centro di un piatto con fondo di vetro #1.5 H. Premere delicatamente per far aderire tutta la superficie al vetro evitando bolle d'aria, quindi rimuovere lo strato protettivo rimanente del nastro staccandolo.
- Seguire le istruzioni nel passaggio 3.2.
- Preparazione della camera per esperimenti con cellule in confinamento
NOTA: Per confinare con precisione le cellule, le microparticelle monodisperse con un diametro noto possono essere utilizzate come distanziatori tra i due vetri di copertura.- Aggiungere 10 μm di perle di polistirene alle cellule sospese ad una concentrazione di 104 perline/μL.
- Mettere una goccia di 10 μL di soluzione contenente celle e perline su un vetro di copertura di 22 x 60 mm.
- Molto delicatamente, inserire il campione con un altro vetro di copertura 22 x 60 mm in modo tale che la goccia si diffonda in tutta l'area e non si osservino bolle. Per posizionare delicatamente il vetro di copertura superiore (evitare che cada bruscamente), è conveniente usare una pinza.
- Poiché il campione può asciugarsi, si consiglia di eseguire rapidamente la preparazione.
5. Impostazione della trappola ottica per le misurazioni intracellulari
NOTA: i seguenti passaggi sono ottimizzati per una piattaforma commerciale di pinzette ottiche comprendente un modulo di micromanipolazione ottica basato sulla deflessione acusto-ottica (AOD) e un sensore di forza ottica basato sul rilevamento diretto delle variazioni del momento luminoso (Figura 2, riferimento12,40,49). I dettagli e i componenti ottici del set-up sono riportati nella Figura 2F. Per osservare la deformazione indotta dalla forza durante le manipolazioni ottiche della pinzetta, un microscopio confocale a disco rotante Nipkow è accoppiato alla porta sinistra del microscopio invertito per l'imaging a fluorescenza a doppio colore. Senza la mancanza di generalità, questo protocollo può essere applicato con qualsiasi sistema OT dinamico dotato di misurazioni della forza diretta basate sul rilevamento del momento leggero. Sono disponibili procedure dettagliate passo-passo per costruire trappole a gradiente ottico costruite in casa per applicazioni in vivo50. Quelli basati sulla modulazione AOD si distinguono per eventuali esperimenti con trappole multiple e misurazioni veloci51,52. Esistono diversi protocolli per costruire uno strumento basato sul momento luminoso36,39,40,53 e qualsiasi altra modalità di imaging (contrasto di interferenza differenziale, fluorescenza a campo largo, ecc.) può essere impiegata.
- Avviamento pinzette ottiche
- Al fine di ottimizzare la stabilità della potenza di uscita, accendere il laser a una potenza considerevolmente elevata (ad esempio, 3 W) almeno 30 minuti prima dell'esperimento.
- Accendere il modulo elettronico della micromanipolazione ottica e delle unità di misurazione della forza.
NOTA: Applicare tutte le misure di sicurezza laser e utilizzare solo apparecchiature approvate dal consiglio istituzionale. Non utilizzare mai gli oculari del microscopio ottico quando il laser è acceso. Utilizzare sempre occhiali di protezione IR approvati (OD7 nell'intervallo 950-1080 nm), bloccare la luce laser IR con l'otturatore nella porta di epifluorescenza 2 e non eseguire il software di intrappolamento ottico fino a quando non si completa l'allineamento del sensore di forza ottica dopo il passaggio 5.3. In generale, non utilizzare un campione altamente riflettente, poiché la retroriflessione potrebbe causare danni al laser. - Controllare la potenza della trappola con l'HWP rotante (Figura 2F) all'ingresso del modulo di micromanipolazione ottica.
NOTA: il modulo di micromanipolazione ottica commerciale utilizzato in questo protocollo incorpora già questa funzione. Per i sistemi di intrappolamento ottico costruiti in casa, integrare questo strumento per il controllo dell'alimentazione in modo da poter utilizzare potenze laser più elevate e più stabili.
- Utilizzare una microcamera vuota per la calibrazione
- Tagliare un quadrato di 1 x 1 cm su uno scotch biadesivo e fissarlo su un vetrino per microscopio di 1 mm di spessore.
- Aggiungere acqua nel quadrato e chiuderlo dall'alto con un vetro di copertura #1,5 (22 x 22 mm). Si consiglia di aggiungere un volume leggermente superiore di acqua, ad esempio 30-40 μL per evitare bolle all'interno della camera coperta. Pulire delicatamente la camera di calibrazione in caso di fuoriuscita di acqua.
- Allineamento del sensore ottico di forza
- Metti una goccia d'acqua sull'obiettivo di immersione in acqua 60x/1.2. Posizionare la camera di calibrazione sul palco con il vetro di copertura #1.5 rivolto verso l'obiettivo. Concentrati sulla superficie inferiore, dove alla fine si troveranno i campioni di cellule.
- Aggiungere una goccia di olio per immersione sopra il vetrino superiore che copre il campione (Figura 2D). Abbassare attentamente la lente di raccolta dell'unità sensore di forza fino a quando non contatta la goccia d'olio.
NOTA: la goccia deve essere abbastanza grande da coprire l'intera lente che raccoglie la luce laser che emerge dalle trappole. Di solito, 200 μL sono sufficienti per coprire l'intera superficie e fornire un contatto di immersione stabile. Sii prudente ed evita il riempimento eccessivo in quanto potrebbe fuoriuscire nel campione. - Seguendo il protocollo del produttore per l'allineamento del sensore di forza ottica, guardare l'immagine del piano campione sulla fotocamera ausiliaria che verrà utilizzata per posizionare gli OT (AUX, Figura 2F). Molto delicatamente, abbassare il sensore di forza ottica fino a quando l'arresto del campo (FS, Figura 2F-G) appare coniugato sul piano del campione. Ciò garantirà adeguate misurazioni della forza diretta dal rilevamento invariante del campione delle variazioni del momento luminoso40.
NOTA: chiudere fs abbastanza in modo che la sua immagine diventi più piccola del campo visivo (FOV), quindi visibile. Prestare particolare attenzione e non spingere la lente di raccolta del sensore di forza ottica contro il campione. La posizione verticale del sensore di forza ottica può in alternativa essere determinata dall'analisi della distribuzione della luce di intrappolamento al BFP per i coni di luce con apertura numerica definita (NA). - Assicurarsi che non ci siano bolle d'aria nella goccia d'olio; questi possono influenzare direttamente le misurazioni della forza. Per verificare la presenza di bolle d'aria, posizionare la lente di Bertrand (BL, Figura 2G) e osservare il percorso di imaging attraverso l'oculare. Se sono visibili sporcizia o bolle d'aria o è necessario più olio (Figura S1A), pulire la lente e la camera con tessuto privo di polvere e ripetere la procedura nei passaggi 5.3.2 e 5.3.3. Un percorso ottico senza ostacoli è illustrato nella Figura S1B.
- Utilizzando le viti laterali posizionate sul supporto del sensore di forza ottica, centrare l'FS nel FOV. Per una maggiore precisione, aprire l'FS in modo che riempia quasi il FOV visibile sulla telecamera ausiliaria (AUX, Figura 2F).
6. Ottimizzazione della pinzetta ottica
NOTA: La misurazione della forza diretta si basa esclusivamente sul cambiamento del momento luminoso derivante dalla forza esercitata sulla particella intrappolata e quindi, a differenza dei metodi indiretti, la rigidità della trappola non deve essere calibrata prima di ogni esperimento. La conversione specifica dello strumento del fattore di deflessione/forza (α; pN/V, riferimento41) è calibrata dal produttore ed è quindi invariante sperimentale. Tuttavia, poiché lo spot laser viene manipolato su un'area di 70 μm x 70 μm, i passaggi da 6,2 a 6,5 sono fondamentali per garantire un intrappolamento ottimale e la stabilità di potenza. I seguenti passaggi sono forniti nel software del produttore in modo che gli OT vengano ottimizzati sull'area di lavoro in modo semi-automatico.
- Avviare il software OT e il software di acquisizione per fotocamera AUX.
- Sottrarre la linea di base della tensione iniziale facendo clic sul passaggio Passaggio 1: Offset elettronico nel sottomenu Calibrazione sistema del software di guida delle pinzette ottiche.
- Per eseguire l'appiattimento della potenza di trappola nell'area di lavoro OT, impostare la potenza della trappola a metà del suo massimo ruotando di conseguenza l'HWP. Non modificare la potenza della trappola cambiando l'uscita laser, ma con l'HWP rotante (Figura 2F). Fare clic su Passaggio 2: Avviare la routine automatizzata per l'appiattimento dell'alimentazione della trappola.
NOTA: questo è un passaggio fondamentale per compensare la variazione della potenza della trappola nell'area di lavoro degli OT (Figura S1D). Una routine di successo porta la variazione della potenza della trappola al 2% nell'area di lavoro degli OT e converge dopo 2 minuti. - Per eseguire la calibrazione della posizione della trappola, rimuovere il filtro IR in modo che la luce del laser sia visibile sulla fotocamera. Individuate il punto IR impostando il piano dell'immagine focalizzato sulla superficie inferiore della microcamera. Ottieni il punto IR più piccolo possibile regolando il piano dell'immagine (posizione dell'obiettivo) e il contrasto dell'istogramma nel software di acquisizione AUX della fotocamera. Se necessario, ridurre la potenza della trappola ottica ruotando l'HWP (Figura 2F). Fare clic su Passaggio 3: Posizione per avviare la calibrazione automatica della routine o del posizionamento dell'intrappola.
NOTA: questa routine consente la corrispondenza precisa delle coordinate di posizione dell'OT nell'AUX della telecamera con gli angoli di sterzata AOD. Una routine di successo genera la mappatura angolo-posizione in pochi secondi. - Compensazione iniziale del momentum
NOTA: il movimento della trappola ottica attraverso il campione provoca variazioni nella distribuzione del momento luminoso alla BFP (Figura S1E, F). Ciò porta a cambiamenti di segnale indipendenti dalla forza relativi alla posizione del laser sull'area di lavoro, anche se la potenza della trappola è stata appiattita come nel passaggio 6.3. La conseguenza è una variazione della forza basale dovuta alla posizione (indipendente da una forza effettiva che agisce sul tallone intrappolato otticamente) che deve essere corretta prima di ogni esperimento.- Impostare la potenza trappola che verrà utilizzata negli esperimenti, ruotando l'HWP (Figura 2F).
- Fate clic sull'opzione Offset globale (Global Offset ) nel sottomenu Strumenti (Tools ). Verrà aperto l'assistente Offset Cancel del software delle pinzette ottiche che corregge la linea di base del momentum iniziale.
- Fate clic su Offset | Compensare per correggere il momentum iniziale della variante di posizione.
NOTA: se nessuna modifica influisce sul percorso ottico durante le settimane in corso, le mappe di appiattimento della potenza della trappola (passaggio 6.3) e di posizione (passaggio 6.4) rimarranno invarianti. Raccomandiamo quindi di utilizzare sempre la stessa combinazione di elementi ottici (specchi dicroici, filtri, ecc.) che possono influenzare il percorso della trappola laser o di eseguire una nuova routine di appiattimento della potenza della trappola. Per quanto riguarda la compensazione del momento iniziale (fase 6.5), il produttore della piattaforma OT fornisce una calibrazione al volo che deve essere modificata per ogni nuova potenza di intrappolamento e sessione sperimentale. Le fasi 6.3 e 6.4 devono essere eseguite sul vetrino di taratura vuoto descritto al punto 5.2. In un campione contenente cellule o altri oggetti, il passaggio 6.5 deve essere effettuato privo di oggetti che possano alterare la diffusione della luce nell'area di lavoro degli OT.
- Facoltativamente, intrappolare una microsfera e spostare la trappola a una velocità nota durante la registrazione del segnale di forza. Ad esempio, impostare la trappola per eseguire un'oscillazione triangolare: il segnale di forza registrato sarà un segnale quadrato.
NOTA: il valore della forza deve aumentare linearmente con la velocità, in base alla forza di trascinamento che agisce sul tallone. Questo test serve come controllo positivo che le misurazioni della forza vengano eseguite correttamente38. In alternativa, il sensore di forza ottica può essere utilizzato per ottenere la rigidità di intrappolamento ottico, κ [pN/μm], e il fattore di calibrazione della posizione, β [μm/V], dall'analisi spettrale di potenza35. Con un corretto allineamento, il fattore di calibrazione invariante fornito dal produttore è α = κ·β [pN/V].- Avviare una lettura della forza in tempo reale facendo clic su Plot 1 nel sottomenu Misure nel software del produttore. Ciò fornirà una lettura dell'attuale forza e potenza di intrappolamento ottico.
- Aprite la finestra di dialogo Parametri oscillazione dal sottomenu Strumenti . Impostate una forma d'onda a spazio triangolare rispettivamente negli anelli di selezione Forma (Shape) e Tipo (Type). Ad esempio, impostare un'ampiezza di 10 μm e una frequenza di 3 Hz. Ciò si tradurrà in una forza viscosa di circa 1 pN su una microperla con un diametro di 1 μm38.
- Nella finestra AUX della fotocamera, fare clic con il pulsante destro del mouse sulla microperla e selezionare Avvia oscillazione. La lettura della forza diventerà un segnale di forza quadrato con plateau a ±1 pN.
- Fare clic con il pulsante destro del mouse sulla microperla e selezionare Interrompi oscillazione.
7. Microscopia confocale a disco rotante
- Accendere il microscopio confocale a disco rotante e le apparecchiature accessorie, i motori laser integrati e le telecamere di acquisizione.
- Avviare il software di imaging.
- Impostare i canali di imaging per la colorazione Hoechst del nucleo e GFP per la membrana plasmatica cellulare.
- Attivare le linee laser di eccitazione a 405 nm e 488 nm.
- Aggiungere un dicroico multibanda per riflettere l'eccitazione al campione e che consenta alla luce emessa di passare alle telecamere.
- Dividere l'emissione di fluorescenza con uno specchio dicroico a bordo passante lungo 500 nm.
- Utilizzare i filtri di emissione DAPI/BFP (~445 nm) e GFP (~521 nm) rispettivamente davanti alle due telecamere di acquisizione. Fare riferimento alla Figura 2F,G.
- Impostare il tempo di esposizione su 100 ms per ogni canale.
- Impostare l'emissione laser per ottenere una potenza di 5 mW sul piano del campione. Per misurare la potenza, utilizzare un misuratore di potenza commerciale.
- Impostare il protocollo di imaging. Per evitare il sanguinamento spettrale dal canale Hoechst al canale GFP, i due coloranti devono essere ripresi in sequenza.
NOTA: se esiste una sincronizzazione hardware tra gli AOD della trappola ottica e l'acquisizione della fotocamera, assicurarsi che la polarità del trigger sia impostata correttamente. In caso di dubbio, consultare il responsabile della struttura o il produttore del microscopio.
8. Esecuzione degli esperimenti di indentazione del nucleo
NOTA: Spegnere sempre le trappole ottiche, sia utilizzando il software che chiudendo l'otturatore sulla porta di epifluorescenza 2, quando si solleva il modulo del sensore di forza e si cambia il campione. In caso contrario, potrebbero verificarsi gravi danni agli elementi ottici e allo sperimentatore. Prestare attenzione alla distanza laterale tra il supporto della lente e il bordo inferiore della parabola quando si cercano le celle per evitare di urtare la lente nel piatto di fase/ coltura (Figura 2).
- Posizionare il campione al microscopio e seguire il passaggio 5.3 di questo protocollo.
- Utilizzando l'HWP rotante (Figura 2F), impostare la potenza della trappola a 200 mW come valore di partenza se la rigidità del nucleo o della struttura intracellulare studiata non è nota. Tradurre l'area di lavoro degli OT (utilizzando lo stadio del microscopio) in un luogo privo di cellule per compensare la linea di base del momento iniziale fino al passaggio 6.5.
NOTA: a seconda della rigidità della struttura subcellulare, il valore della potenza della trappola deve essere regolato su valori inferiori o superiori per ottenere una profondità di rientro simile. - Utilizzando il controller software dello stadio del microscopio, cercare una cella con una o due perline attraverso la microscopia a campo luminoso trasmessa (Figura 3A).
- Definite una traiettoria trappola.
- Aprite la finestra di dialogo Traiettoria nel sottomenu Strumenti (Tools ) e scegliete Spostamento (Displacement) nell'anello del selettore Tipo traiettoria (Trajectory Type ).
- Nel foglio numerico, scrivi lo spostamento e il tempo di ogni passo successivo della traiettoria. Ecco due esempi.
- Per un esperimento di rilassamento dello stress, programmare carichi trapezoidali, come mostrato nella Figura 3B. Nella tabella S1 sono state applicate due rientranze trapezoidali con una distanza di viaggio di 5 μm; velocità di 5 μm/s; tempo di attesa prima della ritrattazione: 10 s.
- Per un esperimento di rientranza ripetitiva a velocità costante per ottenere una routine triangolare senza tempo di permanenza sul nucleo, impostare l'ampiezza della traiettoria, ad esempio 5 μm, e il tempo per il passo, ad esempio, 2 s per una velocità di 2,5 μm/s. Nella tabella S2, questo viene applicato otto volte alla stessa velocità.
NOTA: Questi valori devono essere determinati per ogni tipo di cellula ed esperimento, ma i seguenti parametri di una routine trapezoidale catturano le dinamiche più importanti nell'esperimento qui presentato. Il tempo di attesa dovrebbe essere sufficiente affinché il nucleo mostri il suo completo rilassamento da stress dopo l'indentazione
- Intrappolamento di una microsfera
- Impostare il piano dell'immagine leggermente sopra il tallone con il controller software dello stadio del microscopio.
- Attivare le trappole utilizzando il software OTs e fare clic sul tallone nella finestra di imaging AUX della fotocamera (calibrato dopo il passaggio 6.4). Il confinamento riuscito del tallone da parte della trappola ottica ridurrà fortemente il movimento del tallone.
- Fai clic e trascina il tallone attraverso il citoplasma e posizionalo a una distanza di ~ 2 μm dall'involucro nucleare (Figura 3A). Assicurarsi che la traiettoria sia impostata in modo che la rientranza del tallone sia perpendicolare alla membrana nucleare.
- Facoltativamente, se necessario per le misurazioni della posizione del tallone rispetto alla trappola, eseguire la scansione della trappola attraverso il tallone per determinare la rigidità di intrappolamento, k [pN / μm] 54, quindi Δxbead = -F / k (vedere Discussione). Il modulo di micromanipolazione ottica utilizzato in questo protocollo ha una routine integrata per questo scopo.
- Aprite la finestra di dialogo Scansione particelle nel sottomenu Strumenti .
- Selezionare la trap che si desidera scansionare e Alta frequenza come metodo di scansione. Selezionate la direzione (x o y) della traiettoria di rientro per la misurazione della scansione del tallone.
- Apparirà una finestra con la misurazione della rigidità di intrappolamento. Nel grafico, trascinare i due cursori per selezionare l'area di abbondanza lineare corrispondente a F = -kx. L'adattamento lineare alla porzione di dati selezionata verrà aggiornato automaticamente.
NOTA: Impostare la posizione iniziale del tallone lontano dalla membrana cellulare (~5 μm), poiché le deflessioni del momento leggero all'interfaccia medio-cella influenzano l'appropriatezza delle misurazioni della forza. Se il nucleo si trova troppo vicino alla membrana cellulare, prova a far rientrare il nucleo dal sito opposto. Scartare la cella se non è possibile.
- Avviare l'acquisizione delle immagini facendo clic sul pulsante di acquisizione nel software di imaging.
- Avviare la posizione di trappola e il salvataggio dei dati di misurazione della forza facendo clic su Dati | Salva nella finestra di lettura forzata in tempo reale (aperta come nel passaggio 6.6.1).
NOTA: La trap ottica è dotata di un ingresso trigger che può essere collegato all'uscita di temporizzazione della telecamera. Pertanto, i dati di immagine e forza sono sincronizzati hardware e l'elettronica è in grado di mappare i cicli di trappola con il numero di fotogrammi delle immagini durante l'acquisizione. - Avviate la traiettoria caricata in precedenza facendo clic con il pulsante destro del mouse sul tallone e selezionando Avvia traiettoria (Start Trajectory).
- Attendere che la traiettoria sia terminata e che il sistema si stabilizzi.
- Salvataggio dei dati di misurazione della forza di arresto della trappola. Apparirà una finestra di dialogo di salvataggio dei dati.
NOTA: per ottimizzare l'archiviazione dei dati, i dati possono essere decimati selezionando il parametro di decimazione in questa finestra di dialogo (10, 100 o 1000). - Interrompere l'acquisizione delle immagini e tracciare i risultati nel software di post-elaborazione di scelta dell'utente.
- Se la microsfera viene persa durante la routine e il nucleo non può essere rientrato (Figura S2), scartare la misurazione e aumentare la potenza. Si noti che il passaggio 6.5 deve essere ripetuto. Nelle nostre mani, almeno il 95% delle routine viene completato con successo senza perdere il tallone dalla trappola.
Risultati
Microiniezione di perline di intrappolamento:
Microsfere iniettate nell'embrione di zebrafish unicellulare si diffondono su tutto il cappuccio animale durante la morfogenesi. Per una visualizzazione più chiara, abbiamo ripetuto il protocollo di iniezione con microsfere fluorescenti rosse e abbiamo scattato immagini volumetriche con il nostro microscopio confocale in diverse fasi dello sviluppo. Nella Figura 4A-D, le perle iniettate sono visualizzate nel citoplasma delle cellule staminali progenitrici in vivo a 5 hfp. Più tardi, le microsfere sono apparse sparse su tutto l'embrione a 24 hpf (Figura 4E). Gli embrioni in entrambe le fasi si sono sviluppati normalmente e i tassi di sopravvivenza erano paragonabili agli embrioni di controllo non iniettati o finti iniettati (vedere Figura S3). Ciò è coerente con altri studi che riportano una sopravvivenza imperturbabile del pesce zebra iniettato con perline fino a 5 giorni dopo la fecondazione55.
Il nostro microscopio confocale a disco rotante è compatibile con la microscopia a fluorescenza multicanale. Nella Figura 5A, mostriamo cellule staminali isolate con una o due perle nel citoplasma. Più etichette fluorescenti possono essere utilizzate per studiare diversi aspetti della cellula (Figura 5B). La morfologia nucleare può essere tracciata con un colorante Hoechst o utilizzando un'espressione di mRNA H2A::mCherry, mentre la membrana nucleare interna può essere analizzata con Lap2b-eGFP12. La dinamica della corteccia actomiosina, così come i livelli di calcio intracellulare, possono essere osservati con una linea transgenica My12.1::eGFP56 e Calbryte-520, rispettivamente. Il protocollo che è stato descritto qui mira a confrontare la meccanica del nucleo cellulare di cellule wildtype immobilizzate su substrati adesivi (in seguito denominati sospensione) e nel confinamento meccanico. Le cellule staminali isolate confinate in microcamere di altezza 10 μm hanno mostrato un parziale dispiegamento della membrana nucleare interna (INM) e un successivo aumento della contrattilità dell'actomiosina12. Nella Figura 5C, vengono mostrate cellule confinate con una o due perle nel citoplasma. Il confinamento riuscito sarà visibile attraverso cellule appiattite ed espanse con una sezione trasversale più ampia del nucleo. La membrana nucleare è ulteriormente dispiegata in cellule confinate e dovrebbe apparire levigata rispetto alle cellule in sospensione (Figura 5C).
Analisi forza-tempo e forza-deformazione
L'analisi dei risultati ottenuti dipende fortemente dal campione indagato e dalla questione di interesse e quindi non possono essere generalizzati qui. Ad esempio, un modo comune per analizzare la misurazione dell'indentazione consiste nell'estrarre un modulo di Young adattando un modello hertz modificato ai dati di rientro forzato57. Tuttavia, l'ipotesi per un tale trattamento deve essere attentamente valutata e potrebbe non essere sempre adeguatamente giustificata (come la struttura studiata è isotropa, omogenea, con elasticità lineare e rientranze più piccole del raggio del tallone). Consideriamo quindi solo misure indipendenti dal modello che consentono di confrontare il comportamento meccanico della struttura studiata tra diversi scenari sperimentali.
Come punto di partenza, la misurazione della pendenza della curva forza-spostamento a una certa profondità di rientranza fornisce una misura di una rigidità strutturale indipendente dal modello58 del nucleo. Questo valore può quindi essere raccolto da più campioni e confrontato tra diverse impostazioni sperimentali e perturbazioni del campione.
Misurazione dell'indentazione
Nelle righe seguenti, ci concentriamo sulla risposta meccanica del nucleo cellulare durante la deformazione cellulare in confinamento. Gli esperimenti nella fase 8 di questo protocollo in genere portano a picchi di forza fino a 200 pN per profondità di indentazione di circa 2-3 μm. Tuttavia, questi valori possono essere in gran parte diversi, a seconda del tipo di cellula e delle condizioni sperimentali, con nuclei più morbidi che portano a una forza inferiore per una data rientranza. È quindi necessario misurare con precisione la deformazione nucleare, insieme alla forza, per un'accurata caratterizzazione meccanica del nucleo cellulare. In questa sezione, otterremo la rigidità nucleare della cella da misurazioni di rientranza della forza rappresentativa.
Nella Figura 6, mostriamo le deformazioni dei lati distale e prossimale di un nucleo in una cellula sospesa e confinata. Si può osservare un ricco comportamento meccanico. In una tipica cella sospesa su un substrato adesivo, il nucleo era fortemente rientrato dal tallone, ma anche leggermente spostato in caso di eventi di spinta ripetitivi. Abbiamo misurato la rientranza del tallone sul nucleo analizzando i chimografi ottenuti dall'imaging a fluorescenza dei nuclei cellulari colorati di Hoechst. I kymograph sono stati facilmente calcolati utilizzando il plug-in Multi Kymograph di Fiji lungo la direzione di rientro (Figura 6A, B) e importati in Matlab (versione 2021, Mathworks) per ulteriori elaborazioni. Una funzione di passo è stata adattata al profilo di intensità grezza con l'obiettivo di tracciare i bordi delimitanti del nucleo lungo la traiettoria della routine di rientranza. Come si può vedere, contiene informazioni accurate sul cambiamento di forma nucleare (Figura 6 e Figura S2). Abbiamo usato la seguente curva a doppio sigmoide come versione analitica di una funzione step:
 (Equazione 1)
(Equazione 1)
Qui, x1 e x2 denotano i bordi distali e prossimali del nucleo, mentre A e B sono i valori massimi e di grigio di sfondo del canale blu (colorante di Hoechst) dell'immagine (Figura 6B). La larghezza del bordo è stata considerata (e0 = 0,25 mm). Mentre il bordo del nucleo prossimale rientrato (x2) ha seguito la traiettoria applicata dalla routine di trappola ottica dopo il contatto microsfera-nucleo, il bordo distale opposto (x1) mostra dinamiche di rilassamento come previsto per un materiale viscoelastico come il citoplasma (Figura 6D). Al contrario, i nuclei in cellule confinate in microcamere alte 10 μm non mostrano tale comportamento di traslocazione del nucleo al momento della rientranza all'interno della cellula (Figura 6B,D). Mostrato anche nella Figura 6D, i bordi posteriori dei nuclei rimangono inalterati dalla spinta del tallone dal lato prossimale, molto probabilmente a causa delle forze più forti derivanti dalla contrattilità cellulare e dall'attrito che agiscono contro la forza di rientranza. Per ottenere la corretta profondità di deformazione, lo spostamento x1 è stato sottratto dalla misura rientrata x2: Δx = x2 - x1 (vedi anche Figura 6D).
Forzare l'analisi dei dati
La forza che causa la deformazione nucleare è stata misurata dal cambiamento del momento della luce originato dalla microperla intrappolata otticamente (Figura 7A). La forza all'applicazione di traiettorie trapezoidali (passo 8.4.3, Figura 7B) inizialmente aumentava linearmente fino a quando la trappola smetteva di muoversi, ma poi si rilassava a un valore di stato stazionario. Questo comportamento indicava un materiale viscoelastico che mostrava moduli di perdita e stoccaggio. Subito dopo l'evento di indentazione, la forza ha raggiunto un valore di picco, Fp, seguito da un rilassamento dello stress (Figura 7C):
 (Equazione 2)
(Equazione 2)
dove F0 è la forza immagazzinata per la componente elastica e f(t) è una funzione di rilassamento senza dimensione. Abbiamo analizzato questo comportamento in tre modi:
1. Considerare un solido lineare standard con un rilassamento esponenziale dello stress, cioè f(t) = e-t/τ, schematicamente rappresentato nell'inserto della Figura 7C.
2. Usando un decadimento generale, doppio esponenziale:
F(t) = A + B1e-t/τ1 + B2e-t/τ2.
3. Usando una legge di potenza seguita da un decadimento esponenziale59:
f(t) = t-pe-t/τ, montato nella figura 7C.
Mentre l'adattamento per il modello 1 può essere eseguito in modo semplice, si consiglia di stimare le ipotesi iniziali per (τ1, τ2) e (p, τ) per i modelli 2 e 3, rispettivamente. Questo può essere eseguito, rispettivamente, adattando le linee sui dati in scale logaritmica-versus-lineare (Figura 7D, a sinistra) e logaritmica-versus-logaritmica (Figura 7D, a destra). La tabella S3 riepiloga i risultati per l'esempio analizzato nella Figura 7. Nella sezione seguente, considereremo la combinazione di una legge di potenza e una legge esponenziale per la caratterizzazione della meccanica del nucleo cellulare.
Relazione di spostamento della forza
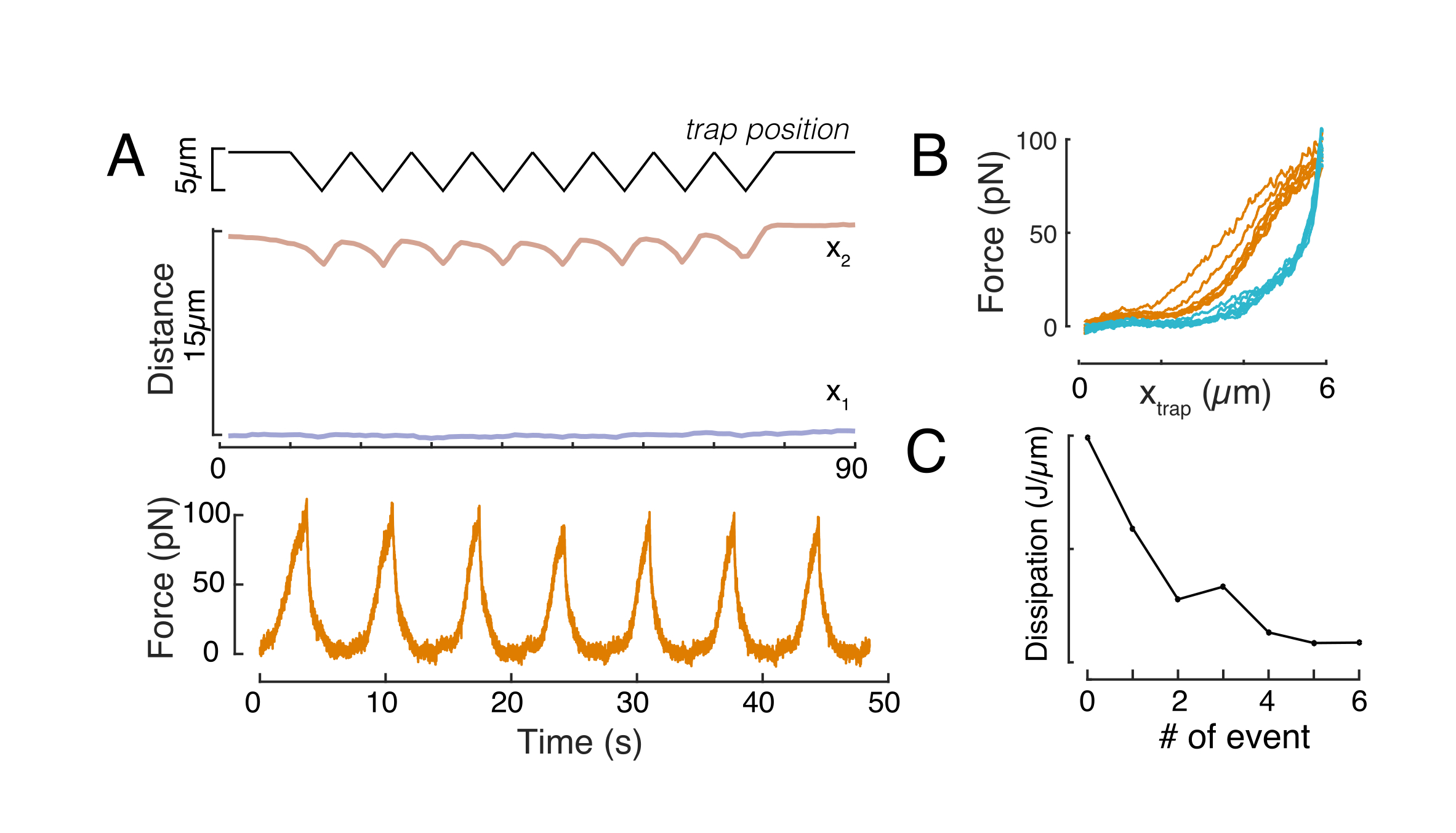
Allo stesso modo, il set-up sperimentale descritto può essere utilizzato per ottenere la relazione forza-spostamento di più eventi di indentazione. Eseguendo routine triangolari (passo 8.4.4, Figura 8A), è possibile correlare la forza alla deformazione e tracciare una curva forza-rientro. Un risultato esemplare è mostrato nella Figura 8B, in cui una linea di base piatta ha cambiato dolcemente pendenza una volta che il tallone è entrato in contatto con il nucleo. Identificare il vero punto di contatto nei dati rumorosi è una sfida e bisogna fare attenzione per vedere se la regione di contatto è adatta ai modelli elastici60. In questo particolare esperimento, si è anche potuto vedere che le rientranze successive si traducono in curve con punti di contatto più profondi, indicativi di un recupero troppo lento della forma nucleare dopo la retrazione del tallone e un cambiamento nel ciclo isteretico definito dalle proprietà del materiale viscoelastico del nucleo (Figura 8C). Pertanto, il ricercatore dovrebbe essere consapevole se ciò accade e incorporarlo nella pipeline analitica o limitare il numero di misurazioni successive in modo tale che questo effetto non modifichi la misurazione.
Meccanica del nucleo in cellule in sospensione e confinamento inferiore a 10 μm
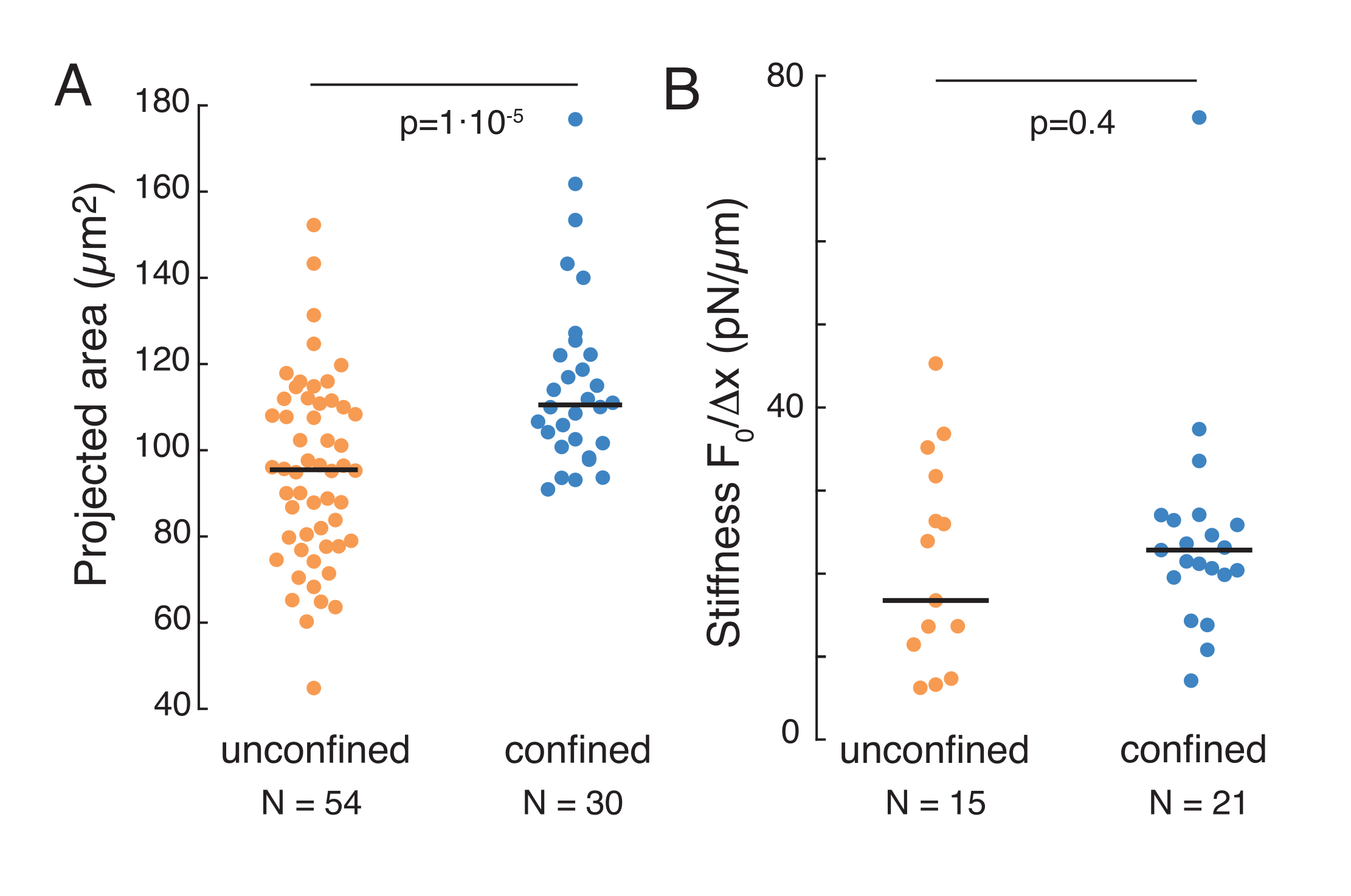
Il suddetto approccio è stato utilizzato per analizzare le dinamiche del rilassamento dello stress del nucleo in cellule sospese su substrati adesivi e cellule confinate. I nostri risultati mostrano che il confinamento si traduce in un'espansione dell'area prevista (Figura 9A), ma in un cambiamento insignificante nella rigidità nucleare (Figura 9B). Abbiamo misurato un rilassamento simile con τ = 6,08 ± 1,1 s (non confinato) e τ = 4,00 ± 0,6 s (confinamento), che indica una rapida dissipazione viscoelastica, seguita da un valore di forza immagazzinata che corrisponde al modulo elastico del nucleo. Al fine di tenere conto delle variazioni sperimentali, che possono essere prodotte da diverse condizioni iniziali nelle routine di indentazione, le forze memorizzate misurate sono state normalizzate alla profondità di rientro, come  . Questo parametro tiene conto della rigidità del nucleo e descrive la forza, o lo stress, necessaria per una certa rientranza. Abbiamo ottenuto una rigidità simile sotto confinamento e in celle non confinate: = 20,1 ± 12,6 pN/μm e = 24,6 ± 13,6 pN/μm (deviazione media ± standard), rispettivamente.
. Questo parametro tiene conto della rigidità del nucleo e descrive la forza, o lo stress, necessaria per una certa rientranza. Abbiamo ottenuto una rigidità simile sotto confinamento e in celle non confinate: = 20,1 ± 12,6 pN/μm e = 24,6 ± 13,6 pN/μm (deviazione media ± standard), rispettivamente.

Figura 1: Microiniezione di embrioni di zebrafish allo stadio di una cellula (zigote). (A) Piastra di iniezione: per l'iniezione viene utilizzata una piastra di iniezione di forma triangolare. La piastra è composta per l'1% da agarosio ultrapuro in E3 (mezzo dell'uovo). Le viste superiore e laterale sono mostrate sulla destra. (B) Posizionamento dell'embrione: orientare delicatamente gli embrioni usando un pennello e orientare in modo che l'una cellula sia chiaramente visibile e facilmente accessibile con l'ago. Suggeriamo di orientare gli embrioni con la cellula situata nel lato opposto dell'ago, come mostrato nello schizzo. (C) Procedura di iniezione nell'embrione monocellulare: perforare con l'ago il corion che circonda l'embrione e la singola cellula. Assicurarsi che la punta dell'ago sia all'interno della cellula e rilasciare la pressione per l'iniezione. (D) Incubare gli embrioni a 28-31 °C fino a quando non si sviluppano fino allo stadio di blastula (sfera) (4 hpf). Eseguire il protocollo di isolamento cellulare e la colorazione cellulare (fase 2) e preparare la camera di intrappolamento ottico con celle isolate in sospensione e/o confinamento combinate con il corrispondente rivestimento superficiale del substrato (fase 3). Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: Preparazione dell'apparecchio ottico a pinzetta. (A) Strati di rivestimento spin-coating di PDMS con un'altezza definita su piatti con fondo di vetro. La caduta del PDMS si diffonderà uniformemente a causa della forza centrifuga. (B) Preparazione della camera campione fuori dallo strato PDMS. 1: tagliare un quadrato con un bisturi, 2: rivestire il pozzo interno con concanavalina A (ConA), lavare e seminare le cellule; 3: coprire con una diapositiva di vetro o coprire lo slip per sigillare il pozzo. (C) Immagine del taglio quadrato con un bisturi e rimozione del PDMS bene con pinza. (D) Montare la lente di raccolta del sensore di forza ottica sopra la camera di intrappolamento. Una goccia di olio per immersione funge da mezzo di immersione tra la lente di raccolta e il coperchio superiore in vetro. Schema non scalabile. Prestare attenzione mentre si abbassa la lente di raccolta per non toccare il coperchio di vetro della piatta campione. (E) Immagine dell'unità di rilevamento della forza a contatto con il campione. (F) Schema dell'assetto sperimentale. Il modulo di micromanipolazione ottica utilizza un raggio laser a onda continua (5W, λ = 1064 nm) con controllo della potenza attraverso una piastra a mezza onda (HWP) e uno splitter a fascio polarizzatore (BS). Dopo essere stato modulato con una coppia di AOD, viene accoppiato alla porta di epifluorescenza superiore di un microscopio invertito. Il raggio laser viene quindi riflesso da uno specchio dicroico a passaggio corto (IR-DM) da 950 nm, che consente la trasmissione dell'eccitazione e dell'emissione di fluorescenza. Il laser di intrappolamento viene guidato nella porta di epifluorescenza posteriore del microscopio (torretta superiore). Gli OT sono creati sul piano focale di un obiettivo ad immersione in acqua (60x, NA = 1,2). Il sensore di forza ottica viene sottoposto dalla torretta del microscopio e cattura la luce laser che emerge dagli OT con una lente ad alta NA, ad immersione in olio. Allo stesso tempo, il sensore di forza consente l'illuminazione del campo luminoso. L'unità confocale a disco rotante è accoppiata alla porta sinistra. È dotato di due motori laser integrati (ILE) che controllano sette laser di eccitazione a fluorescenza e due telecamere sCMOS retroilluminate, consentendo l'imaging a doppio fluoroforo in parallelo Abb: TI, Transilluminator; FS, field stop; AOD, deflettore acustooptical; HWP, piastra a mezza onda; CAM, fotocamera (G) Fotografia dell'apparecchiatura di intrappolamento ottico. Il cerchio rosso indica la lente di Bertrand, che può essere commutata manualmente nel percorso ottico. Fare clic qui per visualizzare una versione più grande di questa figura.
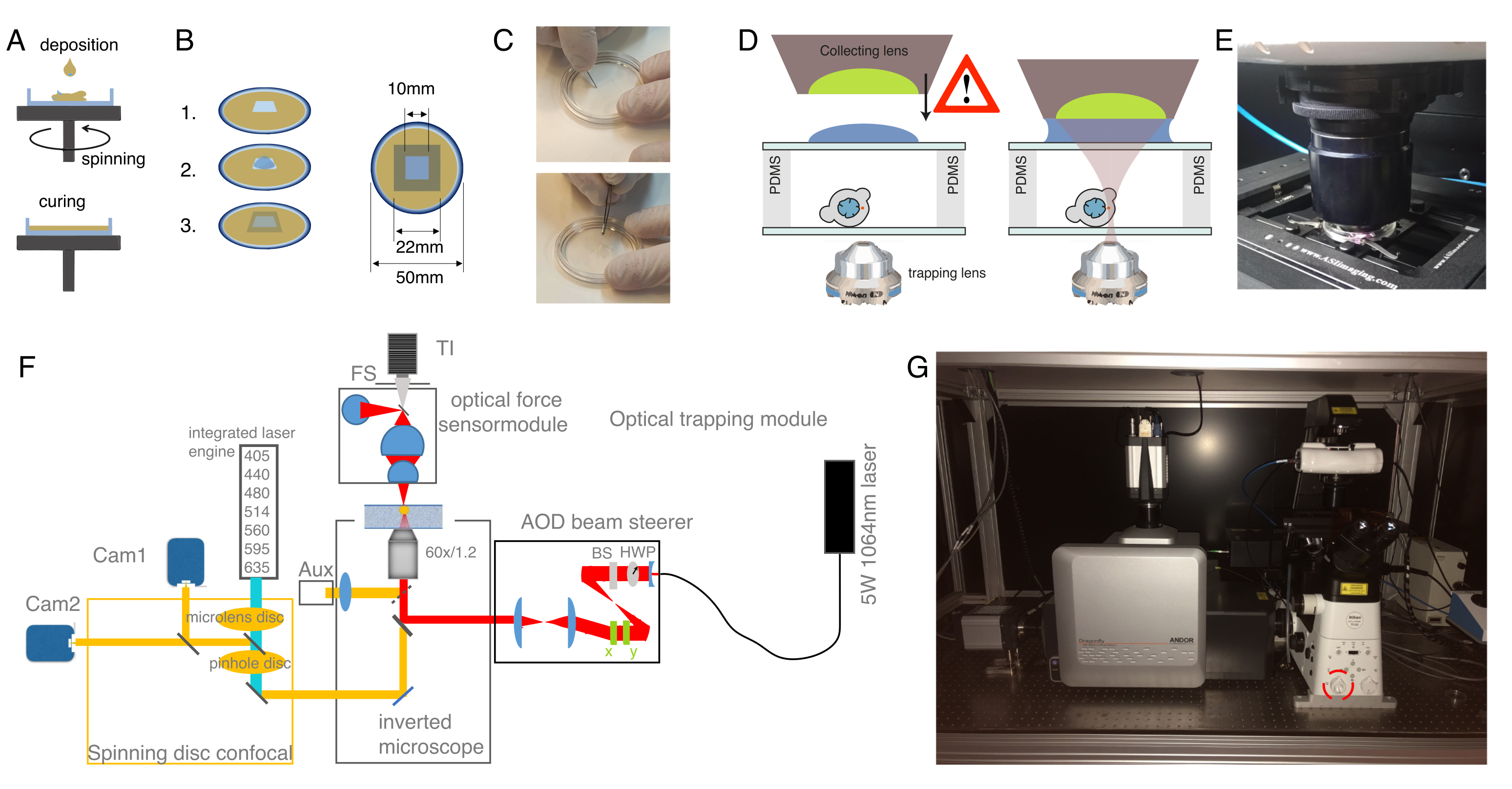{kind=link}

Figura 3: Scelta dei campioni e dei parametri giusti. (A) Immagine rappresentativa di una cellula staminale progenitrice di zebrafish isolata con una singola microsfera posizionata abbastanza vicino al nucleo per eseguire l'esperimento di indentazione. Barra della scala = 10 μm. (B) Traiettoria della trappola esemplare; profondità di indentazione 5 μm; velocità di indentazione = 5 μm/s; tempo di rilassamento 10 s. Fare clic qui per visualizzare una versione più grande di questa figura.
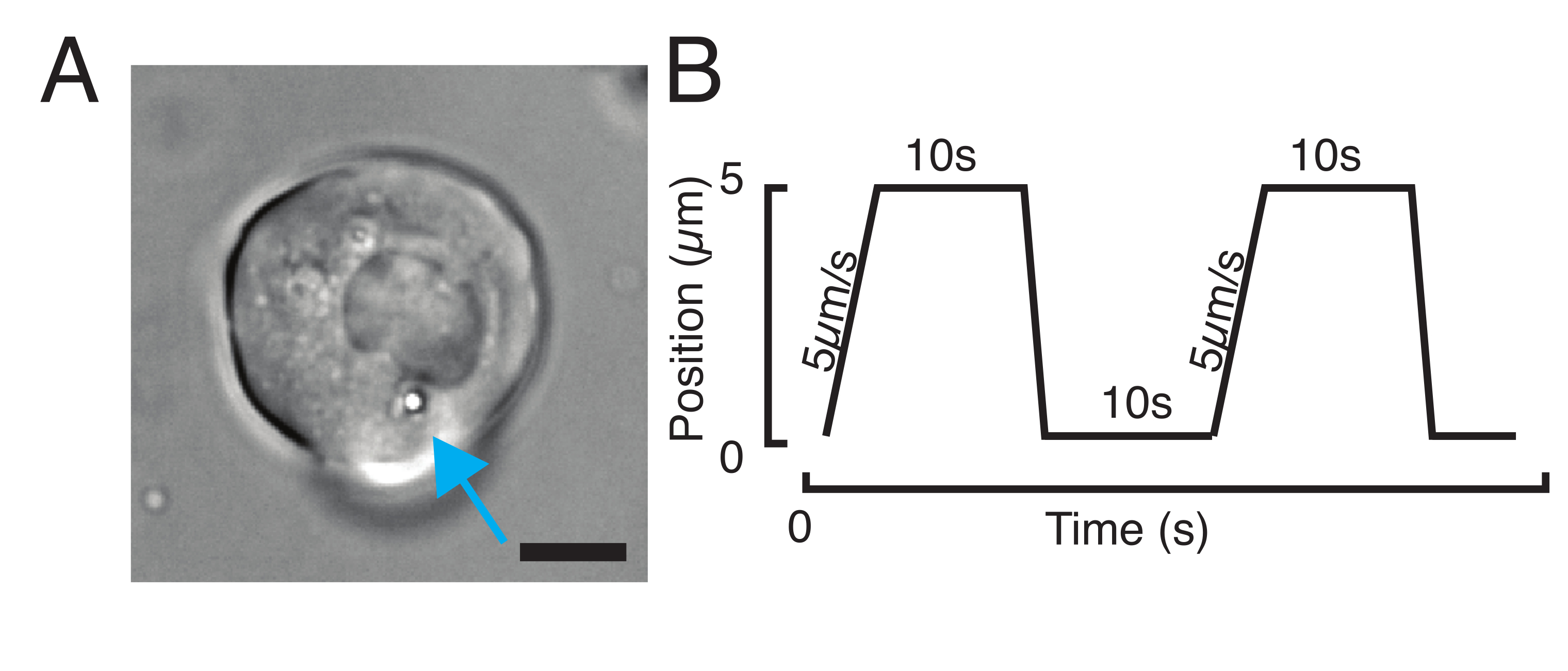{kind=link}

Figura 4: Localizzazione di microsfere all'interno di embrioni di zebrafish durante lo sviluppo. 0,5 nL di 1 μm di perle fluorescenti rosse vengono iniettate insieme all'mRNA GPI-GFP (100 pg/embrione, membrana plasmatica) negli embrioni WT per visualizzare le localizzazioni delle perline. (A-D) Distribuzione della microsfera 5 h dopo l'iniezione all'interno di un embrione montato in agarosio allo 0,75%. (A) Immagine a campo luminoso e fluorescenza. Le perle sono disperse omogeneamente attraverso il tessuto embrionale come si vede in una micrografia confocale. (B) Proiezione massima della fluorescenza confocale z-stack. Le perline sono codificate a colori dal viola al giallo in base alla loro posizione z nella pila di immagini. Il viola/magenta corrisponde alle perle/cellule più esterne (EVL; strato epiteliale avvolgente; o cellule staminali progenitrici situate vicino alla superficie EVL), il giallo corrisponde alle perle interne (cellule profonde progenitrici), come mostrato nello schizzo a destra. (C) Taglio e proiezione massima di una sottofia di (B) corrispondente alla regione nella scatola arancione: una grande frazione di celle profonde contiene 1-2 perline. (D) Taglio e proiezione massima di una sottofia di (B) corrispondente alla scatola magenta: alcune celle EVL contengono 1-2 perline. (E) Immagine di campo luminoso e proiezione massima di uno z-stack di un embrione da 24 hpf montato in agarosio allo 0,75% e anestetizzato con tricaina. Gli embrioni sono stati pre-incubati con tricaina per 15 minuti. Da sinistra a destra: microsfere (diametro 1 μm), GPI-GFP e sovrapposizione dell'immagine. Le perle distribuite su tutto il corpo dell'embrione. Dimensione della barra di scala indicata in ciascun pannello. Fare clic qui per visualizzare una versione più grande di questa figura.
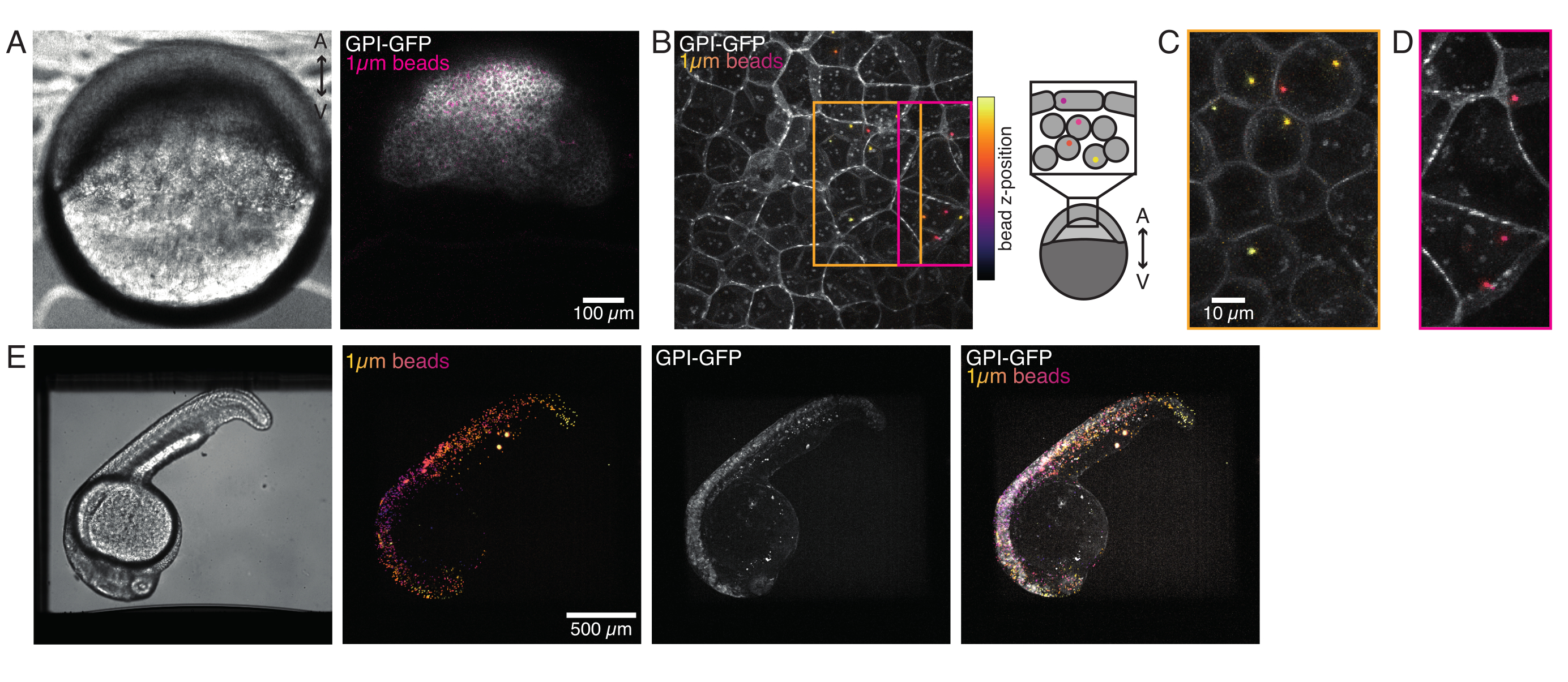{kind=link}

Figura 5: Cellule staminali progenitrici di zebrafish isolate con etichettatura diversa. (A) Immagine al microscopio ottico a trasmissione di cellule in sospensione con 1 (in alto) o 2 (in basso) perline iniettate. Le frecce ciano puntano alle perline. (B) Immagini confocali fluorescenti di cellule in sospensione con colorazioni diverse. In alto a sinistra: Lap2b-eGFP (membrana nucleare interna, 80 pg/embrione) e H2A-mCherry. In alto a destra: GPI-GFP (membrana plasmatica, 100 pg/embrione) e DNA-Hoechst (colorato come descritto nel paragrafo 2). In basso a sinistra: MyI12.1-eGFP (linea transgenica) e DNA-Hoechst. In basso a destra: Calbryte488 e DNA-Hoechst (macchiato come descritto nel paragrafo 2). (C) Immagine al microscopio ottico a trasmissione di cellule confinate con 1 (in alto) o 2 (in basso) perline iniettate. Le frecce ciano puntano alle perline. Barre di scala = 10 μm. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 6: Stima della deformazione nucleare da filmati di dischi rotanti. (A,B) Time-lapse di un esperimento di indentazione del nucleo in (A) una cella sospesa e (B) una cellula confinata. Scala bar 10 μm. Le istantanee rappresentative di un nucleo marcato con Hoechst sono mostrate 5 s prima, durante e 5 s dopo la rientranza con una microsfera intrappolata otticamente (punta di freccia bianca). Kymografi lungo il segmento di rientro (linea rossa, pannello di destra). x1 e x2 sono i confini distali e prossimali (vicino al tallone) del nucleo durante l'esperimento di indentazione estratto dall'adattamento del profilo di intensità all'equazione 1. (C) Profili di intensità lungo il segmento di rientro per tre diversi fotogrammi (prima, durante e dopo l'indentazione) e montati sull'equazione 1 per valutare le posizioni distale, x1 e prossimale, x2, dei bordi del nucleo. (D) Traiettorie rappresentative di x1(t) in blu e x2(t) in ambra durante un esperimento di indentazione di cellule sospese e confinate (10 μm). Le aree ombreggiate indicano la rientranza, la distanza tra x1 e x2 indica il diametro del nucleo. Fare clic qui per visualizzare una versione più grande di questa figura.
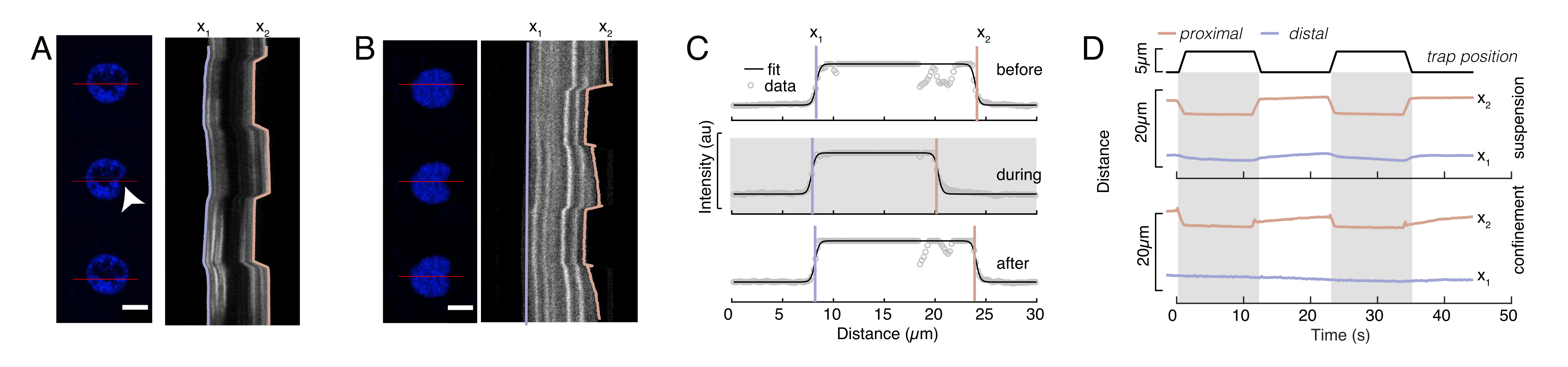{kind=link}

Figura 7: Elaborazione forzata del segnale. (A) Schema di una microsfera intrappolata otticamente che deforma il nucleo cellulare al momento dell'indentazione. La membrana nucleare e le forze ottiche sono indicate dalle frecce nere. La variazione della quantità di moto del fascio è indicata dalla freccia verde Pout. (B) Traiettoria della trappola (in alto) e forza (in basso) sperimentate dalla microsfera intrappolata otticamente durante un ripetuto esperimento di indentazione nucleare. (C) Decadimento del rilassamento della forza dopo il picco di forza alla massima profondità di rientro. L'inserto mostra uno schema di solido lineare standard la cui dinamica si avvicina alle osservazioni fenomenologiche qui. (D) A sinistra: logaritmo della forza normalizzata rispetto al tempo. Le aree ombreggiate indicano la porzione di dati utilizzata per adattarsi al doppio decadimento esponenziale (linee rosse). A destra: logaritmo della forza normalizzata contro il logaritmo del tempo. L'area ombreggiata indica la porzione di dati utilizzata per adattarsi alla legge di potenza. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 8: Routine di rientro forzato con spostamenti di trappole triangolari. (A) Traiettoria rappresentativa di x1(t) in blu e x2(t) in ambra durante un esperimento di rientranza triangolare effettuato su una cellula in altezza di confinamento di 10 μm. In alto: posizione della trappola. Al centro: analisi della forma del nucleo. La distanza tra x1 e x2 indica il diametro del nucleo. In basso: Segnale di forza. (B) Posizione forza vs trappola per otto rientranze consecutive. (C) Evoluzione della dissipazione, derivata dall'isteresi tra la parte di avvicinamento e di prelievo della curva f-d, del nucleo per ogni successivo evento di indentazione. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 9. Proprietà nucleari delle cellule in sospensione (superficie adesiva) e confinamento da routine trapezoidali. (A) Area proiettata del nucleo da cellule in sospensione e sotto confinamento di 10 μm. La barra nera rappresenta la mediana. (B) Rigidità nucleare delle cellule in sospensione e sotto confinamento. La barra nera rappresenta la mediana. Valori P derivati dal test Kruskal-Wallis utilizzando MatLab. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Tabella supplementare 1: Traiettoria trapezoidale definita dal software delle pinzette ottiche. La prima (seconda) riga è la distanza x (y) in cui la trappola verrà spostata linearmente. Nella terza riga, la durata di un determinato passaggio è impostata in secondi. Questa traiettoria è composta da sette punti e corrisponde al trapezio caricato due volte contro il nucleo in Figura 7B. Fare clic qui per scaricare questa tabella.
Tabella supplementare 2: Traiettoria triangolare definita dal software delle pinzette ottiche. Analogamente alla Tabella 2, questa traiettoria è composta da 16 punti, corrispondenti a otto eventi di indentazione ad una profondità di 5 μm e una velocità di 2,5 μm/s. Fare clic qui per scaricare questa tabella.
Tabella supplementare 3: Parametri di adattamento per i dati nella Figura 7. IG: ipotesi iniziale. Fare clic qui per scaricare questa tabella.
Figura supplementare S1: Allineamento del sensore di forza ottica e compensazione della linea di base del momento. (A) Field stop ripreso dalla telecamera ausiliaria (AUX, Figura 2) attraverso l'obiettivo Bertrand. Una bolla d'aria appare visibile nell'olio di immersione, che non è visibile attraverso l'oculare. (B) Percorso ottico pulito. Per un allineamento accurato, aprire l'arresto del campo e farlo coincidere con il cono di luce NA = 1.2. (C) Immagine del piano campione. Il quadrato rosso indica l'area di lavoro OT. Barra della scala: 20 μm. (D) Potenza della trappola misurata attraverso il FOV, lungo doppie frecce bianche indicate in C. In rosso, trappola la variazione di potenza quando non viene applicata alcuna correzione. In blu, la potenza della trappola è stata corretta su tutto il campo visivo. (E) Componente X della linea di base della quantità di moto lungo lo stesso intervallo. In rosso, traccia non corretta. In blu, traccia corretta per la potenza della trappola. In verde, traccia corretta per la linea di base del momento utilizzando Global Offset Compensation nel software del produttore. (F) Come in E, per la componente Y. Si noti che durante il normale funzionamento, i componenti ombreggiati vengono utilizzati per la meccanica e le misurazioni della forza, ad esempio la componente di forza x durante il movimento lungo la coordinata x e la componente di forza y durante il movimento lungo l'asse y. Dopo aver implementato tutte le correzioni, si ottiene un rumore RMSD di <0,5 pN. Fare clic qui per scaricare questo file.
Figura supplementare S2: Una routine fallita a causa di trappole deboli. (A) Kymograph che mostra una rientranza del nucleo da una routine fallita. Solo brevi deformazioni transitorie sono visibili a causa di una fuga del tallone dalla trappola. È importante sottolineare che il laser di intrappolamento si muove ancora senza perline per completare la traiettoria predefinita (linea tratteggiata verde). Barra della scala = 10 μm. (B) In alto: posizione della trappola rispetto al tempo. Al centro: risultato del tracciamento dei bordi del bordo del nucleo prossimale e distale rientrato. Si noti che il bordo distale non si muove senza la rientranza come comunemente osservato per le routine completate su cellule isolate su substrati adesivi. In basso: Forza contro tempo che mostra la perdita della microsfera indicata da una riduzione del rumore termico e da un improvviso calo a forza zero. Fare clic qui per scaricare questo file.
Figura supplementare S3: Sopravvivenza degli embrioni iniettati. Gli embrioni iniettati con 1 μm di perline e 100 pg/embrione di mRNA alle concentrazioni delineate nel protocollo sono stati confrontati con embrioni non iniettati e non mostrano differenze significative 24 ore dopo la fecondazione. La deviazione media e standard di tre esperimenti indipendenti con N > 21 embrioni per condizione per ciascun esperimento. Fare clic qui per scaricare questo file.
Discussione
In questo protocollo, descriviamo un metodo unico per interrogare le proprietà meccaniche del nucleo cellulare all'interno delle cellule viventi. A differenza di altre tecniche di spettroscopia di forza, l'intrappolamento ottico non invasivo ci ha permesso di disaccoppiare il contributo della membrana cellulare e del citoscheletro dalla rigidità nucleare cellulare. È importante sottolineare che la micromanipolazione ottica è compatibile con la microscopia multimodale, che consentirà allo sperimentatore di studiare diversi processi coinvolti nella meccanobiologia nucleare cellulare. Come risultato rappresentativo, abbiamo usato la colorazione DNA-Hoechst per misurare la deformazione del nucleo dopo l'indentazione eseguita da forze dell'ordine di diverse centinaia di picoNewton.
Potenziali applicazioni del nostro metodo al di là degli esempi delineati in questo protocollo
La possibilità di estrarre informazioni meccaniche quantitative da misurazioni all'interno di cellule viventi senza perturbazioni esterne consente una pletora di opportunità senza precedenti che stanno appena iniziando ad essere esplorate. Pertanto, il protocollo presentato della nostra piattaforma di micromanipolazione ottica può essere esteso a esperimenti più complessi con grande versatilità. I deflettori acusto-ottici (AOD) possono generare più trappole ottiche per misurazioni sincrone della forza in diverse posizioni cellulari, nonché possono essere utilizzati per la microreologia attiva in un'ampia gamma di frequenze51,61. Come è stato accennato, la risposta di forza al momento dell'indentazione può superare la massima forza di intrappolamento, portando a una fuga del tallone dalla trappola ottica. In questo caso, è possibile configurare un force feedback con l'AOD per bloccare la forza ottica. Tutto sommato, molteplici approcci microreologici, come il rilassamento dello stress descritto in questo protocollo, ma anche la microreologia attiva o la conformità allo scorrimento, possono essere ottenuti sperimentalmente con questa piattaforma e analizzati a fondo da nuovi pacchetti software61,62,63,64,65 . Inoltre, l'applicazione delle forze non è limitata al nucleo, ma potrebbe in linea di principio essere effettuata per misurare diverse strutture intracellulari e in tessuti complessi, come dimostrato per intrappolare i globuli rossi che fluiscono all'interno di vasi sanguigni intatti66,67 o intrappolare e deformare cloroplasti e mitocondri68 . La calibrazione del momento luminoso è indipendente dalla forma e dalle dimensioni dell'oggetto intrappolato, consentendo quindi misurazioni di forza diretta su qualsiasi sonda di forza con forma arbitraria38,39. L'uso di microsfere iniettate ci ha permesso di applicare forze elevate sul nucleo con una potenza laser relativamente bassa rispetto alla manipolazione diretta delle strutture cellulari69,70,71. Tuttavia, data una differenza di indice di rifrazione sufficientemente elevata, non è necessaria alcuna sonda di forza applicata esternamente e gli organelli intracellulari possono essere manipolati direttamente senza perline iniettate (osservazioni e riferimenti non pubblicati70).
Potenziali modifiche del nostro metodo per estendere le applicazioni
Diverse dimensioni di microsfere possono essere iniettate a seconda dell'esperimento, ma i relativi controlli devono essere fatti. Ad esempio, per studiare le cellule nelle fasi successive possono essere iniettate perline più piccole. Ciò ridurrà la forza massima che può essere esercitata dalla trappola ottica (come mostrato nel riferimento55). Perle più grandi possono essere iniettate per esercitare forze più elevate, ma queste potrebbero influenzare lo sviluppo dell'embrione a seconda delle loro dimensioni o dello stadio di interesse. Negli esperimenti in cui l'iniezione di microsfere non è un'opzione, vari organelli che mostrano differenze negli indici di rifrazione rispetto a quelli del citoplasma possono ancora essere manipolati otticamente, dando origine a forze ottiche misurabili dai cambiamenti del momento luminoso42. Come accennato in precedenza, questi metodi sono stati impiegati da Bambardekar et al. per deformare le giunzioni cellula-cellula nell'embrione di Drosophila70. Allo stesso modo, il nucleo della cellula ha un indice di rifrazione inferiore rispetto al mezzo circostante44, che consente una rientranza senza perline (osservazioni non pubblicate e riferimento72) anche se con una forza di intrappolamento inferiore. Pertanto, il nucleo non può essere intrappolato facilmente e sfugge alla trappola.
Il distanziatore PDMS rivestito di spin è fabbricato tramite un metodo comodo e veloce, ma potrebbe essere fuori dalla portata dei laboratori senza accesso a una struttura di micro / nanofabbricazione o laboratori di ingegneria. Pertanto, il distanziatore può essere facilmente assemblato da nastro da laboratorio o parafilm (passaggio 4). Il protocollo può anche essere adattato producendo canali microfluidici che automatizzano la consegna di singole celle in pozzi di misura predefiniti o in una camera con un'altezza definita per stimare l'effetto di confinamento all'interno dello stesso campione. Tuttavia, tali dispositivi microfluidici devono essere progettati in modo da adattarsi allo spazio tra l'obiettivo del microscopio e la lente di raccolta del sensore di forza ottica, di circa 2 mm (vedere punto 3). Si noti che il sensore di forza ottica deve essere posizionato all'altezza appropriata in modo che nessuna aberrazione ottica da sfocatura influisca sulla misurazione del momento del fotone.
Altre modifiche potrebbero includere il cambiamento dei reporter biologici. Abbiamo scoperto che la fluorescenza di Hoechst sanguina spettralmente nel canale GFP e quindi favoriamo la combinazione con l'istone marcato con mCherry come marcatore nucleare per la misurazione simultanea in due canali fluorescenti. In alternativa, la deformazione nucleare può essere facilmente tracciata con un'etichetta mirata alla membrana nucleare interna come Lap2b-GFP (Figura 2).
L'indentazione sul nucleo cellulare era dell'ordine di 2-3 micron, che potevamo misurare con precisione mediante l'analisi dell'immagine della microscopia confocale a disco rotante limitata alla diffrazione. Nel caso di nuclei più rigidi o forze più piccole, l'indentazione sarà a malapena misurabile usando questo approccio. Tuttavia, le pinzette ottiche calibrate con forza assoluta possono anche essere calibrate per le misurazioni di posizione del tallone intrappolato in situ utilizzando l'interferometria BFP con precisione nanometrica51. Utilizzando questo approccio, il segnale di tensione e il sensore di forza ottica possono essere tradotti nella posizione della sonda intrappolata attraverso il parametro β [nm/V], mentre il parametro invariante α [pN/V] produce valori di forza attraverso la suddetta calibrazione del momento luminoso41 (vedi sotto per i dettagli).
Risoluzione dei problemi
Abbiamo scoperto che durante l'esperimento potrebbero verificarsi le seguenti sfide:
Non si forma alcuna trappola stabile e la microsfera sfugge facilmente
Qualsiasi sporcizia sull'obiettivo del microscopio o su un collare di correzione disallineato potrebbe portare al fallimento di una trappola stabile. Se non si trova una soluzione immediata, misurare la funzione point-spread della lente dell'obiettivo. Se il campione di interesse è in profondità all'interno di un tessuto otticamente denso, la messa a fuoco laser potrebbe subire gravi aberrazioni ottiche che portano a un intrappolamento instabile (questo effetto è solitamente trascurabile nelle cellule isolate, ma diventa più evidente nei tessuti più spessi). Per un'elevata rigidità, la forza di ripristino del nucleo potrebbe superare la forza di fuga della trappola, in modo tale che la microsfera venga persa e la routine di indentazione fallisca. Inizialmente, il bordo della membrana nucleare prossimale alla trappola ottica diventa difficilmente rientrato (Figura S2A). Quando ciò si verifica, il laser di intrappolamento non è più influenzato dalla forza e dal movimento browniano, che porta a una caduta di forza a zero e una diminuzione del rumore del segnale (Figura S2B). Nel caso in cui ciò accada, la potenza del laser può essere aumentata per avere una trappola più forte, l'ampiezza della traiettoria trapezoidale che spinge il tallone nel nucleo può essere ridotta o la posizione iniziale della microsfere intrappolata può essere spostata più lontano dal nucleo.
La cellula si muove durante la stimolazione
Se le cellule non sono sufficientemente attaccate, la trappola del gradiente ottico muoverà le cellule durante l'esecuzione della routine di indentazione intracellulare, in modo tale che le forze e la meccanica sottostante del nucleo siano artefatte. Per evitare lo spostamento dell'intera cellula, si consiglia di aumentare la concentrazione di molecole di adesione cellulare sulla superficie, ad esempio ConA.
Compensazione iniziale del momentum
Se nella piattaforma OT non è disponibile una routine iniziale di compensazione del momento (fase 6.5), è necessario correggere un segnale di base artificiale indipendente dalla forza. Questo è visibile come una pendenza sulla curva di forza anche senza perline intrappolate (Figura S1E). Per eseguire la correzione, la stessa traiettoria deve essere eseguita senza una perlina, al di fuori della cella esattamente nella stessa posizione. Per questo, spostare la cella lontano dalla trappola usando il controllo del palcoscenico. Come riferimento, l'offset di forza cambia 5 pN attraverso il FOV a 200 mW nel nostro sistema; quindi, diventa trascurabile per traiettorie brevi. In alternativa, è possibile utilizzare una fase di scansione piezoelettrica per spostare le cellule sul campione, lasciando costante la posizione del laser.
Passaggi critici del protocollo presentato
Le microsfere devono essere iniettate allo stadio destro di 1 cellula per garantire la massima distribuzione sull'embrione. Le perline non devono essere fluorescenti in modo che la luce non perda nei canali fluorescenti utilizzati per l'imaging. Ad esempio, anche le tipiche perle rosso-fluorescenti sono chiaramente visibili nel canale blu utilizzato per l'imaging del nucleo cellulare dopo la colorazione di Hoechst a causa della loro luminosità (eccitazione: 405 nm; emissione: 445 nm). L'attacco stabile della cellula al substrato è fondamentale per prevenire lo spostamento laterale durante la routine di rientranza. Se la cellula si muove durante la routine, le forze sono sottovalutate. Se ciò dovesse accadere frequentemente, ottimizzare il protocollo di allegato. Per le cellule di coltura tissutale, altre proteine di adesione cellulare, come la fibronectina, il collagene o la poli-L-lisina portano ad un attaccamento soddisfacente (osservazioni non pubblicate). Durante il confinamento, le cellule sono sottoposte a un improvviso e grave stress meccanico. Ciò può causare danni alle cellule e spesso lo sperimentatore incontrerà cellule scoppiate se la procedura non viene eseguita con attenzione. Inoltre, se l'altezza di confinamento è troppo piccola, tutte le cellule soffriranno di rottura dell'involucro nucleare o danni irreversibili. Per mitigarli, abbassare più lentamente il coverslip superiore e/o aumentare la spaziatura tra il coverslip.
Limiti della tecnica e suggerimenti per superarli
Una chiara limitazione della tecnica è la penetrazione della luce laser in sezioni profonde del tessuto, che porta ad aberrazioni e intrappolamento instabile. Pertanto, un limite inferiore di profondità di penetrazione dipende dalla chiarezza del campione, dalla correzione dell'aberrazione che può essere impiegata73 e dalla potenza laser applicata. Va tenuto conto del fatto che una maggiore potenza laser porta all'eccitazione termica del campione in prossimità della microsfera. Tuttavia, il riscaldamento del campione originato dallo spot laser a lunghezza d'onda di 1064 nm è ridotto al minimo per evitare uno stress plausibile legato al calore sui nostri campioni biologici74.
Un'altra limitazione è la forza massima che può essere misurata. Anche se il rilevamento diretto del momento della luce consente misurazioni della forza ben oltre il regime di risposta lineare della trappola ottica40,41, la forza massima applicata è nell'ordine di poche centinaia di picoNewton. Ciò è limitato dalla potenza del laser e dalla conseguente soglia di danno del materiale biologico morbido e dalle differenze dell'indice di rifrazione, che normalmente non sono superiori a 0,1 o 0,344. Sono stati proposti diversi metodi per aumentare il limite di rilevamento della forza, ad esempio utilizzando luce strutturata75, microsfere rivestite antiriflesso76, particelle ad alto indice di rifrazione77 o punti quantici altamente drogati78.
Gli OT possono essere utilizzati per misurazioni di posizione su scala nanometrica attraverso l'interferometria BFP, in modo tale che la posizione del tallone all'interno della trappola sia Δx = β Sx, dove Sx è il segnale di tensione del sensore e β [μm/V] può essere calibrato al volo seguendo diversi protocolli35,54. Per un sensore di forza ottica, si può dimostrare che il fattore di conversione invariante tensione-forza α [pN/V] si riferisce direttamente a β e alla rigidità della trappola, k [pN/μm], attraverso α = kβ 37) Negli esperimenti con spostamenti di perline troppo piccoli per essere rilevati dall'imaging ottico, questa strategia può essere utilizzata per integrare le misurazioni della forza con il rilevamento di una posizione ridotta. Un esempio è l'applicazione delle routine sperimentali qui presentate su nuclei molto rigidi, per i quali forze a potenze laser ragionevoli (200-500 mW) non sono sufficienti per indurre valori di indentazione abbastanza grandi. In tal caso, il tallone deve essere portato a contatto con il nucleo e la rigidità di intrappolamento deve essere calibrata prima della misurazione (fase 8.6). La rientranza d del nucleo in funzione della forza può essere determinata indirettamente come:
d = xtrap - F/k
dove xtrap è la posizione della trappola. Diverso dal fattore di quantità di luce invariante α [pN / V], il fattore β [μm / V] deve essere calibrato prima di ogni esperimento poiché dipende da molte variabili locali che determinano la dinamica di intrappolamento, come la dimensione delle particelle, la dimensione dello spot della trappola ottica e gli indici di rifrazione relativi.
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
MK riconosce il sostegno finanziario del Ministero spagnolo dell'Economia e della Competitività attraverso il Programma Nacional (PGC2018-097882-A-I00), FEDER (EQC2018-005048-P), Severo Ochoa programma per i centri di eccellenza in R & S (CEX2019-000910-S; RYC-2016-21062), dalla Fundació Privada Cellex, Fundació Mir-Puig e dalla Generalitat de Catalunya attraverso il programma CERCA e ricerca (2017 SGR 1012), oltre ai finanziamenti attraverso ERC (MechanoSystems) e HFSP (CDA00023/2018). V.R. riconosce il sostegno del Ministero spagnolo della Scienza e dell'Innovazione alla partnership EMBL, al Centro de Excelencia Severo Ochoa, al Plan Nacional di MINECO (BFU2017-86296-P, PID2020-117011GB-I00) e alla Generalitat de Catalunya (CERCA). V.V. riconosce il sostegno del programma di dottorato ICFOstepstone finanziato dal programma di ricerca e innovazione Horizon 2020 dell'Unione europea nell'ambito dell'accordo di sovvenzione Marie Skłodowska-Curie 665884. Ringraziamo Arnau Farré per la lettura critica del manoscritto; Maria Marsal per l'aiuto con l'imaging e il montaggio dell'embrione da 24 hpf e; Senda Jiménez-Delgado per il supporto con microiniezioni di zebrafish.
Materiali
| Name | Company | Catalog Number | Comments |
| #1.5 22 mm cover glasses | Ted Pella | 260148 | |
| #1.5 22x60 mm Coverglasses | Ted Pella | 260152 | |
| #1.5H glass bottom dishes | Willco | GWST-5040 | |
| 10-um beads | Supelco | 72986 | |
| 1-mm glass capillaries | Harvard Apparatus | 30-0020 GC100F-15 | |
| 1-um polystyrene microbeads | Sigma | 89904 | |
| 1-um red-fluorescent beads | ThermoFisher | F8816 | |
| Agar | ThermoFisher | 16500500 | |
| Aqcuisition cameras sCMOS | Andor | Sona-4BV11-UNI | |
| Auxiliary camera (Figure 3, AUX) | Blackfly, FLIR | BFS-U3-200S6M-C | |
| Calbryte 520 | AAT Bioquest | 520 AM | |
| Centrifuge | Eppendorf | 5453000011 | |
| Concanavalin A | Sigma | C5275 | |
| DMEM | Sigma | D2906 | |
| DNA-Hoechst | ThermoFisher | 33342 | |
| Double scotch tape | Biesse Adesivi | ||
| E3 | 5 mM NaCl. 0.17 mM KCl. 0.33 mM CaCl2. 0.33 mM MgSO4 | ||
| Eclipse Ti2 | Nikon | ||
| Forceps | Fine Science Tools | 11252-20 | |
| GPI-GFP | |||
| H2A-mCh | |||
| Image acquisition software | Fusion-Andor | ||
| Immersion Oil | Cargille | Type B: 16484 | |
| IR protection googles | Thorlabs | LG1 | |
| Lap2b-eGFP | |||
| Micro loader pipette | Eppendorf | GELoader | |
| Microinjector | World Precision Instruments | SYS-PV820 | |
| MicroManager 2.0 | |||
| Micromiter slide | ID5243 GXMGRAT-5 5mm/100 divisions | ||
| Mineral oil | Sigma | M3616 | |
| Motorized stage | ASI | ||
| Needle puller | Sutter instrument Co. | Model P-97 | |
| Optical tweezers platform | Impetux Optics | Sensocell | |
| OTs software (LightAce) | Impetux Optics | ||
| PDMS | Sigma | Sylgard 184 | |
| PDMS Curing agent | Sigma | Sylgard 184 | |
| Post processing software (Matlab) | Mathworks | ||
| RNAse free water | Thermofisher | AM9937 | |
| Short-pass dichroic mirror (Figure 3, IR-F) | Semrock | FF01-950/SP-25 | |
| Spin-coater | Specialty Coating Systems | Spincoat G3P-8 | |
| Spinning-disk confocal microscope | Andor | DragonFly 502 | |
| Stereomicroscope | Leica M80 | ||
| Triangular microinjection mold | Adaptive Science Tools | TU1 | |
| Universal oven | Memmert | UNB 200 | |
| Water immersion objective | Nikon | MRD07602 |
Riferimenti
- Chan, C. J., Heisenberg, C. P., Hiiragi, T. Coordination of Morphogenesis and Cell-Fate Specification in Development. Current Biology. 27 (18), 1024-1035 (2017).
- Heller, E., Fuchs, E. Tissue patterning and cellular mechanics. Journal of Cell Biology. 211 (2), 219-231 (2015).
- Heisenberg, C. P., Bellaïche, Y. Forces in tissue morphogenesis and patterning. Cell. 153 (5), 948-962 (2013).
- Petridou, N. I., Spiró, Z., Heisenberg, C. P. Multiscale force sensing in development. Nature Cell Biology. 19 (6), 581-588 (2017).
- Krieg, M., et al. Tensile forces govern germ-layer organization in zebrafish. Nature Cell Biology. 10 (4), 429-436 (2008).
- Ruprecht, V., et al. Cortical contractility triggers a stochastic switch to fast amoeboid cell motility. Cell. 160 (4), 673-685 (2015).
- Shellard, A., Mayor, R. Supracellular migration - Beyond collective cell migration. Journal of Cell Science. 132 (8), (2019).
- Mongera, A., et al. A fluid-to-solid jamming transition underlies vertebrate body axis elongation. Nature. 561 (7723), 401-405 (2018).
- Atia, L., et al. Geometric constraints during epithelial jamming. Nature Physics. 14 (6), 613-620 (2018).
- Turlier, H., Maître, J. -. L. Mechanics of tissue compaction. Seminars in Cell & Developmental Biology. 47-48, 110-117 (2015).
- Ladoux, B., Mège, R. M. Mechanobiology of collective cell behaviours. Nature Reviews Molecular Cell Biology. 18 (12), 743-757 (2017).
- Venturini, V., et al. The nucleus measures shape changes for cellular proprioception to control dynamic cell behavior. Science. 370 (6514), (2020).
- Charras, G., Sahai, E. Physical influences of the extracellular environment on cell migration. Nature Reviews Molecular Cell Biology. 15 (12), 813-824 (2014).
- Kirby, T. J., Lammerding, J. Emerging views of the nucleus as a cellular mechanosensor. Nature Cell Biology. 20 (4), 373-381 (2018).
- Lee, H. P., et al. The nuclear piston activates mechanosensitive ion channels to generate cell migration paths in confining microenvironments. Science Advances. 7 (2), (2021).
- Friedl, P., Wolf, K., Lammerding, J. Nuclear mechanics during cell migration. Current Opinion in Cell Biology. 23 (1), 55-64 (2011).
- Versaevel, M., Riaz, M., Grevesse, T., Gabriele, S. Cell confinement: Putting the squeeze on the nucleus. Soft Matter. 9 (29), 6665-6676 (2013).
- Zuela-Sopilniak, N., et al. Measuring nucleus mechanics within a living multicellular organism: Physical decoupling and attenuated recovery rate are physiological protective mechanisms of the cell nucleus under high mechanical load. Molecular Biology of the Cell. 31 (17), 1943-1950 (2020).
- Kim, D. H., Wirtz, D. Cytoskeletal tension induces the polarized architecture of the nucleus. Biomaterials. 48, 161-172 (2015).
- Lomakin, A. J., et al. The nucleus acts as a ruler tailoring cell responses to spatial constraints. Science. 370 (6514), (2020).
- Hampoelz, B., et al. Microtubule-induced nuclear envelope fluctuations control chromatin dynamics in Drosophila embryos. Development. 138 (16), 3377-3386 (2011).
- Heo, S. J., et al. Differentiation alters stem cell nuclear architecture, mechanics, and mechano-sensitivity. eLife. 5, 1-21 (2016).
- Cosgrove, B. D., et al. Nuclear envelope wrinkling predicts mesenchymal progenitor cell mechano-response in 2D and 3D microenvironments. Biomaterials. 270, 120662 (2021).
- Liu, H., et al. In situ mechanical characterization of the cell nucleus by atomic force microscopy. ACS Nano. 8 (4), 3821-3828 (2014).
- Hobson, C. M., et al. Correlating nuclear morphology and external force with combined atomic force microscopy and light sheet imaging separates roles of chromatin and lamin A/C in nuclear mechanics. Molecular Biology of the Cell. 31 (16), 1788-1801 (2020).
- Pajerowski, J. D., Dahl, K. N., Zhong, F. L., Sammak, P. J., Discher, D. E. Physical plasticity of the nucleus in stem cell differentiation. Proceedings of the National Academy of Sciences of the United States of America. 104 (40), 15619-15624 (2007).
- Rowat, A. C., Lammerding, J., Ipsen, J. H. Mechanical properties of the cell nucleus and the effect of emerin deficiency. Biophysical Journal. 91 (12), 4649-4664 (2006).
- Davidson, P. M., et al. High-throughput microfluidic micropipette aspiration device to probe time-scale dependent nuclear mechanics in intact cells. Lab on a Chip. 19 (21), 3652-3663 (2019).
- Lombardi, M., Zwerger, M., Lammerding, J. Biophysical assays to probe the mechanical properties of the interphase cell nucleus: Substrate strain application and microneedle manipulation. Journal of Visualized Experiments: JoVE. (55), (2011).
- Luo, T., Mohan, K., Iglesias, P. A., Robinson, D. N. Molecular mechanisms of cellular mechanosensing. Nature Materials. 12 (11), 1064-1071 (2013).
- Dahl, K. N., Engler, A. J., Pajerowski, J. D., Discher, D. E. Power-law rheology of isolated nuclei with deformation mapping of nuclear substructures. Biophysical Journal. 89 (4), 2855-2864 (2005).
- Guilluy, C., et al. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nature Cell Biology. 16 (4), 376-381 (2014).
- Bustamante, C. J., Wang, M. D. Optical tweezers in single-molecule biophysics. Nature Reviews Methods Primers. , 1-29 (2021).
- Svoboda, K., Block, S. M. Force and velocity measured for single kinesin molecules. Cell. 77 (5), 773-784 (1994).
- Berg-Sørensen, K., Flyvbjerg, H. Power spectrum analysis for optical tweezers. Review of Scientific Instruments. 75 (3), 594-612 (2004).
- Smith, S. B., Cui, Y., Bustamante, C. Optical-trap force transducer that operates by direct measurement of light momentum. Methods in Enzymology. 361 (1994), 134-162 (2003).
- Farré, A., Montes-Usategui, M. A force detection technique for single-beam optical traps based on direct measurement of light momentum changes. Optics Express. 18 (11), 11955 (2010).
- Català, F., Marsà, F., Montes-Usategui, M., Farré, A., Martín-Badosa, E. Extending calibration-free force measurements to optically-trapped rod-shaped samples. Scientific Reports. 7, 1-10 (2017).
- Bui, A. A. M., et al. Calibration of force detection for arbitrarily shaped particles in optical tweezers. Scientific Reports. 8 (1), 1-12 (2018).
- Farré, A., Marsà, F., Montes-Usategui, M. Beyond the hookean spring model: Direct measurement of optical forces through light momentum changes. Methods in Molecular Biology. 1486, (2017).
- Farré, A., Marsà, F., Montes-Usategui, M. Optimized back-focal-plane interferometry directly measures forces of optically trapped particles. Optics Express. 20 (11), 12270 (2012).
- Jun, Y., Tripathy, S. K., Narayanareddy, B. R. J., Mattson-Hoss, M. K., Gross, S. P. Calibration of optical tweezers for in vivo force measurements: How do different approaches compare. Biophysical Journal. 107 (6), 1474-1484 (2014).
- Mas, J., Farré, A., Sancho-Parramon, J., Martín-Badosa, E., Montes-Usategui, M. Force measurements with optical tweezers inside living cells. Optical Trapping and Optical Micromanipulation XI. 9164, (2014).
- Schürmann, M., Scholze, J., Müller, P., Guck, J., Chan, C. J. Cell nuclei have lower refractive index and mass density than cytoplasm. Journal of Biophotonics. 9 (10), 1068-1076 (2016).
- Rosen, J. N., Sweeney, M. F., Mably, J. D. Microinjection of zebrafish embryos to analyze gene function. Journal of Visualized Experiments: JoVE. (25), (2009).
- Westerfield, M. . The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish. (Danio rerio), 5th Edition. , (2007).
- Schubert, R., et al. Assay for characterizing the recovery of vertebrate cells for adhesion measurements by single-cell force spectroscopy. FEBS Letters. 588 (19), 3639-3648 (2014).
- Koschwanez, J. H., Carlson, R. H., Meldrum, D. R. Thin PDMS films using long spin times or tert-butyl alcohol as a solvent. PLoS One. 4 (2), 2-6 (2009).
- Das, R., et al. Mechanical stretch inhibition sensitizes proprioceptors to compressive stresses. bioRxiv. , (2021).
- Chardès, C., Clement, R., Blanc, O., Lenne, P. F. Probing cell mechanics with bead-free optical tweezers in the drosophila embryo. Journal of Visualized Experiments: JoVE. (141), (2018).
- Staunton, J. R., Blehm, B., Devine, A., Tanner, K. In situ calibration of position detection in an optical trap for active microrheology in viscous materials. Optics Express. 25 (3), 1746 (2017).
- Bola, R., Treptow, D., Marzoa, A., Montes-Usategui, M., Martin-Badosa, E. Acousto-holographic optical tweezers. Optics Letters. 45 (10), 2938-2941 (2020).
- Thalhammer, G., Obmascher, L., Ritsch-Marte, M. Direct measurement of axial optical forces. Optics Express. 23 (5), 6112 (2015).
- Vermeulen, K. C., et al. Calibrating bead displacements in optical tweezers using acousto-optic deflectors. Review of Scientific Instruments. 77 (1), 1-6 (2006).
- Dzementsei, A., Barooji, Y. F., Ober, E. A., Oddershede, L. B. Foregut organ progenitors and their niche display distinct viscoelastic properties in vivo during early morphogenesis stages. bioRxiv. , 1-35 (2021).
- Behrndt, M., et al. Forces driving epithelial spreading in zebrafish gastrulation. Science. 338 (6104), 257-260 (2012).
- Krieg, M., et al. Atomic force microscopy-based mechanobiology. Nature Reviews Physics. , (2018).
- A-Hassan, E., et al. Relative microelastic mapping of living cells by atomic force microscopy. Biophysical Journal. 74 (3), 1564-1578 (1998).
- Khalilgharibi, N., et al. Stress relaxation in epithelial monolayers is controlled by the actomyosin cortex. Nature Physics. 15, (2019).
- Crick, S. L., Yin, F. C. Assessing micromechanical properties of cells with atomic force microscopy: importance of the contact point. Biomechanics and Modeling in Mechanobiology. 6 (3), 199-210 (2007).
- Hurst, S., Vos, B. E., Betz, T. Intracellular softening and fluidification reveals a mechanical switch of cytoskeletal material contributions during division. bioRxiv. , 425761 (2021).
- Kaplan, J. L., Bonfanti, A., Kabla, A. RHEOS.jl - A Julia package for rheology data analysis. arXiv. 4, 1-5 (2020).
- Bonfanti, A., Kaplan, J. L., Charras, G., Kabla, A. Fractional viscoelastic models for power-law materials. Soft Matter. 16 (26), 6002-6020 (2020).
- Rivas-Barbosa, R., Escobedo-Sánchez, M. A., Tassieri, M., Laurati, M. i-Rheo: determining the linear viscoelastic moduli of colloidal dispersions from step-stress measurements. Physical Chemistry Chemical Physics: PCCP. 22 (7), 3839-3848 (2020).
- Tassieri, M., et al. i-Rheo: Measuring the materials' linear viscoelastic properties "in a step". Journal of Rheology. 60 (4), 649-660 (2016).
- Zhong, M. C., Wei, X. B., Zhou, J. H., Wang, Z. Q., Li, Y. M. Trapping red blood cells in living animals using optical tweezers. Nature Communications. 4, 1767-1768 (2013).
- Harlepp, S., Thalmann, F., Follain, G., Goetz, J. G. Hemodynamic forces can be accurately measured in vivo with optical tweezers. Molecular Biology of the Cell. 28 (23), 3252-3260 (2017).
- Bayoudh, S., Mehta, M., Rubinsztein-Dunlop, H., Heckenberg, N. R., Critchley, C. Micromanipulation of chloroplasts using optical tweezers. Journal of Microscopy. 203 (2), 214-222 (2001).
- Favre-Bulle, I. A., Stilgoe, A. B., Rubinsztein-Dunlop, H., Scott, E. K. Optical trapping of otoliths drives vestibular behaviours in larval zebrafish. Nature Communications. 8 (1), 630 (2017).
- Bambardekar, K., Clément, R., Blanc, O., Chardès, C., Lenne, P. F. Direct laser manipulation reveals the mechanics of cell contacts in vivo. Proceedings of the National Academy of Sciences of the United States of America. 112 (5), 1416-1421 (2015).
- Ferro, V., Chuai, M., McGloin, D., Weijer, C. J. Measurement of junctional tension in epithelial cells at the onset of primitive streak formation in the chick embryo via non-destructive optical manipulation. Development (Cambridge). 147 (3), (2020).
- Hörner, F., et al. Holographic optical tweezers-based in vivo manipulations in zebrafish embryos. Journal of Biophotonics. 10 (11), 1492-1501 (2017).
- Zhong, M. -. C., Wang, Z. -. Q., Li, Y. -. M. Aberration compensation for optical trapping of cells within living mice. Applied Optics. 56 (7), 1972 (2017).
- Català, F., Marsà, F., Montes-Usategui, M., Farré, A., Martín-Badosa, E. Influence of experimental parameters on the laser heating of an optical trap. Scientific Reports. 7 (1), 1-9 (2017).
- Taylor, M. A., Waleed, M., Stilgoe, A. B., Rubinsztein-Dunlop, H., Bowen, W. P. Enhanced optical trapping via structured scattering. Nature Photonics. 9 (10), 669-673 (2015).
- Bormuth, V., et al. Optical trapping of coated microspheres. Optics Express. 16 (18), 13831-13844 (2008).
- Sudhakar, S., et al. Germanium nanospheres for ultraresolution picotensiometry of kinesin motors. Science. 371 (6530), (2021).
- Shan, X., et al. Optical tweezers beyond refractive index mismatch using highly doped upconversion nanoparticles. Nature Nanotechnology. 16 (5), 531-537 (2021).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati