
Method Article
Mesures de force directe de la mécanique subcellulaire en confinement à l’aide de pinces optiques
Dans cet article
Résumé
Nous présentons ici un protocole pour étudier les propriétés mécaniques intracellulaires de cellules embryonnaires isolées de poisson-zèbre en confinement tridimensionnel avec mesure directe de la force par un piège optique.
Résumé
Au cours du développement d’un organisme multicellulaire, une seule cellule fécondée se divise et donne naissance à de multiples tissus aux fonctions diverses. La morphogenèse tissulaire va de pair avec des changements moléculaires et structurels au niveau de la cellule unique qui entraînent des variations des propriétés mécaniques subcellulaires. En conséquence, même au sein d’une même cellule, différents organites et compartiments résistent différemment aux contraintes mécaniques; et les voies de mécanotransduction peuvent réguler activement leurs propriétés mécaniques. La capacité d’une cellule à s’adapter au microenvironnement de la niche tissulaire est donc en partie due à la capacité de détecter et de répondre aux contraintes mécaniques. Nous avons récemment proposé un nouveau paradigme de mécanosensation dans lequel la déformation et le positionnement nucléaires permettent à une cellule d’évaluer l’environnement physique 3D et confèrent à la cellule un sentiment de proprioception pour décoder les changements de forme cellulaire. Dans cet article, nous décrivons une nouvelle méthode pour mesurer les forces et les propriétés matérielles qui façonnent le noyau cellulaire à l’intérieur des cellules vivantes, illustrées sur les cellules adhérentes et les cellules confinées mécaniquement. Les mesures peuvent être effectuées de manière non invasive avec des pièges optiques à l’intérieur des cellules, et les forces sont directement accessibles grâce à la détection sans étalonnage de la quantité de mouvement lumineuse. Cela permet de mesurer la mécanique du noyau indépendamment des déformations de la surface cellulaire et de permettre la dissection des voies de mécanotransduction extéroceptives et interoceptives. Il est important de noter que l’expérience de piégeage peut être combinée à la microscopie optique pour étudier la réponse cellulaire et la dynamique subcellulaire à l’aide de l’imagerie par fluorescence du cytosquelette, des ions calcium ou de la morphologie nucléaire. La méthode présentée est simple à appliquer, compatible avec les solutions commerciales pour les mesures de force, et peut facilement être étendue pour étudier la mécanique d’autres compartiments subcellulaires, par exemple les mitochondries, les fibres de stress et les endosomes.
Introduction
La morphogenèse tissulaire est un processus complexe dans lequel les signaux biochimiques et les forces physiques sont coordonnés spatio-temporellement. Dans l’embryon en développement, des gradients de facteurs de signalisation biochimiques dictent la spécification du devenir et assurent une structure tissulaire correcte1,2. Dans le même temps, les forces intrinsèques et extrinsèques jouent un rôle dans la construction de l’architecture de l’embryon3,4. L’influence de la mécanique du cortex cellulaire dans ce contexte a été largement étudiée5,6. L’interconnexion étroite entre les processus mécanochimiques au cours de la morphogenèse repose sur les propriétés des cellules individuelles pour détecter et répondre aux forces mécaniques dans leur microenvironnement tissulaire. Les cellules décodent ainsi les signaux mécaniques via la présence d’éléments subcellulaires et moléculaires sensibles à la force qui transduisent l’information mécanique dans des voies de signalisation spécifiques contrôlant le comportement cellulaire, le destin cellulaire et la mécanique cellulaire.
Une caractéristique des processus de développement est que les cellules s’organisent en groupes pour construire des structures multicellulaires. En tant que telles, les cellules individuelles se réarrangent rarement et se déplacent seules, mais sont associées à un sociotope serré dans lequel elles montrent un comportement collectif tel que la migration supracellulaire7, les transitions (dé)bloquantes8,9 ou le compactage des blastocystes10. Les forces mécaniques générées à l’intérieur et entre les cellules servent d’indices importants pour instruire la dynamique collective des cellules7,11. Mais même lorsque les cellules se déplacent seules, comme les cellules progénitrices qui se faufilent entre les feuilles de tissus ou les niches tissulaires étroites, elles subissent des forces mécaniques anisotropes étendues lorsqu’elles naviguent dans un environnement tridimensionnel. Ces contraintes mécaniques sur les cellules ont des conséquences profondes sur le comportement cellulaire12,13. Plusieurs mécanismes ont été étudiés qui convergent vers le noyau en tant qu’élément majeur de mécanotransduction14,15, en tant qu’élément mécanique passif ou actif lors de la migration dans un environnement tissulaire 3D dense15,16.
Nous avons récemment proposé un mécanisme qui équipe les cellules pour mesurer les déformations de forme en utilisant le noyau comme mécano-jauge intracellulaire élastique12. Le noyau, étant le plus grand organite d’une cellule, subit de grandes déformations lorsque les cellules se polarisent, migrent ou changent de forme sous un étirement mécanique, un confinement ou un stress osmotique16,17,18,19. Nous avons constaté que l’étirement de l’enveloppe nucléaire avec le positionnement intracellulaire du noyau fournit aux cellules des informations sur l’ampleur et le type de déformation cellulaire (comme la compression cellulaire par rapport au gonflement cellulaire). L’étirement du noyau est associé à un déploiement de la membrane nucléaire interne (INM), qui favorise l’activité lipase cPLA2 (cytosolique phospholipase A2) dépendante du calcium à l’INM, suivie de la libération d’acide arachidonique (AA) et de l’activation rapide de la myosine II au niveau du cortex cellulaire. Cela conduit à une augmentation de la contractilité cellulaire et de la migration des cellules amiboïdes au-dessus d’un seuil de contractilité corticale6. La réponse mécanosensible à la déformation cellulaire se produit en moins d’une minute et est réversible lors de la libération du confinement, ce qui suggère que le noyau agit comme une jauge de contrainte pour la proprioception cellulaire régulant le comportement adaptatif des cellules dans des conditions de stress mécanique. Cette voie mécanosensible s’est avérée active dans les cellules souches progénitrices dérivées d’embryons de poisson-zèbre, à la fois dans les cellules pluripotentes et les cellules engagées dans la lignée12 et est conservée dans différentes espèces et lignées cellulaires20.
En plus des propriétés nucléaires en tant que mécanocapteur cellulaire, l’architecture et la mécanique nucléaires sont intrinsèquement régulées au cours du développement et en réponse à la spécification du devenir cellulaire21, d’où l’ajustement de la mécanosensibilité cellulaire22,23. La conséquence pourrait être un changement dans la conformité nucléaire qui permet des changements morphologiques et des transitions d’un état prémigratoire à un état migratoire et vice versa8.
Plusieurs techniques pour mesurer la mécanique du noyau cellulaire ont été appliquées, telles que la microscopie à force atomique24,25, l’aspiration par micropipette26,27, la technologie microfluidique28 et les micro-aiguilles29. Cependant, beaucoup de ces techniques sont invasives dans le sens où la cellule entière doit être déformée, ce qui limite la mesure des caractéristiques mécaniques et des réponses dépendantes de la force du noyau lui-même. Pour contourner la déformation simultanée de la surface cellulaire et de son cortex cellulaire mécanosensible30, des noyaux isolés ont été étudiés dans divers contextes31,32. Cependant, il n’est pas exclu que l’isolement nucléaire soit associé à une modification des propriétés mécaniques du noyau et de leur régulation (référence24 et observations propres non publiées).
Les pinces optiques (OT) sont une technologie polyvalente qui a permis une pléthore d’expériences en mécanobiologie cellulaire et a joué un rôle déterminant dans notre compréhension de la façon dont les machines moléculaires convertissent l’énergie chimique en énergie mécanique33,34. Les pinces optiques utilisent un faisceau laser étroitement focalisé pour exercer des forces optiques sur les particules diélectriques qui ont un indice de réfraction supérieur à celui du milieu environnant33. De telles forces peuvent être de l’ordre de centaines de pico-Newtons et entraîner un confinement efficace de la particule dans le foyer du piège laser, permettant la manipulation de la particule piégée en trois dimensions. L’utilisation de la lumière présente un avantage important en ce sens que la mesure peut être effectuée de manière non invasive à l’intérieur des cellules vivantes. Les manipulations optiques sont en outre limitées au foyer piège du faisceau laser. Par conséquent, la manipulation peut être effectuée sans stimuler les membranes cellulaires environnantes et ne perturbe pas le cortex actinique ou les processus mécanosensibles au niveau de la membrane plasmique, tels que l’activation dépendante de la force des canaux ioniques.
La difficulté de l’approche par pince optique est de déterminer avec précision les forces appliquées à la microsphère en utilisant des approches classiques qui reposent sur l’étalonnage de force indirecte basé sur le théorème d’équipartition ou l’utilisation de forces de traînée de Stokes définies pour mesurer une force d’échappement dépendante de la puissance laser35. Bien que ces méthodes soient simples à mettre en œuvre dans une expérience in vitro, elles ne peuvent généralement pas être traduites dans un environnement cellulaire. Plusieurs stratégies ont été introduites sur le terrain qui reposent sur un étalonnage direct de la force, dérivé des premiers principes de conservation de la quantité de mouvement36,37. Contrairement à d’autres approches de spectroscopie de force, les mesures de force sont déduites d’un échange local de moment lumineux avec la particule piégée de forme arbitraire38,39. Dans notre installation expérimentale, les changements de quantité de mouvement lumineux résultant des forces optiques sont directement mesurés sans qu’il soit nécessaire d’effectuer un étalonnage in situ des pièges40,41,42,43. Ainsi, les mesures deviennent possibles dans un environnement visqueux tel que l’intérieur de la cellule ou même dans un tissu, et les forces peuvent être facilement quantifiées jusqu’au niveau pN.
Dans ce protocole, nous décrivons un test pour manipuler mécaniquement des organites ou des structures intracellulaires et évaluer quantitativement leurs propriétés mécaniques par une pince optique. Cette configuration est intégrée dans un microscope fluorescent à disque rotatif permettant l’imagerie parallèle du comportement cellulaire ou de la dynamique intracellulaire. Le test permet de caractériser les propriétés mécaniques de compartiments cellulaires spécifiques, tels que le noyau, tout en étudiant simultanément la mécanoréponse possible et l’activation des voies de signalisation moléculaire à la suite de la déformation elle-même. De plus, le piégeage optique des microbilles injectées dans les cellules permet une augmentation de la force d’indentation grâce à un indice de réfraction considérablement plus élevé de la bille de polystyrène (n = 1,59) par rapport au contraste de réfraction intrinsèque44 du noyau (n ~ 1,35) par rapport au cytoplasme (n ~ 1,38). La stratégie présentée peut être facilement adaptée à l’étude d’autres structures intracellulaires et organites, ainsi qu’à d’autres approches impliquant la microrhéologie active, l’utilisation de plusieurs pièges optiques pour sonder simultanément les mêmes structures subcellulaires / différentes, et des mesures ciblant la mécanobiologie cellulaire dans l’embryon vivant.
Protocole
Tous les protocoles utilisés ont été approuvés par le Comité institutionnel d’éthique des soins et de l’utilisation des animaux (PRBB-IACUEC) et mis en œuvre conformément aux réglementations nationales et européennes. Toutes les expériences ont été réalisées conformément aux principes des 3R. Le poisson-zèbre (Danio rerio) a été maintenu comme décrit précédemment.
1. Préparation de cellules souches progénitrices embryonnaires primaires isolées du poisson-zèbre
- Préparation de micropipette et d’agarose
REMARQUE: Pour un protocole complet de micro-injection d’embryons de poisson zèbre, voir la référence45.- À l’aide d’un extracteur de micropipette, tirez un capillaire en verre de 1,0 mm pour obtenir deux aiguilles45. Conservez les aiguilles inutilisées dans une boîte de Petri de 150 mm fixée à un coussin de pâte à modeler ou dans un anneau de ruban adhésif de laboratoire à l’envers pour protéger la pointe mince des dommages pendant le transport.
- Fondre 1% d’agarose ultrapure dans E3 (5 mM NaCl, 0,17 mM KCl, 0,33 mM CaCl2, 0,33 mM MgSO4) dans un micro-ondes standard de cuisine / laboratoire pendant 10 s. Chauffer le mélange à plusieurs reprises pendant de courtes périodes (quelques secondes) jusqu’à ce que l’agarose fonde.
- Lorsque l’agarose est complètement fondue, laissez-la refroidir brièvement, puis versez-la dans une boîte de Petri de 10 cm. Ajouter lentement le moule de micro-injection triangulaire (voir Tableau des matériaux) sur le dessus de l’agarose en évitant l’apparition de bulles. Ne poussez pas le moule en vous assurant qu’il reste sur la surface de l’agarose.
- Lorsque l’agarose se solidifie complètement, retirez le moule triangulaire très lentement en exerçant une force douce pour éviter toute rupture dans l’agarose. La plaque peut être conservée à l’envers à 4 °C pendant 2 à 4 semaines.
- 30 min avant la microinjection, sortez la plaque du réfrigérateur et ajoutez l’E3 préavertissé à 28 °C pour la laisser se stabiliser à température ambiante.
- Préparation du mélange d’injection
- Pour préparer le mélange injectable, diluer 1 μm de microbilles (polystyrène, non fluorescent) dans un rapport de 1:5 dans de l’eau sans RNase.
- Préparer l’ARNm pour l’expression transitoire de marqueurs fluorescents ou l’expression de constructions de gènes recombinants et/ou la co-injection de morpholino à la concentration souhaitée.
REMARQUE: Un mélange d’injection typique pour la co-injection de microbilles avec 100 pg d’ARNm par embryon à marquer, par exemple, le noyau avec H2A-mCherry est: 1 μL de perles + 1 μL d’ARNm (la concentration en stock est de 1 μg / μL) + 2,5 μL d’eau sans ARN + 0,5 μL de rouge phénol (solution mère 0,5%, le rouge de phénol n’est pas obligatoire; il est utilisé pour une meilleure visualisation de la goutte injectée mais de l’injection non marquée drop est également visible pour un expérimentateur expérimenté). L’injection d’ARN peut également être utile pour sélectionner des embryons injectés. Des microbilles fluorescentes peuvent être injectées, au lieu de non fluorescentes, pour les visualiser.
- Chargement et étalonnage de l’aiguille de micro-injection
- Allumez le micro-injecteur à l’aide de l’option Time-Gated . Ce réglage est très important pour calibrer correctement le volume d’injection. Réglez le temps de contrôle à environ 500 ms.
- Chargez 3 μL du mélange d’injection dans l’aiguille à l’aide d’une pipette à micro-chargeur.
- Insérez l’aiguille dans le micromanipulateur et scellez hermétiquement. Vérifiez si le micromanipulateur est en bonne position et a suffisamment de liberté pour se déplacer dans la direction x-y sur la plaque d’injection.
- Mesurez la taille de la goutte à l’aide d’une lame micrométrique (5 mm/100 divisions) avec une goutte d’huile minérale sur le dessus45 et éjectez une goutte du mélange d’injection directement dans l’huile minérale.
- Recadrez l’aiguille avec des pinces pointues à un angle raide pour générer une pointe pointue pointue. Ajustez la taille de la goutte à 0,1 mm, correspondant à 0,5 nL de matériau injecté.
REMARQUE: Si en coupant l’aiguille, ce volume est dépassé, il est recommandé de refaire la procédure d’étalonnage avec une nouvelle aiguille. Le temps de contrôle du micro-injecteur peut être légèrement ajusté pour correspondre au volume de chute; cependant, des temps de contrôle courts correspondent à un grand diamètre d’aiguille, ce qui peut endommager les embryons.
- Microinjection d’embryons de poisson-zèbre au stade unicellulaire
- Recueillir les embryons de poisson zèbre peu de temps après la fécondation pour la micro-injection du mélange de billes directement dans l’embryon au stade unicellulaire (zygote) avant la première division cellulaire.
REMARQUE: Cela garantit une bonne distribution des microsphères et un rendement suffisamment élevé de blastomères isolés avec au moins une microsphère par cellule aux stades de développement ultérieurs dans lesquels des expériences sont effectuées (stade blastula-gastrula). Les expériences d’indentation peuvent toujours être effectuées s’il y a deux sphères dans la cellule, mais les cellules qui n’ont pas de perles doivent être exclues (même si l’indentation sans sphères est possible). Des souches de type sauvage AB ont été utilisées dans ce protocole, mais toute autre souche, par exemple TL, peut être utilisée. - Placer des embryons d’une cellule (zygote) dans un moule d’agarose de forme triangulaire préavertissé à 1 %, comme le montre la figure 1A, à l’aide d’une pipette Pasteur en plastique.
- Retirez le milieu supplémentaire avec la même pipette pour éviter que les embryons ne flottent autour. Poussez doucement les embryons dans le moule triangulaire via une brosse. Gardez un peu d’espace entre les embryons pour faciliter l’orientation correcte (Figure 1B).
- Alignez doucement les embryons avec une brosse afin qu’ils soient orientés latéralement, la seule cellule du zygote étant clairement visible, comme le montre la figure 1B. Une orientation idéale pour la micro-injection est atteinte lorsque la cellule de l’embryon est orientée vers la direction de l’aiguille (injection via le pôle animal de l’embryon) ou dans le sens opposé face à la cellule jaune (injection via le pôle végétal de l’embryon), comme le montre la figure 1C.
- Tenez le plat d’une main et utilisez l’autre main pour positionner la pointe de l’aiguille à l’aide du contrôleur de micromanipulateur. Abaissez la pointe de l’aiguille vers les embryons.
- Percez le chorion et entrez dans l’embryon unicellulaire avec l’aiguille tout en surveillant la procédure à travers le stéréomicroscope. Assurez-vous de l’emplacement correct de l’aiguille et, après l’injection, de l’emplacement correct de la goutte injectée, comme illustré à la figure 1C.
- Répétez l’opération pour tous les embryons : déplacez l’aiguille vers le haut, faites glisser le plat avec les embryons jusqu’à ce que l’embryon suivant soit centré, abaissez l’aiguille et injectez-la.
- Une fois que l’ensemble des embryons est injecté, retirez les embryons de la moisissure d’agarose / boîte de Petri en rinçant un peu d’E3 et mettez-les dans une nouvelle boîte de Petri à l’aide d’une pipette Pasteur en plastique. Il est recommandé de placer suffisamment de milieu sur la plaque d’injection pour éviter le dessèchement des embryons pendant la procédure de micro-injection.
- Répétez la procédure jusqu’à ce que le nombre souhaité d’embryons soit injecté. Les embryons doivent être à un stade cellulaire pour assurer une propagation maximale et homogène des perles.
REMARQUE: Cette procédure est optimisée pour les embryons de blastula précoces et doit probablement être optimisée si différents stades de développement doivent être étudiés. - Placer les embryons injectés à l’intérieur d’un incubateur à une température de 28-31 °C pendant environ 4 h ou jusqu’au stade souhaité (figure 1D) avant de procéder au protocole de culture cellulaire primaire.
REMARQUE: En option, laissez les embryons se développer au-delà du stade de blastula (ou du temps de mesure souhaité) pour assurer la survie et exclure les artefacts de toxicité. Au stade larvaire, montez des larves anesthésiées avec de la tricaïne dans 0,75% d’agarose et imagez la distribution des microsphères dans divers tissus. Pour faire une solution mère, mélanger : 400 mg de poudre de tricaïne dans 97,9 mL d’eau distillée, environ 2,1 mL de 1 M de tris-base (pH 9), et ajuster à pH 7. Cette solution peut être conservée à 4 °C. Pour utiliser la tricaïne comme anesthésique, diluer 4,2 mL de solution mère dans 100 mL de milieu de l’œuf (ou milieu souhaité); dans ce cas, E3 a été utilisé. Consultez la référence46 pour plus de détails.
- Recueillir les embryons de poisson zèbre peu de temps après la fécondation pour la micro-injection du mélange de billes directement dans l’embryon au stade unicellulaire (zygote) avant la première division cellulaire.
2. Préparation et coloration unicellulaires
- Placer les embryons au stade de la sphère (4 hpf, heures après la fécondation) dans un plat en verre à l’aide d’une pipette Pasteur en plastique. Sélectionnez les embryons qui sont positifs pour le signal des perles injectées, et qui expriment la protéine fluorescente en cas d’injection d’ARNm. Certains embryons peuvent présenter un regroupement élevé de perles et peuvent être exclus.
- Déchorionate manuellement les embryons à l’aide de pinces. Transférer environ 10 à 15 embryons dans des récipients de réaction de 1,5 mL à l’aide d’une pipette Pasteur en verre.
REMARQUE: Lorsque les embryons sont décholonnés, ils se fixent au plastique et l’utilisation de verrerie est requise. Comme alternative à la plaque de verre, une boîte de Petri en plastique avec une fine couche de 1% d’agarose peut être utilisée. La décection manuelle doit être préférée au traitement enzymatique à la pronase pour prévenir les dommages protéolytiques aux protéines de surface cellulaire et les changements potentiels dans les propriétés mécaniques des cellules et des tissus, évitant ainsi des temps de récupération prolongés47.
- Déchorionate manuellement les embryons à l’aide de pinces. Transférer environ 10 à 15 embryons dans des récipients de réaction de 1,5 mL à l’aide d’une pipette Pasteur en verre.
- Retirer le milieu E3 et ajouter 500 μL de milieu de culture tissulaire préchauffé indépendant du CO2 (DMEM-F12; avec de la L-glutamine et 15 mM de HEPES, sans bicarbonate de sodium et rouge de phénol complété par 10 unités de pénicilline et 10 mg/L de streptomycine).
REMARQUE: N’utilisez pas de milieux dépendants du CO2 à moins d’utiliser un incubateur de microscope. L’utilisation, par exemple, de RPMI dans des conditions tamponnées de carbonate provoque des changements dans le pH du milieu et peut affecter la survie cellulaire. Un autre aspect clé est d’éviter les milieux de culture qui contiennent du sérum. Le sérum peut contenir de l’acide lysophosphatidique (LPA), un puissant activateur de la voie Rho/ROCK, capable de contrôler la contractilité cellulaire et la motilité dans les cellules souches progénitrices6. L’osmolarité du milieu doit être maintenue à 300 mOsm pour éviter les problèmes osmotiques qui pourraient interférer avec la morphologie ou la mécanique nucléaire12. - Dissocier manuellement les cellules en secouant doucement le tube. Assurez-vous que le contenu du tube devient trouble sans gros morceaux visibles par l’œil. Évitez la formation de bulles pour minimiser les dommages et la perte de cellules.
- Centrifuger à 200 x g pendant 3 min. La pastille doit être clairement visible.
- Retirez le surnageant et suivez l’une des étapes détaillées ci-dessous.
- Si aucune coloration n’est nécessaire, ajoutez 500 μL de DMEM. Remettez en suspension doucement avec une pipette de 200 μL en ciblant un jet de liquide sur la pastille. N’exercez pas de force de cisaillement excessive sur les cellules. La mousse indique des dommages aux cellules.
- Pour marquer le noyau avec des colorants à ADN tels que Hoechst, mélanger 0,5 μL d’ADN-Hoechst (stock 2 mg/mL) dans 1 000 μL de DMEM pour obtenir 1 μg/mL de concentration finale. Ajouter 500 μL de cette solution de coloration aux cellules et remettre en suspension doucement. Incuber pendant 7 min dans l’obscurité.
- Pour colorer les cellules avec un indicateur de calcium chimique fluorescent Calbryte-520, ajoutez Calbryte-520 à une concentration de 5 μM dans le DMEM. Incuber pendant 20 min dans l’obscurité.
REMARQUE : Les protocoles indiqués aux étapes 2.5.2 et 2.5.3 ont été optimisés pour ces produits spécifiques. D’autres colorations peuvent être effectuées en utilisant les protocoles indiqués par le fabricant.
- Centrifuger à nouveau en utilisant les mêmes réglages qu’à l’étape 2.4; prélever le surnageant et remettre doucement en suspension les cellules (pour éviter la formation de grappes) dans 50 μL de DMEM pour les échantillons en suspension ou 20 μL de DMEM pour les cellules en confinement.
3. Préparation des chambres de piégeage optiques à l’aide d’espacements entreposés au polydiméthylsiloxane (PDMS)
REMARQUE: Les mesures de force optique basées sur la détection de la quantité de mouvement lumineux nécessitent la capture de toute la lumière sortant des pièges optiques40. Pour la robustesse du facteur d’étalonnage invariant α (pN/V), la distribution de la lumière au plan focal arrière (BFP) du capteur de force optique doit avoir une correspondance précise avec le moment cinétique du photon. Cela détermine la distance entre la surface de la lentille collectrice et le plan de piégeage à environ 2 mm, ce qui correspond à la hauteur maximale des chambres de piégeage optique.
- PDMS spin-coating des plats de fond en verre n ° 1.5.
REMARQUE: La recette suivante est fournie pour environ 40 plats. La microchambre résultante aura des hauteurs différentes selon que les expériences doivent être menées sur des cellules en suspension ou confinées (Figure 1D).- Mélanger 9 mL du polymère de base PDMS et 1 mL d’agent de durcissement PDMS dans un tube conique de 50 mL. Mélangez activement les deux produits pour assurer une distribution correcte de l’agent de durcissement.
- Dégazez le mélange pour éviter les bulles à l’aide d’une pompe à vide. Introduisez le tube conique dans une bouteille sous vide et évacuez la chambre. Attendez qu’aucune bulle ne soit présente dans le mélange.
REMARQUE: Ouvrez lentement le vide pour éviter la formation de mousse et les déversements du PDMS hors du tube de faucon. - Placez le plat inférieur en verre sur le mandrin de l’enrobeur rotatif (Figure 2A). Soyez doux pour ne pas gratter, prendre les empreintes digitales ou salir le plat. Protégez la boîte de spin-coater des fuites PDMS avec du papier d’aluminium.
- Pour les chambres OT pour les expériences sur cellules en suspension, ajoutez environ 250 μL de mélange PDMS au centre de la parabole inférieure et faites-la tourner à 750 tr/min pendant 1 min. La hauteur de la couche PDMS sera d’environ 50 μm48.
- Pour les chambres OT pour les expériences sur cellules confinées, ajoutez une petite goutte de PDMS (environ 50 μL) et faites-la tourner à 4 000 tr/min pendant 5 min. La hauteur de la couche PDMS sera de 10 μm environ. Pour obtenir un protocole détaillé sur la façon d’obtenir différentes épaisseurs PDMS, voir référence48.
- Durcir les plats à fond de verre revêtus de PDMS à 70 °C pendant 1 h.
- Coupez un carré de 1 x 1 cm sur la couche PDMS avec un scalpel et décollez-le avec une pince à épiler (Figure 2C). Dans le cas de cellules confinées, laver les débris de PDMS avec de l’isopropanol.
- Revêtement de chambre pour des expériences avec des cellules légèrement fixées en suspension
- Ajouter 100 μL de concanavaline A (ConA) à 0,5 mg/mL pour couvrir toute la surface de la cavité carrée et laisser incuber pendant 30 min.
REMARQUE: ConA est une lectine qui se lie aux sucres de surface cellulaire et couple des cellules individuelles sur la surface du verre de couverture. - Retirez la goutte ConA et rincez soigneusement la surface avec un milieu DMEM sans rayer la surface traitée ConA.
- Ajouter 30 μL de l’échantillon préalablement préparé (étape 2.6) dans le puits et remettre doucement en suspension pour se débarrasser des grappes de cellules.
- Fermez la cavité en plaçant doucement un verre de couverture de 22 x 22 mm #1.5 sur le dessus des jantes PDMS (évitez de le laisser tomber brusquement, utilisez une pince si possible, Figure 2B, C).
REMARQUE: Toute épaisseur de couvercle fonctionnerait pour le couvercle supérieur en verre (la lentille collectrice a une distance de travail de 2 mm).
- Ajouter 100 μL de concanavaline A (ConA) à 0,5 mg/mL pour couvrir toute la surface de la cavité carrée et laisser incuber pendant 30 min.
- Préparation de la chambre pour des expériences avec des cellules en confinement
- Placer une goutte de solution de 10 μL contenant des cellules (étape 2.6) dans la cavité carrée (figure 2B).
- Très doucement, sandwichez l’échantillon avec un verre de couverture de 22 x 22 mm de sorte que la goutte se propage dans toute la zone et qu’aucune bulle ne soit observée. Encore une fois, il est pratique d’utiliser des pinces, comme le montre la figure 2C, pour empêcher le verre de couverture de tomber brusquement.
4. Options alternatives pour l’espacement des chambres OT
REMARQUE: Ces étapes peuvent être suivies si aucun atelier de microfabrication ou de spin coater n’est disponible.
- Préparation de la chambre pour des expériences avec des cellules en suspension
REMARQUE: Dans le cas où aucun enduit de spin n’est disponible, une entretoise peut être fabriquée à l’aide de ruban adhésif scotch double face normal (environ 100 μm de hauteur).- Coupez un morceau de scotch double face avec un trou carré d’environ 10 cm x 10 cm au centre (mêmes dimensions que dans PDMS, Figure 2B).
- Retirez l’une des couches protectrices du ruban adhésif en le décollant et placez le côté découvert du ruban au centre d’un plat à fond de verre #1,5 H. Appuyez doucement pour que toute la surface adhère au verre tout en évitant les bulles d’air, puis retirez la couche protectrice restante du ruban en la décollant.
- Suivez les instructions de l’étape 3.2.
- Préparation de la chambre pour des expériences avec des cellules en confinement
REMARQUE: Pour confiner précisément les cellules, des microparticules monodispersées d’un diamètre connu peuvent être utilisées comme entretoises entre les deux verres de couverture.- Ajouter 10 μm de billes de polystyrène aux cellules en suspension à une concentration de 104 billes/μL.
- Mettez une goutte de solution de 10 μL contenant des cellules et des billes sur un verre de couverture de 22 x 60 mm.
- Très doucement, sandwichez l’échantillon avec un autre verre de couverture de 22 x 60 mm de sorte que la goutte se propage dans toute la zone et qu’aucune bulle ne soit observée. Pour positionner doucement le verre du couvercle supérieur (éviter qu’il ne tombe brusquement), il est pratique d’utiliser une pince.
- Comme l’échantillon peut se dessécher, il est recommandé d’effectuer la préparation rapidement.
5. Mise en place du piège optique pour les mesures intracellulaires
REMARQUE: Les étapes suivantes sont optimisées pour une plate-forme de pinces optiques commerciale comprenant un module de micromanipulation optique basé sur la déviation acousto-optique (AOD) et un capteur de force optique basé sur la détection directe des changements de moment lumineux (Figure 2, référence12,40,49). Les détails et les composants optiques de la configuration se trouvent à la figure 2F. Pour observer la déformation induite par la force lors des manipulations de la pince optique, un microscope confocal à disque rotatif Nipkow est couplé au port gauche du microscope inversé pour l’imagerie par fluorescence bicolore. Sans manque de généralité, ce protocole peut être appliqué avec n’importe quel système d’OT dynamique équipé de mesures de force directe basées sur la détection de moment lumineux. Des procédures détaillées étape par étape sont disponibles pour construire des pièges à gradient optiques maison pour des applications in vivo50. Ceux basés sur la modulation AOD se distinguent par d’éventuelles expériences avec des pièges multiples et des mesures rapides51,52. Plusieurs protocoles pour construire un instrument basé sur la quantité de mouvement lumineux existent dans la littérature36,39,40,53, et toute autre modalité d’imagerie (contraste d’interférence différentiel, fluorescence à grand champ, etc.) peut être utilisée.
- Démarrage de la pince à épiler optique
- Afin d’optimiser la stabilité de la puissance de sortie, allumez le laser à une puissance considérablement élevée (par exemple, 3 W) au moins 30 minutes avant l’expérience.
- Allumez le module électronique des unités de micromanipulation optique et de mesure de force.
REMARQUE: Appliquez toutes les mesures de sécurité laser et n’utilisez que l’équipement approuvé par le conseil d’administration de l’établissement. N’utilisez jamais les oculaires du microscope optique lorsque le laser est allumé. Utilisez toujours des lunettes de protection IR approuvées (OD7 dans la gamme 950-1080 nm), bloquez la lumière laser IR avec l’obturateur dans le port d’épifluorescence 2 et n’exécutez pas le logiciel de piégeage optique avant d’avoir terminé l’alignement du capteur de force optique après l’étape 5.3. En général, n’utilisez pas un échantillon hautement réfléchissant, car la rétroréflexion pourrait endommager le laser. - Contrôlez la puissance du piège avec le HWP rotatif (Figure 2F) à l’entrée du module de micromanipulation optique.
REMARQUE: Le module de micromanipulation optique commercial utilisé dans ce protocole intègre déjà cette fonctionnalité. Pour les systèmes de piégeage optique faits maison, intégrez cet outil de contrôle de la puissance afin que des puissances laser plus élevées et plus stables puissent être utilisées.
- Utilisez une microchambre vide pour l’étalonnage
- Coupez un carré de 1 x 1 cm sur un ruban adhésif double face et fixez-le sur une lame de microscope de 1 mm d’épaisseur.
- Ajoutez de l’eau dans le carré et fermez-le par le haut avec un verre de couverture #1.5 (22 x 22 mm). Il est conseillé d’ajouter un volume d’eau légèrement plus élevé, par exemple 30 à 40 μL, pour éviter les bulles à l’intérieur de la chambre couverte. Essuyez doucement la chambre d’étalonnage en cas de déversement d’eau.
- Alignement du capteur de force optique
- Mettez une gouttelette d’eau sur l’objectif d’immersion dans l’eau 60x/1.2. Placez la chambre d’étalonnage sur la scène avec le verre de couverture #1.5 face à l’objectif. Concentrez-vous sur la surface inférieure, où se trouveront éventuellement les échantillons de cellules.
- Ajouter une gouttelette d’huile d’immersion sur le dessus de la lame de verre supérieure recouvrant l’échantillon (Figure 2D). Abaissez soigneusement la lentille collectrice de l’unité de capteur de force jusqu’à ce qu’elle entre en contact avec la gouttelette d’huile.
REMARQUE: La gouttelette doit être suffisamment grande pour couvrir toute la lentille qui recueille la lumière laser sortant des pièges. Habituellement, 200 μL sont suffisants pour couvrir toute la surface et fournir un contact d’immersion stable. Soyez prudent et évitez le remplissage excessif car il pourrait s’infiltrer dans l’échantillon. - En suivant le protocole du fabricant pour l’alignement du capteur de force optique, regardez l’image du plan d’échantillonnage sur la caméra auxiliaire qui sera utilisée pour positionner les OT (AUX, Figure 2F). Très doucement, abaissez le capteur de force optique jusqu’à ce que l’arrêt de champ (FS, Figure 2F-G) apparaisse conjugué sur le plan de l’échantillon. Cela garantira des mesures de force directe appropriées à partir de la détection invariante de l’échantillon des changements de moment lumineux40.
REMARQUE: Fermez suffisamment le FS pour que son image devienne plus petite que le champ de vision (FOV), donc visible. Soyez très prudent et ne poussez pas la lentille de collecte du capteur de force optique contre l’échantillon. La position verticale du capteur de force optique peut également être déterminée à partir de l’analyse de la distribution de la lumière de piégeage au BFP pour les cônes de lumière avec une ouverture numérique définie (NA). - Assurez-vous qu’il n’y a pas de bulles d’air dans la gouttelette d’huile; ceux-ci peuvent affecter directement les mesures de force. Pour vérifier la présence de bulles d’air, mettez la lentille Bertrand en place (BL, Figure 2G) et observez le trajet d’imagerie à travers l’oculaire. Si des bulles de saleté ou d’air sont visibles ou si plus d’huile est nécessaire (figure S1A), nettoyez la lentille et la chambre avec du tissu de lentille sans poussière et répétez la procédure décrite aux étapes 5.3.2 et 5.3.3. Un chemin optique dégagé est représenté à la figure S1B.
- À l’aide des vis latérales placées sur le support du capteur de force optique, centrez le FS dans le champ de vision. Pour plus de précision, ouvrez le FS de sorte qu’il remplisse presque le champ de vision visible sur la caméra auxiliaire (AUX, Figure 2F).
6. Optimisation de la pince optique
REMARQUE: La mesure de la force directe repose uniquement sur le changement de moment lumineux résultant de la force exercée sur la particule piégée, et donc, contrairement aux méthodes indirectes, la rigidité du piège n’a pas besoin d’être étalonnée avant chaque expérience. La conversion spécifique à l’instrument du facteur de déviation/force (α; pN/V, référence41) est calibrée par le fabricant et est donc invariante expérimentalement. Cependant, étant donné que le point laser est manipulé sur une surface de 70 μm x 70 μm, les étapes 6.2 à 6.5 sont essentielles pour assurer un piégeage optimal et une stabilité de puissance optimale. Les étapes suivantes sont fournies dans le logiciel du fabricant afin que les OT soient optimisés sur la zone de travail de manière semi-automatique.
- Lancez le logiciel OTs et le logiciel d’acquisition pour l’appareil photo AUX.
- Soustrayez la ligne de base de tension initiale en cliquant sur l’étape Étape 1 : Décalage électronique dans le sous-menu Calibration du système du logiciel d’entraînement de la pince optique.
- Pour effectuer un aplatissement de la puissance du piège sur la zone de travail OT, réglez la puissance du piège à la moitié de son maximum en faisant pivoter le HWP en conséquence. Ne modifiez pas la puissance du piège en changeant la sortie laser, mais avec le HWP rotatif (Figure 2F). Cliquez sur Étape 2: Puissance pour lancer la routine automatisée pour l’aplatissement de l’alimentation par piège.
REMARQUE : Il s’agit d’une étape critique pour compenser la variation de la puissance du piège dans la zone de travail des OT (Figure S1D). Une routine réussie réduit la variation de puissance du piège à 2% dans la zone de travail des OT et converge après 2 minutes. - Pour effectuer l’étalonnage de la position du piège, retirez le filtre IR afin que la lumière du laser soit visible sur la caméra. Trouvez le point IR en réglant le plan d’image focalisé sur la surface inférieure de la microchambre. Obtenez le plus petit spot IR possible en réglant le plan d’image (position de l’objectif) et le contraste de l’histogramme dans le logiciel d’acquisition AUX de la caméra. Si nécessaire, réduisez la puissance du piège optique en faisant pivoter le HWP (Figure 2F). Cliquez sur Étape 3 : Position pour démarrer l’étalonnage automatisé de la routine ou du positionnement des pièges.
REMARQUE: Cette routine permet la correspondance précise des coordonnées de position de l’OT dans l’APPAREIL de caméra aux angles de braquage AOD. Une routine réussie génère la cartographie angle-position en quelques secondes. - Compensation initiale du momentum
REMARQUE : Le mouvement du piège optique à travers l’échantillon provoque des variations dans la distribution lumière-moment au niveau du BFP (Figure S1E, F). Cela conduit à des changements de signal indépendants de la force liés à la position du laser sur la zone de travail, même si la puissance du piège a été aplatie comme à l’étape 6.3. La conséquence est une variation de la base de force due à la position (indépendante d’une force réelle agissant sur la perle piégée optiquement) qui doit être corrigée avant chaque expérience.- Réglez la puissance de piège qui sera utilisée dans les expériences, en faisant pivoter le HWP (Figure 2F).
- Cliquez sur l’option Décalage global dans le sous-menu Outils . Cela ouvrira l’assistant Offset Cancel du logiciel de pince à épiler optique qui corrige la ligne de base de momentum initiale.
- Cliquez sur Décalage | Compenser pour corriger l’élan initial de la variante de position.
REMARQUE: Si aucune modification n’affecte le chemin optique au cours des semaines en cours, les cartes d’aplatissement de la puissance de piégeage (étape 6.3) et de position (étape 6.4) resteront invariantes. Nous recommandons donc de toujours utiliser la même combinaison d’éléments optiques (miroirs dichroïques, filtres, etc.) susceptibles d’affecter le trajet du piège laser ou d’effectuer une nouvelle routine d’aplatissement de la puissance du piège. En ce qui concerne la compensation initiale de l’élan (étape 6.5), le fabricant de la plate-forme OTs fournit un étalonnage à la volée qui doit être modifié pour chaque nouvelle puissance de piégeage et session expérimentale. Les étapes 6.3 et 6.4 doivent être effectuées sur la lame d’étalonnage vide décrite à l’étape 5.2. Dans un échantillon contenant des cellules ou d’autres objets, l’étape 6.5 doit être effectuée sans objets susceptibles de modifier la diffusion de la lumière dans la zone de travail des OT.
- En option, piégez une microsphère et déplacez le piège à une vitesse connue tout en enregistrant le signal de force. Par exemple, réglez le piège pour effectuer une oscillation triangulaire : le signal de force enregistré sera un signal carré.
REMARQUE: La valeur de la force doit augmenter linéairement avec la vitesse, en fonction de la force de traînée agissant sur le cordon. Ce test sert de contrôle positif que les mesures de force sont effectuées correctement38. Alternativement, le capteur de force optique peut être utilisé pour obtenir la rigidité de piégeage optique, κ [pN/μm], et le facteur d’étalonnage de position, β [μm/V], à partir d’une analyse spectrale de puissance35. Sous alignement correct, le facteur d’étalonnage invariant fourni par le fabricant est α = κ·β [pN/V].- Lancez une lecture de force en temps réel en cliquant sur le graphique 1 dans le sous-menu Mesures du logiciel du fabricant. Cela fournira une lecture de la force et de la puissance de piégeage optique actuelles.
- Ouvrez la boîte de dialogue Paramètres d’oscillation dans le sous-menu Outils . Définissez une forme de forme d’onde d’espace triangulaire dans les anneaux de sélection Forme et Type, respectivement. À titre d’exemple, définissez une amplitude de 10 μm et une fréquence de 3 Hz. Il en résultera une force visqueuse d’environ 1 pN sur une microbille d’un diamètre de 1 μm38.
- Dans la fenêtre AUX de l’appareil photo, cliquez avec le bouton droit de la souris sur la microbille et sélectionnez Démarrer l’oscillation. La lecture de force deviendra un signal de force carré avec des plateaux à ±1 pN.
- Faites un clic droit sur la microbille et sélectionnez Arrêter d’osciller.
7. Microscopie confocale à disque rotatif
- Allumez le microscope confocal à disque rotatif et l’équipement accessoire, les moteurs laser intégrés et les caméras d’acquisition.
- Lancez le logiciel d’imagerie.
- Définir des canaux d’imagerie pour la coloration Hoechst du noyau et GFP pour la membrane plasmique cellulaire.
- Activez les lignes des lasers d’excitation 405 nm et 488 nm.
- Ajoutez un dichroïque multibande pour réfléchir l’excitation à l’échantillon et qui permet à la lumière émise de passer aux caméras.
- Divisez l’émission de fluorescence avec un miroir dichroïque à bord de passage de 500 nm de long.
- Utilisez les filtres d’émission DAPI/BFP (~445 nm) et GFP (~521 nm) devant les deux caméras d’acquisition, respectivement. Reportez-vous à la Figure 2F,G.
- Réglez le temps d’exposition à 100 ms pour chaque canal.
- Réglez l’émission laser pour obtenir une puissance de 5 mW au niveau du plan d’échantillonnage. Pour mesurer la puissance, utilisez un capteur de puissance commercial.
- Définissez le protocole d’imagerie. Pour éviter le saignement spectral du canal Hoechst dans le canal GFP, les deux colorants doivent être imagés séquentiellement.
REMARQUE : S’il existe une synchronisation matérielle entre les AOD du piège optique et l’acquisition de la caméra, assurez-vous que la polarité du déclencheur est correctement configurée. En cas de doute, consultez votre gestionnaire d’installations ou votre fabricant de microscopes.
8. Réalisation des expériences d’indentation du noyau
REMARQUE: Éteignez toujours les pièges optiques - à la fois à l’aide d’un logiciel et en fermant l’obturateur sur le port d’épifluorescence 2 - lorsque vous soulevez le module de capteur de force et changez l’échantillon. Sinon, de graves dommages aux éléments optiques et à l’expérimentateur pourraient se produire. Faites attention à la distance latérale entre le support de lentille et le bord inférieur de la parabole lorsque vous recherchez des cellules afin d’éviter de heurter la lentille dans la scène/la boîte de culture (Figure 2).
- Placez l’échantillon dans le microscope et suivez l’étape 5.3 de ce protocole.
- À l’aide du HWP rotatif (Figure 2F), réglez la puissance du piège à 200 mW comme valeur de départ si la rigidité du noyau ou de la structure intracellulaire étudiée n’est pas connue. Traduisez la zone de travail des OT (à l’aide de l’étage du microscope) en un endroit exempt de cellules afin de compenser la base de mouvement initiale jusqu’à l’étape 6.5.
REMARQUE: Selon la rigidité de la structure subcellulaire, la valeur de puissance du piège doit être ajustée à des valeurs inférieures ou supérieures pour obtenir une profondeur d’indentation similaire. - À l’aide du contrôleur logiciel de l’étage de microscope, recherchez une cellule avec une ou deux perles grâce à la microscopie à fond clair transmise (Figure 3A).
- Définissez une trajectoire de piège.
- Ouvrez la boîte de dialogue Trajectoire dans le sous-menu Outils et choisissez Déplacement dans l’anneau de sélection Type de trajectoire .
- Dans la feuille numérique, écrivez le déplacement et le temps de chaque étape de trajectoire suivante. Voici deux exemples.
- Pour une expérience de relaxation du stress, programmez des charges trapézoïdales, comme illustré à la figure 3B. Dans le tableau S1, deux indentations trapézoïdales ont été appliquées avec une distance de déplacement de 5 μm; vitesse de 5 μm/s; temps d’attente avant rétractation : 10 s.
- Pour une expérience d’indentation répétitive à vitesse constante afin d’obtenir une routine triangulaire sans temps d’arrêt sur le noyau, définissez l’amplitude de la trajectoire, par exemple, 5 μm, et le temps du pas, par exemple, 2 s pour une vitesse de 2,5 μm / s. Dans le tableau S2, cela est appliqué huit fois à la même vitesse.
REMARQUE: Ces valeurs doivent être déterminées pour chaque type de cellule et expérience, mais les paramètres suivants d’une routine trapézoïdale capturent la dynamique la plus importante de l’expérience présentée ici. Le temps d’attente doit être suffisant pour que le noyau montre sa relaxation complète du stress après l’indentation
- Piéger une microsphère
- Réglez le plan de l’image légèrement au-dessus de la perle avec le contrôleur logiciel de l’étage du microscope.
- Activez les pièges à l’aide du logiciel OTs et cliquez sur le cordon dans la fenêtre d’imagerie AUX de la caméra (calibrée à l’étape 6.4). Un confinement réussi de la perle par le piège optique réduira fortement le mouvement de la perle.
- Cliquez et faites glisser la bille sur le cytoplasme et placez-la à une distance d’environ 2 μm de l’enveloppe nucléaire (Figure 3A). Assurez-vous que la trajectoire est définie de manière à ce que l’indentation de la perle soit perpendiculaire à la membrane nucléaire.
- Éventuellement, si nécessaire pour les mesures de position de la perle par rapport au piège, scannez le piège à travers la perle pour déterminer la rigidité du piégeage, k [pN/μm]54, donc Δxbead = -F/k (voir Discussion). Le module de micromanipulation optique utilisé dans ce protocole dispose d’une routine intégrée à cet effet.
- Ouvrez la boîte de dialogue Analyse des particules dans le sous-menu Outils .
- Sélectionnez l’interruption que vous souhaitez analyser et Haute fréquence comme méthode d’analyse. Sélectionnez la direction (x ou y) de la trajectoire d’indentation pour la mesure de balayage des perles.
- Une fenêtre apparaîtra avec la mesure de la rigidité du piégeage. Dans le graphique, faites glisser les deux curseurs pour sélectionner la zone de recouvrement linéaire correspondant à F = -kx. L’ajustement linéaire à la partie de données sélectionnée sera actualisé automatiquement.
REMARQUE: Réglez la position initiale de la perle loin de la membrane cellulaire (~ 5 μm), car les déviations lumière-moment à l’interface milieu-cellule affectent la pertinence des mesures de force. Si le noyau est situé trop près de la membrane cellulaire, essayez d’indenter le noyau du site opposé. Jetez la cellule si ce n’est pas possible.
- Démarrez l’acquisition d’images en cliquant sur le bouton d’acquisition dans le logiciel d’imagerie.
- Démarrez l’enregistrement des données de mesure de la position du piège et de la force en cliquant sur Données | Enregistrez dans la fenêtre de lecture de force en temps réel (ouverte comme à l’étape 6.6.1).
REMARQUE: Le piège optique est équipé d’une entrée de déclenchement qui peut être connectée à la sortie de synchronisation de la caméra. Ainsi, les données d’image et de force sont synchronisées matériellement et l’électronique est capable de cartographier les cycles de piégeage avec le nombre d’images des images lors de l’acquisition. - Lancez la trajectoire précédemment chargée en cliquant avec le bouton droit de la souris sur le cordon et en sélectionnant Démarrer la trajectoire.
- Attendez que la trajectoire soit terminée et que le système se stabilise.
- Arrêtez l’enregistrement des données de mesure de la force de piégeage. Une boîte de dialogue d’enregistrement des données apparaîtra.
REMARQUE : Pour optimiser le stockage des données, les données peuvent être décimées en sélectionnant le paramètre décimant dans cette boîte de dialogue (10, 100 ou 1000). - Arrêtez l’acquisition d’images et tracez les résultats dans le logiciel de post-traitement au choix de l’utilisateur.
- Si la microsphère est perdue pendant la routine et que le noyau ne peut pas être indenté (figure S2), abandonnez la mesure et augmentez la puissance. Notez que l’étape 6.5 doit être répétée. Dans nos mains, au moins 95% des routines sont terminées avec succès sans perdre la perle du piège.
Résultats
Microinjection de perles de piégeage:
Les microsphères injectées dans l’embryon de poisson zèbre unicellulaire se sont répandues sur l’ensemble de la calotte animale au cours de la morphogenèse. Pour une visualisation plus claire, nous avons répété le protocole d’injection avec des microbilles fluorescentes rouges et pris des images volumétriques avec notre microscope confocal à différents stades de développement. Dans la figure 4A-D, les billes injectées sont visualisées dans le cytoplasme des cellules souches progénitrices in vivo à 5 hfp. Plus tard, des microsphères sont apparues réparties sur l’ensemble de l’embryon à 24 hpf (Figure 4E). Les embryons aux deux stades se sont développés normalement et les taux de survie étaient comparables à ceux des embryons témoins non injectés ou injectés de manière simulée (voir la figure S3). Ceci est cohérent avec d’autres études qui rapportent une survie imperturbable du poisson-zèbre injecté dans des perles jusqu’à 5 jours après la fécondation55.
Notre microscope confocal à disque rotatif est compatible avec la microcopie à fluorescence multicanal. Dans la figure 5A, nous montrons des cellules souches isolées avec une ou deux perles dans le cytoplasme. Plusieurs étiquettes fluorescentes peuvent être utilisées pour étudier différents aspects de la cellule (Figure 5B). La morphologie nucléaire peut être suivie avec un colorant Hoechst ou à l’aide d’une expression d’ARNm H2A::mCherry, tandis que la membrane nucléaire interne peut être analysée avec Lap2b-eGFP12. La dynamique du cortex actomyosin, ainsi que les niveaux de calcium intracellulaire, peuvent être observés avec une incubation my12.1::eGFP transgénique56 et Calbryte-520, respectivement. Le protocole qui a été décrit ici vise à comparer la mécanique du noyau cellulaire des cellules de type sauvage immobilisées sur des substrats adhésifs (plus tard appelés suspension) et en confinement mécanique. Les cellules souches isolées confinées dans des microchambres de 10 μm de hauteur ont présenté un déploiement partiel de la membrane nucléaire interne (INM) et une augmentation subséquente de la contractilité de l’actomyosine12. Dans la figure 5C, des cellules confinées avec une ou deux perles dans le cytoplasme sont montrées. Un confinement réussi sera visible via des cellules aplaties et élargies avec une section transversale plus large du noyau. La membrane nucléaire est ensuite dépliée dans des cellules confinées et devrait apparaître lissée par rapport aux cellules en suspension (Figure 5C).
Analyse force-temps et force-déformation
L’analyse des résultats obtenus dépend fortement du spécimen étudié et de la question d’intérêt et ne peut donc pas être généralisée ici. À titre d’exemple, une façon courante d’analyser la mesure de l’indentation consiste à extraire le module de Young en ajustant un modèle Hertz modifié aux données d’indentation de force57. Cependant, l’hypothèse d’un tel traitement doit être soigneusement évaluée et peut ne pas toujours être correctement justifiée (par exemple, la structure étudiée étant isotrope, homogène, avec une élasticité linéaire et des indentations inférieures au rayon de la perle). Nous ne considérons donc ici que des mesures indépendantes du modèle qui permettent de comparer le comportement mécanique de la structure étudiée entre différents scénarios expérimentaux.
Comme point de départ, la mesure de la pente de la courbe force-déplacement à une certaine profondeur d’indentation fournit une mesure d’une rigidité structurelle indépendante du modèle58 du noyau. Cette valeur peut ensuite être collectée à partir de plusieurs échantillons et comparée entre différents contextes expérimentaux et perturbations d’échantillons.
Mesure de l’indentation
Dans les lignes suivantes, nous nous concentrons sur la réponse mécanique du noyau cellulaire lors de la déformation cellulaire en confinement. Les expériences de l’étape 8 de ce protocole conduisent généralement à des pics de force allant jusqu’à 200 pN pour des profondeurs d’indentation d’environ 2-3 μm. Cependant, ces valeurs peuvent être très différentes, selon le type de cellule et les conditions expérimentales, avec des noyaux plus mous conduisant à une force plus faible pour une indentation donnée. Il est donc nécessaire de mesurer avec précision la déformation nucléaire, ainsi que la force, pour une caractérisation mécanique précise du noyau cellulaire. Dans cette section, nous obtiendrons la rigidité nucléaire de la cellule à partir de mesures d’indentation de force représentatives.
Dans la figure 6, nous montrons les déformations des côtés distal et proximal d’un noyau dans une cellule en suspension et confinée. Un comportement mécanique riche peut être observé. Dans une cellule suspendue typique sur un substrat adhésif, le noyau était fortement indenté par la perle, mais aussi légèrement déplacé lors d’événements de poussée répétitifs. Nous avons mesuré l’indentation des billes sur le noyau en analysant les kymographes obtenus à partir de l’imagerie par fluorescence des noyaux cellulaires colorés par Hoechst. Les kymographes ont été facilement calculés à l’aide du plug-in Multi Kymograph de Fidji le long de la direction d’indentation (Figure 6A, B) et importés dans Matlab (version 2021, Mathworks) pour un traitement ultérieur. Une fonction de pas a été ajustée au profil d’intensité brute dans le but de suivre les bords délimitants du noyau le long de la trajectoire de la routine d’indentation. Comme on peut le voir, il contient des informations précises sur le changement de forme nucléaire (Figure 6 et Figure S2). Nous avons utilisé la courbe double sigmoïde suivante comme version analytique d’une fonction pas à pas :
 (Équation 1)
(Équation 1)
Ici, x1 et x2 désignent les bords distaux et proximaux du noyau, tandis que A et B sont les valeurs de gris maximum et d’arrière-plan du canal bleu (colorant Hoechst) de l’image (Figure 6B). La largeur du bord a été prise en compte (e0 = 0,25 mm). Alors que le bord du noyau proximal en retrait (x2) suivait la trajectoire appliquée par la routine de piège optique après le contact microsphère-noyau, le bord distal opposé (x1) affiche la dynamique de relaxation comme prévu pour un matériau viscoélastique tel que le cytoplasme (Figure 6D). En revanche, les noyaux dans les cellules confinées dans des microchambres de 10 μm de haut ne présentent pas un tel comportement de translocation du noyau lors de l’indentation à l’intérieur de la cellule (Figure 6B, D). Également illustré à la figure 6D, les bords arrière des noyaux restent inchangés par la perle poussant du côté proximal, probablement en raison de forces plus fortes résultant de la contractilité cellulaire et du frottement agissant contre la force d’indentation. Afin d’obtenir la profondeur de déformation correcte, le déplacement x1 a été soustrait de la mesure en retrait x2: Δx = x2 - x1 (voir aussi la figure 6D).
Analyse des données de force
La force à l’origine de la déformation nucléaire a été mesurée à partir du changement de moment lumineux provenant de la microbille piégée optiquement (figure 7A). La force lors de l’application de trajectoires trapézoïdales (étape 8.4.3, figure 7B) a d’abord augmenté linéairement jusqu’à ce que le piège cesse de bouger, puis s’est détendue à une valeur stable. Ce comportement indiquait un matériau viscoélastique présentant des modules de perte et de stockage. Juste après l’événement d’indentation, la force a atteint une valeur de crête, Fp, suivie d’une relaxation de contrainte (Figure 7C) :
 (Équation 2)
(Équation 2)
où F0 est la force stockée pour le composant élastique et f(t) est une fonction de relaxation sans dimension. Nous avons analysé ce comportement de trois manières :
1. Considérant un solide linéaire standard avec une relaxation exponentielle des contraintes, c’est-à-dire f(t) = e-t/τ, représenté schématiquement dans l’encart de la figure 7C.
2. Utilisation d’une désintégration générale à double exponentielle :
F(t) = A + B1e-t/τ1 + B2e-t/τ2.
3. Utilisation d’une loi de puissance suivie d’une désintégration exponentielle59 :
f(t) = t-pe-t/τ, ajusté à la figure 7C.
Bien que l’ajustement pour le modèle 1 puisse être effectué facilement, nous recommandons d’estimer les suppositions initiales pour (τ1, τ2) et (p, τ) pour les modèles 2 et 3, respectivement. Cela peut être effectué, respectivement, en ajustant des lignes sur les données dans des échelles logarithmiques versus linéaires (Figure 7D, à gauche) et logarithmiques contre logarithmiques (Figure 7D, à droite). Le tableau S3 résume les résultats de l’exemple analysé à la figure 7. Dans la section suivante, nous examinerons la combinaison d’une loi de puissance et d’une loi exponentielle pour la caractérisation de la mécanique du noyau cellulaire.
Relation de déplacement de force
De même, la configuration expérimentale décrite peut être utilisée pour obtenir la relation force-déplacement de plusieurs événements d’indentation. En effectuant des routines triangulaires (étape 8.4.4, Figure 8A), il est possible de relier la force à la déformation et de tracer une courbe force-indentation. Un résultat exemplaire est illustré à la figure 8B, dans laquelle une ligne de base plate changeait de pente en douceur une fois que la perle est entrée en contact avec le noyau. Identifier le véritable point de contact dans les données bruyantes est un défi, et il faut prendre soin de voir si la région de contact est adaptée aux modèles élastiques60. Dans cette expérience particulière, on a également pu voir que les indentations ultérieures entraînent des courbes avec des points de contact plus profonds, ce qui indique une récupération trop lente de la forme nucléaire après la rétraction des billes et une modification du cycle hystérétique défini par les propriétés du matériau viscoélastique du noyau (Figure 8C). Ainsi, le chercheur doit être conscient si cela se produit et l’intégrer dans le pipeline analytique, ou restreindre le nombre de mesures ultérieures de sorte que cet effet ne modifie pas la mesure.
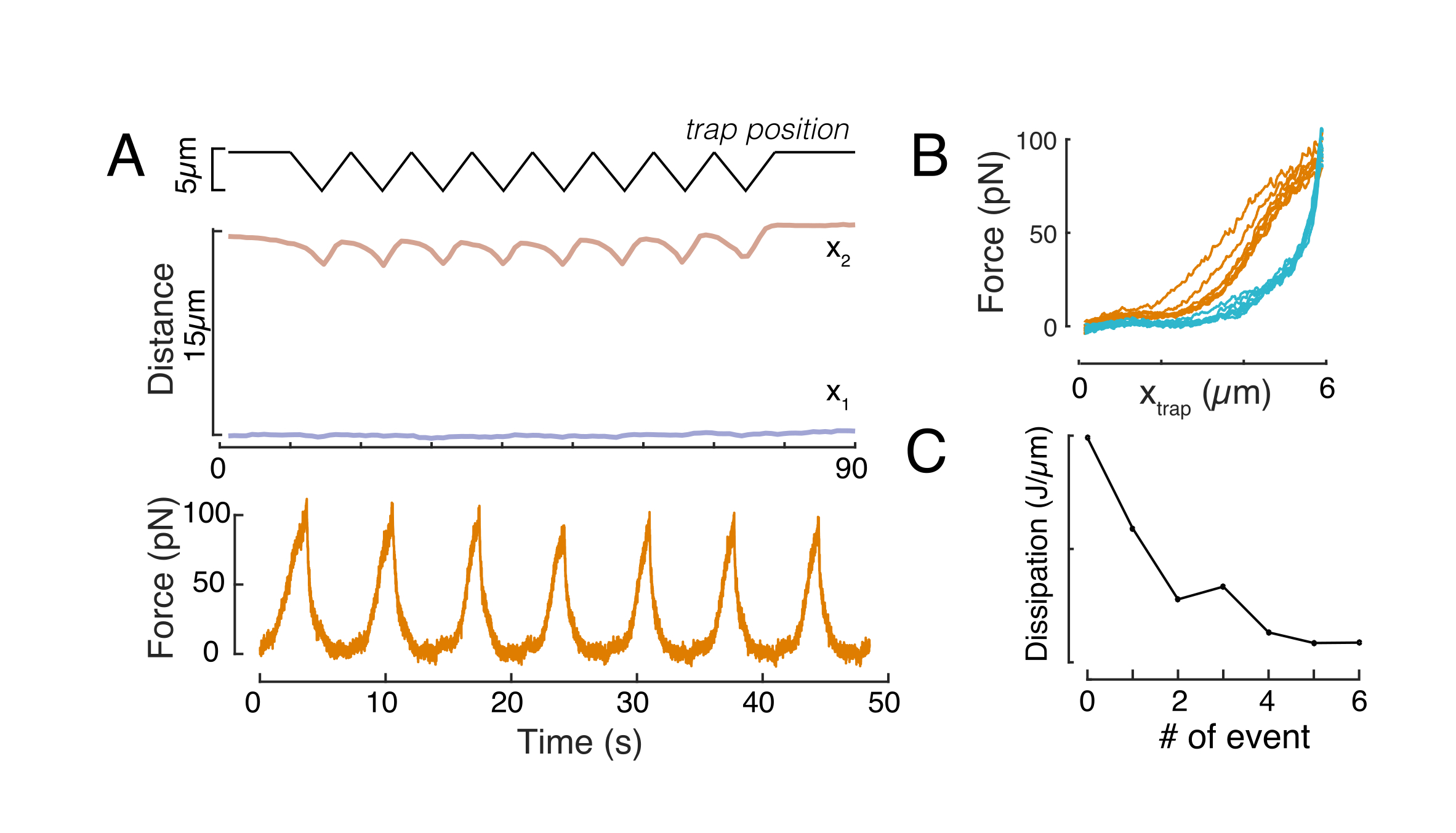
Mécanique du noyau dans les cellules en suspension et sous confinement de 10 μm
L’approche susmentionnée a été utilisée pour analyser la dynamique de la relaxation du stress du noyau dans les cellules en suspension sur des substrats adhésifs et des cellules confinées. Nos résultats montrent que le confinement entraîne une expansion de la surface projetée (Figure 9A), mais un changement insignifiant de la rigidité nucléaire (Figure 9B). Nous avons mesuré une relaxation similaire avec τ = 6,08 ± 1,1 s (non confiné) et τ = 4,00 ± 0,6 s (confinement), ce qui indique une dissipation viscoélastique rapide, suivie d’une valeur de force stockée qui correspond au module élastique du noyau. Afin de tenir compte des variations expérimentales, qui peuvent être produites par différentes conditions initiales dans les routines d’indentation, les forces stockées mesurées ont été normalisées à la profondeur d’indentation, comme  . Ce paramètre tient compte de la rigidité du noyau et décrit la force, ou la contrainte, nécessaire pour une certaine indentation. Nous avons obtenu une rigidité similaire sous confinement et dans des cellules non confinées : = 20,1 ± 12,6 pN/μm et = 24,6 ± 13,6 pN/μm (écart-type moyen ±), respectivement.
. Ce paramètre tient compte de la rigidité du noyau et décrit la force, ou la contrainte, nécessaire pour une certaine indentation. Nous avons obtenu une rigidité similaire sous confinement et dans des cellules non confinées : = 20,1 ± 12,6 pN/μm et = 24,6 ± 13,6 pN/μm (écart-type moyen ±), respectivement.

Figure 1: Microinjection d’embryons de poisson zèbre au stade unicellulaire (zygote). (A) Plaque d’injection: une plaque d’injection de forme triangulaire est utilisée pour l’injection. La plaque est composée de 1% d’agarose ultrapure dans E3 (milieu de l’œuf). Les vues supérieure et latérale sont affichées à droite. (B) Positionnement de l’embryon: orienter doucement les embryons à l’aide d’une brosse et s’orienter de manière à ce que la cellule soit clairement visible et facilement accessible avec l’aiguille. Nous suggérons d’orienter les embryons avec la cellule située du côté opposé de l’aiguille, comme le montre le croquis. (C) Procédure d’injection dans l’embryon au stade unicellulaire: percer le chorion entourant l’embryon et la cellule unique avec l’aiguille. Assurez-vous que le bout de l’aiguille est à l’intérieur de la cellule et relâchez la pression pour injecter. D) Incuber les embryons à 28-31 °C jusqu’à ce qu’ils se développent jusqu’au stade de blastula (sphère) (4 hpf). Effectuer le protocole d’isolement cellulaire et la coloration cellulaire (étape 2) et préparer la chambre de piégeage optique avec des cellules isolées en suspension et/ou en confinement combinées avec le revêtement de surface du substrat correspondant (étape 3). Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2: Préparation de l’appareil de préadolescent optique. (A) Couches de PDMS à revêtement rotatif d’une hauteur définie sur des plats de fond en verre. La goutte PDMS s’étalera uniformément en raison de la force centrifuge. B) Préparation de la chambre d’échantillonnage à partir de la couche PDMS. 1: couper un carré avec un scalpel, 2: recouvrir le puits intérieur de concanlarine A (ConA), laver et ensemencer les cellules; 3: couvrir avec une glissière en verre ou un couvercle pour sceller le puits. (C) Image de la coupe carrée avec un scalpel et du retrait du puits PDMS avec une pince. (D) Montage de la lentille collectrice du capteur de force optique au-dessus de la chambre de piégeage. Une goutte d’huile d’immersion sert de milieu d’immersion entre la lentille collectrice et le couvercle supérieur en verre. Schématique pas à l’échelle. Soyez prudent lorsque vous abaissez la lentille collectrice pour ne pas toucher le couvercle en verre du plat d’échantillon. E) Image de l’unité de détection de force en contact avec l’échantillon. (F) Schéma du dispositif expérimental. Le module de micromanipulation optique utilise un faisceau laser à ondes continues (5W, λ = 1064 nm) avec contrôle de puissance à travers une plaque demi-onde (HWP) et un séparateur de faisceau polarisant (BS). Après avoir été modulé avec une paire d’AOD, il est couplé au port d’épifluorescence supérieur d’un microscope inversé. Le faisceau laser est ensuite réfléchi par un miroir dichroïque à passage court de 950 nm (IR-DM), permettant la transmission de l’excitation et de l’émission de fluorescence. Le laser de piégeage est guidé dans l’orifice d’épifluorescence arrière du microscope (tourelle supérieure). Les OT sont créés au plan focal d’un objectif à immersion dans l’eau (60x, NA = 1,2). Le capteur de force optique est soumis par la tourelle du microscope et capture la lumière laser émergeant des OT avec une lentille à haute NA à immersion dans l’huile. Dans le même temps, le capteur de force permet un éclairage en champ lumineux. L’unité confocale à disque rotatif est couplée au port gauche. Il est équipé de deux moteurs laser intégrés (ILE) qui contrôlent sept lasers d’excitation de fluorescence et de deux caméras sCMOS rétro-éclairées, permettant une imagerie à double fluorophore en parallèle Abb: TI, Transilluminator; FS, arrêt sur le terrain; AOD, déflecteur acustooptique; HWP, plaque demi-onde; CAM, caméra (G) Photographie de l’équipement de piégeage optique. Le cercle rouge indique l’objectif bertrand, qui peut être commuté manuellement dans le chemin optique. Veuillez cliquer ici pour voir une version agrandie de cette figure.
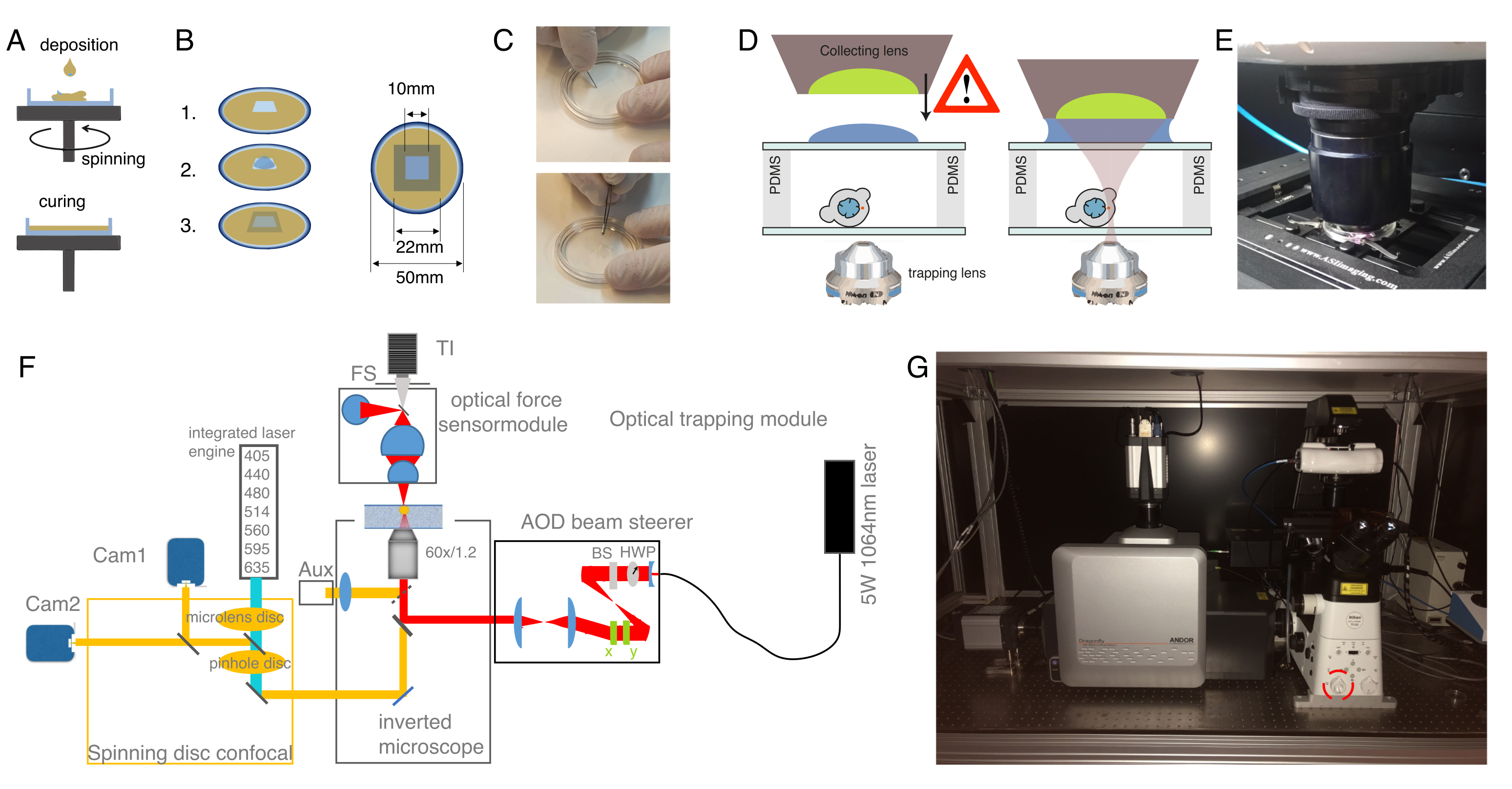{kind=link}

Figure 3 : Choix des bons échantillons et paramètres. (A) Image représentative d’une cellule souche progénitrice isolée du poisson-zèbre avec une seule microsphère positionnée suffisamment près du noyau pour effectuer l’expérience d’indentation. Barre d’échelle = 10 μm. (B) Trajectoire exemplaire du piège; profondeur d’indentation 5 μm; vitesse d’indentation = 5 μm/s; temps de détente 10 s. Veuillez cliquer ici pour voir une version agrandie de cette figure.
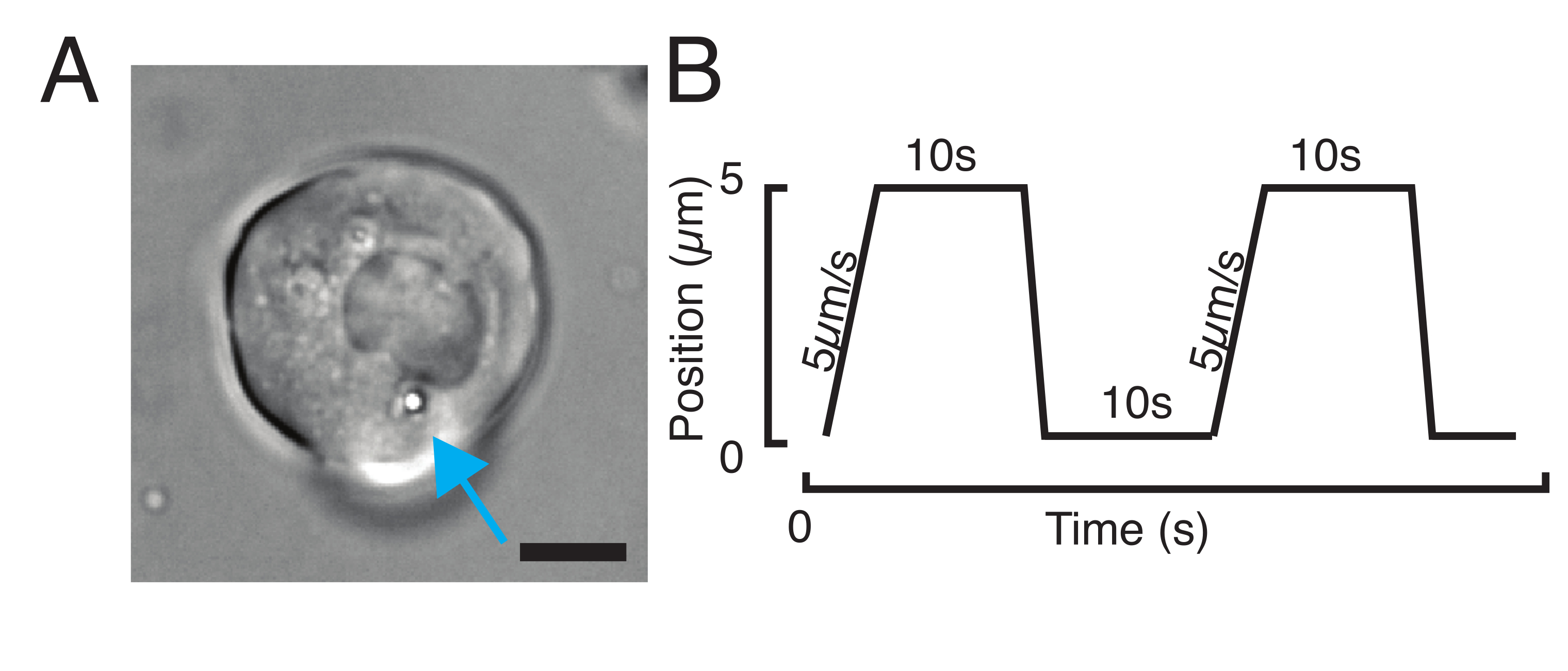{kind=link}

Figure 4 : Localisation des microbilles à l’intérieur des embryons de poisson-zèbre pendant le développement. 0,5 nL de billes fluorescentes rouges de 1 μm sont injectées avec l’ARNm GPI-GFP (100 pg/embryon, membrane plasmique) dans les embryons WT pour visualiser les localisations des billes. (A-D) Distribution de la microsphère 5 h après l’injection à l’intérieur d’un embryon monté dans 0,75% d’agarose. (A) Image en champ clair et en fluorescence. Les perles sont dispersées de manière homogène dans le tissu embryonnaire, comme on le voit dans une micrographie confocale. (B) Projection maximale de la fluorescence confocale z-stack. Les perles sont codées par couleur du violet au jaune en fonction de leur position z dans la pile d’images. Le violet/magenta correspond aux perles/cellules les plus externes (EVL; couche enveloppante épithéliale; ou cellules souches progénitrices situées près de la surface de la LVE), le jaune correspond aux perles internes (cellules profondes progénitrices), comme le montre l’esquisse à droite. (C) Coupe et projection maximale d’une sous-pile de (B) correspondant à la région dans la boîte orange: une grande fraction de cellules profondes contient 1-2 perles. (D) Coupe et projection maximale d’une sous-pile de (B) correspondant à une boîte magenta: certaines cellules EVL contiennent 1-2 perles. (E) Image en champ clair et projection maximale d’une pile z d’un embryon de 24 hpf monté dans de l’agarose à 0,75% et anesthésié avec de la tricaïne. Les embryons ont été pré-incubés avec de la tricaïne pendant 15 min. De gauche à droite : microsphères (1 μm de diamètre), GPI-GFP et chevauchement d’images. Les perles réparties sur tout le corps de l’embryon. Dimension de la barre d’échelle indiquée dans chaque panneau. Veuillez cliquer ici pour voir une version agrandie de cette figure.
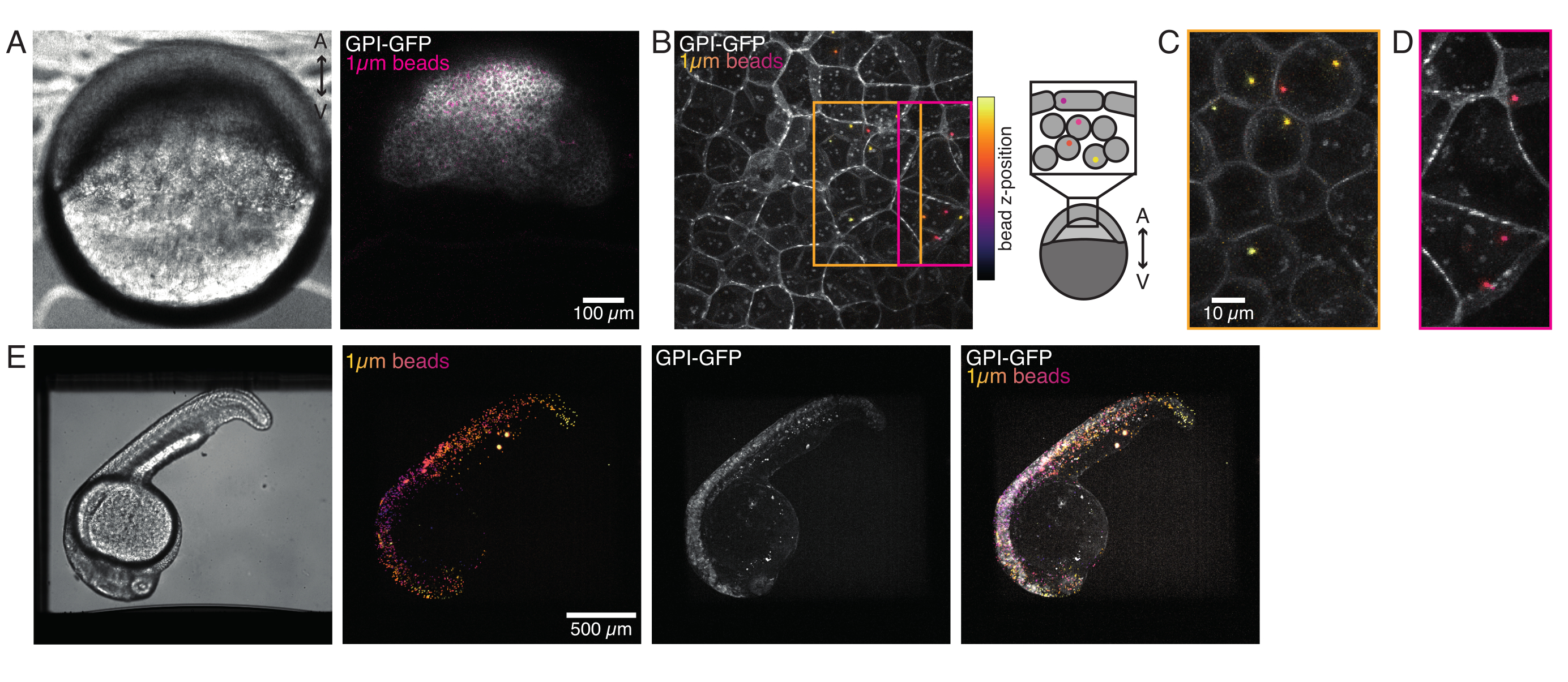{kind=link}

Figure 5 : Cellules souches progénitrices isolées du poisson-zèbre avec un marquage différent. (A) Image de microscopie à lumière à transmission de cellules de suspension avec 1 (en haut) ou 2 (en bas) perles injectées. Des flèches cyan pointent vers les perles. (B) Images confocales fluorescentes de cellules de suspension avec différentes colorations. En haut à gauche : Lap2b-eGFP (membrane nucléaire interne, 80 pg/embryon) et H2A-mCherry. En haut à droite : GPI-GFP (membrane plasmique, 100 pg/embryon) et DNA-Hoechst (coloré comme décrit à la rubrique 2). En bas à gauche : MyI12.1-eGFP (lignée transgénique) et DNA-Hoechst. En bas à droite : Calbryte488 et DNA-Hoechst (coloré comme décrit à la rubrique 2). (C) Image de microscopie à lumière à transmission de cellules confinées avec 1 (en haut) ou 2 (en bas) perles injectées. Des flèches cyan pointent vers les perles. Barres d’échelle = 10 μm. Veuillez cliquer ici pour voir une version plus grande de cette figure.
{kind=link}

Figure 6: Estimation de la déformation nucléaire à partir de films de disques en rotation. (A, B) Time-lapse d’une expérience d’indentation du noyau dans (A) une cellule en suspension et (B) une cellule confinée. Barre d’échelle 10 μm. Des instantanés représentatifs d’un noyau marqué hoechst sont montrés 5 s avant, pendant et 5 s après l’indentation avec une microsphère piégée optiquement (pointe de flèche blanche). Kymographes le long du segment d’indentation (ligne rouge, panneau de droite). x1 et x2 sont les limites distales et proximales (proches de la perle) du noyau pendant l’expérience d’indentation extraites de l’ajustement du profil d’intensité à l’équation 1. (C) Profils d’intensité le long du segment d’indentation pour trois images différentes (avant, pendant et après l’indentation) et ajustés à l’équation 1 pour évaluer les positions distales, x1 et proximales, x2, des bords du noyau. (D) Trajectoires représentatives de x1(t) en bleu et x2(t) en ambre lors d’une expérience d’indentation de cellules en suspension et confinées (10 μm). Les zones ombrées indiquent l’indentation, la distance entre x1 et x2 indique le diamètre du noyau. Veuillez cliquer ici pour voir une version agrandie de cette figure.
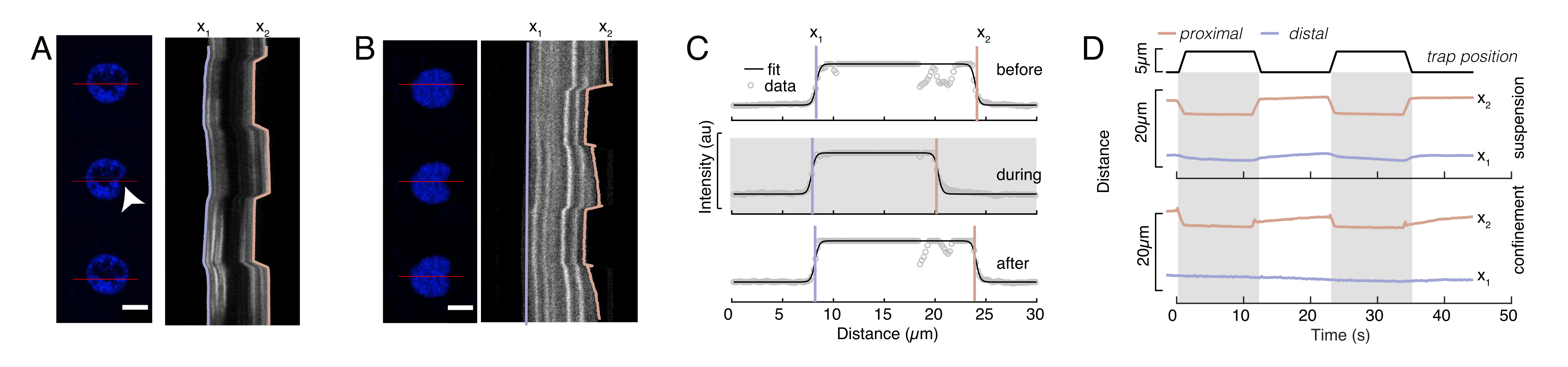{kind=link}

Figure 7: Traitement du signal de force. (A) Schéma d’une microsphère piégée optiquement déformant le noyau cellulaire lors de l’indentation. La membrane nucléaire et les forces optiques sont indiquées par les flèches noires. Le changement de moment cinétique du faisceau est indiqué par la flèche verte Pout. (B) Trajectoire du piège (en haut) et force (en bas) subies par la microsphère piégée optiquement au cours d’une expérience répétée d’indentation nucléaire. (C) Désintégration de relaxation de la force après le pic de force à la profondeur d’indentation maximale. L’encadré montre un schéma de solide linéaire standard dont la dynamique se rapproche des observations phénoménologiques ici. (D) Gauche : logarithme de la force normalisée par rapport au temps. Les zones ombrées indiquent la partie de données utilisée pour s’adapter à la double désintégration exponentielle (lignes rouges). Droite : logarithme de la force normalisée versus logarithme du temps. La zone ombrée indique la partie de données utilisée pour s’adapter à la loi de puissance. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 8 : Routine d’indentation de force avec déplacements de pièges triangulaires. (A) Trajectoire représentative de x1(t) en bleu et x2(t) en ambre lors d’une expérience d’indentation triangulaire prise sur une cellule à une hauteur de confinement de 10 μm. En haut : Position du piège. Milieu : Analyse de la forme du noyau. La distance entre x1 et x2 indique le diamètre du noyau. En bas: Signal de force. (B) Force vs position du piège pendant huit indentations consécutives. (C) Évolution de la dissipation, dérivée de l’hystérésis entre la partie approche et retrait de la courbe f-d, du noyau pour chaque événement d’indentation ultérieur. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Graphique 9. Propriétés nucléaires des cellules en suspension (surface adhésive) et confinement des routines trapézoïdales. (A) Surface projetée du noyau à partir de cellules en suspension et sous confinement de 10 μm. La barre noire représente la médiane. B) Rigidité nucléaire des cellules en suspension et en confinement. La barre noire représente la médiane. Valeurs P dérivées du test de Kruskal-Wallis à l’aide de MatLab. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Tableau supplémentaire 1 : Trajectoire trapézoïdale définie par le logiciel de pinces optiques. La première (deuxième) ligne est la distance x (y) à laquelle le piège sera déplacé linéairement. Sur la troisième ligne, la durée d’une étape donnée est définie en secondes. Cette trajectoire est composée de sept points et correspond au trapèze chargé deux fois contre le noyau de la figure 7B. Veuillez cliquer ici pour télécharger ce tableau.
Tableau supplémentaire 2 : Trajectoire triangulaire définie par le logiciel de pince à épiler optique. Analogue au tableau 2, cette trajectoire est composée de 16 points, correspondant à huit événements d’indentation à une profondeur de 5 μm et à une vitesse de 2,5 μm/s. Veuillez cliquer ici pour télécharger ce tableau.
Tableau supplémentaire 3 : Paramètres d’ajustement pour les données de la figure 7. IG : première supposition. Veuillez cliquer ici pour télécharger ce tableau.
Figure supplémentaire S1 : Alignement du capteur de force optique et compensation de la ligne de base de momentum. (A) Arrêt de champ imagé à la caméra auxiliaire (AUX, Figure 2) à travers l’objectif Bertrand. Une bulle d’air apparaît visible dans l’huile d’immersion, qui n’est pas visible à travers l’oculaire. (B) Chemin optique propre. Pour un alignement précis, ouvrez l’arrêt de champ et faites-le coïncider avec le cône de lumière NA = 1,2. (C) Image du plan échantillon. Le carré rouge indique la zone de travail OT. Barre d’échelle: 20 μm. (D) Puissance du piège mesurée à travers le champ de vision, le long des doubles flèches blanches indiquées en C. En rouge, piégez la variation de puissance lorsqu’aucune correction n’est appliquée. En bleu, puissance du piège corrigée sur tout le champ de vision. (E) Composante X de la base de référence du momentum le long de la même plage. En rouge, trace non corrigée. En bleu, trace corrigée pour la puissance du piège. En vert, trace corrigée de la ligne de base de momentum à l’aide de global Offset Compensation dans le logiciel du fabricant. (F) Identique à E, pour la composante Y. Notez qu’en fonctionnement normal, les composants ombrés sont utilisés pour la mécanique et les mesures de force, par exemple, la composante de force x pendant le mouvement le long de la coordonnée x et la composante de force y pendant le mouvement le long de l’axe y. Une fois toutes les corrections implémentées, un bruit RMSD de <0,5 pN est obtenu. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire S2 : Une routine ratée en raison de pièges faibles. (A) Kymograph montrant une indentation du noyau d’une routine ratée. Seules de courtes déformations transitoires sont visibles en raison d’une fuite de la perle du piège. Il est important de noter que le laser de piégeage se déplace toujours sans perle pour compléter la trajectoire prédéfinie (ligne pointillée verte). Barre d’échelle = 10 μm. (B) Haut : Position du piège par rapport au temps. Milieu : Résultat du suivi des bords du noyau proximal et distal en retrait. Notez que le bord distal ne bouge pas sans l’indentation comme on l’observe couramment pour les routines terminées sur des cellules isolées sur des substrats adhésifs. En bas : Force versus temps montrant la perte de la microsphère indiquée par une réduction du bruit thermique et une chute soudaine à zéro force. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire S3 : Survie des embryons injectés. Les embryons injectés avec des billes de 1 μm et 100 pg/embryon d’ARNm aux concentrations indiquées dans le protocole ont été comparés à des embryons non injectés et ne présentent aucune différence significative 24 heures après la fécondation. Moyenne et écart-type de trois expériences indépendantes avec N > 21 embryons par condition pour chaque expérience. Veuillez cliquer ici pour télécharger ce fichier.
Discussion
Dans ce protocole, nous décrivons une méthode unique pour interroger les propriétés mécaniques du noyau cellulaire à l’intérieur des cellules vivantes. Contrairement à d’autres techniques de spectroscopie de force, le piégeage optique non invasif nous a permis de découpler la contribution de la membrane cellulaire et du cytosquelette de la rigidité nucléaire cellulaire. Il est important de noter que la micromanipulation optique est compatible avec la microscopie multimodale, ce qui permettra à l’expérimentateur d’étudier différents processus impliqués dans la mécanobiologie nucléaire cellulaire. En conséquence, nous avons utilisé la coloration ADN-Hoechst pour mesurer la déformation du noyau lors de l’indentation effectuée par des forces de l’ordre de plusieurs centaines de picoNewton.
Applications potentielles de notre méthode au-delà des exemples décrits dans ce protocole
La possibilité d’extraire des informations mécaniques quantitatives à partir de mesures à l’intérieur de cellules vivantes sans perturbations externes permet une pléthore d’opportunités sans précédent qui commencent tout juste à être explorées. Ainsi, le protocole présenté de notre plateforme de micromanipulation optique peut être étendu à des expériences plus complexes avec une grande polyvalence. Les déflecteurs acousto-optiques (AOD) peuvent générer plusieurs pièges optiques pour des mesures de force synchrone à travers différents emplacements de cellules, ainsi que peuvent être utilisés pour la microrhéologie active dans une large gamme de fréquences51,61. Comme cela a été mentionné, la réponse de la force lors de l’indentation peut surmonter la force de piégeage maximale, conduisant à une fuite de la perle du piège optique. Dans ce cas, un retour de force peut être configuré avec l’AOD afin de serrer la force optique. Dans l’ensemble, de multiples approches microrhéologiques, telles que la relaxation du stress décrite dans ce protocole, mais aussi la microrhéologie active ou la conformité au fluage, peuvent être obtenues expérimentalement avec cette plate-forme et analysées en profondeur par de nouveaux progiciels61,62,63,64,65 . En outre, l’application de forces ne se limite pas au noyau mais pourrait en principe être réalisée pour mesurer diverses structures intracellulaires et dans des tissus complexes, comme démontré pour piéger les globules rouges qui circulent à l’intérieur de vaisseaux sanguins intacts66,67 ou piéger et déformer les chloroplastes et les mitochondries68 . L’étalonnage lumière-moment est indépendant de la forme et de la taille de l’objet piégé, ce qui permet des mesures de force directes sur n’importe quelle sonde de force de forme arbitraire38,39. L’utilisation de microsphères injectées nous a permis d’appliquer des forces élevées sur le noyau avec une puissance laser relativement faible par rapport à la manipulation directe des structures cellulaires69,70,71. Cependant, étant donné une différence d’indice de réfraction suffisamment élevée, aucune sonde de force appliquée de l’extérieur n’est nécessaire et les organites intracellulaires peuvent être manipulés directement sans perles injectées (observations non publiées et référence70).
Modifications potentielles de notre méthode pour étendre les applications
Différentes tailles de microbilles peuvent être injectées en fonction de l’expérience, mais les contrôles relatifs doivent être effectués. Par exemple, pour étudier les cellules à des stades ultérieurs, de plus petites perles peuvent être injectées. Cela réduira la force maximale qui peut être exercée par le piège optique (comme indiqué dans la référence55). Des perles plus grosses peuvent être injectées pour exercer des forces plus élevées, mais celles-ci peuvent affecter le développement de l’embryon en fonction de leur taille ou de leur stade d’intérêt. Dans les expériences où l’injection de microbilles n’est pas une option, divers organites présentant des différences d’indices de réfraction par rapport à celui du cytoplasme peuvent encore être manipulés optiquement, ce qui donne lieu à des forces optiques mesurables à partir de changements de moment lumineux42. Comme mentionné ci-dessus, ces méthodes ont été utilisées par Bambardekar et al. pour déformer les jonctions cellule-cellule dans l’embryon de drosophile70. De même, le noyau de la cellule a un indice de réfraction inférieur à celui du milieu environnant44, ce qui permet une indentation sans perles (observations non publiées et référence72) même si avec une force de piégeage plus faible. Ainsi, le noyau ne peut pas être piégé facilement et échappe au piège.
L’entretoise PDMS revêtue de spin est fabriquée via une méthode pratique et rapide, mais peut être hors de portée des laboratoires sans accès à une installation de micro/ nanofabrication ou à des laboratoires d’ingénierie. Ainsi, l’entretoise peut être facilement assemblée à partir d’une bande de laboratoire ou d’un parafilm (étape 4). Le protocole peut également être adapté en fabriquant des canaux microfluidiques qui automatisent la livraison de cellules individuelles dans des puits de mesure prédéfinis ou dans une chambre avec une hauteur définie pour estimer l’effet de confinement dans le même échantillon. Cependant, ces dispositifs microfluidiques doivent être conçus de manière à s’adapter à l’espace entre l’objectif du microscope et la lentille collectrice du capteur de force optique, d’environ 2 mm (voir étape 3). Notez que le capteur de force optique doit être positionné à la hauteur appropriée afin qu’aucune aberration optique due à la défocalisation n’affecte la mesure du moment cinétique des photons.
D’autres modifications pourraient inclure le changement de déclarants biologiques. Nous avons constaté que la fluorescence de Hoechst saigne spectralement dans le canal GFP et nous privilégions donc la combinaison avec l’histone marquée mCherry comme marqueur nucléaire pour la mesure simultanée dans deux canaux fluorescents. Alternativement, la déformation nucléaire peut facilement être suivie avec une étiquette ciblée sur la membrane nucléaire interne telle que Lap2b-GFP (Figure 2).
L’indentation sur le noyau cellulaire était de l’ordre de 2-3 microns, que nous pouvions mesurer avec précision par analyse d’image de microscopie confocale à disque rotatif à diffraction limitée. Pour le cas de noyaux plus rigides ou de forces plus petites, l’indentation sera à peine mesurable en utilisant cette approche. Cependant, les pinces optiques étalonnées par force absolue peuvent également être étalonnées pour les mesures de position de la bille piégée in situ à l’aide de l’interférométrie BFP avec une précision nanométrique51. En utilisant cette approche, le signal de tension et le capteur de force optique peuvent être traduits dans la position de la sonde piégée par le paramètre β [nm/ V], tandis que le paramètre invariant α [pN / V] donne des valeurs de force grâce à l’étalonnage lumière-moment susmentionné41 (voir ci-dessous pour plus de détails).
Dépannage
Nous avons constaté que les défis suivants pouvaient survenir au cours de l’expérience :
Aucun piège stable ne se forme et la microsphère s’échappe facilement
Toute saleté sur l’objectif du microscope ou un collier de correction mal aligné pourrait entraîner la défaillance d’un piège stable. Si aucune solution immédiate n’est trouvée, mesurez la fonction d’étalement ponctuel de la lentille de l’objectif. Si l’échantillon d’intérêt se trouve profondément à l’intérieur d’un tissu optiquement dense, le foyer laser peut subir de graves aberrations optiques conduisant à un piégeage instable (cet effet est généralement négligeable dans les cellules isolées, mais devient plus évident dans les tissus plus épais). Pour une rigidité élevée, la force de restauration du noyau pourrait dépasser la force d’échappement du piège, de sorte que la microsphère est perdue et que la routine d’indentation échoue. Initialement, le bord de la membrane nucléaire proximal au piège optique est à peine indenté (figure S2A). Lorsque cela se produit, le laser de piégeage n’est plus affecté par la force et le mouvement brownien, ce qui entraîne une chute de force à zéro et une diminution du bruit du signal (Figure S2B). Dans le cas où cela se produit, la puissance du laser peut être augmentée pour avoir un piège plus fort, l’amplitude de la trajectoire trapézoïdale poussant la perle dans le noyau peut être réduite, ou la position initiale de la microbille piégée peut être réglée plus loin du noyau.
La cellule se déplace pendant la stimulation
Si les cellules ne sont pas suffisamment attachées, le piège à gradient optique déplacera les cellules tout en effectuant la routine d’indentation intracellulaire, de sorte que les forces et la mécanique sous-jacente du noyau soient artéfactuelles. Pour éviter le déplacement de la cellule entière, nous recommandons d’augmenter la concentration de molécules d’adhésion cellulaire à la surface, par exemple, ConA.
Compensation initiale du momentum
Si une routine initiale de compensation de momentum n’est pas disponible dans la plate-forme DES OT (étape 6.5), un signal de base artificiel et indépendant de la force doit être corrigé. Ceci est visible sous la forme d’une pente sur la courbe de force même sans perle piégée (figure S1E). Pour effectuer la correction, la même trajectoire doit être effectuée sans perle, à l’extérieur de la cellule exactement à la même position. Pour cela, éloignez la cellule du piège à l’aide du contrôle de scène. À titre de référence, le décalage de force change de 5 pN sur le champ de vision à 200 mW dans notre système; ainsi, il devient négligeable pour les trajectoires courtes. Alternativement, un étage de balayage piézoélectrique peut être utilisé pour déplacer les cellules de l’échantillon, laissant la position du laser constante.
Étapes critiques du protocole présenté
Les microsphères doivent être injectées au bon stade, 1 cellule pour assurer une distribution maximale sur l’embryon. Les perles ne doivent pas être fluorescentes afin qu’aucune lumière ne s’échappe dans les canaux fluorescents utilisés pour l’imagerie. Par exemple, même les billes fluorescentes rouges typiques sont clairement visibles dans le canal bleu utilisé pour l’imagerie du noyau cellulaire après la coloration de Hoechst en raison de leur luminosité (excitation: 405 nm; émission: 445 nm). Une fixation stable de la cellule au substrat est essentielle pour éviter le déplacement latéral pendant la routine d’indentation. Si la cellule se déplace pendant la routine, les forces sont sous-estimées. Si cela se produit fréquemment, optimisez le protocole de pièce jointe. Pour les cellules de culture tissulaire, d’autres protéines d’adhésion cellulaire, telles que la fibronectine, le collagène ou la poly-L-lysine, conduisent à un attachement satisfaisant (observations non publiées). Pendant le confinement, les cellules sont soumises à un stress mécanique soudain et sévère. Cela peut causer des dommages aux cellules et souvent l’expérimentateur rencontrera des cellules éclatées si la procédure n’est pas effectuée avec soin. De plus, si la hauteur de confinement est trop petite, toutes les cellules souffriront d’une rupture de l’enveloppe nucléaire ou de dommages irréversibles. Pour les atténuer, abaissez le couvercle supérieur plus lentement et/ou augmentez l’espacement entre le couvercle.
Limites de la technique et suggestions pour les surmonter
Une limitation claire de la technique est la pénétration de la lumière laser dans les sections profondes du tissu, ce qui conduit à des aberrations et à un piégeage instable. Ainsi, une limite inférieure de profondeur de pénétration dépend de la clarté de l’échantillon, de la correction d’aberration qui peut être utilisée73 et de la puissance laser appliquée. Il faut tenir compte du fait qu’une puissance laser plus élevée entraîne une excitation thermique de l’échantillon au voisinage de la microsphère. Cependant, le chauffage de l’échantillon provenant du point laser de longueur d’onde de 1064 nm est minimisé pour éviter un stress plausible lié à la chaleur sur nos échantillons biologiques74.
Une autre limite est la force maximale qui peut être mesurée. Même si la détection directe lumière-moment permet des mesures de force bien au-delà du régime de réponse linéaire du piège optique40,41, la force maximale appliquée est de l’ordre de quelques centaines de picoNewtons. Ceci est limité par la puissance du laser et le seuil de dommages consécutifs du matériel biologique mou et les différences d’indice de réfraction, qui ne sont normalement pas supérieures à 0,1 ou 0,344. Plusieurs méthodes ont été proposées pour augmenter la limite de détection de force, par exemple en utilisant la lumière structurée75, les microsphères revêtues d’antireflet76, les particules à indice de réfraction élevé77 ou les points quantiques hautement dopés78.
Les OT peuvent être utilisés pour les mesures de position à l’échelle nanométrique par interférométrie BFP, de sorte que la position de la bille dans le piège est Δx = β Sx, où Sx est le signal de tension du capteur, et β [μm/V] peut être étalonnée à la volée en suivant différents protocoles35,54. Pour un capteur de force optique, il peut être prouvé que le facteur de conversion invariant tension-force α [pN/V] est directement lié à β et à la rigidité du piège, k [pN/μm], à travers α = kβ 37) Dans les expériences avec des déplacements de billes trop petits pour être détectés à partir de l’imagerie optique, cette stratégie peut être utilisée pour compléter les mesures de force avec une détection de petite position. Un exemple est l’application des routines expérimentales présentées ici sur des noyaux très rigides, pour lesquels des forces à des puissances laser raisonnables (200-500 mW) ne sont pas suffisantes pour induire des valeurs d’indentation suffisamment grandes. Dans ce cas, la bille doit être mise en contact avec le noyau et la rigidité de piégeage doit être étalonnée avant la mesure (étape 8.6). L’indentation d du noyau en fonction de la force peut être indirectement déterminée comme suit:
d = xtrap - F/k
où xtrap est la position du piège. Différent du facteur lumière-moment invariant α [pN/V], le facteur β [μm/V] doit être étalonné avant chaque expérience, car il dépend de nombreuses variables locales déterminant la dynamique de piégeage, telles que la taille des particules, la taille du point de piège optique et les indices de réfraction relatifs.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
MK reconnaît le soutien financier du ministère espagnol de l’Économie et de la Compétitivité à travers le Plan Nacional (PGC2018-097882-A-I00), FEDER (EQC2018-005048-P), programme Severo Ochoa pour les centres d’excellence en R&D (CEX2019-000910-S; RYC-2016-21062), de la Fundació Privada Cellex, de la Fundació Mir-Puig et de la Generalitat de Catalunya par le biais du programme CERCA et Recherche (2017 SGR 1012), en plus du financement par ERC (MechanoSystems) et HFSP (CDA00023/2018). V.R. reconnaît le soutien du ministère espagnol de la Science et de l’Innovation au partenariat EMBL, au Centro de Excelencia Severo Ochoa, au Plan Nacional de MINECO (BFU2017-86296-P, PID2020-117011GB-I00) et à la Generalitat de Catalunya (CERCA). V.V. reconnaît le soutien du programme de doctorat ICFOstepstone financé par le programme de recherche et d’innovation Horizon 2020 de l’Union européenne dans le cadre de la convention de subvention Marie Skłodowska-Curie 665884. Nous remercions Arnau Farré pour la lecture critique du manuscrit ; Maria Marsal pour l’aide à l’imagerie et au montage de l’embryon de 24 hpf et; Senda Jiménez-Delgado pour le soutien avec des micronjections de poisson zèbre.
matériels
| Name | Company | Catalog Number | Comments |
| #1.5 22 mm cover glasses | Ted Pella | 260148 | |
| #1.5 22x60 mm Coverglasses | Ted Pella | 260152 | |
| #1.5H glass bottom dishes | Willco | GWST-5040 | |
| 10-um beads | Supelco | 72986 | |
| 1-mm glass capillaries | Harvard Apparatus | 30-0020 GC100F-15 | |
| 1-um polystyrene microbeads | Sigma | 89904 | |
| 1-um red-fluorescent beads | ThermoFisher | F8816 | |
| Agar | ThermoFisher | 16500500 | |
| Aqcuisition cameras sCMOS | Andor | Sona-4BV11-UNI | |
| Auxiliary camera (Figure 3, AUX) | Blackfly, FLIR | BFS-U3-200S6M-C | |
| Calbryte 520 | AAT Bioquest | 520 AM | |
| Centrifuge | Eppendorf | 5453000011 | |
| Concanavalin A | Sigma | C5275 | |
| DMEM | Sigma | D2906 | |
| DNA-Hoechst | ThermoFisher | 33342 | |
| Double scotch tape | Biesse Adesivi | ||
| E3 | 5 mM NaCl. 0.17 mM KCl. 0.33 mM CaCl2. 0.33 mM MgSO4 | ||
| Eclipse Ti2 | Nikon | ||
| Forceps | Fine Science Tools | 11252-20 | |
| GPI-GFP | |||
| H2A-mCh | |||
| Image acquisition software | Fusion-Andor | ||
| Immersion Oil | Cargille | Type B: 16484 | |
| IR protection googles | Thorlabs | LG1 | |
| Lap2b-eGFP | |||
| Micro loader pipette | Eppendorf | GELoader | |
| Microinjector | World Precision Instruments | SYS-PV820 | |
| MicroManager 2.0 | |||
| Micromiter slide | ID5243 GXMGRAT-5 5mm/100 divisions | ||
| Mineral oil | Sigma | M3616 | |
| Motorized stage | ASI | ||
| Needle puller | Sutter instrument Co. | Model P-97 | |
| Optical tweezers platform | Impetux Optics | Sensocell | |
| OTs software (LightAce) | Impetux Optics | ||
| PDMS | Sigma | Sylgard 184 | |
| PDMS Curing agent | Sigma | Sylgard 184 | |
| Post processing software (Matlab) | Mathworks | ||
| RNAse free water | Thermofisher | AM9937 | |
| Short-pass dichroic mirror (Figure 3, IR-F) | Semrock | FF01-950/SP-25 | |
| Spin-coater | Specialty Coating Systems | Spincoat G3P-8 | |
| Spinning-disk confocal microscope | Andor | DragonFly 502 | |
| Stereomicroscope | Leica M80 | ||
| Triangular microinjection mold | Adaptive Science Tools | TU1 | |
| Universal oven | Memmert | UNB 200 | |
| Water immersion objective | Nikon | MRD07602 |
Références
- Chan, C. J., Heisenberg, C. P., Hiiragi, T. Coordination of Morphogenesis and Cell-Fate Specification in Development. Current Biology. 27 (18), 1024-1035 (2017).
- Heller, E., Fuchs, E. Tissue patterning and cellular mechanics. Journal of Cell Biology. 211 (2), 219-231 (2015).
- Heisenberg, C. P., Bellaïche, Y. Forces in tissue morphogenesis and patterning. Cell. 153 (5), 948-962 (2013).
- Petridou, N. I., Spiró, Z., Heisenberg, C. P. Multiscale force sensing in development. Nature Cell Biology. 19 (6), 581-588 (2017).
- Krieg, M., et al. Tensile forces govern germ-layer organization in zebrafish. Nature Cell Biology. 10 (4), 429-436 (2008).
- Ruprecht, V., et al. Cortical contractility triggers a stochastic switch to fast amoeboid cell motility. Cell. 160 (4), 673-685 (2015).
- Shellard, A., Mayor, R. Supracellular migration - Beyond collective cell migration. Journal of Cell Science. 132 (8), (2019).
- Mongera, A., et al. A fluid-to-solid jamming transition underlies vertebrate body axis elongation. Nature. 561 (7723), 401-405 (2018).
- Atia, L., et al. Geometric constraints during epithelial jamming. Nature Physics. 14 (6), 613-620 (2018).
- Turlier, H., Maître, J. -. L. Mechanics of tissue compaction. Seminars in Cell & Developmental Biology. 47-48, 110-117 (2015).
- Ladoux, B., Mège, R. M. Mechanobiology of collective cell behaviours. Nature Reviews Molecular Cell Biology. 18 (12), 743-757 (2017).
- Venturini, V., et al. The nucleus measures shape changes for cellular proprioception to control dynamic cell behavior. Science. 370 (6514), (2020).
- Charras, G., Sahai, E. Physical influences of the extracellular environment on cell migration. Nature Reviews Molecular Cell Biology. 15 (12), 813-824 (2014).
- Kirby, T. J., Lammerding, J. Emerging views of the nucleus as a cellular mechanosensor. Nature Cell Biology. 20 (4), 373-381 (2018).
- Lee, H. P., et al. The nuclear piston activates mechanosensitive ion channels to generate cell migration paths in confining microenvironments. Science Advances. 7 (2), (2021).
- Friedl, P., Wolf, K., Lammerding, J. Nuclear mechanics during cell migration. Current Opinion in Cell Biology. 23 (1), 55-64 (2011).
- Versaevel, M., Riaz, M., Grevesse, T., Gabriele, S. Cell confinement: Putting the squeeze on the nucleus. Soft Matter. 9 (29), 6665-6676 (2013).
- Zuela-Sopilniak, N., et al. Measuring nucleus mechanics within a living multicellular organism: Physical decoupling and attenuated recovery rate are physiological protective mechanisms of the cell nucleus under high mechanical load. Molecular Biology of the Cell. 31 (17), 1943-1950 (2020).
- Kim, D. H., Wirtz, D. Cytoskeletal tension induces the polarized architecture of the nucleus. Biomaterials. 48, 161-172 (2015).
- Lomakin, A. J., et al. The nucleus acts as a ruler tailoring cell responses to spatial constraints. Science. 370 (6514), (2020).
- Hampoelz, B., et al. Microtubule-induced nuclear envelope fluctuations control chromatin dynamics in Drosophila embryos. Development. 138 (16), 3377-3386 (2011).
- Heo, S. J., et al. Differentiation alters stem cell nuclear architecture, mechanics, and mechano-sensitivity. eLife. 5, 1-21 (2016).
- Cosgrove, B. D., et al. Nuclear envelope wrinkling predicts mesenchymal progenitor cell mechano-response in 2D and 3D microenvironments. Biomaterials. 270, 120662 (2021).
- Liu, H., et al. In situ mechanical characterization of the cell nucleus by atomic force microscopy. ACS Nano. 8 (4), 3821-3828 (2014).
- Hobson, C. M., et al. Correlating nuclear morphology and external force with combined atomic force microscopy and light sheet imaging separates roles of chromatin and lamin A/C in nuclear mechanics. Molecular Biology of the Cell. 31 (16), 1788-1801 (2020).
- Pajerowski, J. D., Dahl, K. N., Zhong, F. L., Sammak, P. J., Discher, D. E. Physical plasticity of the nucleus in stem cell differentiation. Proceedings of the National Academy of Sciences of the United States of America. 104 (40), 15619-15624 (2007).
- Rowat, A. C., Lammerding, J., Ipsen, J. H. Mechanical properties of the cell nucleus and the effect of emerin deficiency. Biophysical Journal. 91 (12), 4649-4664 (2006).
- Davidson, P. M., et al. High-throughput microfluidic micropipette aspiration device to probe time-scale dependent nuclear mechanics in intact cells. Lab on a Chip. 19 (21), 3652-3663 (2019).
- Lombardi, M., Zwerger, M., Lammerding, J. Biophysical assays to probe the mechanical properties of the interphase cell nucleus: Substrate strain application and microneedle manipulation. Journal of Visualized Experiments: JoVE. (55), (2011).
- Luo, T., Mohan, K., Iglesias, P. A., Robinson, D. N. Molecular mechanisms of cellular mechanosensing. Nature Materials. 12 (11), 1064-1071 (2013).
- Dahl, K. N., Engler, A. J., Pajerowski, J. D., Discher, D. E. Power-law rheology of isolated nuclei with deformation mapping of nuclear substructures. Biophysical Journal. 89 (4), 2855-2864 (2005).
- Guilluy, C., et al. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nature Cell Biology. 16 (4), 376-381 (2014).
- Bustamante, C. J., Wang, M. D. Optical tweezers in single-molecule biophysics. Nature Reviews Methods Primers. , 1-29 (2021).
- Svoboda, K., Block, S. M. Force and velocity measured for single kinesin molecules. Cell. 77 (5), 773-784 (1994).
- Berg-Sørensen, K., Flyvbjerg, H. Power spectrum analysis for optical tweezers. Review of Scientific Instruments. 75 (3), 594-612 (2004).
- Smith, S. B., Cui, Y., Bustamante, C. Optical-trap force transducer that operates by direct measurement of light momentum. Methods in Enzymology. 361 (1994), 134-162 (2003).
- Farré, A., Montes-Usategui, M. A force detection technique for single-beam optical traps based on direct measurement of light momentum changes. Optics Express. 18 (11), 11955 (2010).
- Català, F., Marsà, F., Montes-Usategui, M., Farré, A., Martín-Badosa, E. Extending calibration-free force measurements to optically-trapped rod-shaped samples. Scientific Reports. 7, 1-10 (2017).
- Bui, A. A. M., et al. Calibration of force detection for arbitrarily shaped particles in optical tweezers. Scientific Reports. 8 (1), 1-12 (2018).
- Farré, A., Marsà, F., Montes-Usategui, M. Beyond the hookean spring model: Direct measurement of optical forces through light momentum changes. Methods in Molecular Biology. 1486, (2017).
- Farré, A., Marsà, F., Montes-Usategui, M. Optimized back-focal-plane interferometry directly measures forces of optically trapped particles. Optics Express. 20 (11), 12270 (2012).
- Jun, Y., Tripathy, S. K., Narayanareddy, B. R. J., Mattson-Hoss, M. K., Gross, S. P. Calibration of optical tweezers for in vivo force measurements: How do different approaches compare. Biophysical Journal. 107 (6), 1474-1484 (2014).
- Mas, J., Farré, A., Sancho-Parramon, J., Martín-Badosa, E., Montes-Usategui, M. Force measurements with optical tweezers inside living cells. Optical Trapping and Optical Micromanipulation XI. 9164, (2014).
- Schürmann, M., Scholze, J., Müller, P., Guck, J., Chan, C. J. Cell nuclei have lower refractive index and mass density than cytoplasm. Journal of Biophotonics. 9 (10), 1068-1076 (2016).
- Rosen, J. N., Sweeney, M. F., Mably, J. D. Microinjection of zebrafish embryos to analyze gene function. Journal of Visualized Experiments: JoVE. (25), (2009).
- Westerfield, M. . The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish. (Danio rerio), 5th Edition. , (2007).
- Schubert, R., et al. Assay for characterizing the recovery of vertebrate cells for adhesion measurements by single-cell force spectroscopy. FEBS Letters. 588 (19), 3639-3648 (2014).
- Koschwanez, J. H., Carlson, R. H., Meldrum, D. R. Thin PDMS films using long spin times or tert-butyl alcohol as a solvent. PLoS One. 4 (2), 2-6 (2009).
- Das, R., et al. Mechanical stretch inhibition sensitizes proprioceptors to compressive stresses. bioRxiv. , (2021).
- Chardès, C., Clement, R., Blanc, O., Lenne, P. F. Probing cell mechanics with bead-free optical tweezers in the drosophila embryo. Journal of Visualized Experiments: JoVE. (141), (2018).
- Staunton, J. R., Blehm, B., Devine, A., Tanner, K. In situ calibration of position detection in an optical trap for active microrheology in viscous materials. Optics Express. 25 (3), 1746 (2017).
- Bola, R., Treptow, D., Marzoa, A., Montes-Usategui, M., Martin-Badosa, E. Acousto-holographic optical tweezers. Optics Letters. 45 (10), 2938-2941 (2020).
- Thalhammer, G., Obmascher, L., Ritsch-Marte, M. Direct measurement of axial optical forces. Optics Express. 23 (5), 6112 (2015).
- Vermeulen, K. C., et al. Calibrating bead displacements in optical tweezers using acousto-optic deflectors. Review of Scientific Instruments. 77 (1), 1-6 (2006).
- Dzementsei, A., Barooji, Y. F., Ober, E. A., Oddershede, L. B. Foregut organ progenitors and their niche display distinct viscoelastic properties in vivo during early morphogenesis stages. bioRxiv. , 1-35 (2021).
- Behrndt, M., et al. Forces driving epithelial spreading in zebrafish gastrulation. Science. 338 (6104), 257-260 (2012).
- Krieg, M., et al. Atomic force microscopy-based mechanobiology. Nature Reviews Physics. , (2018).
- A-Hassan, E., et al. Relative microelastic mapping of living cells by atomic force microscopy. Biophysical Journal. 74 (3), 1564-1578 (1998).
- Khalilgharibi, N., et al. Stress relaxation in epithelial monolayers is controlled by the actomyosin cortex. Nature Physics. 15, (2019).
- Crick, S. L., Yin, F. C. Assessing micromechanical properties of cells with atomic force microscopy: importance of the contact point. Biomechanics and Modeling in Mechanobiology. 6 (3), 199-210 (2007).
- Hurst, S., Vos, B. E., Betz, T. Intracellular softening and fluidification reveals a mechanical switch of cytoskeletal material contributions during division. bioRxiv. , 425761 (2021).
- Kaplan, J. L., Bonfanti, A., Kabla, A. RHEOS.jl - A Julia package for rheology data analysis. arXiv. 4, 1-5 (2020).
- Bonfanti, A., Kaplan, J. L., Charras, G., Kabla, A. Fractional viscoelastic models for power-law materials. Soft Matter. 16 (26), 6002-6020 (2020).
- Rivas-Barbosa, R., Escobedo-Sánchez, M. A., Tassieri, M., Laurati, M. i-Rheo: determining the linear viscoelastic moduli of colloidal dispersions from step-stress measurements. Physical Chemistry Chemical Physics: PCCP. 22 (7), 3839-3848 (2020).
- Tassieri, M., et al. i-Rheo: Measuring the materials' linear viscoelastic properties "in a step". Journal of Rheology. 60 (4), 649-660 (2016).
- Zhong, M. C., Wei, X. B., Zhou, J. H., Wang, Z. Q., Li, Y. M. Trapping red blood cells in living animals using optical tweezers. Nature Communications. 4, 1767-1768 (2013).
- Harlepp, S., Thalmann, F., Follain, G., Goetz, J. G. Hemodynamic forces can be accurately measured in vivo with optical tweezers. Molecular Biology of the Cell. 28 (23), 3252-3260 (2017).
- Bayoudh, S., Mehta, M., Rubinsztein-Dunlop, H., Heckenberg, N. R., Critchley, C. Micromanipulation of chloroplasts using optical tweezers. Journal of Microscopy. 203 (2), 214-222 (2001).
- Favre-Bulle, I. A., Stilgoe, A. B., Rubinsztein-Dunlop, H., Scott, E. K. Optical trapping of otoliths drives vestibular behaviours in larval zebrafish. Nature Communications. 8 (1), 630 (2017).
- Bambardekar, K., Clément, R., Blanc, O., Chardès, C., Lenne, P. F. Direct laser manipulation reveals the mechanics of cell contacts in vivo. Proceedings of the National Academy of Sciences of the United States of America. 112 (5), 1416-1421 (2015).
- Ferro, V., Chuai, M., McGloin, D., Weijer, C. J. Measurement of junctional tension in epithelial cells at the onset of primitive streak formation in the chick embryo via non-destructive optical manipulation. Development (Cambridge). 147 (3), (2020).
- Hörner, F., et al. Holographic optical tweezers-based in vivo manipulations in zebrafish embryos. Journal of Biophotonics. 10 (11), 1492-1501 (2017).
- Zhong, M. -. C., Wang, Z. -. Q., Li, Y. -. M. Aberration compensation for optical trapping of cells within living mice. Applied Optics. 56 (7), 1972 (2017).
- Català, F., Marsà, F., Montes-Usategui, M., Farré, A., Martín-Badosa, E. Influence of experimental parameters on the laser heating of an optical trap. Scientific Reports. 7 (1), 1-9 (2017).
- Taylor, M. A., Waleed, M., Stilgoe, A. B., Rubinsztein-Dunlop, H., Bowen, W. P. Enhanced optical trapping via structured scattering. Nature Photonics. 9 (10), 669-673 (2015).
- Bormuth, V., et al. Optical trapping of coated microspheres. Optics Express. 16 (18), 13831-13844 (2008).
- Sudhakar, S., et al. Germanium nanospheres for ultraresolution picotensiometry of kinesin motors. Science. 371 (6530), (2021).
- Shan, X., et al. Optical tweezers beyond refractive index mismatch using highly doped upconversion nanoparticles. Nature Nanotechnology. 16 (5), 531-537 (2021).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.