
Method Article
Medições de força direta de mecânica subcelular em confinamento usando pinças ópticas
Neste Artigo
Resumo
Aqui, apresentamos um protocolo para investigar as propriedades mecânicas intracelulares de células isoladas de zebrafish em bidimensionais com medição direta de força por uma armadilha óptica.
Resumo
Durante o desenvolvimento de um organismo multicelular, uma única célula fertilizada se divide e dá origem a múltiplos tecidos com funções diversas. A morfogênese tecidual acompanha mudanças moleculares e estruturais no nível celular único que resultam em variações de propriedades mecânicas subcelulares. Como consequência, mesmo dentro da mesma célula, diferentes organelas e compartimentos resistem de forma diferente aos estresses mecânicos; e as vias de mecanotransdução podem regular ativamente suas propriedades mecânicas. A capacidade de uma célula de se adaptar ao microambiente do nicho tecidual, portanto, deve-se, em parte, à capacidade de sentir e responder a tensões mecânicas. Recentemente propusemos um novo paradigma de mecanosensação no qual a deformação e o posicionamento nuclear permitem que uma célula avalie o ambiente 3D físico e dota a célula com um senso de propriocepção para decodificar mudanças na forma celular. Neste artigo, descrevemos um novo método para medir as forças e propriedades materiais que moldam o núcleo celular dentro das células vivas, exemplificado em células aderentes e células mecanicamente confinadas. As medições podem ser realizadas não invasivamente com armadilhas ópticas dentro das células, e as forças são diretamente acessíveis através da detecção livre de calibração do momento da luz. Isso permite medir a mecânica do núcleo independentemente das deformações da superfície celular e permitir a dissecção de vias de mecanodução exteroceptiva e interoceptiva. É importante ressaltar que o experimento de captura pode ser combinado com microscopia óptica para investigar a resposta celular e a dinâmica subcelular usando imagens de fluorescência do citoesqueleto, íons de cálcio ou morfologia nuclear. O método apresentado é simples de aplicar, compatível com soluções comerciais para medições de força, e pode ser facilmente estendido para investigar a mecânica de outros compartimentos subcelulares, por exemplo, mitocôndrias, fibras de estresse e endossários.
Introdução
A morfogênese tecidual é um processo complexo no qual sinais bioquímicos e forças físicas são coordenados espacialmente. No embrião em desenvolvimento, gradientes de fatores de sinalização bioquímica ditam a especificação do destino e garantem a padronização correta do tecido1,2. Ao mesmo tempo, forças intrínsecas e extrínsecas desempenham um papel na construção da arquitetura do embrião3,4. A influência da mecânica do córtex celular neste contexto tem sido estudada extensivamente5,6. A interconexão apertada entre os processos mecano-químicos durante a morfogênese depende das propriedades de células únicas para sentir e responder às forças mecânicas em seu microambiente tecidual. As células, assim, decodificam sinais mecânicos através da presença de elementos subcelulares e moleculares sensíveis à força que transduzem informações mecânicas em vias específicas de sinalização controlando o comportamento celular, o destino celular e a mecânica celular.
Uma marca dos processos de desenvolvimento é que as células se organizam como grupos para construir estruturas multicelulares. Como tal, as células únicas raramente reorganizam e se movem sozinhas, mas estão associadas a um sociotope apertado no qual mostram comportamentos coletivos como migração supracelular7, (un)transições de interferência8,9 ou compactação blastocisto10. As forças mecânicas geradas dentro e entre as células servem como importantes pistas para instruir a dinâmica celular coletiva7,11. Mas mesmo quando as células se movem sozinhas, como células progenitoras que se espremem entre folhas de tecido ou nichos de tecido estreitos, elas experimentam extensas forças mecânicas anisotrópicas ao navegar em um ambiente tridimensional. Essas tensões mecânicas nas células têm profundas consequências no comportamento celular12,13. Vários mecanismos têm sido investigados que convergem no núcleo como um elemento principal de mecanotransdução14,15, como elemento mecânico passivo ou ativo durante a migração dentro de um ambiente desnso de tecido 3D15,16.
Recentemente propusemos um mecanismo que equipa células para medir deformações de forma usando o núcleo como um medidor de mecano intracelular elástico12. O núcleo, sendo a maior organela de uma célula, sofre grandes deformações quando as células polarizam, migram ou mudam sua forma sob estiramento mecânico, confinamento ou estresse osmótico16,17,18,19. Descobrimos que o estiramento do envelope nuclear junto com o posicionamento intracelular do núcleo fornece às células informações sobre a magnitude e o tipo de deformação celular (como compressão celular versus inchaço celular). O alongamento do núcleo está associado a um desdobramento da membrana nuclear interna (INM), que promove a atividade de lipase dependente de cálcio cPLA2 (fosfolipase citossolica A2) no INM seguido da liberação do Ácido Aracidônico (AA) e rápida ativação da minocina II no córtex celular. Isso leva ao aumento da contratilidade celular e migração de células amoeboides acima de um limiar de contratilidade cortical6. A resposta mecanosensível à deformação celular ocorre em menos de um minuto e é reversível após a liberação do confinamento, sugerindo que o núcleo age como um medidor de tensão para propriocepção celular regulando o comportamento celular adaptativo em condições mecânicas de estresse. Este caminho mecanosensível mostra-se ativo em células-tronco progenitoras derivadas de embriões de zebrafish, tanto em células pluripotentes quanto em linhagem12 e é conservado em diferentes espécies e linhas celulares20.
Além das propriedades nucleares como mecanosensor celular, a arquitetura nuclear e a mecânica são intrinsecamente reguladas durante o desenvolvimento e em resposta à especificação de destino celular21, afinando assim a sensibilidade à mecano celular22,23. A consequência pode ser uma mudança na conformidade nuclear que permite mudanças morfológicas e transições de um pré-aluno para um estado migratório e vice-versa8.
Várias técnicas para medir a mecânica do núcleo celular foram aplicadas, como microscopia de força atômica24,25, aspiração de micropipette26,27, tecnologia microfluida28 e microaícetas29. No entanto, muitas dessas técnicas são invasivas no sentido de que toda a célula deve ser deformada, limitando a medição de características mecânicas e respostas dependentes da força do próprio núcleo. Para contornar a deformação simultânea da superfície celular e seu córtex celular mecanosensível30, núcleos isolados foram estudados em diversos contextos31,32. No entanto, não se pode descartar que o isolamento nuclear esteja associado a uma mudança nas propriedades do núcleo mecânico e sua regulação (referência24 e próprias observações inéditas).
Pinças ópticas (OTs) são uma tecnologia versátil que permitiu uma infinidade de experimentos na mecanobiologia celular e têm sido fundamentais em nossa compreensão de como as máquinas moleculares convertem produtos químicos em energia mecânica333,34. As pinças ópticas usam um raio laser bem focado para exercer forças ópticas em partículas dielétricas que têm um índice de refração superior ao médio circundante33. Tais forças podem ser da ordem de centenas de pico-newtons e resultar em confinamento efetivo da partícula dentro do foco da armadilha laser, permitindo a manipulação da partícula presa em três dimensões. O uso da luz tem uma vantagem importante na medida em que a medida pode ser realizada não invasivamente dentro das células vivas. As manipulações ópticas estão ainda limitadas ao foco da armadilha do raio laser. Assim, a manipulação pode ser realizada sem estimular as membranas celulares circundantes e não perturba os processos do córtex de actina ou mecanosensíveis na membrana plasmática, como a ativação dependente da força dos canais de íons.
A dificuldade da abordagem óptica da pinça é determinar precisamente as forças aplicadas à microesfera usando abordagens clássicas que dependem da calibração da força indireta com base no teorema da equipartição ou no uso de forças definidas de arrasto de Stokes para medir uma força de fuga dependente de laser35. Considerando que esses métodos são simples de implementar em um experimento in vitro, eles geralmente não podem ser traduzidos em um ambiente celular. Várias estratégias foram introduzidas no campo que dependem de uma calibragem direta da força, derivada dos primeiros princípios de conservação do momento36,37. Ao contrário de outras abordagens de espectroscopia de força, as medidas de força são deduzidas de um intercâmbio local de impulso de luz com a partícula presa de forma arbitrária38,39. Em nossa configuração experimental, as mudanças no momento da luz decorrentes de forças ópticas são diretamente medidas sem a necessidade de calibração in situ trap40,41,42,43. Assim, as medidas se tornam possíveis em um ambiente viscoso, como o interior da célula ou mesmo dentro de um tecido, e as forças podem ser facilmente quantificadas até o nível pN.
Neste protocolo, descrevemos um ensaio para manipular mecanicamente organelas ou estruturas intracelulares e avaliar quantitativamente suas propriedades mecânicas por uma configuração óptica de pinça. Esta configuração é integrada a um microscópio fluorescente de disco giratório que permite imagens paralelas do comportamento celular ou dinâmica intracelular. O ensaio permite a caracterização das propriedades mecânicas de compartimentos celulares específicos, como o núcleo, ao mesmo tempo em que estuda a possível mecanoresponse e ativação de vias de sinalização molecular como resultado da própria deformação. Além disso, a captura óptica de microesferas injetadas dentro das células permite um aumento da força de recuo graças a um índice refrativo consideravelmente maior da conta de poliestireno (n = 1,59) em comparação com o contraste refrativo intrínseco44 do núcleo (n ~ 1,35) versus citoplasma (n ~ 1,38). A estratégia apresentada pode ser facilmente adaptada ao estudo de outras estruturas intracelulares e organelas, bem como outras abordagens envolvendo microrrehetologia ativa, uso de múltiplas armadilhas ópticas para sondar as mesmas/diferentes estruturas subcelulares simultaneamente, e medições visando a mecanobiologia celular no embrião vivo.
Protocolo
Todos os protocolos utilizados foram aprovados pelo Comitê De Ética Institucional de Cuidados e Uso de Animais (PRBB-IACUEC) e implementados de acordo com regulamentos nacionais e europeus. Todos os experimentos foram realizados de acordo com os princípios dos 3Rs. Os zebrafish (Danio rerio) foram mantidos como descrito anteriormente.
1. Preparação de células-tronco progenitoras de zebrafish embrionários primários isolados
- Micropipette e preparação de agarose
NOTA: Para um protocolo completo de microinjeção de embriões de zebrafish, consulte referência45.- Com um puxador de micropipette, puxe um capilar de vidro de 1,0 mm para obter duas agulhas45. Armazene as agulhas nãousadas em uma placa de Petri de 150 mm presa a uma almofada de playdough ou em um anel de fita de laboratório de dentro para fora para proteger a ponta fina de danos durante o transporte.
- Derreta 1% de ultrapure agarose em E3 (5 mM NaCl, 0,17 mM KCl, 0,33 mM CaCl2, 0,33 mM MgSO4) em um micro-ondas padrão de cozinha/laboratório para 10 s. Aqueça a mistura repetidamente por curtos períodos de tempo (poucos segundos) até que a agarose derreta.
- Quando a agarose estiver completamente derretida, deixe esfriar brevemente, e depois despeje-a em uma placa de Petri de 10 cm. Adicione lentamente o molde de microinjeção triangular (ver Tabela de Materiais) na parte superior da agarose evitando o aparecimento de bolhas. Não empurre o molde, garantindo que ele permaneça na superfície da agarose.
- Quando a agarose se solidificar completamente, remova o molde triangular muito lentamente, exercendo uma força suave para evitar qualquer quebra na ágarose. A placa pode ser armazenada de cabeça para baixo a 4 °C durante 2-4 semanas.
- 30 min antes da microinjeção, tire o prato da geladeira e adicione E3 pré-eded a 28 °C para deixá-lo estabilizar em temperatura ambiente.
- Preparação da mistura de injeção
- Para preparar a mistura de injeção, diluir microesferas de 1 μm (poliestireno, não fluorescente) na proporção 1:5 na água livre de RNase.
- Prepare mRNA para expressão transitória de marcadores fluorescentes ou expressão de construtos genéticos recombinantes e/ou co-injeção de morfolino na concentração desejada.
NOTA: Uma mistura típica de injeção para a co-injeção de microesferas juntamente com 100 pg de mRNA por embrião para rotular, por exemplo, o núcleo com H2A-mCherry é: 1 μL de contas + 1 μL de mRNA (concentração de estoque é de 1 μg/μL) + 2,5 μL de RNA água livre + 0,5 μL de fenol vermelho (solução de estoque 0,5%, vermelho fenol não é obrigatório; é usado para uma melhor visualização da gota injetada, mas a injeção não rotulada a queda também é visível para um experimentador experiente). A injeção de RNA também pode ser útil para selecionar embriões injetados. Microesferas fluorescentes podem ser injetadas, em vez de não fluorescentes, para visualizá-las.
- Carga e calibração da agulha de microinjeção
- Ligue o microinjetor usando a opção Time-Gated . Esta configuração é muito importante para calibrar o volume de injeção corretamente. Defina o tempo de gating em aproximadamente 500 ms.
- Carregue 3 μL da mistura de injeção na agulha usando uma pipeta micro-carregadeira.
- Insira a agulha no micromanipulador e veda firmemente. Verifique se o micromanipulador está em uma boa posição e tem liberdade suficiente para se mover na direção x-y na placa de injeção.
- Meça o tamanho da gota usando um slide de micrômetro (5 mm/100 divisões) com uma gota de óleo mineral no topo45 e ejetando uma gota da mistura de injeção diretamente no óleo mineral.
- Corte a agulha com fórceps afiados em um ângulo íngreme para gerar uma ponta pontiaguda afiada. Ajuste o tamanho da gota para 0,1 mm, correspondendo a 0,5 nL de material injetado.
NOTA: Se ao cortar a agulha, este volume for excedido, recomenda-se refazer o procedimento de calibração com uma nova agulha. O tempo de gating do microinjetor pode ser ligeiramente ajustado para corresponder ao volume de queda; no entanto, os curtos tempos de gating correspondem a um grande diâmetro da agulha, o que potencialmente danifica os embriões.
- Microinjeção de embriões de zebrafish em estágio unicelular
- Colete embriões de zebrafish logo após a fertilização para microinjeção da mistura de contas diretamente no embrião de estágio de uma célula (zigoto) antes da primeira divisão celular ocorrer.
NOTA: Isso garante a distribuição adequada das microesferas e um alto rendimento suficiente de blastomeres isolados com pelo menos uma microesfera por célula em estágios de desenvolvimento posteriores nos quais os experimentos são realizados (estágio blastula-gastrula). Experimentos de recuo ainda podem ser realizados se houver duas esferas dentro da célula, mas células que não têm contas devem ser excluídas (mesmo que o recuo sem esferas seja possível). Cepas de tipo selvagem AB foram usadas neste protocolo, mas qualquer outra cepa, por exemplo, TL pode ser usada. - Coloque embriões de estágio de uma célula (zigoto) em um molde de 1% de agarose em forma triangular pré-enchido, como mostrado na Figura 1A, usando uma pipeta pasteur de plástico.
- Remova o meio extra com a mesma pipeta para evitar que os embriões flutuam ao redor. Empurre suavemente os embriões para o molde triangular através de um pincel. Mantenha algum espaço entre os embriões para facilitar a orientação correta (Figura 1B).
- Alinhe suavemente os embriões com uma escova para que os embriões sejam orientados lateralmente, com a única célula do zigoto sendo claramente visível, como mostrado na Figura 1B. Uma orientação ideal para a microinjeção é alcançada quando uma célula do embrião está voltada para a direção da agulha (injeção através do polo animal do embrião) ou na direção oposta de frente para a célula da gema (injeção através do polo vegetal do embrião), como mostrado na Figura 1C.
- Segure o prato com uma mão e use a outra mão para posicionar a ponta da agulha usando o controlador micromanipulador. Abaixe a ponta da agulha em direção aos embriões.
- Fure o acorde e digite o embrião de uma célula com a agulha enquanto monitora o procedimento através do estereoscópio. Certifique-se da colocação correta da agulha e, após a injeção, a localização correta da gota injetada, conforme mostrado na Figura 1C.
- Repita para todos os embriões: mova a agulha para cima, deslize o prato com os embriões até que o próximo embrião esteja centrado, abaixe a agulha e injete- a.
- Uma vez que todo o conjunto de embriões é injetado, remova os embriões da placa de molde/petri agarose, lavando um pouco de E3 e coloque-os em uma nova placa de Petri usando uma pipeta Pasteur de plástico. Recomenda-se colocar mídia suficiente na placa de injeção para evitar a secagem de embriões durante o procedimento de microinjeção.
- Repita o procedimento até que o número desejado de embriões seja injetado. Os embriões devem estar em um estágio celular para garantir a disseminação máxima e homogênea das contas.
NOTA: Este procedimento é otimizado para embriões de blastula precoce e provavelmente precisa ser otimizado se diferentes estágios de desenvolvimento forem investigados. - Coloque os embriões injetados dentro de uma incubadora a 28-31 °C por aproximadamente 4h ou até o estágio desejado (Figura 1D) antes de prosseguir com o protocolo para a cultura celular primária.
NOTA: Opcionalmente, deixe que os embriões se desenvolvam além do estágio blastula (ou ponto de tempo de medição desejado) para garantir a sobrevivência e descartar artefatos de toxicidade. Em fases larvas, monte larvas anestesiadas com tricaine em 0,75% de ágarose e imagem da distribuição de microesferas em vários tecidos. Para fazer uma solução de estoque, misture: 400 mg de tricaine em pó em 97,9 mL de água destilada, aproximadamente 2,1 mL de 1 M TRIS-base (pH 9), e ajuste ao pH 7. Esta solução pode ser armazenada a 4 °C. Para usar o tricaine como anestésico, dilui 4,2 mL de solução de estoque em 100 mL de meio de ovo (ou mídia desejada); neste caso, foi usado e3. Consulte referência46 para obter detalhes.
- Colete embriões de zebrafish logo após a fertilização para microinjeção da mistura de contas diretamente no embrião de estágio de uma célula (zigoto) antes da primeira divisão celular ocorrer.
2. Preparação e coloração de células únicas
- Coloque os embriões do estágio da esfera (4 hpf, horas após a fertilização) em um prato de vidro usando uma pipeta Pasteur de plástico. Selecione os embriões positivos para o sinal das contas injetadas, e que expressem a proteína fluorescente no caso da injeção de mRNA. Alguns embriões podem mostrar agrupamento de contas altas e podem ser excluídos.
- Descoriora manualmente os embriões usando fórceps. Transfira aproximadamente 10-15 embriões para recipientes de reação de 1,5 mL usando uma pipeta Pasteur de vidro.
NOTA: Quando os embriões são descorrioados, eles se prendem ao plástico, e o uso de vidro é necessário. Como alternativa à placa de vidro, uma placa de Petri de plástico com uma fina camada de 1% de agarose pode ser usada. A descorção manual deve ser preferida em relação ao tratamento pronase enzimático para evitar danos protelíticos às proteínas da superfície celular e possíveis alterações nas propriedades mecânicas das células e tecidos, impedindo vezes de recuperação prolongada47.
- Descoriora manualmente os embriões usando fórceps. Transfira aproximadamente 10-15 embriões para recipientes de reação de 1,5 mL usando uma pipeta Pasteur de vidro.
- Remova a mídia E3 e adicione 500 μL de meio de cultura de tecido independente de CO2 pré-aquecido (DMEM-F12; com L-glutamina e 15 mM HEPES, sem bicarbonato de sódio e vermelho fenol suplementado com penicilina de 10 unidades e estreptomicina de 10 mg/L).
NOTA: Não use mídia dependente de CO2 a menos que uma incubadora de microscópio seja usada. O uso, por exemplo, de RPMI em condições tamponadas com carbonato causam alterações no pH da mídia e podem afetar a sobrevivência celular. Outro aspecto fundamental é evitar meios culturais que contenham soro. O soro pode conter ácido linfosfísico (LPA), um potente ativador da via Rho/ROCK, capaz de controlar a contratilidade celular e a motilidade nas células-tronco progenitoras6. A osmolaridade do meio deve ser mantida em 300 mOsm para evitar desafios osmóticos que possam interferir na morfologia nuclear ou mecânica12. - Dissociar manualmente as células agitando suavemente o tubo. Certifique-se de que o conteúdo do tubo fique turva sem grandes pedaços visíveis pelo olho. Evite a formação de bolhas para minimizar os danos e perdas de células.
- Centrifugar a 200 x g por 3 min. A pelota deve ser claramente visível.
- Remova o supernatante e siga um dos passos detalhados abaixo.
- Se não for necessário coloração, adicione 500 μL de DMEM. Levemente resuspend com uma pipeta de 200 μL mirando um jato líquido na pelota. Não exerça força excessiva de tesoura nas células. A espuma indica danos às células.
- Para rotular o núcleo com corantes de DNA como Hoechst, misture 0,5 μL de DNA-Hoechst (estoque 2 mg/mL) em 1.000 μL de DMEM para obter 1 μg/mL de concentração final. Adicione 500 μL desta solução de coloração às células e resuspenque suavemente. Incubar por 7 minutos no escuro.
- Para manchar as células com um indicador químico de cálcio fluorescente Calbryte-520, adicione Calbryte-520 a uma concentração de 5 μM no DMEM. Incubar por 20 minutos no escuro.
NOTA: Os protocolos indicados nas etapas 2.5.2 e 2.5.3 foram otimizados para esses produtos específicos. Outras manchas podem ser realizadas utilizando os protocolos indicados pelo fabricante.
- Centrifugar novamente usando as mesmas configurações da etapa 2.4; remova o supernasce e ressuspenque suavemente as células (para evitar a formação de clusters) em 50 μL de DMEM para amostras em suspensão ou 20 μL de DMEM para células em confinamento.
3. Preparação de câmaras ópticas de trapping utilizando espaçamento de polidimetilsiloxano (PDMS)
NOTA: Medições de força óptica baseadas na detecção de impulso de luz requerem a captura de toda a luz emergindo das armadilhas ópticas40. Para a robustez do fator de calibração invariante α (pN/V), a distribuição de luz no plano focal traseiro (BFP) do sensor de força óptica deve ter uma correspondência precisa para o momento do fóton. Isso determina a distância da superfície da lente coletora para o plano de captura para aproximadamente 2 mm, que é a altura máxima das câmaras de trapping óptica.
- PDMS spin-coating de #1.5 pratos de fundo de vidro.
NOTA: A seguinte receita é fornecida para aproximadamente 40 pratos. A microcâmara resultante terá diferentes alturas dependendo se os experimentos devem ser realizados em células suspensas ou confinadas (Figura 1D).- Misture 9 mL do polímero base PDMS e 1 mL de agente de cura PDMS em um tubo cônico de 50 mL. Misture os dois produtos ativamente para garantir a distribuição adequada do agente de cura.
- Desgas a mistura para evitar bolhas usando uma bomba de vácuo. Introduza o tubo cônico em uma garrafa de vácuo e evacue a câmara. Espere até que não haja bolhas presentes na mistura.
NOTA: Abra o vácuo lentamente para evitar espumas e derramamentos do PDMS para fora do tubo falcão. - Coloque o prato de fundo de vidro no mandril do spin-coater (Figura 2A). Seja gentil para não arranhar, impressões digitais, ou sujar o prato. Proteja a caixa de spin-coater dos vazamentos PDMS com papel alumínio.
- Para câmaras OT para experimentos em células em suspensão, adicione aproximadamente 250 μL de mistura PDMS no centro do prato inferior e gire-o a 750 rpm por 1 min. A altura da camada PDMS será de aproximadamente 50 μm.
- Para câmaras OT para experimentos em células confinadas, adicione uma pequena gota de PDMS (aproximadamente 50 μL) e gire-o a 4.000 rpm por 5 min. A altura da camada PDMS será aproximadamente de 10 μm. Para obter um protocolo detalhado sobre como obter diferentes espessuras de PDMS, consulte referência48.
- Cure os pratos de fundo de vidro revestidos de PDMS a 70 °C por 1 h.
- Corte um quadrado de 1 x 1 cm na camada PDMS com um bisturi e retire-o com pinças (Figura 2C). No caso de células confinadas, lave detritos PDMS com isopropanol.
- Revestimento de câmara para experimentos com células levemente ligadas em suspensão
- Adicione 100 μL de Concanavalina A (ConA) a 0,5 mg/mL para cobrir toda a superfície da cavidade quadrada e deixe incubar por 30 minutos.
NOTA: é uma lectina que se liga a açúcares de superfície celular e casa células individuais na superfície da tampa. - Remova a gota e enxágue a superfície cuidadosamente com o meio DMEM sem arranhar a superfície tratada.
- Adicione 30 μL da amostra previamente preparada (etapa 2.6) no poço e levemente resuspend para se livrar de qualquer aglomerado celular.
- Feche a cavidade colocando suavemente um vidro de cobertura de 22 x 22 mm #1.5 em cima das bordas do PDMS (evite deixá-la cair abruptamente, use fórceps, se possível, Figura 2B,C).
NOTA: Qualquer espessura de deslizamento de tampa funcionaria para a tampa superior do vidro (a lente coletora tem uma distância de trabalho de 2 mm).
- Adicione 100 μL de Concanavalina A (ConA) a 0,5 mg/mL para cobrir toda a superfície da cavidade quadrada e deixe incubar por 30 minutos.
- Preparação da câmara para experimentos com células em confinamento
- Coloque uma gota de 10 μL de solução contendo células (passo 2.6) na cavidade quadrada (Figura 2B).
- Muito gentilmente, sanduiche a amostra com um copo de cobertura de 22 x 22 mm de tal forma que a gota se espalhe em toda a área e nenhuma bolha seja observada. Mais uma vez, é conveniente usar fórceps, como mostrado na Figura 2C, para evitar que o vidro de cobertura caia abruptamente.
4. Opções alternativas para espaçamento de câmara OT
NOTA: Estas etapas podem ser seguidas se não houver oficina de microfabização ou spin coater.
- Preparação da câmara para experimentos com células em suspensão
NOTA: Caso não haja um revestimento de giro disponível, um espaçador pode ser feito usando fita adesiva normal de duas faces (aproximadamente 100 μm de altura).- Corte um pedaço de fita adesiva de duas faces com um buraco quadrado de aproximadamente 10 cm x 10 cm no centro (mesmas dimensões do PDMS, Figura 2B).
- Remova uma das camadas protetoras da fita descascando-a e coloque o lado descoberto da fita no centro de um prato de fundo de vidro #1,5 H. Pressione suavemente para obter toda a superfície aderida ao vidro, evitando bolhas de ar e, em seguida, remova a camada protetora restante da fita descascando-a.
- Siga as instruções na etapa 3.2.
- Preparação da câmara para experimentos com células em confinamento
NOTA: Para limitar precisamente as células, micropartículas monodispersas com diâmetro conhecido podem ser usadas como espaçadores entre os dois óculos de cobertura.- Adicione 10 μm de contas de poliestireno às células suspensas a uma concentração de 104 contas/μL.
- Coloque uma gota de 10 μL de solução contendo células e contas em um vidro de cobertura de 22 x 60 mm.
- Muito gentilmente, sanduiche a amostra com outro copo de cobertura de 22 x 60 mm de tal forma que a gota se espalhe em toda a área e nenhuma bolha seja observada. Para posicionar o vidro da tampa superior suavemente (evite que ele caia abruptamente), é conveniente usar fórceps.
- Como a amostra pode secar, recomenda-se realizar a preparação rapidamente.
5. Configuração da armadilha óptica para medições intracelulares
NOTA: As seguintes etapas são otimizadas para uma plataforma de pinça óptica comercial que compreende um módulo de micromanipulação óptica baseado na deflexão acousto-óptica (AOD) e um sensor de força óptica baseado na detecção direta de mudanças de momento de luz (Figura 2, referência12,40,49). Detalhes e componentes ópticos da configuração podem ser encontrados na Figura 2F. Para observar a deformação induzida pela força durante as manipulações ópticas da pinça, um microscópio confocal de disco giratório Nipkow é acoplado à porta esquerda do microscópio invertido para imagens de fluorescência de dupla cor. Sem a falta de generalidade, este protocolo pode ser aplicado com qualquer sistema de OTs dinâmico equipado com medições de força direta com base na detecção de impulso de luz. Procedimentos passo a passo detalhados estão disponíveis para construir armadilhas de gradiente óptico em casa para aplicações in vivo50. Aqueles baseados na modulação AOD destacam-se para eventuais experimentos com múltiplas armadilhas e medições rápidas51,52. Vários protocolos para a construção de um instrumento baseado em impulso leve existem na literatura36,39,40,53, e qualquer outra modalidade de imagem (contraste de interferência diferencial, fluorescência de campo largo, etc.) podem ser empregados.
- Start-up de pinças ópticas
- Para otimizar para a estabilidade de potência de saída, ligue o laser em potência consideravelmente alta (por exemplo, 3 W) pelo menos 30 minutos antes do experimento.
- Ligue o módulo eletrônico das unidades de medição óptica de micromanipulação e força.
NOTA: Aplique todas as medidas de segurança a laser e utilize apenas equipamentos aprovados pelo conselho institucional. Nunca use as oculares do microscópio óptico quando o laser estiver ligado. Use sempre os óculos de proteção IR aprovados (OD7 na faixa de 950-1080 nm), bloqueie a luz laser IR com o obturador na porta de epifluorescência 2 e não execute o software óptico de trapping até terminar o alinhamento do sensor de força óptica após a etapa 5.3. Em geral, não use uma amostra altamente reflexiva, pois o reflexo traseiro pode causar danos ao laser. - Controle a potência da armadilha com o HWP rotativo (Figura 2F) na entrada do módulo de micromanipulação óptica.
NOTA: O módulo de micromanipulação óptica comercial utilizado neste protocolo já incorpora esse recurso. Para sistemas de trapping ópticos construídos em casa, integre esta ferramenta para controle de energia para que potências laser mais altas e mais estáveis possam ser usadas.
- Use uma microcâmara vazia para calibração
- Corte um quadrado de 1 x 1 cm em uma fita de uísque de dois lados e conecte-o a um slide de microscópio de 1 mm de espessura.
- Adicione água no quadrado e feche-a de cima com um copo de cobertura #1.5 (22 x 22 mm). A adição de um volume ligeiramente maior de água, por exemplo, 30-40 μL é aconselhável evitar bolhas dentro da câmara coberta. Limpe a câmara de calibração suavemente em caso de água derramando dela.
- Alinhamento do sensor de força óptica
- Coloque uma gota de água no objetivo de imersão de água 60x/1.2. Coloque a câmara de calibração no palco com o vidro de cobertura #1.5 voltado para o objetivo. Concentre-se na superfície inferior, onde as amostras de células eventualmente estarão.
- Adicione uma gota de óleo de imersão em cima do escorregador superior de vidro que cobre a amostra (Figura 2D). Abaixe cuidadosamente a lente coletora da unidade do sensor de força até que entre em contato com a gota de óleo.
NOTA: A gota deve ser grande o suficiente para cobrir toda a lente que coleta a luz laser emergindo das armadilhas. Normalmente, 200 μL é suficiente para cobrir toda a superfície e fornecer um contato de imersão estável. Seja conservador e evite o excesso de escoamento, pois pode vazar para a amostra. - Seguindo o protocolo do fabricante para o alinhamento do sensor de força óptica, olhe para a imagem do plano de amostra na câmera auxiliar que será usada para posicionar os OTs (AUX, Figura 2F). Muito suavemente, abaixe o sensor de força óptica até que a parada de campo (FS, Figura 2F-G) apareça conjugada no plano de amostra. Isso garantirá medições adequadas de força direta a partir da detecção invariante de amostras de mudanças de impulso de luz40.
NOTA: Feche o FS o suficiente para que sua imagem se torne menor do que o campo de visão (FOV), portanto, visível. Tenha cuidado extra e não empurre a lente coletora do sensor de força óptica contra a amostra. A posição vertical do sensor de força óptica pode ser, alternativamente, determinada a partir da análise da distribuição de luz de trapping no BFP para cones de luz com abertura numérica definida (NA). - Certifique-se de que não há bolhas de ar na gota de óleo; estes podem afetar diretamente as medidas de força. Para verificar se há bolhas de ar, coloque a lente Bertrand no lugar (BL, Figura 2G) e observe o caminho de imagem através da ocular. Se alguma sujeira ou bolhas de ar forem visíveis ou mais óleo for necessário (Figura S1A), limpe a lente e a câmara com tecido de lente sem poeira e repita o procedimento nas etapas 5.3.2 e 5.3.3. Um caminho óptico desobstruído é retratado na Figura S1B.
- Utilizando os parafusos laterais colocados no suporte do sensor de força óptica, centralizar o FS no FOV. Para precisão, abra o FS para que ele quase preencha o FOV visível na câmera auxiliar (AUX, Figura 2F).
6. Otimização óptica da pinça
NOTA: A medição da força direta depende unicamente da mudança do momento da luz decorrente da força exercida sobre a partícula presa e, portanto, em contraste com os métodos indiretos, a rigidez da armadilha não precisa ser calibrada antes de cada experimento. A conversão específica do instrumento de fator de deflexão/força (α; pN/V, reference41) é calibrada pelo fabricante e, portanto, é invariante do experimento. No entanto, como o ponto laser é manipulado sobre uma área de 70 μm x 70 μm, as etapas 6.2-6.5 são fundamentais para garantir a captura ideal e a estabilidade de potência. As etapas a seguir são fornecidas no software do fabricante para que os OTs sejam otimizados sobre a área de trabalho de forma semiautomática.
- Inicie o software OTs e o software de aquisição para a câmera AUX.
- Subtraia a linha de base inicial de tensão clicando no Passo 1: Etapa de deslocamento eletrônico no submenu de calibração do sistema do software de condução de pinças ópticas.
- Para realizar o achatamento de potência da armadilha através da área de trabalho OT, coloque a potência da armadilha em metade do seu máximo, girando o HWP de acordo. Não altere a potência da armadilha alterando a saída do laser, mas com o HWP rotativo (Figura 2F). Clique no Passo 2: Ligue para iniciar a rotina automatizada para achatamento de potência da armadilha.
NOTA: Este é um passo crítico para compensar a variação da potência da armadilha em toda a área de trabalho dos OTs (Figura S1D). Uma rotina bem sucedida reduz a variação de potência da armadilha para 2% através da área de trabalho dos OTs e converge após 2 minutos. - Para realizar a calibração da posição da armadilha, remova o filtro IR para que a luz do laser seja visível na câmera. Encontre o ponto IR definindo o plano de imagem focado na superfície inferior da microcâmara. Obtenha o menor ponto de RI possível afinando o plano de imagem (posição objetiva) e o contraste do histograma no software de aquisição da câmera AUX. Se necessário, reduza a potência da armadilha óptica girando o HWP (Figura 2F). Clique no Passo 3: Posicione-se para iniciar a rotina automatizada ou calibração do posicionamento da armadilha.
NOTA: Esta rotina permite a correspondência precisa das coordenadas de posição do OT na câmera AUX aos ângulos de direção AOD. Uma rotina bem sucedida gera o mapeamento ângulo-a-posição em vários segundos. - Compensação inicial de impulso
NOTA: O movimento da armadilha óptica através da amostra causa variações na distribuição de impulso de luz no BFP (Figura S1E, F). Isso leva a mudanças de sinal independentes da força relacionadas à posição laser sobre a área de trabalho, embora a potência da armadilha tenha sido achatada como na etapa 6.3. A consequência é uma variação na linha de base da força devido à posição (independente de uma força real agindo no bico aprisionado opticamente) que precisa ser corrigida antes de cada experimento.- Coloque a potência da armadilha que será usada nos experimentos, girando o HWP (Figura 2F).
- Clique na opção Deslocamento Global no submenu Ferramentas . Isso abrirá o assistente Offset Cancel do software pinça óptica que corrige a linha de base de impulso inicial.
- Clique em Offset | Compensar para corrigir o momento inicial da variante de posição.
NOTA: Se nenhuma modificação afetar o caminho óptico durante as semanas em curso, os mapas de achatamento de potência da armadilha (etapa 6.3) e posição (passo 6.4) permanecerão invariantes. Por isso, recomendamos sempre usar a mesma combinação de elementos ópticos (espelhos dicrómicos, filtros, etc.) que possam afetar o caminho da armadilha do laser ou para realizar uma nova rotina de achatamento de potência de armadilha. Em relação à compensação inicial de impulso (etapa 6.5), o fabricante da plataforma OTs fornece uma calibração on-the-fly que deve ser alterada para cada nova potência de trapping e sessão experimental. As etapas 6.3 e 6.4 devem ser realizadas no slide de calibração vazio descrito na etapa 5.2. Em uma amostra contendo células ou outros objetos, o passo 6.5 deve ser realizado livre de objetos que podem alterar a dispersão da luz na área de trabalho dos OTs.
- Opcionalmente, presencese uma microesfera e mova a armadilha a uma velocidade conhecida enquanto grava o sinal de força. Por exemplo, defina a armadilha para realizar uma oscilação triangular: o sinal de força gravado será um sinal quadrado.
NOTA: O valor da força deve aumentar linearmente com a velocidade, de acordo com a força de arrasto que atua na conta. Este teste serve como um controle positivo de que as medidas de força estão sendo realizadas corretamente38. Alternativamente, o sensor de força óptica pode ser usado para obter a rigidez óptica de trapping, κ [pN/μm], e o fator de calibração de posição, β [μm/V], a partir da análise espectral de potência35. Sob alinhamento correto, o fator de calibração invariante fornecido pelo fabricante é α = κ·β [pN/V].- Inicie uma leitura de força em tempo real clicando no Plot 1 no submenu Medidas no software do fabricante. Isso fornecerá uma leitura da atual força óptica de captura e potência.
- Abra o diálogo Parâmetros de Oscilação a partir do submenu Ferramentas . Defina uma forma de forma de onda triangular-espacial nos anéis seletor Forma e Tipo, respectivamente. Como exemplo, defina uma amplitude de 10 μm e uma frequência de 3 Hz. Isso resultará em uma força viscosa de aproximadamente 1 pN em uma microesfera com um diâmetro de 1 μm38.
- Na janela AUX da câmera, clique com o botão direito do mouse na microesfera e selecione Iniciar oscilação. A leitura da força se tornará um sinal de força quadrada com platôs a ±1 pN.
- Clique com o botão direito do mouse na microesfera e selecione Parar de Oscilar.
7. Microscopia confocal de disco giratório
- Ligue o microscópio confocal de disco giratório e equipamentos acessórios, os motores laser integrados e as câmeras de aquisição.
- Lance o software de imagem.
- Defina canais de imagem para coloração hoechst do núcleo e GFP para a membrana plasmática celular.
- Ative as linhas de lasers de excitação de 405 nm e 488 nm.
- Adicione um dicroico multiband para refletir a excitação à amostra e que permite que a luz emitida passe para as câmeras.
- Divida a emissão de fluorescência com um espelhodicrómico de borda de passagem longa de 500 nm.
- Use os filtros de emissão DAPI/BFP (~445 nm) e GFP (~521 nm) na frente das duas câmeras de aquisição, respectivamente. Consulte a Figura 2F,G.
- Ajuste o tempo de exposição para 100 ms para cada canal.
- Defina emissão de laser para obter uma potência de 5 mW no plano amostral. Para medir a energia, use um medidor de energia comercial.
- Defina o protocolo de imagem. Para evitar sangramento espectral através do canal Hoechst para o canal GFP, os dois corantes precisam ser imageados sequencialmente.
NOTA: Se existir uma sincronização de hardware entre os AODs da armadilha óptica e a aquisição da câmera, certifique-se de que a polaridade do gatilho esteja configurada corretamente. Em caso de dúvida, consulte o gerente de instalações ou o fabricante de microscópio.
8. Realizando os experimentos de recuo do núcleo
NOTA: Desligue sempre as armadilhas ópticas - tanto usando software quanto fechando o obturador na porta de epifluorescência 2-ao levantar o módulo do sensor de força e alterar a amostra. Caso não, podem ocorrer sérios danos aos elementos ópticos e ao experimentador. Tenha cuidado com a distância lateral entre o suporte da lente e a borda inferior do prato ao procurar células para evitar bater a lente no prato de palco/cultura (Figura 2).
- Coloque a amostra no microscópio e siga o passo 5.3 deste protocolo.
- Utilizando o HWP rotativo (Figura 2F), configure a potência da armadilha para 200 mW como um valor inicial se a rigidez do núcleo ou estrutura intracelular investigada não for conhecida. Traduza a área de trabalho dos OTs (usando o estágio do microscópio) para um lugar livre de células, a fim de compensar a linha de base inicial de impulso através da etapa 6.5.
NOTA: Dependendo da rigidez da estrutura subcelular, o valor de potência da armadilha deve ser ajustado a valores inferiores ou superiores para obter uma profundidade de recuo semelhante. - Usando o controlador de software de estágio do microscópio, procure uma célula com uma ou duas contas através de microscopia de campo brilhante transmitida (Figura 3A).
- Defina uma trajetória de armadilha.
- Abra o diálogo Trajetória no submenu Ferramentas e escolha Deslocamento no anel seletor tipo de trajetória .
- Na folha numérica, escreva o deslocamento e o tempo de cada etapa de trajetória subsequente. Aqui estão dois exemplos.
- Para um experimento de relaxamento do estresse, programe cargas trapezoidais, como mostrado na Figura 3B. Na Tabela S1, foram aplicadas duas indentações trapezoidais com uma distância de viagem de 5 μm; velocidade de 5 μm/s; tempo de espera antes da retração: 10 s.
- Para um experimento de recuo repetitivo em uma velocidade constante para obter uma rotina triangular sem tempo de moradia no núcleo, defina a amplitude da trajetória, por exemplo, 5 μm, e o tempo para a etapa, por exemplo, 2 s para uma velocidade de 2,5 μm/s. Na Tabela S2, isso é aplicado oito vezes na mesma velocidade.
NOTA: Esses valores precisam ser determinados para cada tipo de célula e experimento, mas os seguintes parâmetros de uma rotina trapezoidal capturam a dinâmica mais importante no experimento aqui apresentado. O tempo de espera deve ser suficiente para que o núcleo mostre seu completo relaxamento de estresse após o recuo
- Prendendo uma microsfera
- Defina o plano de imagem ligeiramente acima da conta com o controlador de software de estágio do microscópio.
- Ative armadilhas usando o software OTs e clique na conta na janela de imagem AUX da câmera (calibrada após o passo 6.4). O confinamento bem sucedido da conta pela armadilha óptica reduzirá fortemente o movimento da conta.
- Clique e arraste a conta através do citoplasma e coloque-a a uma distância de ~2 μm do envelope nuclear (Figura 3A). Certifique-se de que a trajetória está definida para que o recuo de contas seja perpendicular à membrana nuclear.
- Opcionalmente, se necessário para as medições de posição da conta em relação à armadilha, escaneie a armadilha através da conta para determinar a rigidez de captura, k [pN/μm]54, assim Δxbead = -F/k (ver Discussão). O módulo óptico de micromanipulação usado neste protocolo tem uma rotina incorporada para este fim.
- Abra a caixa de diálogo de varredura de partículas no submenu Ferramentas .
- Selecione a armadilha deseja escanear e alta frequência como o Método de Digitalização. Selecione a direção (x ou y) da trajetória de recuo para a medição de digitalização de contas.
- Uma janela aparecerá com a medição da rigidez da armadilha. No gráfico, arraste os dois cursores para selecionar a área de trapping linear correspondente a F = -kx. O ajuste linear à porção de dados selecionada será atualizado automaticamente.
NOTA: Defina a posição inicial da conta longe da membrana celular (~5 μm), pois as deflexões de impulso de luz na interface de célula média afetam a adequação das medidas de força. Se o núcleo estiver localizado muito perto da membrana celular, tente indentar o núcleo do local oposto. Descarte a célula se não for possível.
- Inicie a aquisição de imagens clicando no botão de aquisição no software de imagem.
- Inicie a posição da armadilha e force a economia de dados de medição clicando em Data | Economize na janela de leitura de força em tempo real (aberto como na etapa 6.6.1).
NOTA: A armadilha óptica é equipada com uma entrada de gatilho que pode ser conectada à saída de tempo da câmera. Assim, os dados de imagem e força são sincronizados por hardware e o eletrônico é capaz de mapear os ciclos de armadilha com o número de quadros das imagens durante a aquisição. - Inicie a trajetória carregada anteriormente clicando com o botão direito do mouse na conta e selecionando a Trajetória de Início.
- Aguarde até que a trajetória esteja concluída e o sistema se estabilize.
- Pare de medir dados de força de armadilha. Uma caixa de diálogo de economia de dados aparecerá.
NOTA: Para otimizar o armazenamento de dados, os dados podem ser dizimados selecionando o parâmetro dizimador nesta caixa de diálogo (10, 100 ou 1000). - Pare a aquisição de imagens e plote os resultados no software de pós-processamento da escolha do usuário.
- Se a microesfera for perdida durante a rotina e o núcleo não puder ser recuado (Figura S2), descarte a medição e aumente a potência. Note que o passo 6.5 deve ser repetido. Em nossas mãos, pelo menos 95% das rotinas são concluídas com sucesso sem perder a conta da armadilha.
Resultados
Microinjeção de contas de captura:
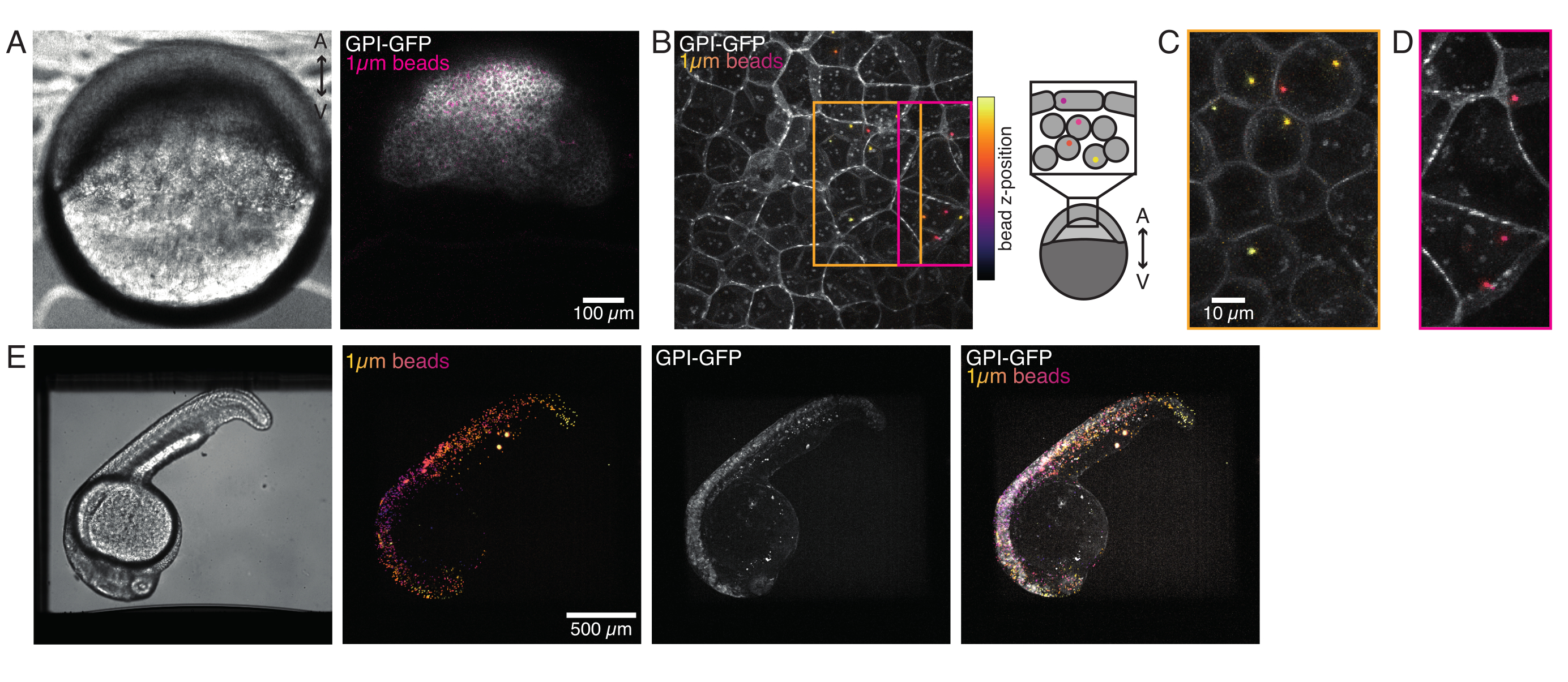
Microesferas injetadas no embrião de zebrafish de uma célula se espalharam por toda a tampa animal durante a morfogênese. Para uma visualização mais clara, repetimos o protocolo de injeção com micróbios fluorescentes vermelhos e pegamos imagens volumétricas com nosso microscópio confocal em diferentes estágios de desenvolvimento. Na Figura 4A-D, as contas injetadas são visualizadas no citoplasma das células-tronco progenitoras in vivo a 5 hfp. Mais tarde, as microesferas apareceram espalhadas por todo o embrião a 24 hpf (Figura 4E). Os embriões em ambos os estágios desenvolveram-se normalmente e as taxas de sobrevivência foram comparáveis com o controle não injetado ou embriões simulados (ver Figura S3). Isso é consistente com outros estudos que relatam sobrevivência não perturbada de zebrafish injetados por contas até 5 dias após a fertilização55.
Nosso microscópio confocal de disco giratório é compatível com microcopia de fluorescência multicanal. Na Figura 5A, mostramos células-tronco isoladas com uma ou duas contas no citoplasma. Vários rótulos fluorescentes podem ser usados para investigar diferentes aspectos da célula (Figura 5B). A morfologia nuclear pode ser rastreada com um corante Hoechst ou usando uma expressão H2A::mCherry mRNA, enquanto a membrana nuclear interna pode ser analisada com Lap2b-eGFP12. A dinâmica do córtex de actomiosina, bem como os níveis intracelulares de cálcio, podem ser observados com uma linha transgênica My12.1::eGFP 56 e Calbryte-520, respectivamente. O protocolo aqui descrito visa comparar a mecânica do núcleo celular de células do tipo selvagem imobilizado em substratos adesivos (posteriormente referidos como suspensão) e em confinamento mecânico. Células-tronco isoladas confinadas em microchambers de 10 μm de altura apresentaram desdobramento parcial da membrana nuclear interna (INM) e um subsequente aumento da contração da actomyosina12. Na Figura 5C, são mostradas células confinadas com uma ou duas contas no citoplasma. O confinamento bem sucedido será visível através de células achatadas e expandidas com uma seção transversal mais ampla do núcleo. A membrana nuclear é ainda mais desdobrada em células confinadas e deve parecer suavizada em comparação com as células em suspensão (Figura 5C).
Análise de deformação de força e força
A análise dos resultados obtidos depende fortemente do espécime investigado e da questão de interesse e, portanto, não podem ser generalizados aqui. Como exemplo, uma maneira comum de analisar a medição do recuo é extrair o módulo de um Young, encaixando um modelo Hertz modificado aos dados de recuo à força57. No entanto, a suposição para tal tratamento precisa ser cuidadosamente avaliada e pode nem sempre ser devidamente justificada (como a estrutura investigada ser isotrópica, homogênea, com elasticidade linear e recuos sendo menores que o raio de contas). Consideramos, portanto, apenas medidas de modelo independentes aqui que permitam que o comportamento mecânico da estrutura investigada seja comparado entre diferentes cenários experimentais.
Como ponto de partida, medir a inclinação da curva de deslocamento de força em uma certa profundidade de recuo fornece uma medida de um modelo de rigidez estrutural independente58 do núcleo. Esse valor pode então ser coletado a partir de várias amostras e comparado entre diferentes configurações experimentais e perturbações de amostras.
Medição de recuo
Nas linhas a seguir, focamos na resposta mecânica do núcleo celular durante a deformação celular no confinamento. Experimentos na etapa 8 deste protocolo normalmente levam a forçar picos de até 200 pN para profundidades de recuo de aproximadamente 2-3 μm. No entanto, esses valores podem ser em grande parte diferentes, dependendo do tipo celular e condições experimentais, com núcleos mais suaves levando a menor força para um dado recuo. É necessário, assim, medir com precisão a deformação nuclear, juntamente com a força, para uma caracterização mecânica precisa do núcleo celular. Nesta seção, obteremos a rigidez nuclear da célula a partir de medidas representativas de recuo de força.
Na Figura 6, mostramos as deformações dos lados distal e proximal de um núcleo em uma célula suspensa e confinada. Um rico comportamento mecânico pode ser observado. Em uma célula suspensa típica em um substrato adesivo, o núcleo foi fortemente recuado pela conta, mas também ligeiramente deslocado em eventos repetitivos de empurrar. Medimos o recuo de contas no núcleo analisando os kymógrafos obtidos a partir de imagens de fluorescência de núcleos celulares manchados por Hoechst. Os kymographs foram facilmente computados usando o plugin Multi Kymograph de Fiji ao longo da direção de recuo (Figura 6A,B) e importados para Matlab (Versão 2021, Mathworks) para posterior processamento. Uma função de passo foi encaixada ao perfil de intensidade bruta com o objetivo de rastrear as bordas delimitantes do núcleo ao longo da trajetória da rotina de recuo. Como pode ser visto, ele carrega informações precisas sobre a mudança nuclear na forma (Figura 6 e Figura S2). Usamos a seguinte curva sigmoide dupla como uma versão analítica de uma função de passo:
 (Equação 1)
(Equação 1)
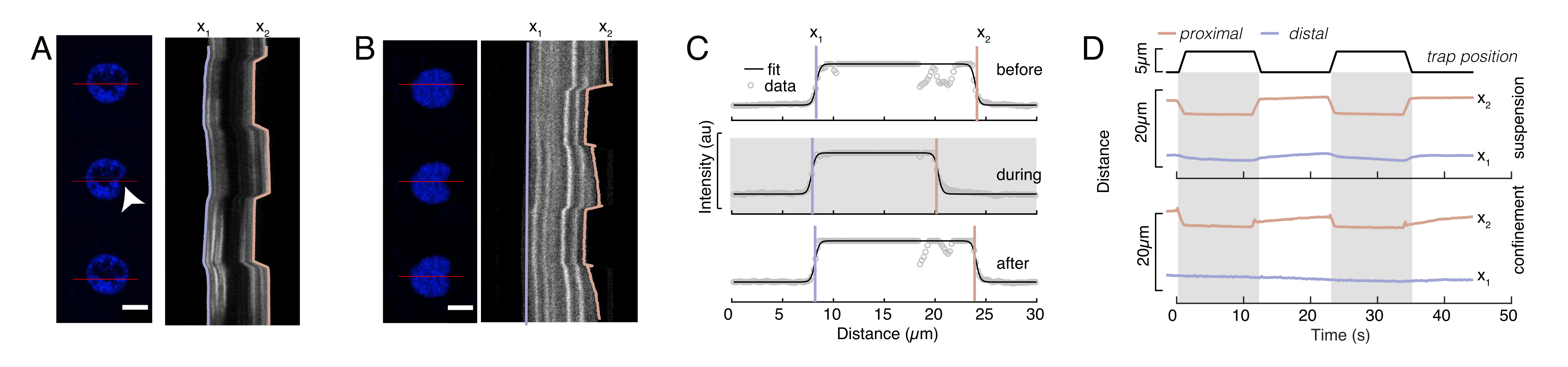
Aqui, x1 e x2 denotam as bordas distal e proximal do núcleo, enquanto A e B são os valores máximos e de fundo cinza do canal azul (corante hoechst) da imagem (Figura 6B). Considerou-se a largura da borda (e0 = 0,25 mm). Enquanto a borda do núcleo proximal (x2) recuada (x2) seguiu a trajetória aplicada pela rotina de armadilha óptica após o contato microesfera-núcleo, a borda oposta e distal (x1) exibe dinâmica de relaxamento como esperado para um material viscoelástico como o citoplasma (Figura 6D). Em contraste, núcleos em células confinadas em microchambers de 10 μm de altura não apresentam tal comportamento de translocação do núcleo após o recuo dentro da célula (Figura 6B,D). Também mostradas na Figura 6D, as bordas traseiras dos núcleos permanecem inalteradas pelo empurrão do lado proximal, provavelmente devido a forças mais fortes decorrentes da contrailidade celular e do atrito agindo contra a força de recuo. Para obter a profundidade de deformação correta, o deslocamento x1 foi subtraído da medida recuada x2: Δx = x2 - x1 (ver também Figura 6D).
Análise de dados de força
A força que causa a deformação nuclear foi medida a partir da mudança no momento da luz originada na microesfera presa opticamente (Figura 7A). A força ao aplicar trajetórias trapezoidais (etapa 8.4.3, Figura 7B) inicialmente aumentou linearmente até que a armadilha parou de se mover, mas depois relaxada a um valor de estado estável. Este comportamento indicou um material viscoelástico exibindo perda e moduli de armazenamento. Logo após o evento de recuo, a força atingiu um valor máximo, Fp, seguido de um relaxamento do estresse (Figura 7C):
 (Equação 2)
(Equação 2)
onde F0 é a força armazenada para o componente elástico e f(t) é uma função de relaxamento sem dimensão. Analisamos esse comportamento de três maneiras:
1. Considerando um sólido linear padrão com relaxamento exponencial do estresse, ou seja, f(t) = e-t/τ, representado esquematicamente no inset da Figura 7C.
2. Usando uma decadência geral, duplamente exponencial:
F(t) = A + B1e-t/τ1 + B2e-t/τ2.
3. Usando uma lei de poder seguida de uma decadência exponencial59:
f(t) = t-pe-t/τ, encaixado na Figura 7C.
Embora o ajuste para o modelo 1 possa ser realizado de forma simples, recomendamos estimar os palpites iniciais para (τ1, τ2) e (p, τ) para os modelos 2 e 3, respectivamente. Isso pode ser realizado, respectivamente, encaixando linhas nos dados em escalas logarítmicas versus lineares (Figura 7D, esquerda) e logarítmicas versus logarítmicas (Figura 7D, direita). Tabela S3 resume os resultados do exemplo analisado na Figura 7. Na seção seguinte, consideraremos a combinação de uma lei de poder e uma lei exponencial para a caracterização da mecânica do núcleo celular.
Relação de deslocamento de força
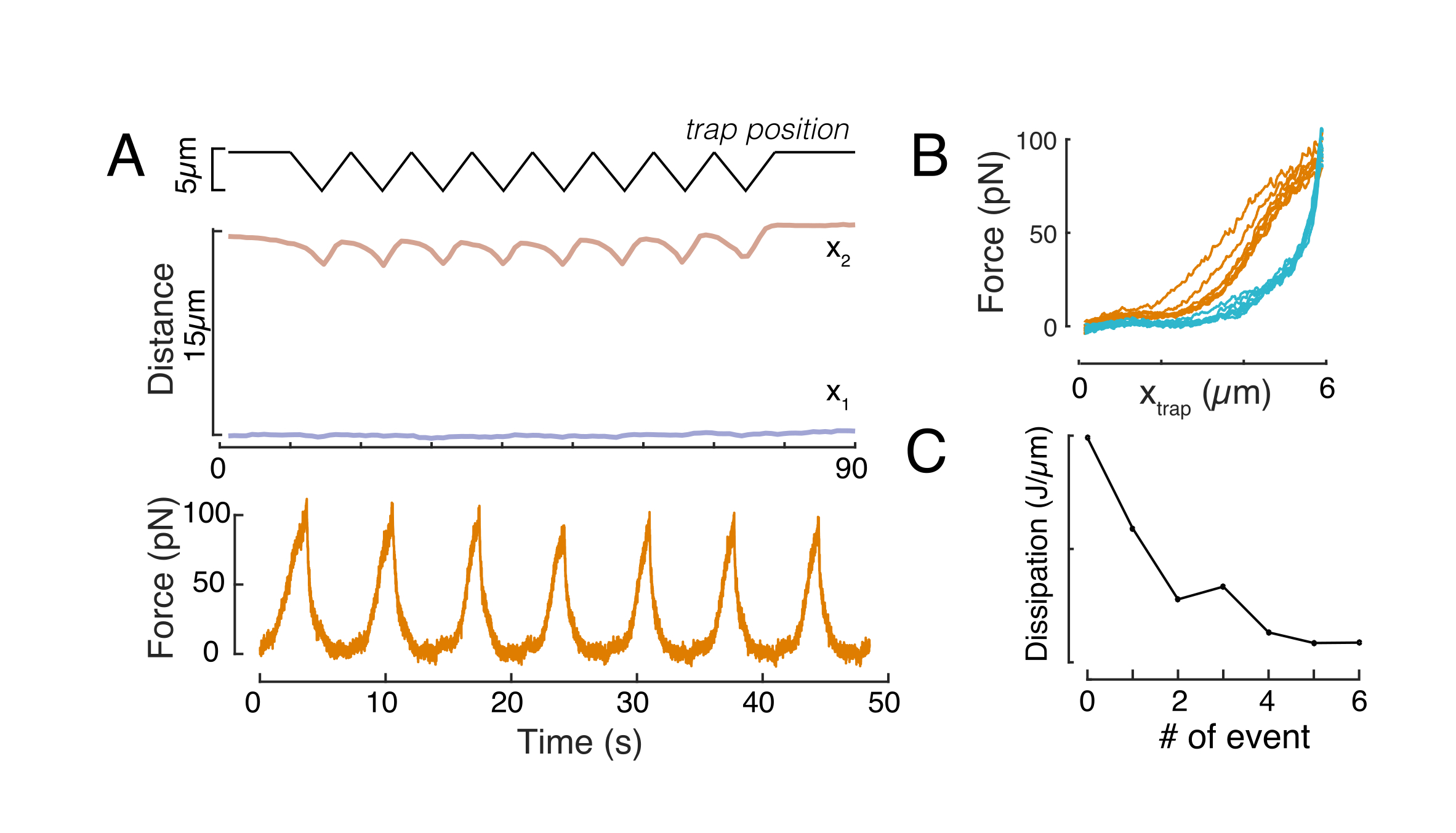
Da mesma forma, a configuração experimental descrita pode ser usada para obter a relação de deslocamento de força de múltiplos eventos de recuo. Ao realizar rotinas triangulares (etapa 8.4.4, Figura 8A), é possível relacionar a força com a deformação e traçar uma curva de recuo à força. Um resultado exemplar é mostrado na Figura 8B, na qual uma linha de base plana mudou suavemente a inclinação uma vez que a esfera entrou em contato com o núcleo. Identificar o verdadeiro ponto de contato nos dados barulhentos é um desafio, e é preciso ter cuidado para ver se a região de contato está adequada aos modelos elásticos60. Neste experimento em particular, também pode-se ver que as subsequentes recuos resultam em curvas com pontos de contato mais profundos, indicativos para recuperação muito lenta da forma nuclear após a retração das contas e uma mudança no ciclo histerético definido pelas propriedades do material viscoelástico do núcleo (Figura 8C). Assim, o pesquisador deve estar ciente se isso acontece e incorporá-lo no pipeline analítico, ou restringir o número de medições subsequentes para que esse efeito não modifique a medição.
Núcleo mecânico em células em suspensão e sob 10 μm de confinamento
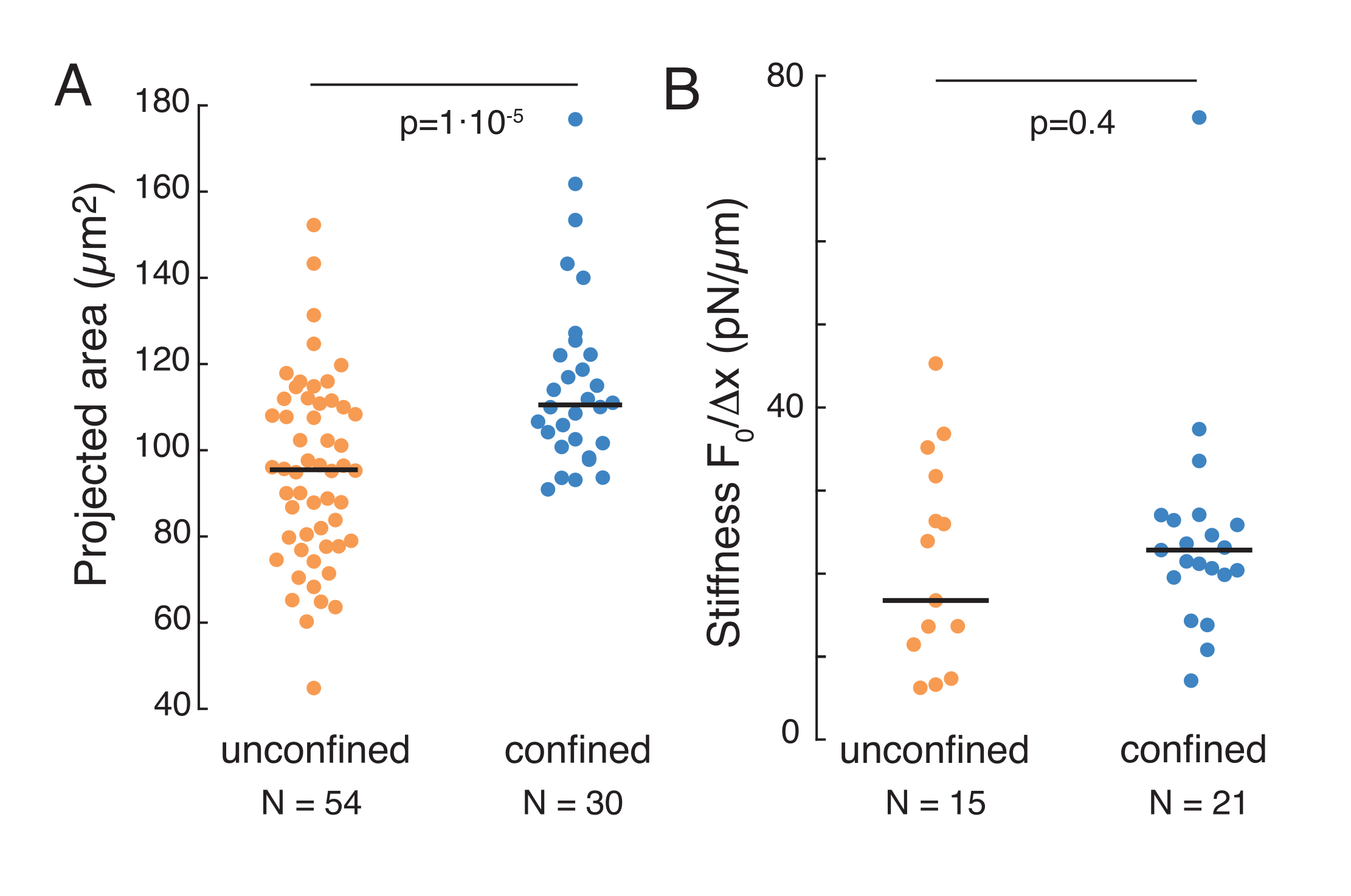
A abordagem supracitada foi utilizada para analisar a dinâmica do relaxamento do estresse do núcleo em células suspensas em substratos adesivos e células confinadas. Nossos resultados mostram que o confinamento resulta em uma expansão da área projetada (Figura 9A), mas mudança insignificante na rigidez nuclear (Figura 9B). Medimos relaxamento semelhante com τ = 6,08 ± 1,1 s (não confinado) e τ = 4,00 ± 0,6 s (confinamento), o que indica rápida dissipação viscoelástica, seguido por um valor de força armazenado que corresponde ao módulo elástico do núcleo. Para explicar as variações experimentais, que podem ser produzidas por diferentes condições iniciais nas rotinas de recuo, as forças armazenadas medidas foram normalizadas até a profundidade do recuo, como  . Este parâmetro explica a rigidez do núcleo e descreve a força, ou o estresse, necessário para um certo recuo. Obtive-se rigidez semelhante sob confinamento e em células não confinadas: = 20,1 ± 12,6 pN/μm e = 24,6 ± 13,6 pN/μm (desvio padrão ± médio), respectivamente.
. Este parâmetro explica a rigidez do núcleo e descreve a força, ou o estresse, necessário para um certo recuo. Obtive-se rigidez semelhante sob confinamento e em células não confinadas: = 20,1 ± 12,6 pN/μm e = 24,6 ± 13,6 pN/μm (desvio padrão ± médio), respectivamente.

Figura 1: Microinjeção de embriões de zebrafish em estágio de uma célula (zigoto) (A) Placa de injeção: uma placa de injeção em forma triangular é usada para a injeção. A placa é feita de 1% de agarose ultrapure na E3 (média do ovo). As visualizações de cima e de lado são mostradas à direita. (B) Posicionamento embrionário: oriente suavemente os embriões usando uma escova e oriente de tal forma que a célula seja claramente visível e facilmente acessível com a agulha. Sugerimos orientar os embriões com a célula localizada no lado oposto da agulha, como mostrado no esboço. (C) Procedimento de injeção no embrião de estágio de uma célula: furar o acorde ao redor do embrião e a única célula com a agulha. Certifique-se de que a ponta da agulha está dentro da célula e libere a pressão para injetar. (D) Incubar os embriões a 28-31 °C até que se desenvolvam até o estágio blastula (esfera) (4 hpf). Realize o protocolo de isolamento celular e a coloração celular (etapa 2) e prepare a câmara óptica de captura com células isoladas em suspensão e/ou confinamento combinado com o revestimento de superfície substrato correspondente (etapa 3). Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: Preparação do aparelho óptico de pinça. (A) Camadas de revestimento de spin de PDMS com altura definida em pratos de fundo de vidro. A queda do PDMS se espalhará uniformemente devido à força centrífuga. (B) Preparação da câmara amostral fora da camada PDMS. 1: corte um quadrado com um bisturi, 2: cubra o poço interno com concanavalina A (ConA), lave e sementes; 3: cubra com um deslizamento de vidro ou deslize de cobertura para selar o poço. (C) Imagem do corte quadrado com um bisturi e remoção do poço PDMS com fórceps. (D) Montagem da lente coletora do sensor de força óptica sobre a câmara de captura. Uma gota de óleo de imersão serve como um meio de imersão entre a lente coletora e a tampa superior do vidro. Esquema para não escalar. Tenha cuidado ao baixar a lente coletora para não tocar na tampa de vidro do prato de amostra. (E) Imagem da unidade de detecção de força em contato com a amostra. (F) Esquema da configuração experimental. O módulo de micromanipulação óptica usa um feixe de laser de onda contínua (5W, λ = 1064 nm) com controle de energia através de uma placa de meia onda (HWP) e um divisor de feixe polarizador (BS). Depois de ser modulado com um par de AODs, ele é acoplado à porta de epifluorescência superior de um microscópio invertido. O raio laser é então refletido por um espelhodicróico de passe curto de 950 nm (IR-DM), permitindo a transmissão da excitação e emissão de fluorescência. O laser de captura é guiado para a porta traseira, epifluorescência do microscópio (torre superior). Os OTs são criados no plano focal de uma lente objetiva de imersão aquática (60x, NA = 1,2). O sensor de força óptica é submetido à torre do microscópio e captura a luz laser emergindo dos OTs com uma lente de alta na, de imersão a óleo. Ao mesmo tempo, o sensor de força permite a iluminação em campo brilhante. A unidade confocal de disco giratório está acoplado à porta esquerda. É equipado com dois motores laser integrados (ILE) que controlam sete lasers de excitação de fluorescência e duas câmeras sCMOS iluminadas para trás, permitindo imagens duplas fluorforeiros em Abb paralelo: TI, Transilluminator; FS, parada de campo; AOD, defletor acustopático; HWP, placa de meia onda; CAM, câmera (G) Fotografia do equipamento óptico de captura. O círculo vermelho indica a lente Bertrand, que pode ser alternada para o caminho óptico manualmente. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: Escolhendo as amostras e parâmetros certos. (A) Imagem representativa de uma célula-tronco progenitora de zebrafish isolada com uma única microesfera posicionada perto o suficiente do núcleo para realizar o experimento de recuo. Barra de escala = 10 μm. (B) Trajetória exemplar da armadilha; profundidade de recuo 5 μm; velocidade de recuo = 5 μm/s; tempo de relaxamento 10 s. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: Localização de microesferas dentro de embriões de zebrafish durante o desenvolvimento. 0,5 nL de contas fluorescentes vermelhas de 1 μm são injetadas juntamente com GPI-GFP mRNA (100 pg/embrião, membrana plasmática) em embriões WT para visualizar localizações de contas. (A-D) Distribuição da microesfera 5h pós-injeção dentro de um embrião montado em 0,75% agarose. (A) Imagem de Brightfield e fluorescência. As contas são dispersas homogêneas através do tecido embrionário, como visto em um micrografo confocal. (B) Projeção máxima de fluorescência confocal z-stack. As contas são codificadas de cor do roxo ao amarelo de acordo com a posição z na pilha de imagens. Roxo/magenta corresponde à maioria das contas/células externas (EVL; camada envolvente epitelial; ou células-tronco progenitoras localizadas perto da superfície EVL), o amarelo corresponde às contas internas (células profundas progenitoras), como mostrado no esboço à direita. (C) Corte e projeção máxima de uma sub pilha de (B) correspondente à região na caixa laranja: uma grande fração de células profundas contém 1-2 contas. (D) Corte e projeção máxima de uma sub pilha de (B) correspondente à caixa magenta: algumas células EVL contêm 1-2 contas. (E) Imagem brightfield e projeção máxima de uma pilha z de um embrião de 24 hpf montado em 0,75% de agarose e anestesiado com tricaine. Os embriões foram pré-incubados com tricaine por 15 minutos. Da esquerda para a direita: microesferas (1 μm de diâmetro), GPI-GFP e sobreposição de imagem. As contas espalhadas por todo o corpo do embrião. Dimensão do scalebar indicado em cada painel. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5: Células-tronco progenitoras de zebrafish isoladas com rotulagem diferente. (A) Imagem de microscopia de luz de transmissão de células de suspensão com 1 (superior) ou 2 (inferior) contas injetadas. Flechas ciano apontam para contas. (B) Imagens confocal fluorescentes de células de suspensão com diferentes manchas. Canto superior esquerdo: Lap2b-eGFP (membrana nuclear interna, 80 pg/embrião) e H2A-mCherry. Superior direito: GPI-GFP (membrana plasmática, 100 pg/embrião) e DNA-Hoechst (manchado como descrito na seção 2). Inferior esquerdo: MyI12.1-eGFP (linha transgênica) e DNA-Hoechst. Canto inferior direito: Calbryte488 e DNA-Hoechst (manchados como descrito na seção 2). (C) Imagem de microscopia de luz de transmissão de células confinadas com 1 (superior) ou 2 (inferior) contas injetadas. Flechas ciano apontam para contas. Barras de escala = 10 μm. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 6: Estimando a deformação nuclear a partir de filmes de discos giratórios. (A,B) Lapso de tempo de um experimento de recuo do núcleo em (A) uma célula suspensa e (B) uma célula confinada. Barra de escala 10 μm. Instantâneos representativos de um núcleo hoechst rotulado são mostrados 5 s antes, durante e 5 s após o recuo com uma microesfera presa opticamente (ponta de flecha branca). Kymographs ao longo do segmento de recuo (linha vermelha, painel direito). x1 e x2 são os limites distal e proximal (próximo à conta) do núcleo durante o experimento de recuo extraído do ajuste do perfil de intensidade para a Equação 1. (C) Perfis de intensidade ao longo do segmento de recuo para três quadros diferentes (antes, durante e depois do encantamento) e instalados na Equação 1 para avaliar as posições distais, x1 e proximal, x2, das bordas do núcleo. (D) Trajetórias representativas de x1(t) em azul e x2(t) em âmbar durante um experimento de recuo de células suspensas e confinadas (10 μm). As áreas sombreadas indicam o recuo, a distância entre x1 e x2 indica o diâmetro do núcleo. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 7: Processamento de sinal de força. (A) Esquema de uma microesfera aprisionada opticamente deformando o núcleo celular após o recuo. Membrana nuclear e forças ópticas são indicadas pelas flechas negras. A mudança no momento do feixe é indicada pelo Pout do Arqueiro Verde. (B) Trajetória de armadilha (superior) e força (inferior) experimentada pela microesfera aprisionada opticamente durante um experimento repetido de recuo nuclear. (C) Forçar a decadência do relaxamento após o pico de força na profundidade máxima do recuo. Inset mostra um esquema de sólido linear padrão cuja dinâmica aproxima as observações fenomenológicas aqui. (D) Esquerda: logaritmo da força normalizada versus tempo. As áreas sombreadas indicam a porção de dados usada para se encaixar na dupla decadência exponencial (linhas vermelhas). Certo: logaritmo da força normalizada contra o logaritmo do tempo. A área sombreada indica a parte de dados usada para se adequar à lei de poder. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 8: Rotina de recuo de força com deslocamentos de armadilha triangular. (A) Trajetória representativa de x1(t) em azul e x2(t) em âmbar durante um experimento de recuo triangular realizado em uma célula na altura de confinamento de 10 μm. Topo: Posição da armadilha. Meio: Análise de forma de núcleo. A distância entre x1 e x2 indica o diâmetro do núcleo. Inferior: Sinal de força. (B) Posição força x armadilha para oito recuos consecutivos. (C) Evolução da dissipação, derivada da histerese entre a abordagem e a parte de retirada da curva f-d, do núcleo para cada evento de recuo subsequente. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 9. Propriedades nucleares de células em suspensão (superfície adesiva) e confinamento de rotinas trapezoidais. (A) Área projetada do núcleo a partir de células em suspensão e sob confinamento de 10 μm. A barra preta representa a mediana. (B) Rigidez nuclear das células em suspensão e sob confinamento. A barra preta representa a mediana. Valores P derivados do teste kruskal-wallis usando MatLab. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Tabela Suplementar 1: Trajetória trapezoidal definida pelo software pinça óptica. A primeira (segunda) linha é a distância x (y) que a armadilha será deslocada linearmente. Na terceira linha, a duração de um dado passo é definida em segundos. Esta trajetória é composta de sete pontos e corresponde ao trapézio carregado duas vezes contra o núcleo na Figura 7B. Clique aqui para baixar esta Tabela.
Tabela Suplementar 2: Trajetória triangular definida pelo software pinça óptica. Análogo à Tabela 2, esta trajetória é composta por 16 pontos, correspondendo a oito eventos de recuo a uma profundidade de 5 μm e uma velocidade de 2,5 μm/s. Clique aqui para baixar esta Tabela.
Tabela Suplementar 3: Encaixe parâmetros para os dados na Figura 7. IG: palpite inicial. Clique aqui para baixar esta Tabela.
Figura Suplementar S1: Alinhamento do sensor de força óptica e compensação da linha de base momentum. (A) Parada de campo imagem na câmera auxiliar (AUX, Figura 2) através da lente Bertrand. Uma bolha de ar aparece visível no óleo de imersão, que não é visível através da ocular. (B) Caminho óptico limpo. Para um alinhamento preciso, abra a parada do campo e faça com que coincida com o cone de luz NA = 1,2. (C) Imagem do plano amostral. O quadrado vermelho indica a área de trabalho OT. Barra de escala: 20 μm. (D) Potência da armadilha medida em todo o FOV, ao longo de setas duplas brancas indicadas em C. Em vermelho, a variação de potência da armadilha quando nenhuma correção é aplicada. Em azul, o poder da armadilha corrigido sobre todo o campo de visão. (E) Componente X da linha de base momentum ao longo da mesma faixa. Em vermelho, traço não corrigido. Em azul, traço corrigido para poder de armadilha. Em verde, traço corrigido para a linha de base momentum usando Compensação Global no software do fabricante. (F) Mesmo que em E, para o componente Y. Observe que, em operação normal, os componentes sombreados são usados para medições mecânicas e de força, por exemplo, componente x força durante o movimento ao longo da coordenada x e o componente de força y durante o movimento ao longo do eixo y. Depois que todas as correções forem implementadas, um ruído RMSD de <0,5 pN é obtido. Clique aqui para baixar este Arquivo.
Figura Suplementar S2: Uma rotina fracassada devido a armadilhas fracas. (A) Kymograph mostrando um recuo do núcleo de uma rotina falha. Apenas deformações curtas e transitórias são visíveis devido a uma fuga da conta da armadilha. É importante ressaltar que o laser de trapping ainda se move sem contas para completar a trajetória predefinida (linha pontilhada verde). Barra de escala = 10 μm. (B) Topo: Posição da armadilha versus tempo. Meio: Resultado de rastreamento de borda da borda do núcleo proximal e distal recuado. Observe que a borda distal não está se movendo sem o recuo como comumente observado para rotinas completas em células isoladas em substratos adesivos. Inferior: Força versus tempo mostrando a perda da microsfera indicada por uma redução no ruído térmico e uma queda repentina para zero de força. Clique aqui para baixar este Arquivo.
Figura Suplementar S3: Sobrevivência de embriões injetados. Os embriões injetados com contas de 1 μm e 100 pg/embrião de mRNA em concentrações descritas no protocolo foram comparados a embriões não injetados e não mostram diferenças significativas 24 h após a fertilização. Desvio médio e padrão de três experimentos independentes com N > 21 embriões por condição para cada experimento. Clique aqui para baixar este Arquivo.
Discussão
Neste protocolo, descrevemos um método único para interrogar as propriedades mecânicas do núcleo celular dentro das células vivas. Diferente de outras técnicas de espectroscopia de força, a captura óptica não invasiva nos permitiu desacoplar a contribuição da membrana celular e do citoesqueleto da rigidez nuclear da célula. É importante ressaltar que a micromanipulação óptica é compatível com microscopia multimodal, que permitirá ao experimentador estudar diferentes processos envolvidos na mecanobiologia nuclear celular. Como resultado representativo, usamos a coloração DNA-Hoechst para medir a deformação do núcleo após o recuo realizado por forças da ordem de várias centenas de picoNewton.
Aplicações potenciais de nosso método além dos exemplos descritos neste protocolo
A possibilidade de extrair informações mecânicas quantitativas de medições dentro de células vivas sem perturbações externas permite uma infinidade de oportunidades sem precedentes que estão apenas começando a ser exploradas. Assim, o protocolo apresentado de nossa plataforma óptica de micromanipulação pode ser estendido a experimentos mais complexos com grande versatilidade. Os defletores acousto-ópticos (AOD) podem gerar múltiplas armadilhas ópticas para medições de força síncrona em diferentes locais de células, bem como podem ser usados para microrrehetologia ativa em uma ampla faixa de frequência51,61. Como foi mencionado, a resposta da força após o recuo pode superar a força máxima de captura, levando a uma fuga da conta da armadilha óptica. Neste caso, um feedback de força pode ser configurado com o AOD para fixar a força óptica. Em suma, múltiplas abordagens microrrehelógicas, como o relaxamento do estresse descrito neste protocolo, mas também a microrrehetologia ativa ou conformidade com rastejantes, podem ser obtidas experimentalmente com esta plataforma e analisadas minuciosamente por novos pacotes de software61,62,63,64,65 . Além disso, a aplicação de forças não se limita ao núcleo, mas poderia, em princípio, ser realizada para medir diversas estruturas intracelulares e em tecidos complexos como demonstrado para prender glóbulos vermelhos fluindo dentro de vasos sanguíneos intactos66,67 ou prender e deformar cloroplastos e mitocôndrias68 . A calibração do momento da luz é independente da forma e tamanho do objeto preso, permitindo assim medições de força direta em qualquer sonda de força com forma arbitrária38,39. O uso de microesferas injetadas nos permitiu aplicar altas forças no núcleo com poder laser relativamente baixo em comparação com a manipulação direta das estruturas celulares69,70,71. No entanto, dada uma alta diferença de índice refrativo suficiente, nenhuma sonda de força aplicada externamente é necessária e organelas intracelulares podem ser manipuladas diretamente sem contas injetadas (observações inéditas e referência70).
Modificações potenciais do nosso método para estender as aplicações
Diferentes tamanhos de microesferas podem ser injetados dependendo do experimento, mas os controles relativos devem ser feitos. Por exemplo, estudar células em estágios posteriores contas menores podem ser injetadas. Isso reduzirá a força máxima que pode ser exercida pela armadilha óptica (como mostrado em referência55). Contas maiores podem ser injetadas para exercer forças mais altas, mas estas podem afetar o desenvolvimento de embriões dependendo de seu tamanho ou estágio de interesse. Em experimentos onde a injeção de microesferas não é uma opção, várias organelas que apresentam diferenças de índices refrativos em comparação com a do citoplasma ainda podem ser manipuladas opticamente, dando origem a forças ópticas mensuráveis a partir de mudanças de momento leve42. Como mencionado acima, esses métodos têm sido empregados por Bambardekar et al. para deformar as junções celulares no embrião Drosophila70. Da mesma forma, o núcleo da célula tem um índice de refração menor do que o médio circundante44, o que permite o recuo livre de contas (observações inéditas e referência72) mesmo com menor força de captura. Assim, o núcleo não pode ser preso facilmente e escapa da armadilha.
O espaçador PDMS revestido de spin é fabricado através de um método conveniente e rápido, mas pode estar fora de alcance para laboratórios sem acesso a uma instalação de micro/nanofabização ou laboratórios de engenharia. Assim, o espaçador pode ser facilmente montado a partir de fita de laboratório ou parafilme (passo 4). O protocolo também pode ser adaptado pela fabricação de canais microfluidos que automatizam a entrega de células únicas em poços de medição predefinidos ou em uma câmara com altura definida para estimar o efeito de confinamento dentro do mesmo espécime. No entanto, tais dispositivos microfluidos devem ser projetados para que se encaixem no espaço entre o objetivo do microscópio e a lente coletora do sensor de força óptica, de cerca de 2 mm (ver passo 3). Observe que o sensor de força óptica deve ser posicionado na altura apropriada para que nenhuma aberração óptica de desfoco afete a medição do momento do fóton.
Outras modificações podem incluir a mudança de repórteres biológicos. Descobrimos que a fluorescência hoechst sangra espectralmente no canal GFP e, assim, favorecemos a combinação com histona mCherry-tagged como um marcador nuclear para medição simultânea em dois canais fluorescentes. Alternativamente, a deformação nuclear pode ser facilmente rastreada com um rótulo direcionado à membrana nuclear interna, como Lap2b-GFP (Figura 2).
O recuo no núcleo celular era da ordem de 2-3 mícrons, que pudemos medir com precisão pela análise de imagem da microscopia confocal de disco giratório limitada de difração. Para o caso de núcleos mais rígidos ou forças menores, o recuo será pouco mensurável usando esta abordagem. No entanto, as pinças ópticas calibradas por força absoluta também podem ser calibradas para medições de posição da conta presa in situ usando interferometria BFP com precisão de nanômetro51. Usando esta abordagem, o sinal de tensão e o sensor de força óptica podem ser traduzidos para a posição da sonda presa através do parâmetro β [nm/V], enquanto o parâmetro invariante α [pN/V] produz valores de força através da calibração de impulso de luz acima mencionada41 (veja abaixo para detalhes).
Solucionando problemas
Descobrimos que os seguintes desafios poderiam ocorrer durante o experimento:
Nenhuma armadilha estável é formada e a microesfera escapa facilmente
Qualquer sujeira no objetivo do microscópio ou uma coleira de correção desalinhada pode levar a uma falha de uma armadilha estável. Se uma solução imediata não for encontrada, meça a função de propagação de pontos da lente objetiva. Se o espécime de interesse estiver no interior de um tecido opticamente denso, o foco do laser pode experimentar severas aberrações ópticas que levam a armadilhas instáveis (este efeito geralmente é insignificante em células isoladas, mas torna-se mais evidente em tecidos mais grossos). Para a alta rigidez, a força restauradora do núcleo poderia exceder a força de fuga da armadilha, de tal forma que a microesfera se perde e a rotina de recuo falha. Inicialmente, a borda da membrana nuclear proximal à armadilha óptica fica quase sem recuada (Figura S2A). Quando isso ocorre, o laser de trapping não é mais afetado pela força e pelo movimento browniano, o que leva a uma queda de força para zero e uma diminuição do ruído do sinal (Figura S2B). Caso isso aconteça, a potência do laser pode ser aumentada para ter uma armadilha mais forte, a amplitude da trajetória trapezoidal empurrando a conta para o núcleo pode ser reduzida, ou a posição inicial da microesfera presa pode ser definida mais longe do núcleo.
A célula está se movendo durante a estimulação
Se as células não estiverem suficientemente ligadas, a armadilha gradiente óptica moverá as células enquanto executa a rotina de recuo intracelular, de tal forma que as forças e a mecânica subjacente do núcleo sejam artefatos. Para evitar o deslocamento de toda a célula, recomendamos aumentar a concentração de moléculas de adesão celular na superfície, por exemplo,.
Compensação inicial de impulso
Se uma rotina inicial de compensação de impulso não estiver disponível na plataforma OTs (etapa 6.5), um sinal de linha de base artificial e independente da força precisa ser corrigido. Isso é visível como uma inclinação na curva de força mesmo sem contas presas (Figura S1E). Para fazer a correção, a mesma trajetória precisa ser realizada sem uma conta, fora da cela exatamente na mesma posição. Para isso, mova a célula para longe da armadilha usando o controle do palco. Como referência, o deslocamento de força muda 5 pN em todo o FOV a 200 mW em nosso sistema; assim, torna-se insignificante para trajetórias curtas. Alternativamente, um estágio de varredura piezo pode ser usado para mover as células na amostra, deixando a posição do laser constante.
Etapas críticas do protocolo apresentado
Microesferas devem ser injetadas no estágio direito de 1 célula para garantir a distribuição máxima sobre o embrião. As contas não devem ser fluorescentes para que nenhuma luz vaze nos canais fluorescentes usados para imagens. Por exemplo, mesmo as contas típicas vermelha-fluorescentes são claramente visíveis no canal azul usado para a imagem do núcleo celular após a coloração de Hoechst devido ao seu brilho (excitação: 405 nm; emissão: 445 nm). A fixação estável da célula ao substrato é fundamental para evitar o deslocamento lateral durante a rotina de recuo. Se a célula se move durante a rotina, as forças são subestimadas. Caso isso aconteça com frequência, otimize o protocolo de fixação. Para células de cultura tecidual, outras proteínas de adesão celular, como fibronectina, colágeno ou poli-L-lysina levam a um apego satisfatório (observações inéditas). Durante o confinamento, as células são submetidas a um estresse mecânico repentino e severo. Isso pode causar danos às células e, frequentemente, o experimentador encontrará células estouradas se o procedimento não for realizado cuidadosamente. Além disso, se a altura do confinamento for muito pequena, todas as células sofrerão de quebra nuclear ou danos irreversíveis. Para amenizar isso, abaixe a tampa superior mais lentamente e/ou aumente o espaçamento entre o deslizamento de tampas.
Limitações da técnica e sugestões para superá-las
Uma clara limitação da técnica é a penetração da luz laser em seções profundas do tecido, o que leva a aberrações e armadilhas instáveis. Assim, um limite menor de profundidade de penetração depende da clareza da amostra, da correção da aberração que pode ser empregada73 e da potência laser aplicada. Deve-se levar em conta que uma maior potência laser leva à excitação térmica da amostra nas proximidades da microesfera. No entanto, o aquecimento da amostra originada pelo ponto laser de comprimento de onda de 1064 nm é minimizado para evitar estresse plausível relacionado ao calor em nossas amostras biológicas74.
Outra limitação é a força máxima que pode ser medida. Embora a detecção direta de impulso de luz permita medições de força muito além do regime de resposta linear da armadilha óptica40,41, a força máxima aplicada está na ordem de algumas centenas de picoNewtons. Isso é limitado pela potência do laser e pelo limiar de dano consequente do material biológico macio e pelas diferenças de índice refrativo, que normalmente não são maiores que 0,1 ou 0,344. Vários métodos foram propostos para aumentar o limite de detecção de força, por exemplo, usando luz estruturada75, microesferas revestidas anti-reflexivas76, partículas de índice de alta refração77 ou pontos quânticos altamente dopados78.
Os OTs podem ser usados para medições de posição em escala de nanômetros através da interferometria BFP, de modo que a posição da conta dentro da armadilha seja Δx = β Sx, onde Sx é o sinal de tensão do sensor, e β [μm/V] pode ser calibrado em tempo real seguindo diferentes protocolos35,54. Para um sensor de força óptica, pode-se provar que o fator de conversão invariante de tensão para força α [pN/V] se relaciona diretamente com β e a rigidez da armadilha, k [pN/μm], através de α = kβ 37) Em experimentos com deslocamentos de contas que são muito pequenos para serem detectados a partir de imagens ópticas, essa estratégia pode ser usada para complementar as medições de força com pequenas detecção de posição. Um exemplo é a aplicação das rotinas experimentais apresentadas aqui em núcleos muito rígidos, para os quais forças a potências laser razoáveis (200-500 mW) não são suficientes para induzir valores de recuo grandes o suficiente. Nesse caso, a conta precisa ser trazida em contato com o núcleo e a rigidez de trapping deve ser calibrada antes da medição (etapa 8.6). O recuo do núcleo em função da força pode ser indiretamente determinado como:
d = xtrap - F/k
onde xtrap é a posição da armadilha. Diferente do fator de impulso de luz invariante α [pN/V], o fator β [μm/V] precisa ser calibrado antes de cada experimento, uma vez que depende de muitas variáveis locais determinando a dinâmica de captura, como o tamanho da partícula, tamanho do ponto de armadilha óptico e índices relativos de refração.
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
MK reconhece apoio financeiro do Ministério da Economia e Competitividade espanhola através do Plano Nacional (PGC2018-097882-A-I00), FEDER (EQC2018-005048-P), Severo Ochoa programa para Centros de Excelência em P&D (CEX2019-000910-S; RYC-2016-21062), da Fundació Privada Cellex, Fundació Mir-Puig, e da Generalitat de Catalunya através do programa CERCA e Pesquisa (2017 SGR 1012), além de financiamento através da ERC (MechanoSystems) e HFSP (CDA00023/2018). A V.R. reconhece o apoio do Ministério da Ciência e Inovação espanhol à parceria da EMBL, do Centro de Excelencia Severo Ochoa, do Plano Nacional da MINECO (BFU2017-86296-P, PID2020-117011GB-I00) e da Generalitat de Catalunya (CERCA). A V.V. reconhece o apoio do Programa de Doutorado icfostepstone financiado pelo programa de pesquisa e inovação Horizon 2020 da União Europeia sob o acordo de subvenção de Marie Skłodowska-Curie 665884. Agradecemos a Arnau Farré pela leitura crítica do manuscrito; Maria Marsal para a ajuda com a imagem e montagem do embrião de 24 hpf e; Senda Jiménez-Delgado para apoio com micronjeções de zebrafish.
Materiais
| Name | Company | Catalog Number | Comments |
| #1.5 22 mm cover glasses | Ted Pella | 260148 | |
| #1.5 22x60 mm Coverglasses | Ted Pella | 260152 | |
| #1.5H glass bottom dishes | Willco | GWST-5040 | |
| 10-um beads | Supelco | 72986 | |
| 1-mm glass capillaries | Harvard Apparatus | 30-0020 GC100F-15 | |
| 1-um polystyrene microbeads | Sigma | 89904 | |
| 1-um red-fluorescent beads | ThermoFisher | F8816 | |
| Agar | ThermoFisher | 16500500 | |
| Aqcuisition cameras sCMOS | Andor | Sona-4BV11-UNI | |
| Auxiliary camera (Figure 3, AUX) | Blackfly, FLIR | BFS-U3-200S6M-C | |
| Calbryte 520 | AAT Bioquest | 520 AM | |
| Centrifuge | Eppendorf | 5453000011 | |
| Concanavalin A | Sigma | C5275 | |
| DMEM | Sigma | D2906 | |
| DNA-Hoechst | ThermoFisher | 33342 | |
| Double scotch tape | Biesse Adesivi | ||
| E3 | 5 mM NaCl. 0.17 mM KCl. 0.33 mM CaCl2. 0.33 mM MgSO4 | ||
| Eclipse Ti2 | Nikon | ||
| Forceps | Fine Science Tools | 11252-20 | |
| GPI-GFP | |||
| H2A-mCh | |||
| Image acquisition software | Fusion-Andor | ||
| Immersion Oil | Cargille | Type B: 16484 | |
| IR protection googles | Thorlabs | LG1 | |
| Lap2b-eGFP | |||
| Micro loader pipette | Eppendorf | GELoader | |
| Microinjector | World Precision Instruments | SYS-PV820 | |
| MicroManager 2.0 | |||
| Micromiter slide | ID5243 GXMGRAT-5 5mm/100 divisions | ||
| Mineral oil | Sigma | M3616 | |
| Motorized stage | ASI | ||
| Needle puller | Sutter instrument Co. | Model P-97 | |
| Optical tweezers platform | Impetux Optics | Sensocell | |
| OTs software (LightAce) | Impetux Optics | ||
| PDMS | Sigma | Sylgard 184 | |
| PDMS Curing agent | Sigma | Sylgard 184 | |
| Post processing software (Matlab) | Mathworks | ||
| RNAse free water | Thermofisher | AM9937 | |
| Short-pass dichroic mirror (Figure 3, IR-F) | Semrock | FF01-950/SP-25 | |
| Spin-coater | Specialty Coating Systems | Spincoat G3P-8 | |
| Spinning-disk confocal microscope | Andor | DragonFly 502 | |
| Stereomicroscope | Leica M80 | ||
| Triangular microinjection mold | Adaptive Science Tools | TU1 | |
| Universal oven | Memmert | UNB 200 | |
| Water immersion objective | Nikon | MRD07602 |
Referências
- Chan, C. J., Heisenberg, C. P., Hiiragi, T. Coordination of Morphogenesis and Cell-Fate Specification in Development. Current Biology. 27 (18), 1024-1035 (2017).
- Heller, E., Fuchs, E. Tissue patterning and cellular mechanics. Journal of Cell Biology. 211 (2), 219-231 (2015).
- Heisenberg, C. P., Bellaïche, Y. Forces in tissue morphogenesis and patterning. Cell. 153 (5), 948-962 (2013).
- Petridou, N. I., Spiró, Z., Heisenberg, C. P. Multiscale force sensing in development. Nature Cell Biology. 19 (6), 581-588 (2017).
- Krieg, M., et al. Tensile forces govern germ-layer organization in zebrafish. Nature Cell Biology. 10 (4), 429-436 (2008).
- Ruprecht, V., et al. Cortical contractility triggers a stochastic switch to fast amoeboid cell motility. Cell. 160 (4), 673-685 (2015).
- Shellard, A., Mayor, R. Supracellular migration - Beyond collective cell migration. Journal of Cell Science. 132 (8), (2019).
- Mongera, A., et al. A fluid-to-solid jamming transition underlies vertebrate body axis elongation. Nature. 561 (7723), 401-405 (2018).
- Atia, L., et al. Geometric constraints during epithelial jamming. Nature Physics. 14 (6), 613-620 (2018).
- Turlier, H., Maître, J. -. L. Mechanics of tissue compaction. Seminars in Cell & Developmental Biology. 47-48, 110-117 (2015).
- Ladoux, B., Mège, R. M. Mechanobiology of collective cell behaviours. Nature Reviews Molecular Cell Biology. 18 (12), 743-757 (2017).
- Venturini, V., et al. The nucleus measures shape changes for cellular proprioception to control dynamic cell behavior. Science. 370 (6514), (2020).
- Charras, G., Sahai, E. Physical influences of the extracellular environment on cell migration. Nature Reviews Molecular Cell Biology. 15 (12), 813-824 (2014).
- Kirby, T. J., Lammerding, J. Emerging views of the nucleus as a cellular mechanosensor. Nature Cell Biology. 20 (4), 373-381 (2018).
- Lee, H. P., et al. The nuclear piston activates mechanosensitive ion channels to generate cell migration paths in confining microenvironments. Science Advances. 7 (2), (2021).
- Friedl, P., Wolf, K., Lammerding, J. Nuclear mechanics during cell migration. Current Opinion in Cell Biology. 23 (1), 55-64 (2011).
- Versaevel, M., Riaz, M., Grevesse, T., Gabriele, S. Cell confinement: Putting the squeeze on the nucleus. Soft Matter. 9 (29), 6665-6676 (2013).
- Zuela-Sopilniak, N., et al. Measuring nucleus mechanics within a living multicellular organism: Physical decoupling and attenuated recovery rate are physiological protective mechanisms of the cell nucleus under high mechanical load. Molecular Biology of the Cell. 31 (17), 1943-1950 (2020).
- Kim, D. H., Wirtz, D. Cytoskeletal tension induces the polarized architecture of the nucleus. Biomaterials. 48, 161-172 (2015).
- Lomakin, A. J., et al. The nucleus acts as a ruler tailoring cell responses to spatial constraints. Science. 370 (6514), (2020).
- Hampoelz, B., et al. Microtubule-induced nuclear envelope fluctuations control chromatin dynamics in Drosophila embryos. Development. 138 (16), 3377-3386 (2011).
- Heo, S. J., et al. Differentiation alters stem cell nuclear architecture, mechanics, and mechano-sensitivity. eLife. 5, 1-21 (2016).
- Cosgrove, B. D., et al. Nuclear envelope wrinkling predicts mesenchymal progenitor cell mechano-response in 2D and 3D microenvironments. Biomaterials. 270, 120662 (2021).
- Liu, H., et al. In situ mechanical characterization of the cell nucleus by atomic force microscopy. ACS Nano. 8 (4), 3821-3828 (2014).
- Hobson, C. M., et al. Correlating nuclear morphology and external force with combined atomic force microscopy and light sheet imaging separates roles of chromatin and lamin A/C in nuclear mechanics. Molecular Biology of the Cell. 31 (16), 1788-1801 (2020).
- Pajerowski, J. D., Dahl, K. N., Zhong, F. L., Sammak, P. J., Discher, D. E. Physical plasticity of the nucleus in stem cell differentiation. Proceedings of the National Academy of Sciences of the United States of America. 104 (40), 15619-15624 (2007).
- Rowat, A. C., Lammerding, J., Ipsen, J. H. Mechanical properties of the cell nucleus and the effect of emerin deficiency. Biophysical Journal. 91 (12), 4649-4664 (2006).
- Davidson, P. M., et al. High-throughput microfluidic micropipette aspiration device to probe time-scale dependent nuclear mechanics in intact cells. Lab on a Chip. 19 (21), 3652-3663 (2019).
- Lombardi, M., Zwerger, M., Lammerding, J. Biophysical assays to probe the mechanical properties of the interphase cell nucleus: Substrate strain application and microneedle manipulation. Journal of Visualized Experiments: JoVE. (55), (2011).
- Luo, T., Mohan, K., Iglesias, P. A., Robinson, D. N. Molecular mechanisms of cellular mechanosensing. Nature Materials. 12 (11), 1064-1071 (2013).
- Dahl, K. N., Engler, A. J., Pajerowski, J. D., Discher, D. E. Power-law rheology of isolated nuclei with deformation mapping of nuclear substructures. Biophysical Journal. 89 (4), 2855-2864 (2005).
- Guilluy, C., et al. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nature Cell Biology. 16 (4), 376-381 (2014).
- Bustamante, C. J., Wang, M. D. Optical tweezers in single-molecule biophysics. Nature Reviews Methods Primers. , 1-29 (2021).
- Svoboda, K., Block, S. M. Force and velocity measured for single kinesin molecules. Cell. 77 (5), 773-784 (1994).
- Berg-Sørensen, K., Flyvbjerg, H. Power spectrum analysis for optical tweezers. Review of Scientific Instruments. 75 (3), 594-612 (2004).
- Smith, S. B., Cui, Y., Bustamante, C. Optical-trap force transducer that operates by direct measurement of light momentum. Methods in Enzymology. 361 (1994), 134-162 (2003).
- Farré, A., Montes-Usategui, M. A force detection technique for single-beam optical traps based on direct measurement of light momentum changes. Optics Express. 18 (11), 11955 (2010).
- Català, F., Marsà, F., Montes-Usategui, M., Farré, A., Martín-Badosa, E. Extending calibration-free force measurements to optically-trapped rod-shaped samples. Scientific Reports. 7, 1-10 (2017).
- Bui, A. A. M., et al. Calibration of force detection for arbitrarily shaped particles in optical tweezers. Scientific Reports. 8 (1), 1-12 (2018).
- Farré, A., Marsà, F., Montes-Usategui, M. Beyond the hookean spring model: Direct measurement of optical forces through light momentum changes. Methods in Molecular Biology. 1486, (2017).
- Farré, A., Marsà, F., Montes-Usategui, M. Optimized back-focal-plane interferometry directly measures forces of optically trapped particles. Optics Express. 20 (11), 12270 (2012).
- Jun, Y., Tripathy, S. K., Narayanareddy, B. R. J., Mattson-Hoss, M. K., Gross, S. P. Calibration of optical tweezers for in vivo force measurements: How do different approaches compare. Biophysical Journal. 107 (6), 1474-1484 (2014).
- Mas, J., Farré, A., Sancho-Parramon, J., Martín-Badosa, E., Montes-Usategui, M. Force measurements with optical tweezers inside living cells. Optical Trapping and Optical Micromanipulation XI. 9164, (2014).
- Schürmann, M., Scholze, J., Müller, P., Guck, J., Chan, C. J. Cell nuclei have lower refractive index and mass density than cytoplasm. Journal of Biophotonics. 9 (10), 1068-1076 (2016).
- Rosen, J. N., Sweeney, M. F., Mably, J. D. Microinjection of zebrafish embryos to analyze gene function. Journal of Visualized Experiments: JoVE. (25), (2009).
- Westerfield, M. . The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish. (Danio rerio), 5th Edition. , (2007).
- Schubert, R., et al. Assay for characterizing the recovery of vertebrate cells for adhesion measurements by single-cell force spectroscopy. FEBS Letters. 588 (19), 3639-3648 (2014).
- Koschwanez, J. H., Carlson, R. H., Meldrum, D. R. Thin PDMS films using long spin times or tert-butyl alcohol as a solvent. PLoS One. 4 (2), 2-6 (2009).
- Das, R., et al. Mechanical stretch inhibition sensitizes proprioceptors to compressive stresses. bioRxiv. , (2021).
- Chardès, C., Clement, R., Blanc, O., Lenne, P. F. Probing cell mechanics with bead-free optical tweezers in the drosophila embryo. Journal of Visualized Experiments: JoVE. (141), (2018).
- Staunton, J. R., Blehm, B., Devine, A., Tanner, K. In situ calibration of position detection in an optical trap for active microrheology in viscous materials. Optics Express. 25 (3), 1746 (2017).
- Bola, R., Treptow, D., Marzoa, A., Montes-Usategui, M., Martin-Badosa, E. Acousto-holographic optical tweezers. Optics Letters. 45 (10), 2938-2941 (2020).
- Thalhammer, G., Obmascher, L., Ritsch-Marte, M. Direct measurement of axial optical forces. Optics Express. 23 (5), 6112 (2015).
- Vermeulen, K. C., et al. Calibrating bead displacements in optical tweezers using acousto-optic deflectors. Review of Scientific Instruments. 77 (1), 1-6 (2006).
- Dzementsei, A., Barooji, Y. F., Ober, E. A., Oddershede, L. B. Foregut organ progenitors and their niche display distinct viscoelastic properties in vivo during early morphogenesis stages. bioRxiv. , 1-35 (2021).
- Behrndt, M., et al. Forces driving epithelial spreading in zebrafish gastrulation. Science. 338 (6104), 257-260 (2012).
- Krieg, M., et al. Atomic force microscopy-based mechanobiology. Nature Reviews Physics. , (2018).
- A-Hassan, E., et al. Relative microelastic mapping of living cells by atomic force microscopy. Biophysical Journal. 74 (3), 1564-1578 (1998).
- Khalilgharibi, N., et al. Stress relaxation in epithelial monolayers is controlled by the actomyosin cortex. Nature Physics. 15, (2019).
- Crick, S. L., Yin, F. C. Assessing micromechanical properties of cells with atomic force microscopy: importance of the contact point. Biomechanics and Modeling in Mechanobiology. 6 (3), 199-210 (2007).
- Hurst, S., Vos, B. E., Betz, T. Intracellular softening and fluidification reveals a mechanical switch of cytoskeletal material contributions during division. bioRxiv. , 425761 (2021).
- Kaplan, J. L., Bonfanti, A., Kabla, A. RHEOS.jl - A Julia package for rheology data analysis. arXiv. 4, 1-5 (2020).
- Bonfanti, A., Kaplan, J. L., Charras, G., Kabla, A. Fractional viscoelastic models for power-law materials. Soft Matter. 16 (26), 6002-6020 (2020).
- Rivas-Barbosa, R., Escobedo-Sánchez, M. A., Tassieri, M., Laurati, M. i-Rheo: determining the linear viscoelastic moduli of colloidal dispersions from step-stress measurements. Physical Chemistry Chemical Physics: PCCP. 22 (7), 3839-3848 (2020).
- Tassieri, M., et al. i-Rheo: Measuring the materials' linear viscoelastic properties "in a step". Journal of Rheology. 60 (4), 649-660 (2016).
- Zhong, M. C., Wei, X. B., Zhou, J. H., Wang, Z. Q., Li, Y. M. Trapping red blood cells in living animals using optical tweezers. Nature Communications. 4, 1767-1768 (2013).
- Harlepp, S., Thalmann, F., Follain, G., Goetz, J. G. Hemodynamic forces can be accurately measured in vivo with optical tweezers. Molecular Biology of the Cell. 28 (23), 3252-3260 (2017).
- Bayoudh, S., Mehta, M., Rubinsztein-Dunlop, H., Heckenberg, N. R., Critchley, C. Micromanipulation of chloroplasts using optical tweezers. Journal of Microscopy. 203 (2), 214-222 (2001).
- Favre-Bulle, I. A., Stilgoe, A. B., Rubinsztein-Dunlop, H., Scott, E. K. Optical trapping of otoliths drives vestibular behaviours in larval zebrafish. Nature Communications. 8 (1), 630 (2017).
- Bambardekar, K., Clément, R., Blanc, O., Chardès, C., Lenne, P. F. Direct laser manipulation reveals the mechanics of cell contacts in vivo. Proceedings of the National Academy of Sciences of the United States of America. 112 (5), 1416-1421 (2015).
- Ferro, V., Chuai, M., McGloin, D., Weijer, C. J. Measurement of junctional tension in epithelial cells at the onset of primitive streak formation in the chick embryo via non-destructive optical manipulation. Development (Cambridge). 147 (3), (2020).
- Hörner, F., et al. Holographic optical tweezers-based in vivo manipulations in zebrafish embryos. Journal of Biophotonics. 10 (11), 1492-1501 (2017).
- Zhong, M. -. C., Wang, Z. -. Q., Li, Y. -. M. Aberration compensation for optical trapping of cells within living mice. Applied Optics. 56 (7), 1972 (2017).
- Català, F., Marsà, F., Montes-Usategui, M., Farré, A., Martín-Badosa, E. Influence of experimental parameters on the laser heating of an optical trap. Scientific Reports. 7 (1), 1-9 (2017).
- Taylor, M. A., Waleed, M., Stilgoe, A. B., Rubinsztein-Dunlop, H., Bowen, W. P. Enhanced optical trapping via structured scattering. Nature Photonics. 9 (10), 669-673 (2015).
- Bormuth, V., et al. Optical trapping of coated microspheres. Optics Express. 16 (18), 13831-13844 (2008).
- Sudhakar, S., et al. Germanium nanospheres for ultraresolution picotensiometry of kinesin motors. Science. 371 (6530), (2021).
- Shan, X., et al. Optical tweezers beyond refractive index mismatch using highly doped upconversion nanoparticles. Nature Nanotechnology. 16 (5), 531-537 (2021).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoExplore Mais Artigos
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados