
Method Article
Ein standardisiertes Verfahren zur Überwachung schädlicher Algenblüten in Chile durch Metabarcoding-Analyse
In diesem Artikel
Zusammenfassung
Dieses Protokoll führt Schritte der Metabarcoding-Analyse ein, die auf 16S rRNA- und 18S rRNA-Gene abzielen, um schädliche Algenblüten und ihr assoziiertes Mikrobiom in Meerwasserproben zu überwachen. Es ist ein leistungsfähiges molekulares Werkzeug, erfordert aber mehrere Verfahren, die hier Schritt für Schritt visuell erklärt werden.
Zusammenfassung
Das Monitoring schädlicher Algenblüten (HABs) wurde weltweit durchgeführt, und Chile, ein für seine Fischerei und Aquakultur bekanntes Land, nutzt zu diesem Zweck seit Jahrzehnten intensiv Mikroskopie- und Toxinanalysen. Molekularbiologische Methoden wie Hochdurchsatz-DNA-Sequenzierung und bakterielle Assemblage-basierte Ansätze werden gerade erst im chilenischen HAB-Monitoring eingeführt, und die Verfahren sind noch nicht standardisiert. Hier werden schrittweise 16S rRNA und 18S rRNA Metabarcoding Analysen zur Überwachung chilenischer HABs vorgestellt. Nach einer neueren Hypothese spielt die algen-bakterielle mutualistische Assoziation eine kritische synergetische oder antagonistische Beziehung, die für die Initiierung, Aufrechterhaltung und Regression der Blüte verantwortlich ist. Daher kann die Überwachung von HAB aus algenbakterieller Perspektive ein breiteres Verständnis der HAB-Mechanismen und die Grundlage für die Frühwarnung liefern. Die Metabarcoding-Analyse ist eines der am besten geeigneten molekularbasierten Werkzeuge für diesen Zweck, da sie massive algenbakterielle taxonomische Informationen in einer Probe nachweisen kann. Die visuellen Verfahren der Probenahme bis zur Metabarcodierungsanalyse hierin enthalten spezifische Anweisungen, die darauf abzielen, Fehler zu reduzieren und zuverlässige Daten zu sammeln.
Einleitung
Es ist bekannt, dass viele marine Phytoplanktonarten endogene Toxine produzieren, und wenn sich diese Arten in ausreichender Anzahl ansammeln, sind sie schädlich für die Meeresumwelt. Solche schädlichen Algenblüten (HABs) werden heute an den Küsten der meisten Kontinente der Welt beobachtet1. Toxisches Phytoplankton sammelt sich zuerst im Muschelgewebe an und führt bei der Verdauung zu Krankheit und Tod in höheren trophischen Ebenen von Organismen, einschließlich des Menschen. In der Folge haben diese Ereignisse schwerwiegende Auswirkungen auf die lokale Wirtschaft, die Sozioökonomie und die öffentliche Gesundheit2. Der Schaden für die Weltwirtschaft, der durch HABs verursacht wird, wird auf Millionen bis Milliarden von Dollar pro Jahr geschätzt3. Chile ist eines von vielen Ländern, die unter häufigen HABs leiden.
Chile ist ein Land mit langem Land, das sich über 4.300 km nördlich und südlich erstreckt. Das langgestreckte westliche Land blickt auf den Pazifischen Ozean, was natürlich die Chancen erhöht, dass Chile HABs erlebt. Vor allem im Süden Chiles gibt es viele weltberühmte Lachs-Aquakulturen, und jedes Mal, wenn ein HAB in der Region auftritt, führen die Algentoxine dazu, dass massiver Zuchtlachse krank wird undstirbt 4,5,6. In Chile war das Jahr, in dem die HAB die Wirtschaft am härtesten traf, 2016 mit einem geschätzten jährlichen Verlust von 800 Millionen US-Dollar7,8. Die verursachenden toxischen Algenarten variieren je nach Jahr und Standort. Für den Fall 2016 verursachte ein Komplex von Alexandrium catanella und Pseudochattonella verruclosa eine weit verbreitete HAB in den meisten Teilen der südlichen chilenischen Küste7,8. Die jüngste HAB in Chile trat mit der ursächlichen Algenart Heterosigma Akashiwo im März 2021 in Camanchaca im Süden Chiles auf, wo das Gebiet über eine große Lachsfarm verfügt.
Chile führt die Küstenüberwachung seit vielen Jahren mit zwei Hauptmethoden durch; Beobachtung des Meerwassers mit Mikroskopen zur regelmäßigen Identifizierung toxischer Algenarten und Messung des Toxingehalts in Schalentieren durch biochemische Assays9. Die Früherkennung von toxischen Algen- und Toxinwerten in Schalentieren verhindert HABs nicht; Diese Analysen können jedoch sofortige Gegenmaßnahmen auslösen und Schäden für lokale Gemeinschaften reduzieren. Um die Wirksamkeit dieser Strategie weiter zu stärken, wurde kürzlich unser konventionelles chilenisches HAB-Monitoringprogramm um eine molekularbasierte Analysemethode zum Nachweis von Algen und verwandten Bakteriengemeinschaften in Meerwasserproben erweitert. Insbesondere wurde eine Massive Parallel Sequencing-Methode unter Verwendung von Metabarcoding ausgewählt, die auf 16S rRNA- und 18S rRNA-Gene abzielt. Obwohl diese Technik komplizierte Verfahren und teure Maschinen und Reagenzien erfordert, handelt es sich um eine fortschrittliche Technologie, die Tausende von Algen- und Bakteriengattungen / -arten in einer Meerwasserprobe gleichzeitig nachweisen kann.
Es wird spekuliert, dass die Ursachen von HAB vielfältig sind, wie Temperatur und Jahreszeit, aber es ist unmöglich, sie zu verallgemeinern. Dies liegt daran, dass HAB-Arten und -Häufigkeiten von der Region und den raumzeitlichen Bedingungen abhängen, einschließlich natürlicher Phänomene wie geografische Einzigartigkeit, aufkeimende Nährstoffmischung und Elementabfluss vom Land aufgrund von Erosion2,10,11,12. Zusätzlich beeinflussen künstliche Faktoren wie Eutrophierung lokale HABs12,13. Aufgrund des komplexen Multifaktors ist es nicht einfach, eine genaue HAB-Vorhersage zu treffen. In den letzten Jahren gibt es die Ansicht, dass spezifische Bakterienpopulationen mit der Entwicklung von HABs als einem der Faktoren in Verbindung gebracht werden können, und die Forschung zur Unterstützung dieser Hypothese wurde zunehmend offensichtlich14,15,16,17,18. Molekularbiologische Techniken werden im Allgemeinen verwendet, um die bakterielle Assemblage zu untersuchen; eine solche standardisierte Methode ist im chilenischen HAB-Monitoring9jedoch noch nicht etabliert. Um die Algen-Bakterien-Assoziation mit HABs zu untersuchen, ist es unerlässlich, gleichzeitig Metabarcoding-Analysen für die aktuellen chilenischen Küstenüberwachungsprogramme durchzuführen. Daher stellt dieses Protokoll unser chilenisches HAB-Überwachungsprogramm visuell vor und konzentriert sich auf ein schrittweises Verfahren zur Analyse des Nachweises von Algen- und Bakterienarten in Meerwasserproben mittels Metabarcoding-Analyse.
Das vollständige Protokoll, das unser chilenisches HAB-Überwachungsprogramm beschreibt, ist in Yarimizu et al.9verfügbar. Es umfasst die Verfahren der Meerwasserprobenahme, des mikroskopischen Algenartennachweises, der Algen-Bakterien-Gendetektion, der Pigmentanalyse, der meteorologischen Datenerfassung sowie physikalischer und chemischer Wassereigenschaftstests. Das schrittweise Protokoll der 16S rRNA- und 18S rRNA-Metabarcoding-Analyse für den Algen- und Bakterienspeziesnachweis ist als Preprint19verfügbar. Dieses Protokoll demonstriert insbesondere Metabarcoding-Analyseschritte, da es der komplizierteste Teil und das Highlight unseres HAB-Monitoring-Programms ist. Dieses Protokoll beinhaltet auch eine Einführung in das Programm und den mikroskopischen Nachweis von Algenarten. Bei der Analyse von Algenarten durch Metabarcoding ist es wichtig, gleichzeitig eine Mikroskopie durchzuführen, um die Ergebnisse der beiden Methoden zu überprüfen. Dieses Protokoll beinhaltet nicht die Verwendung von Software für die Taxonomiezuweisung, aber die Datenbankempfehlung wird am Ende des folgenden Abschnitts kurz erläutert.
Protokoll
1. Probenentnahme und Vorbehandlung
- Sammeln Sie ungefähr 3 l Wasserprobe von der Zielstelle.
- Filtern Sie 1 l Wasserprobe für die 16S rRNA-Analyse durch eine Tandemfiltration (1 μm und 0,2 μm porengroße Membran), um frei lebende und angebundene Bakterien zu trennen.
- Filtern Sie eine weitere 1 L Wasserprobe für die 18S-rRNA-Analyse (Phytoplankton-Detektion) durch eine einzige Filtration mit einer 0,2 μm-Membran.
HINWEIS: Die Filtration der Wasserprobe muss innerhalb von 12 Stunden nach der Probenahme abgeschlossen sein. - Schneiden Sie die gefilterte Membran mit einer sterilen chirurgischen Schere in zwei Hälften und wickeln Sie sie mit Alufolie ein. Bei -20 °C bis zu 1 Monat lagern oder mit dem nächsten Schritt fortfahren.
- Extrahieren Sie DNA mit der Chelex-Methode wie beschrieben9. Bei -20 °C bis zu 1 Monat lagern.
2. Mikroskopische Analyse
- 1 mL der Wasserprobe mit einer Pipette auf einen 1 mL Gitterschieber übertragen.
- Beobachten Sie die Probe unter einem Mikroskop.
- Erfassen Sie Namen und Menge der Phytoplanktonarten.
3. 16S rRNA und 18S rRNA Metabarcoding Analyse
HINWEIS: Dieser Prozess besteht aus sieben Abschnitten: Vorbereitung, erste PCR-Amplicon-Generierung, erste Amplicon-Bereinigung, Indexierung durch zweite PCR, zweite PCR-Amplicon-Verifizierung und -Bereinigung, DNA-Konzentrationsanpassung sowie DNA-Denaturierung und -Sequenzierung. Der gesamte Prozess dauert mindestens 5 Tage (40 h) durch ein erfahrenes Laborpersonal. Siehe die Tabelle der Materialien für Produktnummern und Hersteller.
- Präparat
- Reinigen Sie Pipetten und laminaren Haubenschrank mit 70% Ethanol, gefolgt von UV-Exposition für 30 min häufig. Zu verwendende Materialien sterilisieren.
- DNA-Proben bei Raumtemperatur auftauen, bei 100 x g für 2 min zentrifugieren und 100 μL jedes Probenüberstands in 8-Röhrchenstreifen überführen.
- Erste PCR-Amplicon-Generation
HINWEIS: Führen Sie die folgenden Verfahren in einem Laminarhaubenschrank aus. Primer immer von 1 μM Stamm auf die Zielkonzentration mit PCR-Wasser verdünnen, um eine Primerkontamination zu vermeiden.- Erste PCR-Mastermischung in einem sterilen 1,5-ml-Röhrchen für Reaktionen zubereiten (Tabelle 1, Tabelle 2).
- Aliquot 22,5 μL der Mastermischung in einem 8-Röhrchen-Streifen und 2,5 μL DNA-Probe hinzufügen. Verwenden Sie 2,5 μL PCR-Wasser zur Negativkontrolle.
- Führen Sie den ersten PCR-Zyklus durch (Tabelle 3).
- Bereiten Sie 100 ml 2% iges Agarose-TBE-Gel mit 10 μL 1x Nukleinsäuregelfleck vor.
- Laden Sie eine Mischung aus 1,5 μL 1x DNA-Beladungsfarbstoff und 4 μL PCR-Produkt auf das Agarosegel. Laden Sie auch 100 bp DNA-Leiter auf das Gel.
- Elektrophorese bei 100 V für 30 min durchführen.
- Stellen Sie sicher, dass sich unter einer UV-Lichtbildaufnahme ein Band im Bereich von 500-600 bp befindet. Primer-Dimer-Band ist rund 80 bp.
HINWEIS: Meereswasserproben enthalten PCR-Inhibitoren. Fehlende Amplikone können manchmal durch Verdünnen der Proben 1:100 oder 1:1000 mit PCR-Wasser gelöst werden. - Erste PCR-Produkte bis zum nächsten Schritt bei -20 °C lagern.
ACHTUNG: Überschreiten Sie nicht eine Woche Lagerung.
- Erste PCR-Amplicon-Bereinigung
HINWEIS: Dieser Abschnitt kann außerhalb eines laminaren Haubenschranks durchgeführt werden.- Verwenden Sie ein DNA-Cleanup-System für magnetische Kügelchen, um PCR-Reaktionsrückstände, einschließlich Primer-Dimer-Produkte, zu entfernen.
- Übertragen Sie 20 μL jedes gereinigten ersten PCR-Produkts auf eine neue 96-Well-Platte. Verschließen Sie die Platte mit einer Mikrodichtungsfolie. Bei -20 °C lagern, bis der nächste Schritt abgeschlossen ist.
ACHTUNG: Überschreiten Sie nicht mehr als eine Woche Lagerung.
- Indexierung durch zweite PCR
HINWEIS: In diesem Abschnitt werden gereinigte erste PCR-Produkte mit verschiedenen Index-Primer-Kombinationen verstärkt.- Verdünnen Sie alle Index-1- und Index-2-Primer (Tabelle 4) auf 1 μM mit PCR-Wasser in 8-Röhrchen-PCR-Streifen, die in einem laminaren Haubenschrank platziert sind.
HINWEIS: Die schritte in diesem Abschnitt können außerhalb eines laminaren Haubenschranks durchgeführt werden, da Indexprimer spezifisch für den ersten PCR-Reaktionsüberhangadapter sind. - Position Index 1 Primer in einer horizontalen Zeile Index 2 Primer in einer vertikalen Zeile (Tabelle 5).
- In einer neuen 96-Well-Platte werden zu jeder Vertiefung 12,5 μL Heißstartformulierung hinzugefügt.
- Fügen Sie 2,5 μL jeder Indexprimer (1 μM) zu jeder Vertiefung sowie in Tabelle 4 mit einer Mehrkanalpipette hinzu.
- Fügen Sie 7,5 μL gereinigtes erstes PCR-Produkt hinzu.
- Mischen Sie vorsichtig, indem Sie 10 Mal nach oben und unten pipettieren. Decken Sie die Platte mit einer Mikrodichtungsfolie ab.
- Führen Sie den zweiten PCR-Zyklus durch (Tabelle 3).
- Halten Sie die Platte bei -20 °C.
ACHTUNG: Überschreiten Sie nicht eine Woche Lagerung.
- Verdünnen Sie alle Index-1- und Index-2-Primer (Tabelle 4) auf 1 μM mit PCR-Wasser in 8-Röhrchen-PCR-Streifen, die in einem laminaren Haubenschrank platziert sind.
- Zweite PCR-Amplicon-Verifizierung und -Bereinigung
- Verwenden Sie einen Fragmentanalysator und das zugehörige Reagenz. Wirbel- und Spin-Down-Reagenz vor Gebrauch.
- Lassen Sie den Probenpuffer und das DNA-Screenband bei Raumtemperatur 30 Minuten lang ausgleichen. Legen Sie dann das DNA-Bildschirmband in einen Fragmentanalysator.
- Mischen Sie 2 μL Probenpuffer und 3 μL zweites PCR-Amplikon in neue 8 Röhrchenstreifen. Setzen Sie die 8 Röhrchenstreifen in den Fragmentanalysator ein. Drücken Sie Ausführen, um zu starten.
ACHTUNG: Die Bildung von Luftblasen muss vermieden werden. - Stellen Sie sicher, dass die zweiten PCR-Amplikone ungefähr 613 bp (600 - 630 bp) für die rRNA-Gene 16S und 18S betragen.
- Reinigen Sie zweite PCR-Produkte mit einem DNA-Cleanup-System für magnetische Kügelchen.
- Anpassung der DNA-Konzentration
- Messen Sie die DNA-Konzentration in den gereinigten zweiten PCR-Produkten mit einem Nukleinsäure-Quantifizierungsspektrophotometer.
- Berechnen Sie die Zielgenkonzentration in nM, wobei die durchschnittliche endgültige Bibliotheksgröße als 613 bp für 16S- und 18S-rRNA-Gene berücksichtigt wird:

- Verdünnen Sie jedes gereinigte zweite PCR-Produkt mit sterilem PCR-Wasser auf 4 nM in einer neuen 0,2 ml 96 Wellplatte.
HINWEIS: Die Platte kann hier bei -20 °C gelagert werden. Andernfalls müssen die übrigen Verfahren ohne Unterbrechung durchgeführt werden. - Aliquot 3 μL von jedem 4 nM Sekunden PCR-Produkt und mischen Sie alles in einem neuen sterilen 1,5 ml Röhrchen als gepoolte Bibliothek. Halten Sie das Röhrchen immer bei 4 °C oder im Eisbad.
- Messen Sie die Konzentration der gepoolten Bibliothek zur Bestätigung mit einem Nukleinsäurequantifizierungsspektrophotometer. Stellen Sie die Konzentration auf 4 nM ein, wenn sie höher als 4 nM ist.
HINWEIS: Überkonzentrierte DNA produziert überschätzte Lesezahlen, was die Analyse behindert.
- DNA-Denaturierung und -Sequenzierung
HINWEIS: In diesem Abschnitt wird das Illumina MiSeq-System mit spezifischen Reagenzien verwendet. Siehe Produktnummern in der Materialtabelle.
HINWEIS: Folgen Sie genau der Zeit. Tauen Sie alle Reagenzien mit Ausnahme der Kartuschen am Tag der Sequenzierung auf.- Einen Tag vor der Sequenzierung eine vorgefüllte gebrauchsfertige Reagenzienkartusche von -20 °C entnehmen und bei 4 °C zum Auftauen lagern.
- Stellen Sie einen für 1,5 ml Zentrifugenröhrchen geeigneten Wärmeblock auf 96 °C ein.
- Den Hybridisierungspuffer auf Eis legen.
- Verdünnen Sie NaOH in molekularer Qualität von 1 N auf 0,2 N in einem neuen Röhrchen mit PCR-Wasser.
- Verdünnen Sie eine gebrauchsfertige Kontrollbibliothek mit TE-Puffer (pH 8,0) von 10 nM-Material auf 4 nM in einem neuen Röhrchen (d. h. 2 μL Steuerbibliothek + 3 μL TE-Puffer).
- Mischen Sie 16 μL der 4 nM gepoolten Probenbibliothek mit 4 μL der 4 nM Steuerbibliothek in einem neuen Röhrchen mit der Bezeichnung "1".
- Mischen Sie 10 μL Probe in Röhrchen "1" mit 10 μL 0,2 N NaOH in einem neuen Röhrchen mit der Bezeichnung "2".
- Die Röhre 2 für 5 s schwenken, kurz herunterdrehen und bei Raumtemperatur 5 min inkubieren.
- 980 μL Hybridisierungspuffer in Röhrchen 2 geben.
- Mischen Sie in einem neuen Röhrchen mit der Bezeichnung "3" 260 μL die Probe aus Röhrchen 2 mit 390 μL Hybridisierungspuffer. Mischen Sie, indem Sie die Röhre umkehren.
- Tube 3 bei 96 °C für 2 min inkubieren und sofort für maximal 2 min auf Eis legen.
- Nehmen Sie die Patrone aus dem 4 °C warmen Kühlschrank.
- Richten Sie das Probenblatt für die Sequenzierung mit jedem entsprechenden Index 1 und Index 2 Adapter wie in Tabelle 6 ein.
- Entfernen Sie die Durchflusszelle aus dem MiSeq v3-Kit. Reinigen Sie die Durchflusszelle vorsichtig mit sterilem PCR-Wasser in molekularer Qualität.
HINWEIS: Gießen Sie kein Wasser auf die Kapillarpunkte der Durchflusszelle. Wischen Sie Wasser vorsichtig von der Durchflusszelle mit nicht faserigem Papier ab. - Laden Sie das volle Volumen von Röhrchen 3 (650 μL) in die Kartusche ein.
- Wählen Sie in der Gerätebetriebssoftware die Sequenzierung aus und folgen Sie den Anweisungen. Einsetzen von Durchflusszelle, Einarbeitungspuffer und Kartusche. Musterblatt einlegen. Drücken Sie Ausführen, um die Reaktion zu starten.
HINWEIS: Dieser Sequenzlauf dauert 3-5 Tage. Die Daten werden automatisch auf die Basespace-Plattform hochgeladen. Rohdaten werden in zwei Ordnern auf dem Computer gespeichert: Analyseordner mit fastq-Dateien und Ausgabe mit bcl-Dateien und JPEG-Bildern.
4.Taxonomische Zuordnung zu Sequenzierungsdaten
- FASTQ-Dateiverarbeitung
HINWEIS: DADA2-Paket23 von R24 ist eine der empfohlenen Datenbanken zur Verarbeitung von FASTQ Dateien. Der Leitfaden ist unter [https://github.com/mickeykawai/exec_dada2] verfügbar. Diese Richtlinie wurde durch die Übernahme der aktuellen Version des DADA2-Pipeline-Tutorials [https://benjjneb.github.io/dada2/tutorial.html] erstellt.- Verarbeiten Sie rohe FASTQ-Paired-End-Sequenzen zum Trimmen, Qualitätsfiltern, Dereplizieren und Zählen eindeutiger Sequenzen, Probeninferenz, Zusammenführen zu Contigs und Entfernen von chimären Sequenzen.
- Verwenden Sie die folgenden DADA2-spezifizierten Parameter für 16S rRNA- und 18S rRNA-Genfragmente; trimLefts=0, 0, truncLens=[Überprüfen Sie die Sequenzqualität und stellen Sie sie auf eine geeignete Länge].
HINWEIS: Standardwerte für die anderen Parameter sind maxN=0, maxEE=2,2, trancQ=2.
- Taxonomie-Zuweisung
HINWEIS: Die SILVA rRNA-Datenbank wird für die Taxonomiezuweisung für DADA2 empfohlen [silva_nr_v132_train_set.fa.gz ist als Standard festgelegt].- Entfernen Sie Chloroplasten und mitochondriale OTUs für die 16S rRNA-Genfragmentanalyse. Entfernen Sie Singletons aus der 16S rRNA- und 18S rRNA-Analyse.
- Rarifizieren Sie alle Proben auf gleichmäßige Sequenzierungstiefe basierend auf der Probe mit der niedrigsten Sequenzierungstiefe.
- Notieren Sie die Anzahl der Lesevorgänge, die zugewiesenen OTUs und auf welcher Ebene, die gefilterten Lesevorgänge und die Anzahl der ausgeschlossenen Singletons.
- Statistische Analyse
- Führen Sie statistische Analysen mikrobieller Daten mit R-Packungen von phyloseq25 und vegan26 durch.
Ergebnisse
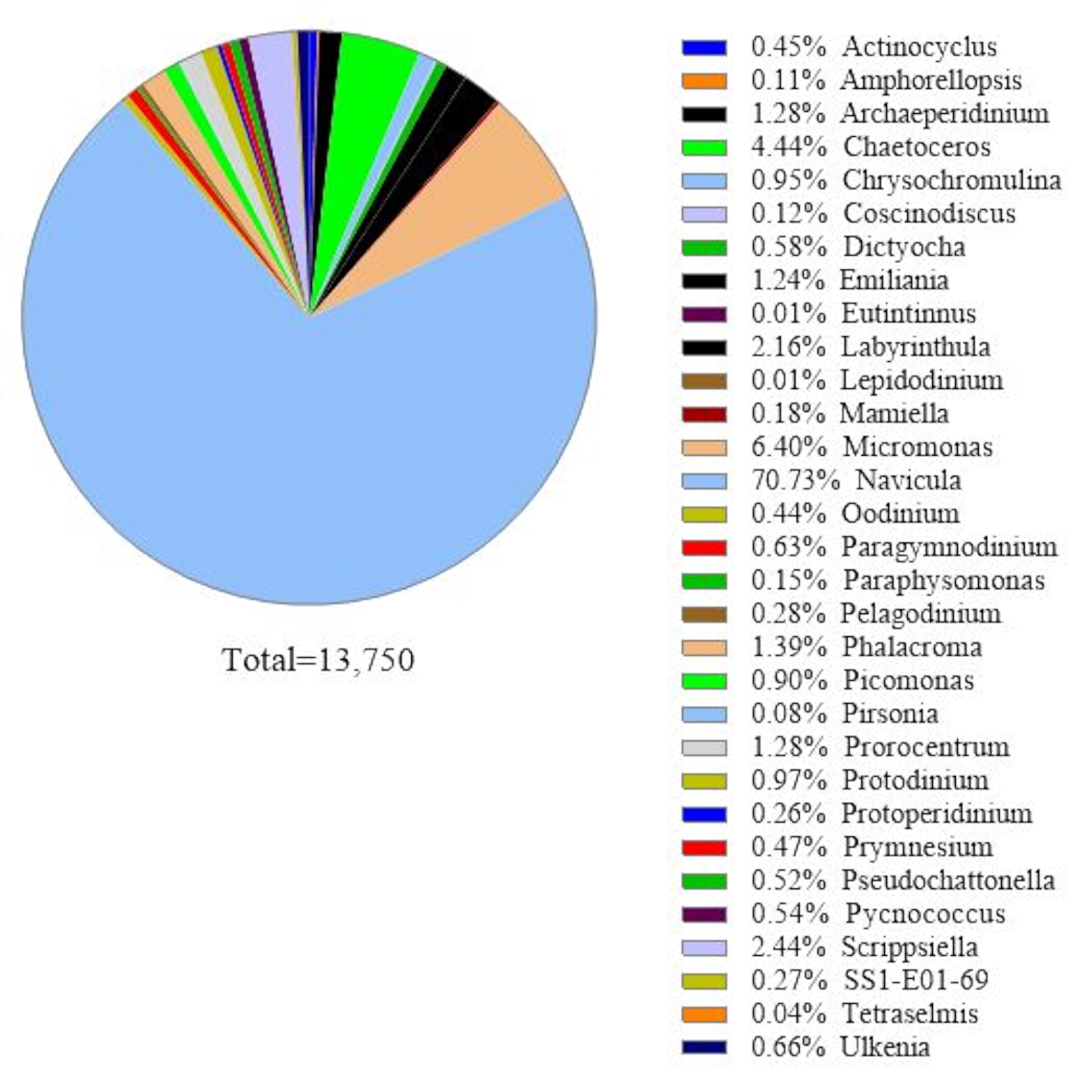
Dieses Protokoll verwendet eine 18S rRNA-Gen-Metabarcoding-Analyse, um Algenarten in Meerwasserproben zu identifizieren. Ein repräsentatives Ergebnis ist in Abbildung 1dargestellt, in dem das am 19. Februar 2019 in Metri, Puerto Montt, Chile (−41.597; −72.7056) gesammelte Meerwasser mit diesem Protokoll analysiert wurde. Das Ergebnis zeigte insgesamt 13.750 Reads mit über 30 Algenarten in der Meerwasserprobe. Die dominante Alge in dieser Probe war Navicula spp. mit einer relativen Häufigkeit von 70,77%. Außerdem wurde eine ausreichende Häufigkeit für Micromonas (6,40%), Chaetoceros spp. (4,44%), Scripsiella spp. (2,44%) und Prorocentrum spp.beobachtet. (1.28%). Pseudochattonella spp.,einer der höchsten toxischen Algenerreger der chilenischen HAB, wurde mit 0,52% aus dieser Meerwasserprobe nachgewiesen.
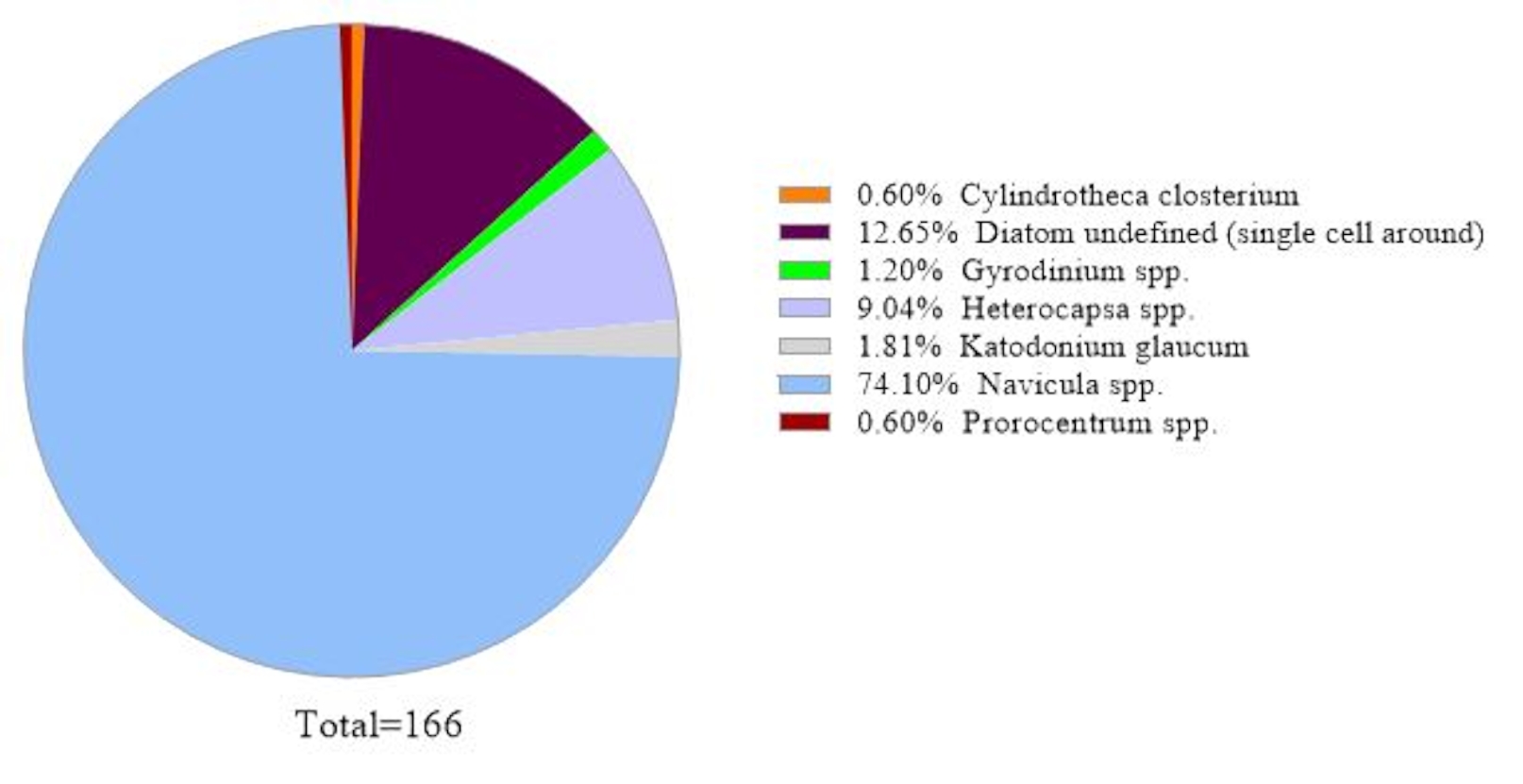
Um die Zuverlässigkeit der Daten zu überprüfen, wurden die durch 18S rRNA-Genanalyse identifizierten Algenarten mit denen verglichen, die durch Mikroskopie in derselben Meerwasserprobe erhalten wurden (Abbildung 2). In Übereinstimmung mit der 18S rRNA-Genanalyse zeigte die Mikroskopie, dass die dominante Spezies Navicula sppwar. mit einer relativen Häufigkeit von 74,1% und Prorocentrum spp. (0,60%) als Nebenart. Umgekehrt wurde Heterocapsa spp. (9,04%), das aus mikroskopischer Beobachtung dokumentiert wurde, nicht durch 18S rRNA-Genanalyse in dieser Probe identifiziert. Es gab 12,6% der kleinen um nicht identifizierbaren Phytoplanktonzellen, die durch Mikroskopie aufgezeichnet wurden. Dies könnte Micromonassein, nach den 18S rRNA-Ergebnissen.
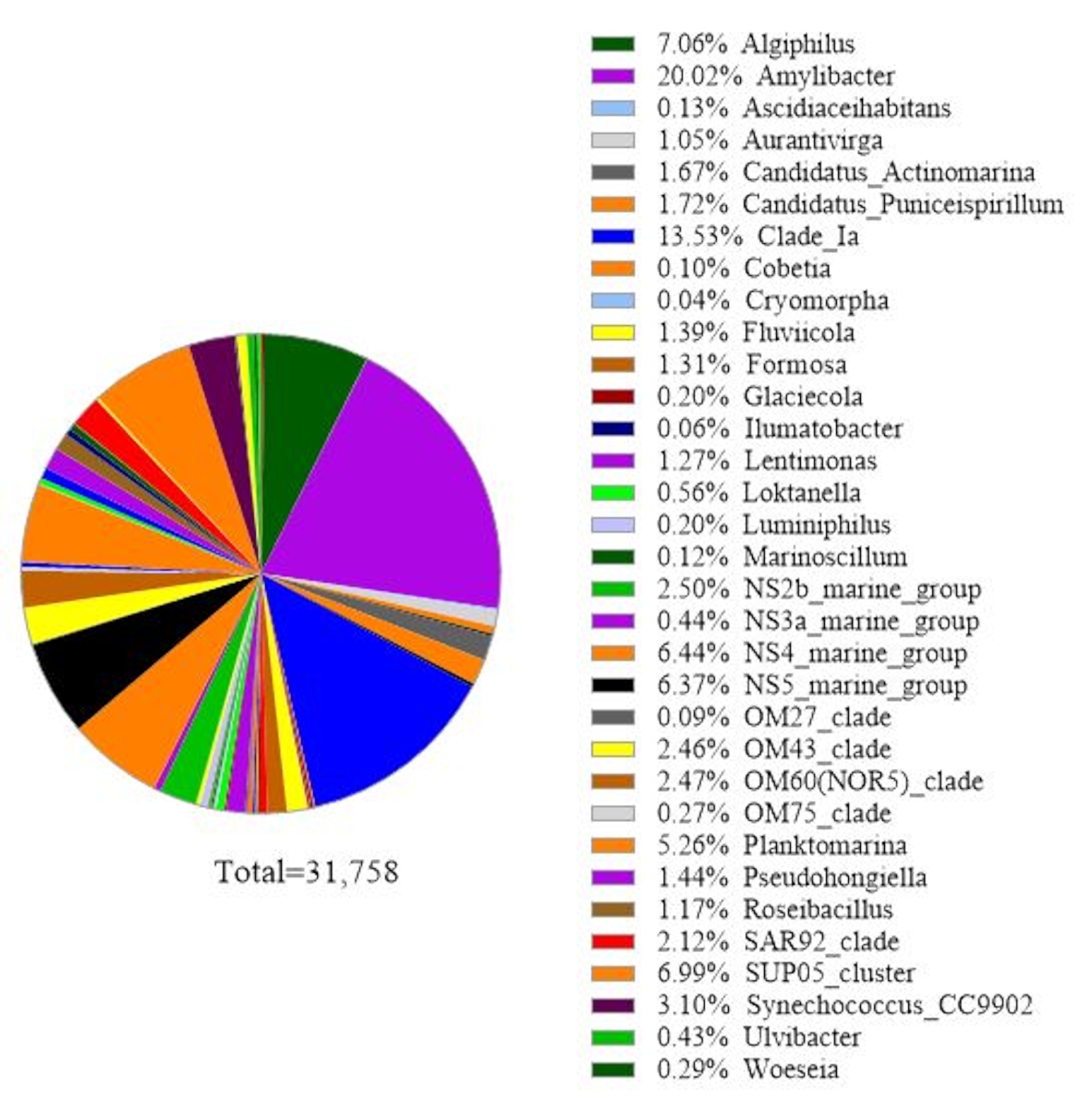
Das Protokoll verwendet eine 16S rRNA-Gen-Metabarcoding-Analyse, um Bakterienarten in Meerwasserproben zu identifizieren. Ein repräsentatives Ergebnis ist in Abbildung 3dargestellt, in dem das gleiche Meerwasser, das für die 18S rRNA-Genanalyse verwendet wurde, mit der 16S rRNA-Genanalyse analysiert wurde. Das Ergebnis zeigte insgesamt 31.758 Messwerte mit über 30 Bakterienarten in der Meerwasserprobe. Es sollte skizziert werden, dass diese Meerwasserprobe durch Tandemfiltermembranen (1 μm und 0,2 μm Porengrößen) geleitet wurde, um frei lebende Bakterien von gebundenen Bakterien zu trennen. Dann wurden die von jeder Filtermembran erfassten Zellen für die DNA-Extraktion behandelt, gefolgt von der 16S rRNA-Genanalyse. Das repräsentative Ergebnis in Abbildung 3 zeigt Bakterienarten, die aus einer 0,2 μm porengroßen Membran identifiziert wurden und als freilebende Bakterien definiert sind. Die dominierenden freilebenden Bakterien waren Amylibacter spp. mit einer relativen Häufigkeit von 20,02%, gefolgt von Clade Ia (13,53%) und Aligiphilus spp. (7.06%). Der Rest der bakterienarten, die aus dieser Meerwasserprobe nachgewiesen wurden, war relativ gleichmäßig verteilt. Die gleiche Analyse kann für Zellen durchgeführt werden, die von einer 1 μm porengroßen Membran als Nachweis von Attach-Bakterienspezies eingefangen wurden.
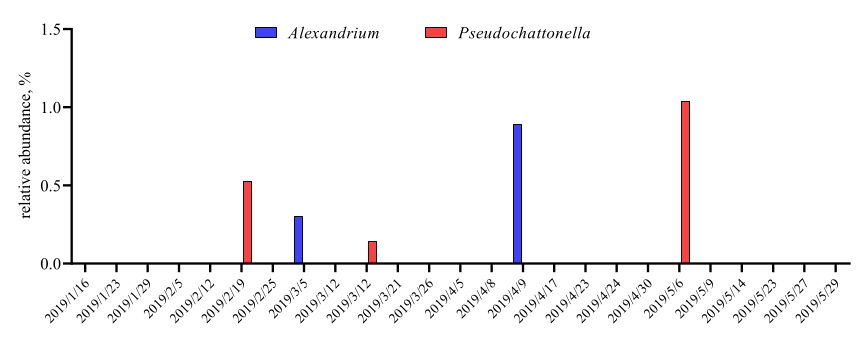
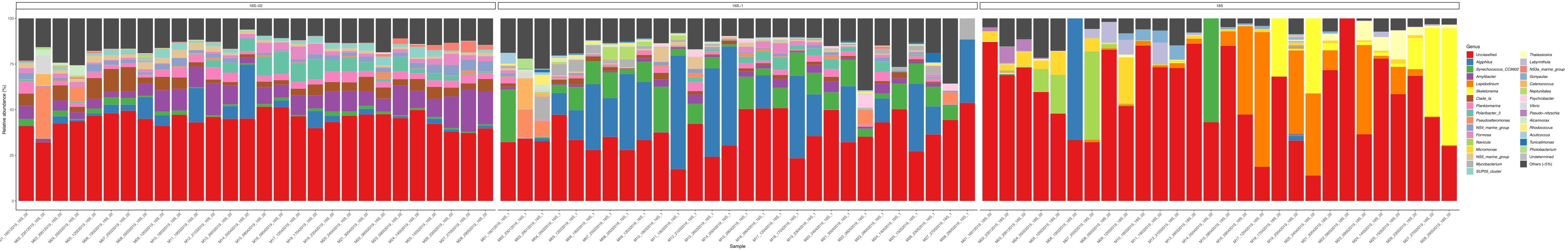
Wenn diese Metabarcoding-Assays zu geplanten Zeitpunkten über einen bestimmten Studienzeitraum durchgeführt werden, können die Ergebnisse als Zeitreihenanalyse zusammengefasst werden. Eine Möglichkeit, dies zu tun, besteht darin, die relative Häufigkeit bestimmter Algen- und Bakterienarten als Funktion der Zeit darzustellen, um ein einzigartiges Wachstumsmuster zu finden. Abbildung 4 zeigt eine repräsentative Zeitreihendarstellung von Alexandrium und Pseudochattonella in Metri, Chile. Eine andere Möglichkeit, eine Zeitreihen-Metabarcoding-Analyse zusammenzufassen, besteht darin, alle identifizierten Algen und Bakterien als Funktion der Zeit darzustellen und die Populationsänderung bestimmter Organismengruppen darzustellen. Abbildung 5 und Abbildung 6 fassen die relative Häufigkeit aller Bakteriengattungen bzw. -ordnungen zusammen, die aus dem Meerwasser von Metri über fünf Monate nachgewiesen werden.
Tabelle 1: Inhalt des ersten PCR-Master-Mixes: Die Tabelle zeigt den Master-Mix-Gehalt pro Reaktion für 16S rRNA- und 18S rRNA-Analysen. Die Primersequenzen sind in Tabelle 3 aufgeführt. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 2: Primer-Sequenzen: Die Primer für die erste PCR sind für 16S rRNA- und 18S rRNA-Analysen gelistet. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 3: PCR-Zyklen: Die thermischen Zyklen für die erste PCR und die zweite PCR sind aufgeführt. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 4: Indexsequenzen: Index 1 (i7) und Index 2 (i5) Primer, die für die zweite PCR verwendet werden sollen, sind aufgeführt. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 5: Beispiel für die Positionierung von Index 1 und Index 2: Um Fehler zu reduzieren, müssen Indexprimer zuerst in der Position platziert werden, und aliquot von jedem wird mit einer Mehrkanalpipette auf eine 96-Well-Platte übertragen. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 6: Beispiel für ein Musterblatt: Vor der Sequenzierung muss ein Probenblatt erstellt werden, das den Adaptern Index 1 und Index 2 entspricht. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.

Abbildung 1: Repräsentatives Ergebnis der 18S rRNA Metabarcoding-Analyse: Algenarten, die in einer Meerwasserprobe aus Metri, Los Lagos, Chile, am 19. Februar 2019 vorhanden sind, wurden durch 18S rRNA Metabarcoding Assay identifiziert. Die Sequenzen, die "unbekannt" zugeordnet wurden, wurden eliminiert und die relative Häufigkeit jeder identifizierten Art wurde aufgetragen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Repräsentatives Ergebnis der mikroskopischen Analyse: Die Algenarten wurden am 19. Februar 2019 aus einer Wasserprobe aus Metri, Los Lagos, Chile, mittels Mikroskopie identifiziert. Die Menge jeder Art wurde manuell gezählt und aufgetragen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: Repräsentatives Ergebnis der 16S rRNA Metabarcoding-Analyse: Bakterienarten, die am 19. Februar 2019 in einer Wasserprobe aus Metri, Los Lagos, Chile, vorhanden waren, wurden mittels 16S rRNA Metabarcoding Assay identifiziert. Die relative Häufigkeit jeder identifizierten Art wurde aufgetragen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 4: Repräsentative Zeitreihendarstellung von Alexandrium spp. und Pseudochattonella spp., erhalten aus 18S rRNA metabarcodingin Metri, Chile: Abbildung nachgedruckt von Yarimizu et al9. Die beiden toxischen Algenarten Alexandrium und Pseudochattonella wurden im Laufe der Zeit selektiv durch 18S rRNA-Analyse überwacht, und die relative Häufigkeit wurde als Funktion des Zeitpunkts aufgetragen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 5: Repräsentative Genus-Zeitreihen-Diagramm aus 16S rRNA und 18S rRNA Metabarcoding: Alle bakteriellen und eukaryotischen Gattungen, die im Wasser von Metri, Chile, identifiziert wurden, wurden über fünf Monate überwacht. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 6: Zeitreihendiagramm repräsentativer Ordnung, erhalten aus 16S rRNA und 18S rRNA Metabarcoding: Alle bakteriellen und eukaryotischen Ordnungen, die im Wasser von Metri, Chile, identifiziert wurden, wurden über fünf Monate überwacht. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Tabelle S1 und Tabelle S2:
Tabelle S1: Rohdaten für Abbildung 5 und Abbildung 6. Tabelle S2: Ergebnis der QC-Prüfung der Rohdaten von Abbildung 5 und Abbildung 6. Bitte klicken Sie hier, um diese Tabellen herunterzuladen.
Diskussion
Dieses Protokoll führte erfolgreich eine 18S rRNA- und 16S rRNA-Gen-Metabarcoding-Analyse durch, um Algen- und Bakterienarten in Meerwasserproben zur Überwachung chilenischer HABs zu identifizieren. Die dominanten Algen und einige kleinere Algen, die durch dieses Protokoll nachgewiesen wurden, stimmten mit denen überein, die durch Mikroskopie erhalten wurden, was die Zuverlässigkeit des Protokolls bestätigte. Der Höhepunkt des Protokolls ist, dass die Analyse Alexandrium spp. und Pseudochattonella spp.,die beiden problematischsten HAB verursachenden Arten im Süden Chiles, aus dem Meerwasser von Metri auch in geringer Häufigkeit nachgewiesen hat. Insbesondere Pseudochattonella spp. sind schwer zu identifizierende Arten unter dem Mikroskop, da die Zellen klein sind und unter Licht oder Verwendung von Fixiermitteln leicht gerissen werdenkönnen 27,28. Metabarcoding kann Algenarteninformationen liefern, die in Meerwasserproben, die die Mikroskopie nicht identifizieren kann, kaum vorhanden sind. Das Protokoll erkannte auch über 30 Bakterienarten. Obwohl zu diesem Zeitpunkt nicht bekannt ist, wie diese Bakterien mit dem Algenwachstum zusammenhängen, wird die massive Datenerhebung von Algen-Bakterienarten in der Zeitreihenanalyse möglicherweise so wichtige Entdeckungen liefern. Daher wird das Hinzufügen von Metabarcoding-Analysen zu den herkömmlichen chilenischen HAB-Überwachungsprogrammen zweifellos die derzeitige Effizienz der HAB-Überwachung stärken. Tatsächlich kann dieses visuelle Protokoll für die Metabarcoding-Analyse nicht nur für die Algen-Bakterien-Überwachung des chilenischen Wassers von Vorteil sein, sondern auch für die Küstenüberwachungsprogramme in anderen von HAB betroffenen Gebieten weltweit.
Obwohl dieses Protokoll die oben genannten Vorteile bot, sollten die Nachteile der Methode diskutiert werden. Wie im visuellen Protokoll zu sehen ist, ist die Metabarcoding-Methode zeitaufwendig und komplex und erfordert teure Maschinen und Reagenzien. Das Laborpersonal muss speziell geschult werden, sonst verschwendet es Material, Arbeit und Zeit. Darüber hinaus muss der Algennachweis durch Metabarcoding mit der Mikroskopie gepaart werden, um zu überprüfen, ob dieselben dominanten Spezies aus den beiden orthogonalen Methoden identifiziert werden29. Die Mikroskopie ist zum größten Teil ein nicht-invasives Werkzeug, um Algenarten zu identifizieren, was bedeutet, dass es schwieriger ist, Fehler zu machen, wenn die Algenarten einzigartige und scheinbare Formen haben. Natürlich kann es zu menschlichem Versagen kommen, wenn die Arten sehr ähnliche Formen haben. Auf der anderen Seite, da die Metabarcoding-Analyse mehrere Schritte erfordert, erhöht sie natürlich die Fehler. Es könnte sich um eine verwechselte Probe, eine falsche Reagenzienzugabe oder das Fehlen einiger Verfahren handeln. Daher ist es wichtig, die Metabarcoding-Ergebnisse mit denen der Mikroskopie zu vergleichen. Schließlich ist das Protokoll nur qualitativ anwendbar, und die Ergebnisse müssen für die relative Häufigkeit berechnet werden. Wie Lamb et al. im Jahr 2019 feststellten, ist das derzeitige Metabarcoding als quantitative Leistung immer noch begrenzt, und es ist zusätzliche Forschung erforderlich, bevor Metabarcoding sicher für quantitative Anwendungen genutzt werden kann30.
Dieses Protokoll wurde für 140 - 170 Proben pro Durchlauf aus dem 16S Metagenomic Sequencing Library Preparation Manual von Illumina Inc. optimiert. Das optimierte Protokoll wurde viele Male getestet und weiter modifiziert, um diese endgültige Version herauszugeben. Daher wird dringend empfohlen, jeden Schritt genau zu befolgen. Der kritischste Teil des Protokolls ist, dass besondere Sorgfalt erforderlich ist, um eine Probenkontamination zu vermeiden. Die Pipetten und die Laminar-Flow-Haube müssen jederzeit mit 70% Alkohol und UV-Strahlung gereinigt werden, und sterilisierbare Materialien sollten autoklaviert werden. Die Primer und Reagenzien sollten bei jedem neuen Durchlauf direkt aus dem Vorrat verdünnt werden, oder die Wiederverwendung von Reagenzien und die Exposition gegenüber mehreren Verdünnungen können eine Probenkontamination verursachen. Das Protokoll gibt an, wann der Vorgang außerhalb der Laminar-Flow-Haube durchgeführt werden kann. Sofern nicht anders, sollten Proben in einer gereinigten Laminar-Flow-Haube behandelt werden. Wenn Sie mit mehreren 8-Rohr-Streifen gleichzeitig arbeiten, wird empfohlen, den ersten 8-Rohr-Streifen zu bearbeiten und zu verschließen, bevor Sie zum nächsten 8-Rohr-Streifen übergehen. Wenn Sie den Deckel lange offen lassen, kann dies zu einer Probenkontamination führen, da die rRNA-Gene 16S und 18S in der Umgebung sehr allgegenwärtig sind. Die angegebene Zeit, wie Mischzeit und Inkubationszeit, sollte wie genau beschrieben befolgt werden, da die beste Dauer für jeden Schritt basierend auf den mehreren Testläufen ausgewählt wurde. Überschüssige oder unzureichende Zeit kann beispielsweise die Probenausbeute verringern. Der Prozess sollte nur an dem Teil gestoppt werden, von dem das Protokoll sagt, dass er gestoppt werden kann. Es ist wichtig, eine Negativkontrolle zu haben, da sie bestätigt, dass die positiven Ergebnisse keine künstlichen falsch positiven Ergebnisse sind. Schließlich wird dringend empfohlen, die Tage zu planen, da es mindestens fünf Tage dauert, um alle Schritte zu bearbeiten, und einige Teile nicht bis zum nächsten Halteschild gestoppt werden können.
Die Einschränkung der taxonomischen Zuordnung für sequenzierte Daten wird hier kurz erwähnt. Neu entdeckte Nukleotidsequenzen für Bakterien- und Algenarten werden täglich in der Datenbank aktualisiert. Während gut untersuchte Algen- und Bakterienarten zuverlässig registriert werden, werden in der Datenbank auch viele Sequenzen für unbekannte Arten aktualisiert. Es zeigt, dass die Auflösung der registrierten Nukleotidsequenzen nicht immer für die Artidentifizierung ausreicht, sondern nur für die Gattungsidentifizierung. Daher muss die mikroskopische Artidentifizierung orthogonal durchgeführt werden. Die Einrichtung einer Open-Access-Pipeline für diese Arbeit hat einen wesentlichen Vorteil, da sie mit einem großen Satz sequenzierter Daten gleichzeitig umgehen und effizient untersuchen kann, wenn ein Fehler auftritt. Darüber hinaus erfolgt die Ausgabe von DADA2 in einer Text- oder CSV-Datei, was die weitere statistische Analyse erleichtert. Auf der anderen Seite ist ein großer Server erforderlich, um taxonomische Zuweisungen durchzuführen, insbesondere wenn ein Dataset groß ist. Um die Pipeline einzurichten, werden Ingenieure benötigt, um die Pipeline einzurichten, und die Fachleute müssen Parameter, Betrieb, Linux und Bioinformatik verstehen.
Neben dem Umfang als HAB-Monitoring-Tool kann die Nutzung dieses Protokolls für investigative Forschungszwecke erweitert werden. Zum Beispiel gibt es laufende Debatten zwischen der chilenischen Regierung und lokalen Fischern zusammen mit Umweltfriedensverbänden über die erhöhte Anzahl lokaler HAB31,32. Die Regierung behauptet, dass dies auf ein Naturphänomen wie die globale Erwärmung und El Niño zurückzuführen ist, während die späteren Parteien behaupten, dass die Lachsaquakultur die Ursache ist. Lachs ist keine einheimische Art Chiles und war vor einem halben Jahrhundert nicht in chilenischem Wasser. Die chilenische Regierung zu dieser Zeit zielte darauf ab, die Wirtschaft zu steigern und Arbeitsplätze für arme Gebiete zu schaffen, indem sie ein Lachsgeschäftentwickelte 33. Mit der Intervention aus Übersee für Kapitalgewinn machte Chile großen Erfolg in der Entwicklung der Aquakultur im Stil von Gehegen, und die Zahl der Lachse hat in den letzten Jahrzehnten bemerkenswert zugenommen31,32,33. In der Folge wurde eine große Menge an Futter für Lachs ins Meer geworfen, was die Einheimischen zu der Vermutung veranlasste, dass die Lachsaquakultur der Grund für die häufigen lokalen HABs31,32ist. Die Wahrheit ist jetzt unbekannt, aber sie muss schließlich verstanden werden, damit Strategien zum Schutz der lokalen Meeresumwelt, der Wirtschaft und der menschlichen Gesundheit vor HABs entwickelt werden können. Die molekulare Analyse der lokalen Algen-Bakterien-Beziehung kann zu einer schrittweisen Klärung für ein solches Thema beitragen. Zum Beispiel kann die Technik die Algen- und Bakterienarten durchsuchen, die zuvor nicht im ozeanischen Zielgebiet vorhanden waren, aber in den letzten Jahren dramatisch zugenommen haben, was der Studie von Sakai et al. zur Entdeckung einer nicht aufgezeichneten Population von Yamato-Salamandern (Hynobius vandenburghi) durch GIS- und eDNA-Analyse34ähnelt. Daher hat dieses Metabarcoding-Protokoll über das HAB-Monitoring-Tool hinaus das Potenzial, für andere potenzielle Kunden verwendet zu werden.
Offenlegungen
Die Autoren erklären keinen Interessenkonflikt. Die Geldgeber spielten keine Rolle bei der Gestaltung der Studie, bei der Erhebung, Analyse oder Interpretation von Daten; beim Schreiben des Manuskripts oder bei der Entscheidung, die Ergebnisse zu veröffentlichen.
Danksagungen
Diese Studie wurde durch den Zuschuss (JPMJSA1705) für eine Studie zur Wissenschafts- und Technologieforschungspartnerschaft für die Überwachung von Algen für nachhaltige Entwicklung in Chile (SATREPS-MACH) unterstützt. Wir danken Dr. Sandra Rios (Universidad de Los Lagos) für die Erlaubnis, einen Filmclip zu verwenden. Wir danken Neal Andrew Holland für sein fleißiges Korrekturlesen des Manuskripts. Wir danken den Mitgliedern der Laborgruppe von CREAN-IFOP in Puerto Montt, Chile, für die Beratung bei der Identifizierung von Phytoplankton.
Materialien
| Name | Company | Catalog Number | Comments |
| 1.5 mL single tube | ThermoFisher | Q32856 | sterile |
| 1.5 mL tubes | ThermoFisher | 2150N | sterile |
| 8-strip 0.2 mL PCR tubes and flat cap | ThermoFisher | AB0451 & 4323032 | sterile |
| 96-well 0.2 mL PCR plate | ThermoFisher | N8010560 | |
| AccuRuler 100 bp DNA Ladder | MaestroGen | 02001-500 | |
| Chelex 100 Chelating Resin | Bio-Rad | 1432832 | |
| Chemical Duty Vacuum Pressure Pump | MilliporeSigma | WP6122050 | |
| D1000 Reagents | Agilent Technologies | 5067-5583 | |
| D1000 sample buffer | Agilent Technologies | 5067-5602 | |
| DNA loading dye (Blue/Orange Loading Dye 6X) | Promega | G1881 | |
| Ethanol Molecular Biology Grade | E7023 | Merck KGaA | |
| Filtration device | ThermoFisher | 09-740-36H, 09-740-23E | |
| Fluorescent DNA concentration measurement kit (Qubit dsDNA HS Assay kit) | Thermo Fisher Scientific | Q32851 | |
| Fluorometer (Qubit4 Fluorometer) | ThermoFisher | Q33238 | |
| Fragment analysis verification matrix (D1000 ScreenTape) | Agilent Technologies | 5067-5582 | |
| Fragment Analyzer (Agilent Tapestation 4150) | Agilent | G2992AA | |
| GelRed Nucleic Acid Gel Stain | Biotium | 41002 | |
| Generic bench vortexer (Vortex Genie 2) | Scientific Industries | SI-0236 | |
| Heat block | |||
| High performance sequencing-grade DNA polymerase (KAPA HiFi Hotstart ReadyMix), 2X | Roche | KK2602 | |
| HT1 Hybridization buffer | Illumina | 20015892 | |
| Illumina Nextera XT v2 Index Kit set A, B, C and D | Illumina | FC-131-2001, -2002, -2003, -2004 | |
| Laminar hood cabinet | Biobase Co. | Biobase PCR-800 PCR Cabinet | |
| Loading tips (Specific for Agilent TapeStation systems) | Agilent Technologies | 5067-5598 | |
| Magnetic beads DNA cleanup system (ProNex® Size-Selective Purification System) | Promega | NG2002 | |
| Magnetic stand for 96 wells (Magnetic Stand-96) | ThermoFisher | AM10027 | |
| Micro-seal film for 96-well plate | ThermoFisher | 4311971 | sterile |
| MiSeq Reagent Kit v3 | Illumina | MS-102-3003 | includes pre-filled, ready-to-use reagent cartridges. |
| MiSeq system | Illumina | SY-410-1003 | includes pre-installed software |
| Molecular grade nuclease-free water | Integrated DNA Technologies | 11-05-01-04 | |
| Molecular grade sodium hydroxide (NaOH) 1M | Merck KGaA | 1091371000 | |
| Multichannel pipettes | Not specified | Not specified | |
| Multi-Purpose Agarose | Cleaver Scientific | CSL-AG500 | |
| Ow D2 Wide-Gel Electrophoresis System | ThermoFisher | D2 | |
| Ow EC-105 Compact Power Supply | ThermoFisher | 105ECA-115 | |
| Pellet Pestle— Cordless Motor | ThermoFisher | K749540-0000 | |
| PhiX control Kit v3 | Illumina | FC-110-300 | ready-to-use control library for Illumina sequencing |
| Pipette tips | Not specified | Not specified | sterile |
| Pipettes | Not specified | Not specified | 2, 10, 20, 100, 1000 μL |
| SterivexTM GP 0.22 μm filter unit | MilliporeSigma | SVGP01050 | |
| Tabletop centrifuge | Eppendorf 5427R Centrifuge | Eppendorf AG | |
| TBE buffer | ThermoFisher | B52 | |
| TE buffer (pH 8.0) | ThermoFisher | AM9849 | |
| Terr PCR Direct FFPE Kit | Takara Bio USA | 639284 | includes 2X Terra PCR Direct Buffer |
| Terra PCR Direct Polymerase Mix, 1.25 U/μL | Takara Bio USA | 639271 | High performance Inhibitor-tolerant DNA polymerase |
| ThermalCycler (MiniAMP Plus Thermal Cycler) | ThermoFisher | A37835 | |
| TransIlluminator and image capture system | Analytik Jena, AG | GelDoc-ItTS2 Imager | |
| Whatman 1.0 m pore-sized membrane | MilliporeSigma | WHA111110 |
Referenzen
- Hallegraeff, G., Enevoldsen, H., Zingone, A. Global harmful algal bloom status reporting. Harmful Algae. 102, 101992(2021).
- Anderson, D. M. Red Tides. Scientific American. 271 (2), 52-58 (1994).
- Sanseverino, I., Conduto, A. D. S., Pozzoli, L., Dobricic, S., Lettieri, T. Algal Bloom and its Economic Impact. , Publications Office of the European Union. (2016).
- Molinet, C., Niklitschek, E., Seguel, M., Díaz, P. Trends of natural accumulation and detoxification of paralytic shellfish poison in two bivalves from the Norwest Patagonian inland sea. Revista de Biologia Marina Y Oceanografia. 45, 195-204 (2010).
- Mardones, J., Clement, A., Rojas, X., Aparicio, C. Alexandrium catenella during 2009 in Chilean waters, and recent expansion to coastal ocean. Harmful Algae News. 41, 8-9 (2010).
- Alvarez, G., et al. Paralytic shellfish toxins in surf clams Mesodesma donacium during a large bloom of Alexandrium catenella dinoflagellates associated to an intense shellfish mass mortality. Toxins (Basel). 11 (4), (2019).
- Clément, A., et al. Exceptional summer conditions and HABs of Pseudochattonella in Southern Chile create record impacts on salmon farms. Harmful Algae News. 53, 1-3 (2016).
- Trainer, V. L., et al. Pelagic harmful algal blooms and climate change: Lessons from nature's experiments with extremes. Harmful Algae. 91, 101591(2020).
- Yarimizu, K., et al. Protocols for monitoring harmful algal blooms for sustainable aquaculture and coastal fisheries in Chile. International Journal of Environmental Research and Public Health. 17 (20), (2020).
- Roegner, G. C., Hickey, B. M., Newton, J. A., Shanks, A. L., Armstrong, D. A. Wind-induced plume and bloom intrusions into Willapa Bay, Washington. Limnology & Oceanography. 47 (4), 1033-1042 (2002).
- Tweddle, J. F., et al. Relationships among upwelling, phytoplankton blooms, and phycotoxins in coastal Oregon shellfish. Marine Ecology Progress Series. 405, 131-145 (2010).
- Glibert, P. M., Anderson, D. M., Gentien, P., Graneli, E., Sellner, K. G. The global complex phenomena of harmful algae blooms. Oceanography. 18 (2), 130-141 (2005).
- Paredes-Mella, J., Varela, D., Fernández, P., Espinoza-González, O. Growth performance of Alexandrium catenella from the Chilean fjords under different environmental drivers: Plasticity as a response to a highly variable environment. Journal of Plankton Research. 42 (2), 119-134 (2020).
- Azam, F., Malfatti, F. Microbial structuring of marine ecosystems. Nature Reviews Microbiology. 5, 782-791 (2007).
- Amin, S. A., et al. Interaction and signaling between a cosmopolitan phytoplankton and associated bacteria. Nature. 522, 98-101 (2015).
- Berdalet, E., et al. Marine harmful algal blooms, human health, and wellbeing: Challenges and opportunities in the 21st century. Journal of Marine Biology Association. 2015, U. K. 1-31 (2015).
- Ramanan, R., Kim, B. -H., Cho, D. -H., Oh, H. -M., Kim, H. -S. Algae-bacteria interactions: Evolution, ecology, and emerging applications. Biotechnology Advances. 34 (1), 14-29 (2016).
- Seymour, J. R., Amin, S. A., Raina, J. B., Stocker, R. Zooming in on the phycosphere: The ecological interface for phytoplankton-bacteria relationships. Nature Reviews Microbiology. 2, 17065(2017).
- Maruyama, F., et al. 16S and 18S Metabarcoding analysis for Chilean coastal waters harmful algal blooms. protocol.io. , (2020).
- Tanabe, A. S., et al. Comparative study of the validity of three regions of the 18S-rRNA gene for massively parallel sequencing-based monitoring of the planktonic eukaryote community. Molecular Ecology Resources. 16, 402-414 (2016).
- Nishitani, G., et al. Multiple plastids collected by the Dinoflagellate Dinophysis mitra through Kleptoplastidy. Applied and Environmental Microbiology. 78 (3), 813-821 (2012).
- Klindworth, A., et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Research. 41 (1), 1(2013).
- Callahan, B. J., et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nature Methods. 13 (7), 581-583 (2016).
- R, R Team DCore. R: A language and environment for statistical computing. R Foundation for Statistical Computing. , Vienna, Austria. Available from: http://www.R-project.org (2007).
- McMurdie, P. J., Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS One. 8 (4), 61217(2013).
- Dixon, P. VEGAN, a package of R functions for community ecology. Journal of Vegetation Science. 14 (6), 927-930 (2003).
- Dittami, S. M., Riisberg, I., Edvardsen, B. Molecular probes for the detection and identification of ichthyotoxic marine microalgae of the genus Pseudochattonella (Dictyochophyceae, Ochrophyta). Environmental Science and Pollution Research. 20 (10), 6824-6837 (2013).
- Mardones, J. I., et al. Salinity-growth response and ichthyotoxic potency of the Chilean Pseudochattonella verruculosa. Frontiers in Marine Science. 6 (24), (2019).
- Santi, I., Kasapidis, P., Karakassis, I., Pitta, P. A comparison of DNA metabarcoding and microscopy methodologies for the study of aquatic microbial eukaryotes. Diversity. 13 (5), 180(2021).
- Lamb, P. D., et al. How quantitative is metabarcoding: A meta-analytical approach. Molecular Ecology. 28 (2), 420-430 (2019).
- Mascareño, A., et al. Controversies in social-ecological systems: lessons from a major red tide crisis on Chiloe Island, Chile. Ecology and Society. 23, (2018).
- Armijo, J., Oerder, V., Auger, P. -A., Bravo, A., Molina, E. The 2016 red tide crisis in southern Chile: Possible influence of the mass oceanic dumping of dead salmons. Marine Pollution Bulletin. 150, 110603(2020).
- Swanson, H. A. Caught in Comparisons: Japanese Salmon in an Uneven World. , University of California, Santa Cruz. Doctor of Phylosophy thesis (2013).
- Sakai, Y., et al. Discovery of an unrecorded population of Yamato salamander (Hynobius vandenburghi) by GIS and eDNA analysis. Environmental DNA. 1 (3), 281-289 (2019).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten