
Method Article
A Standardized Procedure for Monitoring Harmful Algal Blooms in Chile by Metabarcoding Analysis
In This Article
Summary
This protocol introduces steps of metabarcoding analysis, targeting 16S rRNA and 18S rRNA genes, for monitoring harmful algal blooms and their associated microbiome in seawater samples. It is a powerful molecular-based tool but requires several procedures, which are visually explained here step-by-step.
Abstract
Harmful algae blooms (HABs) monitoring has been implemented worldwide, and Chile, a country famous for its fisheries and aquaculture, has intensively used microscopic and toxin analyses for decades for this purpose. Molecular biological methods, such as high-throughput DNA sequencing and bacterial assemblage-based approaches, are just beginning to be introduced in Chilean HAB monitoring, and the procedures have not yet been standardized. Here, 16S rRNA and 18S rRNA metabarcoding analyses for monitoring Chilean HABs are introduced stepwise. According to a recent hypothesis, algal-bacterial mutualistic association plays a critical synergetic or antagonistic relationship accounting for bloom initiation, maintenance, and regression. Thus, monitoring HAB from algal-bacterial perspectives may provide a broader understanding of HAB mechanisms and the basis for early warning. Metabarcoding analysis is one of the best suited molecular-based tools for this purpose because it can detect massive algal-bacterial taxonomic information in a sample. The visual procedures of sampling to metabarcoding analysis herein provide specific instructions, aiming to reduce errors and collection of reliable data.
Introduction
Many marine phytoplankton species are known to produce endogenous toxins, and when these species accumulate in sufficient numbers, they are harmful to the marine environment. Such Harmful Algal Blooms (HABs) are observed today on the coasts of most continents of the world1. Toxic phytoplankton first accumulate in bivalve tissues, leading to illness and death in higher trophic levels of organisms, including humans, upon digestion. Subsequently, these events bring severe implications to the local economy, socioeconomics, and public health2. The damage to the global economy caused by HABs is estimated to be millions to billions of dollars each year3. Chile is one of many countries suffering from frequent HABs.
Chile is a country of long land, stretching over 4,300 km north and south. The elongated western land faces the Pacific Ocean, which naturally increases the chances of Chile experiencing HABs. Especially in southern Chile, there are many world-famous salmon aquacultures, and every time a HAB occurs in the region, the algal-toxins result in massive farmed salmon falling ill and die4,5,6. In Chile, the year when the HAB hit the economy hardest was 2016, with an estimated annual loss of US $ 800 million7,8. The causative toxic algal species vary by year and location. For the 2016 case, a complex of Alexandrium catanella and Pseudochattonella verruclosa caused a widespread HAB in most of the southern Chilean coast7,8. The most recent HAB in Chile occurred with the causative algal species of Heterosigma Akashiwo in March 2021 at Camanchaca, located in southern Chile, where the area has a large salmon farm.
Chile has been conducting coastal monitoring for many years using two main methods; observing seawater with microscopes to identify toxic algae species regularly and measuring toxin levels in shellfish by biochemical assays9. Early detection of toxic algae and toxin levels in shellfish do not prevent HABs; however, these analyses can trigger immediate countermeasures and reduce damages to local communities. To further strengthen the effectiveness of this strategy, a molecular-based analytical method was recently added to our conventional Chilean HAB monitoring program to detect algae and related bacterial communities in seawater samples. Specifically, a Massive Parallel Sequencing method using metabarcoding that targets 16S rRNA and 18S rRNA genes was chosen. Although this technique requires complicated procedures and expensive machinery and reagents, it is an advanced technology that can detect thousands of algae and bacteria genera/species present in a seawater sample at once.
The causes of HAB are speculated to be various, such as temperature and season, but it is impossible to generalize them. This is because HAB species and frequencies depend on the region and spatiotemporal conditions, involving natural phenomena such as geographic uniqueness, upwelling nutrient mixing, and element runoff from land due to errosion2,10,11,12. Additionally, artificial factors such as eutrophication influence local HABs12,13. Due to the complex multifactor, it is not easy to make an accurate HAB prediction. In recent years, there is a view that specific bacterial populations may be related to the development of HABs as one of the factors, and research to support this hypothesis has been increasingly evident14,15,16,17,18. Molecular biology techniques are generally used to study bacterial assemblage; however, such a standardized method has not yet been established in Chilean HAB monitoring9. To study algal-bacterial association with HABs, it is imperative to concurrently perform metabarcoding analysis for the current Chilean coastal monitoring programs. Thus, this protocol visually introduces our Chilean HAB monitoring program, focusing on a stepwise procedure for analyzing algae and bacteria species detection in seawater samples using metabarcoding analysis.
The full protocol describing our Chilean HAB monitoring program is available in Yarimizu et al.9. It includes the procedures of seawater sampling, microscopic algal species detection, algal-bacterial gene detection, pigment analysis, meteorological data collection, and physical and chemical water property assays. The stepwise protocol of 16S rRNA and 18S rRNA metabarcoding analysis for algal and bacterial species detection is available as a preprint19. This protocol demonstrates particularly metabarcoding analysis steps since it is the most complicated part and the highlight of our HAB monitoring program. This protocol also includes an introduction of the program and microscopy detection of algal species. When analyzing algal species by metabarcoding, it is crucial to simultaneously perform microscopy to verify the results from the two methods. This protocol does not include how to use software for taxonomy assignment, but database recommendation is briefly stated at the end of the following section.
Protocol
1. Sample collection and pretreatment
- Collect approximately 3 L of water sample from the target spot.
- Filter 1 L of water sample for 16S rRNA analysis through a tandem filtration (1 µm and 0.2 µm pore-size membrane) to separate free-living and attached bacteria.
- Filter another 1 L of water sample for 18S-rRNA analysis (phytoplankton detection) through a single filtration with a 0.2 µm membrane.
NOTE: Filtration of the water sample must be completed within 12 h of sampling. - Cut filtered membrane in half with sterile surgical scissors and wrap it with aluminum foil. Store at -20 °C for upto 1 month or proceed to the next step.
- Extract DNA with Chelex method as described9. Store at -20 °C for upto 1 month.
2. Microscope analysis
- Transfer 1 mL of the water sample by a pipette onto a 1 mL grid-slide.
- Observe the sample under a microscope.
- Record names and quantity of phytoplankton species.
3. 16S rRNA and 18S rRNA metabarcoding analysis
NOTE: This process consists of seven sections: preparation, first PCR amplicon generation, first amplicon cleanup, indexation by second PCR, second PCR amplicon verification and cleanup, DNA concentration adjustment, and DNA denaturation and sequencing. The entire process takes a minimum of 5 days (40 h) by an experienced lab personnel. See the Table of Materials for product numbers and manufactures.
- Preparation
- Clean pipettes and laminar hood cabinet with 70% ethanol followed by UV exposure for 30 min frequently. Sterilize materials to be used.
- Thaw DNA samples at room temperature, centrifuge at 100 x g for 2 min, and transfer 100 µL of each sample supernatant to 8-tube strips.
- First PCR amplicon generation
NOTE: Perform the following procedures in a laminar hood cabinet. Always dilute primers from 1 µM stock to the target concentration with PCR grade water to avoid primer contamination.- Prepare first PCR master mix in a sterile 1.5 mL tube for reactions (Table 1, Table 2).
- Aliquot 22.5 µL of the master mix in an 8-tube strip and add 2.5 µL of DNA sample. Use 2.5 µL of PCR grade water for negative control.
- Run the first PCR cycle (Table 3).
- Prepare 100 mL of 2% Agarose-TBE gel containing 10 µL of 1x nucleic acid gel stain.
- Load a mixture of 1.5 µL of 1x DNA loading dye and 4 µL of PCR product on the agarose gel. Also, load 100 bp DNA ladder on the gel.
- Perform electrophoresis at 100 V for 30 min.
- Ensure that there is a band at 500-600 bp range under a UV light image capture. Primer-dimer band is round 80 bp.
NOTE: Marine water samples contain PCR inhibitors. Missing amplicons can sometimes be solved by diluting samples 1:100 or 1:1000 with PCR grade water. - Store first PCR products at -20 °C until the next step.
CAUTION: Do not exceed one week of storage.
- First PCR amplicon cleanup
NOTE: This section can be performed outside a laminar hood cabinet.- Use magnetic beads DNA cleanup system to remove PCR reaction residues, including primer-dimer products.
- Transfer 20 µL of each cleaned first PCR product to a new 96 well plate. Seal the plate with a micro-seal film. Store at -20 °C until the next step proceeds.
CAUTION: Do not exceed more than one week of storage.
- Indexation by second PCR
NOTE: In this section, purified first PCR products will be amplified with various index primer combinations.- Dilute all Index 1 and Index 2 primers (Table 4) to 1 µM with PCR grade water in 8 tube PCR strips placed in a laminar hood cabinet.
NOTE: The steps henceforward in this section can be performed outside of a laminar hood cabinet, as index primers are specific to the first PCR reactions overhang adapter. - Position Index 1 primer in a horizontal row Index 2 primers in a vertical row (Table 5).
- In a new 96-well plate, add 12.5 µL of hot-start-ready formulation to each well.
- Add 2.5 µL of each index primer (1 µM) to each well as in Table 4 using a multichannel pipette.
- Add 7.5 µL of purified first PCR product.
- Mix gently by pipetting up and down 10 times. Cover the plate with a micro-seal film.
- Run the second PCR cycle (Table 3).
- Keep the plate at -20 °C.
CAUTION: Do not exceed one week of storage.
- Dilute all Index 1 and Index 2 primers (Table 4) to 1 µM with PCR grade water in 8 tube PCR strips placed in a laminar hood cabinet.
- Second PCR amplicon verification and cleanup
- Use a fragment analyzer and associated reagent. Vortex and spin down reagent before use.
- Allow the sample buffer and DNA screen tape to equilibrate at room temp for 30 min. Then, place the DNA screen tape in a fragment analyzer.
- Mix 2 µL of sample buffer and 3 µL of second PCR amplicon in new 8 tube strips. Insert the 8 tube strips into the fragment analyzer. Press Run to start.
CAUTION: The formation of air bubbles must be avoided. - Ensure that second PCR amplicons are approximately 613 bp (600 - 630 bp) for both 16S and 18S rRNA genes.
- Purify second PCR products using a magnetic beads DNA cleanup system.
- DNA concentration adjustment
- Measure DNA concentration in the purified second PCR products using a nucleic acid quantification spectrophotometer.
- Calculate target gene concentration in nM, accounting for the average final library size as 613 bp for both 16S and 18S rRNA genes:

- Dilute each purified second PCR product with sterile PCR water to 4 nM in a new 0.2 mL 96 well plate.
NOTE: The plate can be stored at -20 °C here. Otherwise, the rest of procedures must be performed without halting. - Aliquot 3 µL of each 4 nM second PCR product and mix all in a new sterile 1.5 mL tube as a pooled library. Keep the tube at 4 °C or on ice bath at all times.
- Measure the concentration of the pooled library for confirmation using a nucleic acid quantification spectrophotometer. Adjust the concentration to 4 nM if it is higher than 4 nM.
NOTE: Over-concentrated DNA produces overestimated read numbers, hampering analysis.
- DNA denaturation and sequencing
NOTE: This section uses Illumina MiSeq system with specific reagents. See product numbers in Table of Materials.
NOTE: Strictly follow time. Thaw all reagents except cartridges on the day of sequencing.- A day before sequencing, remove a pre-filled ready-to-use reagent cartridge from -20 °C and store at 4 °C for thawing.
- Set a heat block suitable for 1.5 mL centrifuge tubes to 96 °C.
- Place the hybridization buffer on ice.
- Dilute molecular grade NaOH from 1 N to 0.2 N in a new tube with PCR grade water.
- Dilute a ready-to-use control library with TE buffer (pH 8.0) from 10 nM stock to 4 nM in a new tube (i.e., 2 µL of control library + 3 µL of TE buffer).
- Mix 16 µL of 4 nM pooled sample library with 4 µL of 4 nM control library in a new tube labeled "1".
- Mix 10 µL of sample in tube "1" with 10 µL of 0.2 N NaOH in a new tube labeled "2".
- Vortex the tube 2 for 5 s, spin down briefly, and incubate at room temperature for 5 min.
- Add 980 µL of hybridization buffer to tube 2.
- In a new tube labeled "3", mix 260 µL the sample from tube 2 with 390 µL of hybridization buffer. Mix by inverting the tube.
- Incubate tube 3 at 96 °C for 2 min and immediately place it on ice for a maximum 2 min.
- Remove the cartridge from 4 °C refrigerator.
- Set up the sample-sheet for sequencing with each corresponding index 1 and index 2 adapters as in Table 6.
- Remove the flowcell from the MiSeq v3 kit. Gently clean the flowcell with sterile molecular grade PCR water.
NOTE: Do not pour water on the capillary dots of the flowcell. Gently wipe off water from the flowcell with non-fibrous paper. - Load the full volume of tube 3 (650 µL) into the cartridge.
- In instrument operation software, select sequencing and follow the instructions. Insert flowcell, incorporation buffer, and cartridge. Load sample-sheet. Press Run to start the reaction.
NOTE: This sequence run will take 3-5 days. The data will be automatically uploaded to Basespace platform. Raw data will be stored in two folders in computer: Analysis folder containing fastq files and Output containing bcl files and jpeg pictures.
4.Taxonomic assignment to sequencing data
- FASTQ file processing
NOTE: DADA2 package23 of R24 is one of recommended databases to process FASTQ files. The guideline is available at [https://github.com/mickeykawai/exec_dada2]. This guideline was prepared by adopting the recent version of DADA2 pipeline tutorial [https://benjjneb.github.io/dada2/tutorial.html].- Process raw FASTQ paired-end sequences for trimming, quality filtering, dereplication and counting of unique sequences, sample inference, merging into contigs and removing chimeric sequences.
- Use following DADA2 specified parameters for both 16S rRNA and 18S rRNA gene fragments; trimLefts=0, 0, truncLens=[Check the sequence quality and set it to an appropriate length].
NOTE: Default values for the other parameters are maxN=0, maxEE=2,2, trancQ=2.
- Taxonomy assignment
NOTE: SILVA rRNA database is recommended for taxonomy assignment for DADA2 [silva_nr_v132_train_set.fa.gz is set as a default].- Remove chloroplast and mitochondrial OTUs for 16S rRNA gene fragment analysis. Remove singletons from 16S rRNA and 18S rRNA analysis.
- Rarify all samples to even sequencing depth based on the sample having the lowest sequencing depth.
- Record the number of reads, OTUs assigned and at what level, filtered reads, and the number of singletons excluded.
- Statistical analysis
- Perform statistical analysis on microbial data using R packages of phyloseq25 and vegan26.
Results
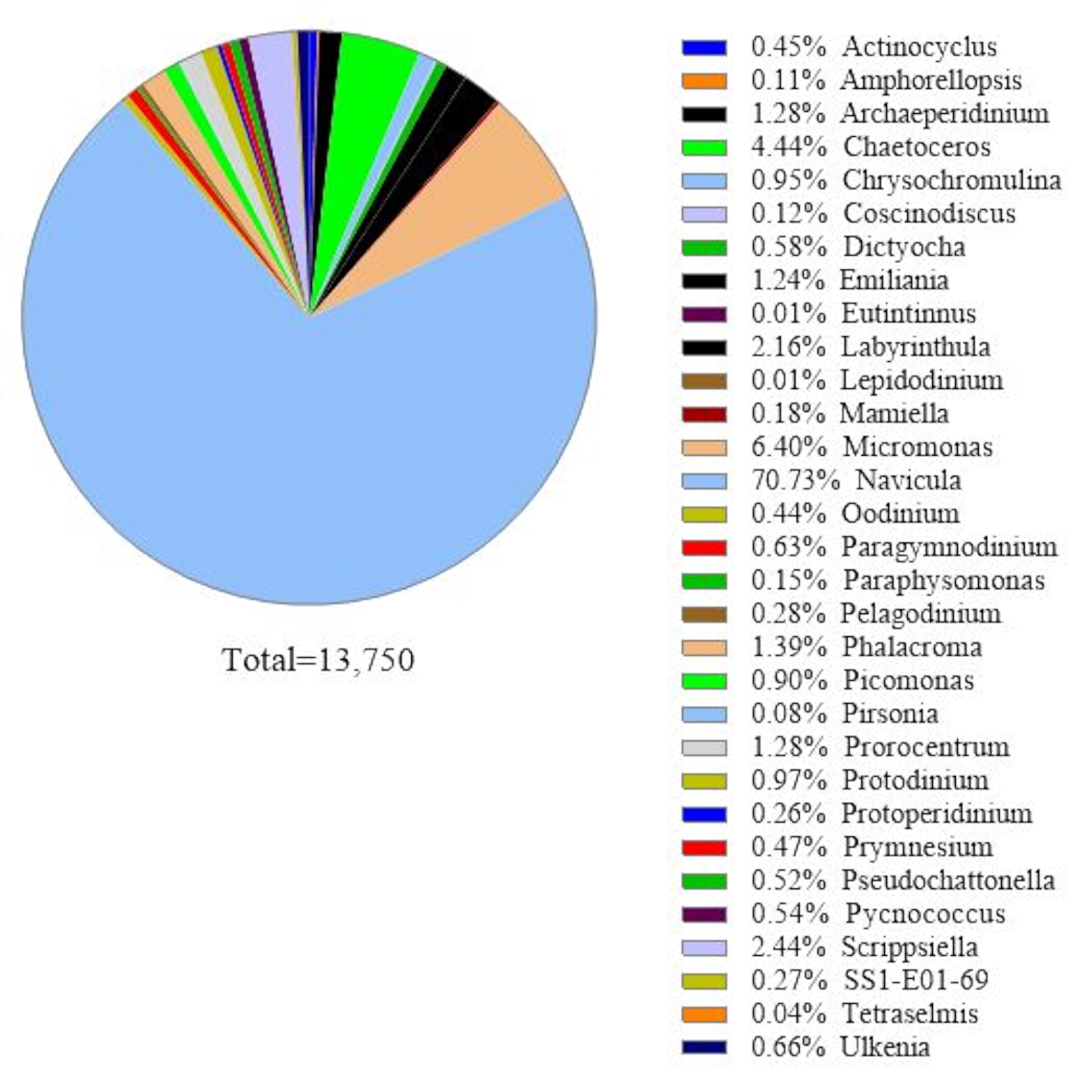
This protocol uses 18S rRNA gene metabarcoding analysis to identify algal species in seawater samples. A representative result is shown in Figure 1, in which the seawater collected from Metri, Puerto Montt, Chile (−41.597; −72.7056) on February 19, 2019, was analyzed with this protocol. The result showed a total of 13,750 reads with over 30 algal species in the seawater sample. The dominant alga in this sample was Navicula spp. with a relative abundance of 70.77%. Also, sufficient abundance was observed for Micromonas (6.40%), Chaetoceros spp. (4.44%), Scripsiella spp. (2.44%), and Prorocentrum spp. (1.28%). Pseudochattonella spp., one of the highest toxic algal causatives of Chilean HAB, was detected with 0.52% from this seawater sample.
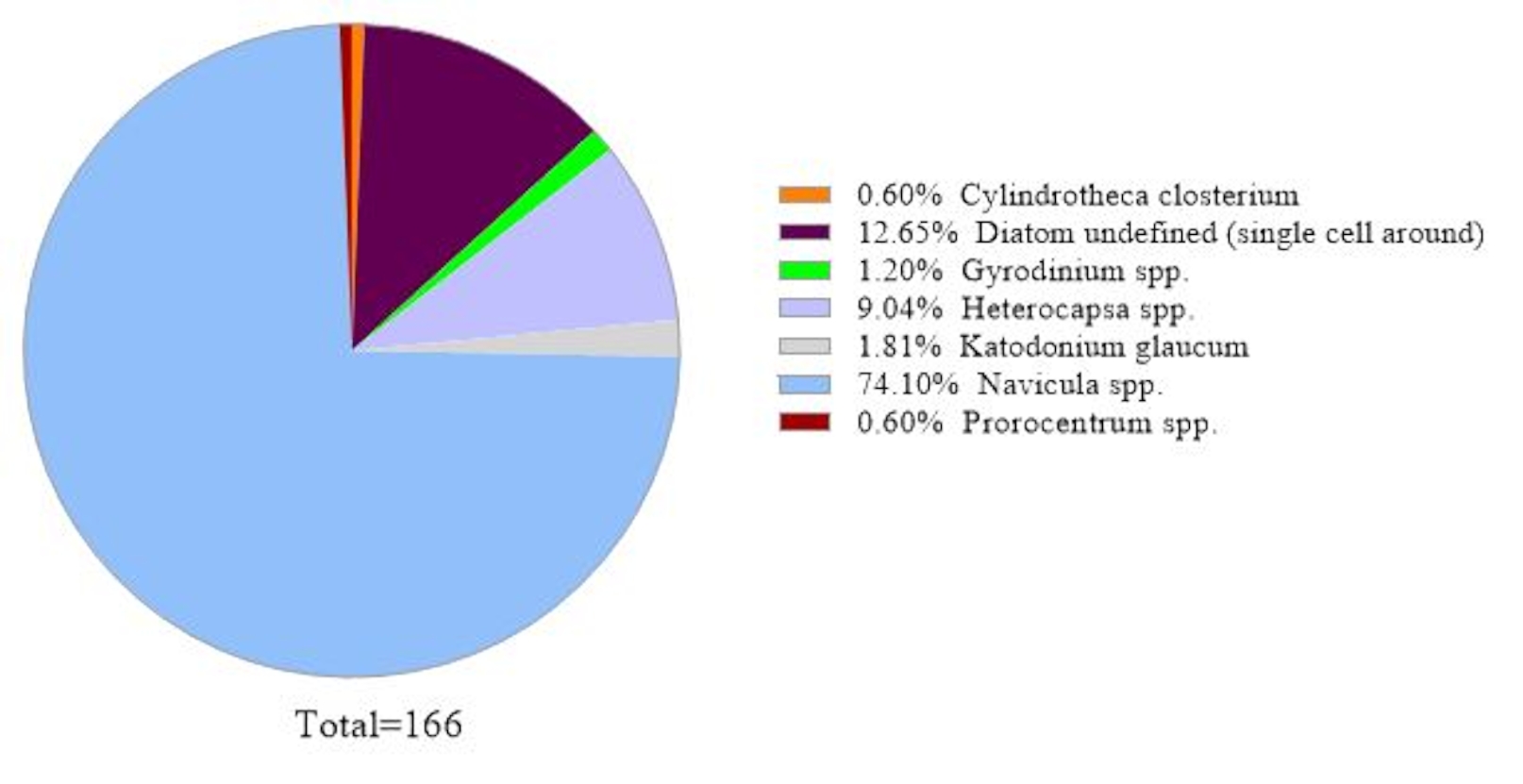
To verify the data reliability, the algal species identified by 18S rRNA gene analysis were compared to those obtained by microscopy in the same seawater sample (Figure 2). Consistent with 18S rRNA gene analysis, the microscopy showed that the dominant species was Navicula spp. with a relative abundance of 74.1% and Prorocentrum spp. (0.60%) as a minor species. Conversely, Heterocapsa spp. (9.04%) documented from microscopic observation was not identified by 18S rRNA gene analysis in this sample. There was 12.6% of small around unidentifiable phytoplankton cells recorded by microscopy. This could be Micromonas, according to the 18S rRNA results.
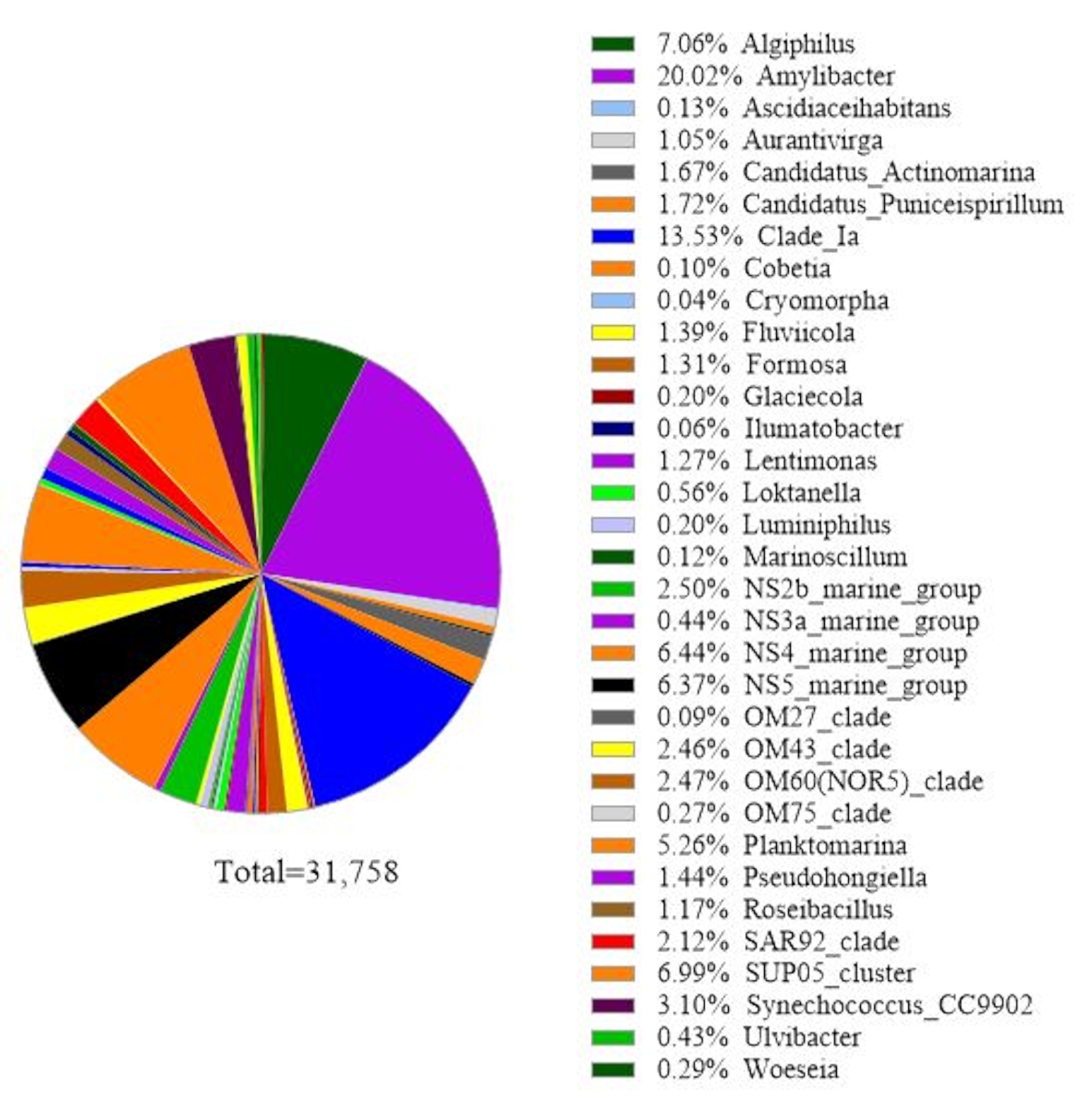
The protocol uses 16S rRNA gene metabarcoding analysis to identify bacterial species in seawater samples. A representative result is shown in Figure 3, in which the same seawater used for 18S rRNA gene analysis was analyzed with 16S rRNA gene analysis. The result showed a total of 31,758 reads with over 30 bacterial species in the seawater sample. It should be outlined that this seawater sample was passed through tandem filter membranes (1 µm and 0.2 µm pore sizes) to separate free-living bacteria from attached bacteria. Then, cells captured by each filter membrane were treated for DNA extraction, followed by the 16S rRNA gene analysis. The representative result in Figure 3 shows bacteria species identified from 0.2 µm pore size membrane, which are defined as free-living bacteria. The dominant free-living bacteria was Amylibacter spp. with a relative abundance of 20.02%, followed by Clade Ia (13.53%) and Aligiphilus spp. (7.06%). The rest of the bacterial species detected from this seawater sample were relatively equally distributed. The same analysis can be done for cells captured by 1 µm pore-sized membrane as attach-bacteria species detection.
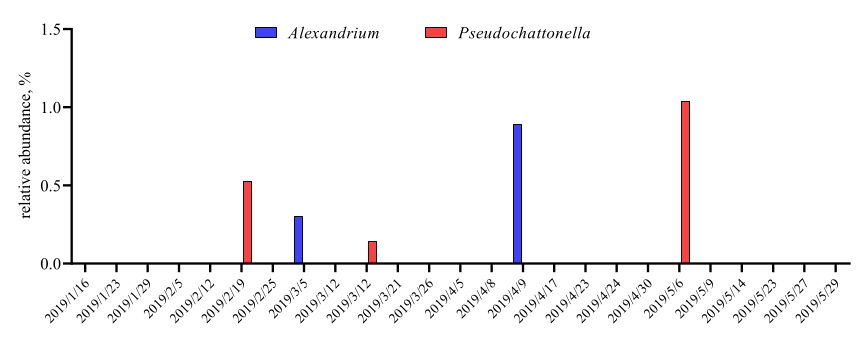
When these metabarcoding assays are performed at scheduled time-points over a certain study period, the results can be summarized as time-series analysis. One way to do it is to plot the relative abundance of particular algal and bacterial species as a function of time to find a unique growth pattern. Figure 4 shows a representative time-series plot of Alexandrium and Pseudochattonella in Metri, Chile. Another way to summarize a time-series metabarcoding analysis is to plot all identified algal and bacterial as a function of time, representing the population change of certain groups of organisms. Figure 5 and Figure 6 summarizes the relative abundance of all the bacterial genus and order, respectively, which are detected from the seawater of Metri over five months.
Table 1: First PCR master mix content: The table shows master mix content per reaction for 16S rRNA and 18S rRNA analyses. The primer sequences are listed in Table 3. Please click here to download this Table.
Table 2: Primer sequences: The primers for first PCR are listed for 16S rRNA and 18S rRNA analyses. Please click here to download this Table.
Table 3: PCR cycles: The thermal cycles for first PCR and second PCR are listed. Please click here to download this Table.
Table 4: Index sequences: Index 1 (i7) and Index 2 (i5) primers to be used for second PCR are listed. Please click here to download this Table.
Table 5: Example of Index 1 and Index 2 positioning: To reduce error, index primers must be placed in the position first, and aliquot of each is transferred to a 96-well plate using multichannel pipette. Please click here to download this Table.
Table 6: Example of sample-sheet: Prior to sequencing, a sample-sheet must be created corresponding to index 1 and index 2 adapters. Please click here to download this Table.

Figure 1: Representative result of 18S rRNA metabarcoding analysis: Algal species present in a seawater sample collected from Metri, Los Lagos, Chile on February 19, 2019, were identified by 18S rRNA metabarcoding assay. The sequences assigned "unknown" were eliminated from, and the relative abundance of each identified species was plotted. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Representative result of microscopic analysis: The algal species were identified from a water sample from Metri, Los Lagos, Chile on February 19, 2019, by microscopy. The quantity of each species was counted manually and plotted. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Representative result of 16S rRNA metabarcoding analysis: Bacterial species present in a water sample from Metri, Los Lagos, Chile on February 19, 2019, were identified by 16S rRNA metabarcoding assay. The relative abundance of each identified species was plotted. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Representative time-series plot of Alexandrium spp. and Pseudochattonella spp. obtained from 18S rRNA metabarcodingin Metri, Chile: Figure reprinted from Yarimizu et al9. The two toxic algal species, Alexandrium and Pseudochattonella were selectively monitored by 18S rRNA analysis over time, and the relative abundance were plotted as a function of time-point. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Representative genus time-series plot obtained from 16S rRNA and 18S rRNA metabarcoding: All bacterial and eukaryotic genus identified in the water of Metri, Chile, monitored over five months. Please click here to view a larger version of this figure.
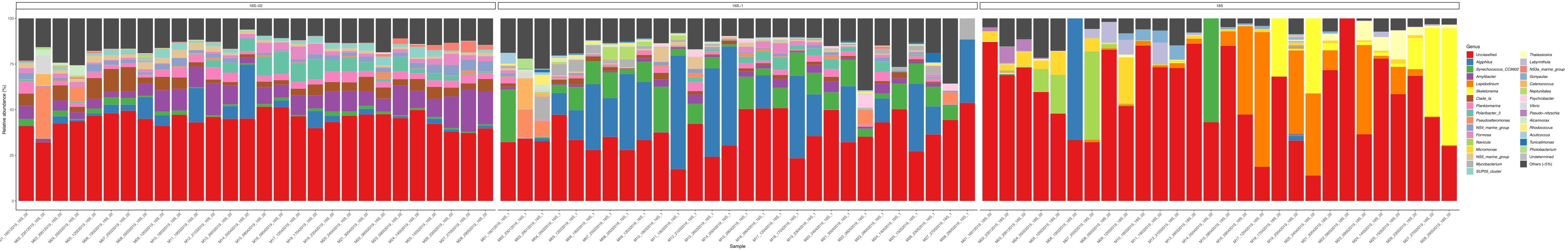{kind=link}

Figure 6: Representative order time-series plot obtained from 16S rRNA and 18S rRNA metabarcoding: All bacterial and eukaryotic orders identified in the water of Metri, Chile, monitored over five months. Please click here to view a larger version of this figure.
{kind=link}
Table S1 and Table S2:
Table S1: Raw data for Figure 5 and Figure 6. Table S2: Result of QC check on raw data of Figure 5 and Figure 6. Please click here to download these Tables.
Discussion
This protocol successfully performed 18S rRNA and 16S rRNA gene metabarcoding analysis to identify algal and bacterial species in seawater samples for monitoring Chilean HABs. The dominant algae and some minor algae detected by this protocol were consistent with those obtained by microscopy, confirming the protocol's reliability. The highlight of the protocol is that the analysis detected Alexandrium spp. and Pseudochattonella spp., the two most problematic HAB causing species in southern Chile, from the seawater of Metri even at a low abundance. Especially, Pseudochattonella spp. are challenging species to identify under a microscope because the cells are small and easily ruptured under light or use of fixatives27,28. Metabarcoding can provide algal species information that is scarcely present in seawater samples that microscopy cannot identify. The protocol also detected over 30 bacterial species. Although how these bacteria are related to algal growth is unknown at this point, massive data collection of algal-bacterial species in the time-series analysis will possibly provide such important discoveries. Thus, adding metabarcoding analysis to the conventional Chilean HAB monitoring programs will undoubtedly strengthen the current HAB monitoring efficiency. In fact, this visual protocol for metabarcoding analysis can be beneficial not only for Chilean water's algal-bacterial monitoring but also for the coast monitoring programs in other HAB-affected areas worldwide.
Although this protocol provided the above-stated advantages, the drawbacks of the method should be discussed. As seen in the visual protocol, the metabarcoding method is time-consuming and complex, requiring expensive machinery and reagents. The laboratory personnel must be specially trained, or it will waste material, labor, and time. In addition, algal detection by metabarcoding must be paired with microscopy to verify that the same dominant species are identified from the two orthogonal methods29. Microscopy is a non-invasive tool, for the most part, to identify algal species, meaning that it is harder to make mistakes when the algal species have unique and apparent shapes. Of course, human error can occur if the species have very similar shapes to one another. On the other hand, since metabarcoding analysis requires multiple steps, it naturally increases errors. It could be a sample mixed up, wrong reagent addition, or missing some procedures. Therefore, it is critical to compare the metabarcoding results with those obtained by microscopy. Lastly, the protocol is applicable only qualitatively, and the results must be calculated for relative abundance. As Lamb et al. stated in 2019, the current metabarcoding as the quantitative performance is still limited, and additional research is required before metabarcoding can be confidently utilized for quantitative applications30.
This protocol was optimized for 140 - 170 samples per run from the 16S Metagenomic Sequencing Library Preparation manual issued by Illumina Inc. The optimized protocol was tested many times and further modified to issue this final version. Therefore, it is highly recommended to follow each step precisely. The most critical part of the protocol is that extra care is required to avoid sample contamination. The pipettes and laminar flow hood must be cleaned with 70% alcohol and UV exposure at all times, and sterilizable materials should be autoclaved. The primers and reagents should be diluted directly from the stock at every new run, or reuse of reagents and exposure to several dilutions can be a sample contamination cause. The protocol specifies when the operation can be done outside of the laminar flow hood. Unless otherwise, samples should be treated in a cleaned laminar flow hood. When dealing with multiple 8-tube strips simultaneously, it is recommended to work on the first 8-tube strip and cap it before moving to the next 8-tube strip. Leaving the lid open for a long time can cause sample contamination as 16S and 18S rRNA genes are highly ubiquitous in surroundings. The stated time, such as mixing time and incubation time, should be followed as exactly described because the best duration for each step was selected based on the multiple test runs. Excess or insufficient time can, for example, reduce sample yield. The process should be halted only at the part that protocol says it can stop. It is crucial to have a negative control as it confirms that the positive outcomes are not artificial false positives. Lastly, it is highly recommended to plan the days because it requires at least five days to process all the steps, and some parts cannot be stopped until the next halt sign.
The limitation of taxonomic assignment for sequenced data are briefly noted here. Newly discovered nucleotide sequences for bacterial and algal species are updated in the databank daily. While well-studied algal and bacterial species are registered reliably, there are also many sequences for unknown species updated in the databank. It indicates that the resolution of registered nucleotide sequences is not always enough for species identification but only for genus identification. Thus, microscopic species identification must be orthogonally performed. Establishing an open-access pipeline for this work has a significant advantage in dealing with a large set of sequenced data at once and efficiently investigating when an error occurs. Additionally, the output of DADA2 is in a text or csv file, which makes it easy to further statistical analysis. On the other hand, a large server is required to perform taxonomic assignments, especially when a dataset is large. To set up the pipeline, engineers are needed to set up the pipeline, and the professionals must understand parameters, operation, Linux, and bioinformatics.
Aside from the scope as a HAB monitoring tool, the use of this protocol can be expanded for investigative research purposes. For example, there are ongoing debates between the Chilean government and local fishermen along with environment peace associations regarding the increased number of local HAB31,32. The government claims that it is due to a natural phenomenon such as global warming and El Niño, while the later parties claim that salmon aquaculture is the cause. Salmon is not an indigenous species of Chile and was not in Chilean water a half-century ago. The Chilean government at that time aimed to grow the economy and create jobs for poor areas by developing a salmon business33. With the intervention from overseas for capital profit, Chile made great success in pen-style aquaculture development, and the number of salmon has increased remarkably in the last several decades31,32,33. Subsequently, a large amount of food for salmon has been thrown into the sea, leading local people to suspect that salmon aquaculture is the reason for the frequent local HABs31,32. The truth is unknown now, but it must be understood eventually so that strategies to protect the local marine environment, economy, and human health from HABs can be devised. The molecular-based analysis on the local algal-bacterial relationship may contribute to a step forward clarification for such a subject. For example, the technique can search the algal and bacterial species that were not present in the target oceanic area before but have increased dramatically in recent years, which is somewhat similar to the study done by Sakai et al. for the discovery of an unrecorded population of Yamato salamander (Hynobius vandenburghi) by GIS and eDNA analysis34. Therefore, beyond the HAB monitoring tool, this metabarcoding protocol has the potential usage for other prospects.
Disclosures
The authors declare no conflict of interest. The funders had no role in the design of the study, in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Acknowledgements
This study was supported by the grant (JPMJSA1705) for a study on Science and Technology Research Partnership for Sustainable Development-Monitoring Algae in Chile (SATREPS-MACH). We thank Dr. Sandra Rios (Universidad de Los Lagos) for allowing us to use a film clip. We thank Neal Andrew Holland for his diligent proofreading of the manuscript. We express our sincere appreciation to the laboratory group members at CREAN-IFOP at Puerto Montt, Chile, for advising us on phytoplankton identification.
Materials
| Name | Company | Catalog Number | Comments |
| 1.5 mL single tube | ThermoFisher | Q32856 | sterile |
| 1.5 mL tubes | ThermoFisher | 2150N | sterile |
| 8-strip 0.2 mL PCR tubes and flat cap | ThermoFisher | AB0451 & 4323032 | sterile |
| 96-well 0.2 mL PCR plate | ThermoFisher | N8010560 | |
| AccuRuler 100 bp DNA Ladder | MaestroGen | 02001-500 | |
| Chelex 100 Chelating Resin | Bio-Rad | 1432832 | |
| Chemical Duty Vacuum Pressure Pump | MilliporeSigma | WP6122050 | |
| D1000 Reagents | Agilent Technologies | 5067-5583 | |
| D1000 sample buffer | Agilent Technologies | 5067-5602 | |
| DNA loading dye (Blue/Orange Loading Dye 6X) | Promega | G1881 | |
| Ethanol Molecular Biology Grade | E7023 | Merck KGaA | |
| Filtration device | ThermoFisher | 09-740-36H, 09-740-23E | |
| Fluorescent DNA concentration measurement kit (Qubit dsDNA HS Assay kit) | Thermo Fisher Scientific | Q32851 | |
| Fluorometer (Qubit4 Fluorometer) | ThermoFisher | Q33238 | |
| Fragment analysis verification matrix (D1000 ScreenTape) | Agilent Technologies | 5067-5582 | |
| Fragment Analyzer (Agilent Tapestation 4150) | Agilent | G2992AA | |
| GelRed Nucleic Acid Gel Stain | Biotium | 41002 | |
| Generic bench vortexer (Vortex Genie 2) | Scientific Industries | SI-0236 | |
| Heat block | |||
| High performance sequencing-grade DNA polymerase (KAPA HiFi Hotstart ReadyMix), 2X | Roche | KK2602 | |
| HT1 Hybridization buffer | Illumina | 20015892 | |
| Illumina Nextera XT v2 Index Kit set A, B, C and D | Illumina | FC-131-2001, -2002, -2003, -2004 | |
| Laminar hood cabinet | Biobase Co. | Biobase PCR-800 PCR Cabinet | |
| Loading tips (Specific for Agilent TapeStation systems) | Agilent Technologies | 5067-5598 | |
| Magnetic beads DNA cleanup system (ProNex® Size-Selective Purification System) | Promega | NG2002 | |
| Magnetic stand for 96 wells (Magnetic Stand-96) | ThermoFisher | AM10027 | |
| Micro-seal film for 96-well plate | ThermoFisher | 4311971 | sterile |
| MiSeq Reagent Kit v3 | Illumina | MS-102-3003 | includes pre-filled, ready-to-use reagent cartridges. |
| MiSeq system | Illumina | SY-410-1003 | includes pre-installed software |
| Molecular grade nuclease-free water | Integrated DNA Technologies | 11-05-01-04 | |
| Molecular grade sodium hydroxide (NaOH) 1M | Merck KGaA | 1091371000 | |
| Multichannel pipettes | Not specified | Not specified | |
| Multi-Purpose Agarose | Cleaver Scientific | CSL-AG500 | |
| Ow D2 Wide-Gel Electrophoresis System | ThermoFisher | D2 | |
| Ow EC-105 Compact Power Supply | ThermoFisher | 105ECA-115 | |
| Pellet Pestle— Cordless Motor | ThermoFisher | K749540-0000 | |
| PhiX control Kit v3 | Illumina | FC-110-300 | ready-to-use control library for Illumina sequencing |
| Pipette tips | Not specified | Not specified | sterile |
| Pipettes | Not specified | Not specified | 2, 10, 20, 100, 1000 μL |
| SterivexTM GP 0.22 μm filter unit | MilliporeSigma | SVGP01050 | |
| Tabletop centrifuge | Eppendorf 5427R Centrifuge | Eppendorf AG | |
| TBE buffer | ThermoFisher | B52 | |
| TE buffer (pH 8.0) | ThermoFisher | AM9849 | |
| Terr PCR Direct FFPE Kit | Takara Bio USA | 639284 | includes 2X Terra PCR Direct Buffer |
| Terra PCR Direct Polymerase Mix, 1.25 U/μL | Takara Bio USA | 639271 | High performance Inhibitor-tolerant DNA polymerase |
| ThermalCycler (MiniAMP Plus Thermal Cycler) | ThermoFisher | A37835 | |
| TransIlluminator and image capture system | Analytik Jena, AG | GelDoc-ItTS2 Imager | |
| Whatman 1.0 m pore-sized membrane | MilliporeSigma | WHA111110 |
References
- Hallegraeff, G., Enevoldsen, H., Zingone, A. Global harmful algal bloom status reporting. Harmful Algae. 102, 101992 (2021).
- Anderson, D. M. Red Tides. Scientific American. 271 (2), 52-58 (1994).
- Sanseverino, I., Conduto, A. D. S., Pozzoli, L., Dobricic, S., Lettieri, T. . Algal Bloom and its Economic Impact. , (2016).
- Molinet, C., Niklitschek, E., Seguel, M., Díaz, P. Trends of natural accumulation and detoxification of paralytic shellfish poison in two bivalves from the Norwest Patagonian inland sea. Revista de Biologia Marina Y Oceanografia. 45, 195-204 (2010).
- Mardones, J., Clement, A., Rojas, X., Aparicio, C. Alexandrium catenella during 2009 in Chilean waters, and recent expansion to coastal ocean. Harmful Algae News. 41, 8-9 (2010).
- Alvarez, G., et al. Paralytic shellfish toxins in surf clams Mesodesma donacium during a large bloom of Alexandrium catenella dinoflagellates associated to an intense shellfish mass mortality. Toxins (Basel). 11 (4), (2019).
- Clément, A., et al. Exceptional summer conditions and HABs of Pseudochattonella in Southern Chile create record impacts on salmon farms. Harmful Algae News. 53, 1-3 (2016).
- Trainer, V. L., et al. Pelagic harmful algal blooms and climate change: Lessons from nature's experiments with extremes. Harmful Algae. 91, 101591 (2020).
- Yarimizu, K., et al. Protocols for monitoring harmful algal blooms for sustainable aquaculture and coastal fisheries in Chile. International Journal of Environmental Research and Public Health. 17 (20), (2020).
- Roegner, G. C., Hickey, B. M., Newton, J. A., Shanks, A. L., Armstrong, D. A. Wind-induced plume and bloom intrusions into Willapa Bay, Washington. Limnology & Oceanography. 47 (4), 1033-1042 (2002).
- Tweddle, J. F., et al. Relationships among upwelling, phytoplankton blooms, and phycotoxins in coastal Oregon shellfish. Marine Ecology Progress Series. 405, 131-145 (2010).
- Glibert, P. M., Anderson, D. M., Gentien, P., Graneli, E., Sellner, K. G. The global complex phenomena of harmful algae blooms. Oceanography. 18 (2), 130-141 (2005).
- Paredes-Mella, J., Varela, D., Fernández, P., Espinoza-González, O. Growth performance of Alexandrium catenella from the Chilean fjords under different environmental drivers: Plasticity as a response to a highly variable environment. Journal of Plankton Research. 42 (2), 119-134 (2020).
- Azam, F., Malfatti, F. Microbial structuring of marine ecosystems. Nature Reviews Microbiology. 5, 782-791 (2007).
- Amin, S. A., et al. Interaction and signaling between a cosmopolitan phytoplankton and associated bacteria. Nature. 522, 98-101 (2015).
- Berdalet, E., et al. Marine harmful algal blooms, human health, and wellbeing: Challenges and opportunities in the 21st century. Journal of Marine Biology Association. 2015, 1-31 (2015).
- Ramanan, R., Kim, B. -. H., Cho, D. -. H., Oh, H. -. M., Kim, H. -. S. Algae-bacteria interactions: Evolution, ecology, and emerging applications. Biotechnology Advances. 34 (1), 14-29 (2016).
- Seymour, J. R., Amin, S. A., Raina, J. B., Stocker, R. Zooming in on the phycosphere: The ecological interface for phytoplankton-bacteria relationships. Nature Reviews Microbiology. 2, 17065 (2017).
- Maruyama, F., et al. 16S and 18S Metabarcoding analysis for Chilean coastal waters harmful algal blooms. protocol.io. , (2020).
- Tanabe, A. S., et al. Comparative study of the validity of three regions of the 18S-rRNA gene for massively parallel sequencing-based monitoring of the planktonic eukaryote community. Molecular Ecology Resources. 16, 402-414 (2016).
- Nishitani, G., et al. Multiple plastids collected by the Dinoflagellate Dinophysis mitra through Kleptoplastidy. Applied and Environmental Microbiology. 78 (3), 813-821 (2012).
- Klindworth, A., et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Research. 41 (1), 1 (2013).
- Callahan, B. J., et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nature Methods. 13 (7), 581-583 (2016).
- McMurdie, P. J., Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS One. 8 (4), 61217 (2013).
- Dixon, P. VEGAN, a package of R functions for community ecology. Journal of Vegetation Science. 14 (6), 927-930 (2003).
- Dittami, S. M., Riisberg, I., Edvardsen, B. Molecular probes for the detection and identification of ichthyotoxic marine microalgae of the genus Pseudochattonella (Dictyochophyceae, Ochrophyta). Environmental Science and Pollution Research. 20 (10), 6824-6837 (2013).
- Mardones, J. I., et al. Salinity-growth response and ichthyotoxic potency of the Chilean Pseudochattonella verruculosa. Frontiers in Marine Science. 6 (24), (2019).
- Santi, I., Kasapidis, P., Karakassis, I., Pitta, P. A comparison of DNA metabarcoding and microscopy methodologies for the study of aquatic microbial eukaryotes. Diversity. 13 (5), 180 (2021).
- Lamb, P. D., et al. How quantitative is metabarcoding: A meta-analytical approach. Molecular Ecology. 28 (2), 420-430 (2019).
- Mascareño, A., et al. Controversies in social-ecological systems: lessons from a major red tide crisis on Chiloe Island, Chile. Ecology and Society. 23, (2018).
- Armijo, J., Oerder, V., Auger, P. -. A., Bravo, A., Molina, E. The 2016 red tide crisis in southern Chile: Possible influence of the mass oceanic dumping of dead salmons. Marine Pollution Bulletin. 150, 110603 (2020).
- Swanson, H. A. . Caught in Comparisons: Japanese Salmon in an Uneven World. , (2013).
- Sakai, Y., et al. Discovery of an unrecorded population of Yamato salamander (Hynobius vandenburghi) by GIS and eDNA analysis. Environmental DNA. 1 (3), 281-289 (2019).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved