
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Entwicklung antiviraler Wirkstoffe mittels Oberflächenplasmonenresonanz
In diesem Artikel
Zusammenfassung
Das vorliegende Protokoll beschreibt neue Werkzeuge für SPR-Bindungsassays zur Untersuchung der CV-N-Bindung an HA, S-Glykoprotein, verwandte Hybrid-Glykane und High-Mannose-Oligosaccharide. SPR wird verwendet, um das KD für die Bindung von entweder dimerem oder monomerem CV-N an diese Glykane zu bestimmen.
Zusammenfassung
Die Oberflächenplasmonenresonanz (SPR) wird verwendet, um die Bindung von Hämagglutinin (HA) an das domänenvertauschte Cyanovirin-N (CV-N)-Dimer zu messen und die Wechselwirkungen zwischen mannosylierten Peptiden und der hochaffinen Bindungsstelle von CV-N zu überwachen. Es wurde berichtet, dass die Virushüllenspitzen gp120, HA und Ebola-Glykoprotein (GP) 1,2 sowohl hoch- als auch niedrigaffine Bindungsstellen an dimeres CVN2 binden. Dimannosyliertes HA-Peptid ist auch an den beiden niedrigaffinen Bindungsstellen an ein konstruiertes Molekül von CVN2 gebunden, das eine hochaffine Stelle für den jeweiligen Liganden trägt und mutiert, um eine stabilisierende Disulfidbindung in der kohlenhydratbindenden Tasche zu ersetzen, wodurch eine multivalente Bindung bestätigt wird. Es wird eine HA-Bindung an eine hochaffine Bindungsstelle des Pseudo-Antikörpers CVN2 bei einer Dissoziationskonstante (KD) von 275 nM gezeigt, die das humane Immundefizienzvirus Typ 1 (HIV-1) durch Oligomerisierung weiter neutralisiert. Die Korrelation der Anzahl der Disulfidbrücken in domänenvertauschtem CVN2, die durch Substitution von Cystinen in polare Restpaare aus Glutaminsäure und Arginin von 4 auf 2 verringert werden, führt zu einer reduzierten Bindungsaffinität zu HA. Unter den stärksten Wechselwirkungen wird Ebola GP1,2 von CVN2 mit zwei hochaffinen Bindungsstellen im unteren nanomolaren Bereich unter Verwendung des Hüllglykans ohne Transmembrandomäne gebunden. In der vorliegenden Studie wird die Bindung des multispezifischen monomeren CV-N an das schwere akute respiratorische Syndrom Coronavirus 2 (SARS-CoV-2) Spike (S) Glykoprotein bei K D = 18,6 μM im Vergleich zu nanomolaren KD an diese anderen Virusspitzen und über seine rezeptorbindende Domäne im mittleren μ-molaren Bereich gemessen.
Einleitung
Tetherin-assoziierte antivirale Aktivität wird durch Interferon-α induziert und umfasst proteinbasierte Tether, die zur Retention von vollständig ausgebildeten Virionen auf infizierten Zelloberflächen führen1. Die Notwendigkeit einer Tetheringlykosylierung bei der Hemmung der Virusfreisetzung bleibt ungewiss, was die Bedeutung von Glykosylierungsmustern auf rekombinant exprimierten Glykanen für In-vitro-Studien 1,2 impliziert, die von der Konformation von (im Falle des Influenzavirus) oberflächenexprimiertem Influenzahämagglutinin HA 3,4 abhängt. . Es wurde festgestellt, dass die Modifikation von Oligosaccharid, das an die N-verknüpfte Glykosylierung gebunden ist, für eine Tetherin-vermittelte Restriktion der HIV-Typ-1-Freisetzung2 ausreicht, während die Dimerisierung eine wesentliche Rolle bei der Verhinderung der Virusfreisetzung spielt, wodurch die Transmembrandomäne oder der Glykosyl-Phosphatidyl-Inositol (GPI)-Anker zum Anbinden der knospenden Virionen beteiligt ist5 . Es werden einzigartige Funktionen für menschliches und murines Tetherin beschrieben, um mehrere behüllte Viren, Retroviren und Filoviren zu blockieren. BST-2/Tetherin ist ein Interferon-induzierbares antivirales Protein der angeborenen Immunität1,6, das mit antiviraler Breitbandaktivität wirkt und durch Hüllglykoproteine5 antagonisiert wird, um entweder Tetherin zu translozieren oder die Struktur von Tetherin 6 zu stören. Zum Beispiel sind oberflächenexprimierte Hüllglykoprotein HA und Neuraminidase auf Influenza-A-Virus für Tetherin-Antagonismus in einer stammspezifischen Weise bekannt7, was die Erkennung von Wirtsrezeptorbindungsstellen erleichtert8. Glykan-Targeting-Antikörper werden in der Stöchiometrie ihrer Wechselwirkungen mit den schnell anpassenden Glykanschilden auf HA untersucht, was zu einer Bindungsaffinität zu Influenza A H3N2 und H1N1 Subtypen4 führt.
Um die Bindungsmechanismen zwischen antiviralen Wirkstoffen und Virushüllenspitzen, d.h. Kohlenhydratliganden, und komplementären immunologischen und spektroskopischen Methoden aufzuklären, werden Mono-, Di- und Tri-Mannose-Einheiten chemisch synthetisiert. Die mannosylierten Peptide werden durch Azido-Glykosylierung von Glykosyl {beta}-Peracetaten zu 1,2-trans-Glykosylaziden-Transformation9 erzeugt, die das typischerweise vorkommende N-Acetylglucosamin und die Oligosaccharide mit hohem Mannosegehalt auf der Oberfläche lebensbedrohlicher Viren nachahmen. Triazol-Bioisoster werden verwendet, um Verknüpfungen nachzuahmen, die den mannosylierten Rest des HA-Peptids10 bilden, und erleichtern ortsspezifische Wechselwirkungen mit antiviralen CV-N-Derivaten um den zweiten N-verknüpften Glykosylierungspunkt auf der HA-Kopfdomäne (HA-Spitze mit 4 N-verknüpften Glykanen N54, N97, N181, N301)8,11,12 . Wechselwirkungen zwischen Glutaminsäure (Glu) und Arginin (Arg) und der resultierende Helix-Dipol zeigten eine gute Stabilität sowohl von Modellpeptiden als auch von Proteinen, werden aber mit SPR visualisiert. Im Vergleich zur Erkennung einer einzelnen chemisch synthetisierten Glykosylierungsstelle auf HA10 durch direkte Hemmung der Rezeptorbindung an die Glykaneinheiten wird gezeigt, dass eine höhere Affinität einer vierfach mutierten Fc-Struktur zu ihrem Rezeptor Effektorfunktionen in vivo hervorruft, was zeigt, dass die nicht verwandte Zusammensetzung von N-verknüpften Glykanen, die an die Fc-Mutante gebunden sind, mechanistisch bestimmt werdensoll 13.
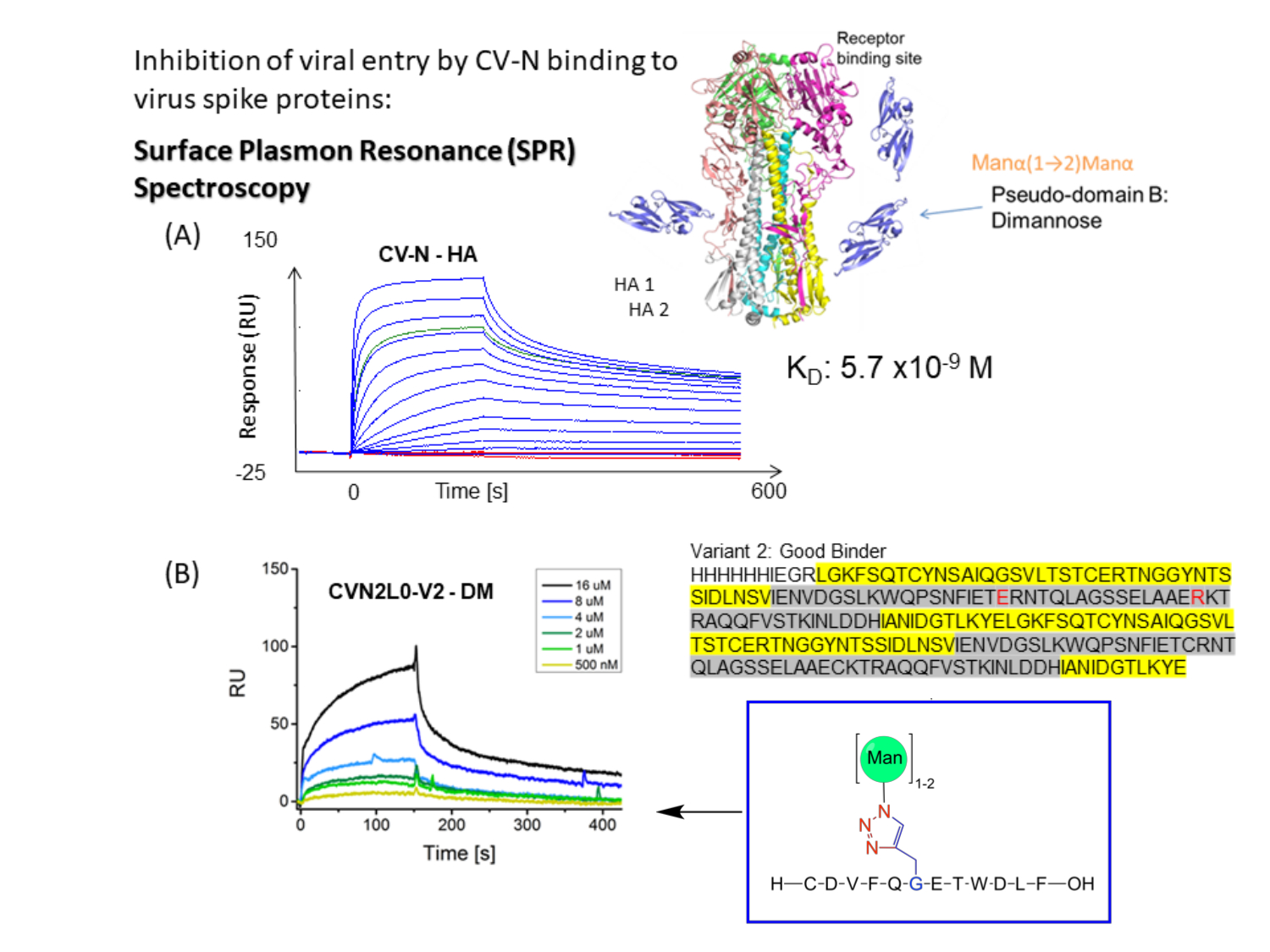
CV-N zeigt antivirale Aktivität gegen HIV 14,15, Influenza 16 und das Ebola-Virus, die durch nanomolare Bindung an hochmannose Oligosaccharidmodifikationen auf Hüllspitzenproteinenvermittelt wird 12,17,18,19. Die Bindung von Influenza HA an eine hochaffine Kohlenhydratbindungsstelle (H) in CV-N oder an zwei Hs in kovalent verknüpftem dimerem CVN2 hat Gleichgewichtsdissoziationskonstanten (K D) = 5,7 nM (Abbildung 1A) bzw. KD = 2,7 nM. Sowohl CV-N als auch CVN2 beherbergen weitere ein oder zwei kohlenhydratbindende Stellen mit niedriger Affinität (L)s 12,17,20,21. Ebola GP1,2 bindet an 2H CVN2 mit Affinitäten im unteren nanomolaren Bereich (KD = 26 nM). CV-N WT-Bindung an Ebola GP1,2 und HA weist Affinitäten von K D = 34 nM bis KD = 5,7 nM auf (A/New York/55/04)12. Lektine wie CV-N, die spezifisch auf hochmannose Glykane auf den Virushüllen abzielen, hemmen die Replikation von Hepatitis-C-Virus, SARS-CoV, Herpesvirus, Marburg-Virus und Masernvirus22.
Das kleine CV-N-Molekül wird seit mehr als 20 Jahren gründlich untersucht, da es funktionalisiert, um eine Vielzahl von Viren zu binden, um den viralen Eintritt zu hemmen16,18. Strukturanalysen und Bindungsaffinitätstests deuten auf eine Vernetzung von zwei Ls in einem domänenvertauschten CVN2-Dimer durch bivalente Bindung im mikromolaren Bereich hin, um die Avidität zu viralen Hüllglykoproteinen zu erhöhen10,19. Die selektive Bindung von Manα1-2Manα an Man(8)-D1D3-Armen und Man(9) umfasst zwei Bindungsstellen unterschiedlicher Affinitäten, die sich auf gegenüberliegenden Proteinprotomern20 befinden und dadurch nanomolare Bindungsaffinitäten erreichen (Abbildung 1B). Daher gilt CVN2 als Pseudo-Antikörper hinsichtlich seiner Anwendung zur Bindung von Epitopen an HIV gp120, ähnlich wie virusneutralisierende Antikörper17. Hier ist der Autor daran interessiert, die mögliche Bindung von CVN2 an den SARS-CoV-2-Spike über seine Rezeptorbindungsdomäne (RBD) zu untersuchen. Bindungskurven des immobilisierten humanen Angiotensin-Converting-Enzyms (ACE)-2 mit dem SARS-CoV-2 RBD ergeben KD = 4,7 nM für diese biologisch relevante Bindungsinteraktion23.
Im Gegensatz dazu erkennen ausgewählte Immunglobulinklassen spezifische und konsistente strukturelle Proteinmuster, die ein Substrat für die Affinitätsreifung in den membranverankerten HA-Regionen vermitteln24. CV-N zeigt eine hochwirksame Aktivität in fast allen Influenza-A- und B-Viren16 und ist ein weitgehend neutralisierendes antivirales Mittel. Unser Wissen ist unvollständig über die Lage von Zielepitopen am Stamm von HA1 und HA2, die möglicherweise epitopische Strukturen für das Glykan-Targeting durch stark neutralisierende Antikörper und im Vergleich zur Lektinbindungbeinhalten 25.

Abbildung 1: Schematische Darstellung des SPR-Bindungsassays für CV-N-zu-Virus-Hüllkurvenspitzen. (A) SPR-Assay für CV-N-Bindung an Liganden: HA-Protein voller Länge (90 kDa). Kinetischer Datensatz (5120, 2560, 1280, 640, 320, 160, 80, 40, 20, 10, 5, 2,5, 0 nM) mit doppelter Bindung an Influenza HA A/New-York/55/04 (H3N2). (B) CVN2L0-Variante V2 bindet an immobilisierten Liganden DM in einem Konzentrationsbereich von 500 nM bis 16 μM. Sequenz: L-Reste sind gelb markiert. H-Rückstände werden grau hervorgehoben. E58 und R73 sind ein Ersatz für Cystein im Wildtyp-Protein und machen V2 zu einer stabilen Proteinfaltung mit drei statt vier Disulfidbindungen Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Während die Glykanabschirmung auf dem membrandistalen HA-Oberteil eine hochaffine Bindung anCV-N12 induziert, wurde eine CVN2-Bindung an HA neben einer Disulfidbrücke des HA-Oberteils weiter an seinen niederaffinen Stellen10,12 beobachtet. Verschiedene polare Wechselwirkungen und Interaktionsstellen werden in der Kohlenhydratbindung durch CV-N identifiziert. Diese Wechselwirkungen werden verifiziert, indem Knock-out-Varianten in der Bindungsstelle erzeugt werden, um Bindungsaffinitäten mit in silico vorhergesagter Glykosylierung zu korrelieren12. Daher zielt das Projekt darauf ab, zuvor getestete chemisch mannosylierte HA-Peptide in Bindungsaffinität und Spezifität mit kurzen Peptidsequenzen aus SARS-bezogenen 2019-nCoV-Spikes und SARS-CoV-2 zu vergleichen, die natürlich durch eine kleine Anzahl verschiedener N-verknüpfter Glykosylierungsstellen und O-verknüpfter Glykosylierung modifiziert vorkommen. Unter Verwendung von Kryo-Elektronenmikroskopie und Bindungsassays berichten Pinto und Mitarbeiter über einen monoklonalen Antikörper, S309, der möglicherweise ein Epitop auf SARS-CoV-2-Spike-Protein erkennt, das ein konserviertes Glykan innerhalb der Sarbecovirus-Untergattung enthält, ohne mit der Rezeptorbindungzu konkurrieren 26. Das Protokoll dieser Studie beschreibt, wie wichtig das Design, die Expression und die Charakterisierung von CV-N-Varianten sind, um zu untersuchen, wie CV-N und CVN2 an glykosylierte Proteine und synthetische mannosylierte Peptide unter Verwendung der SPR-Technologie10,12 binden.
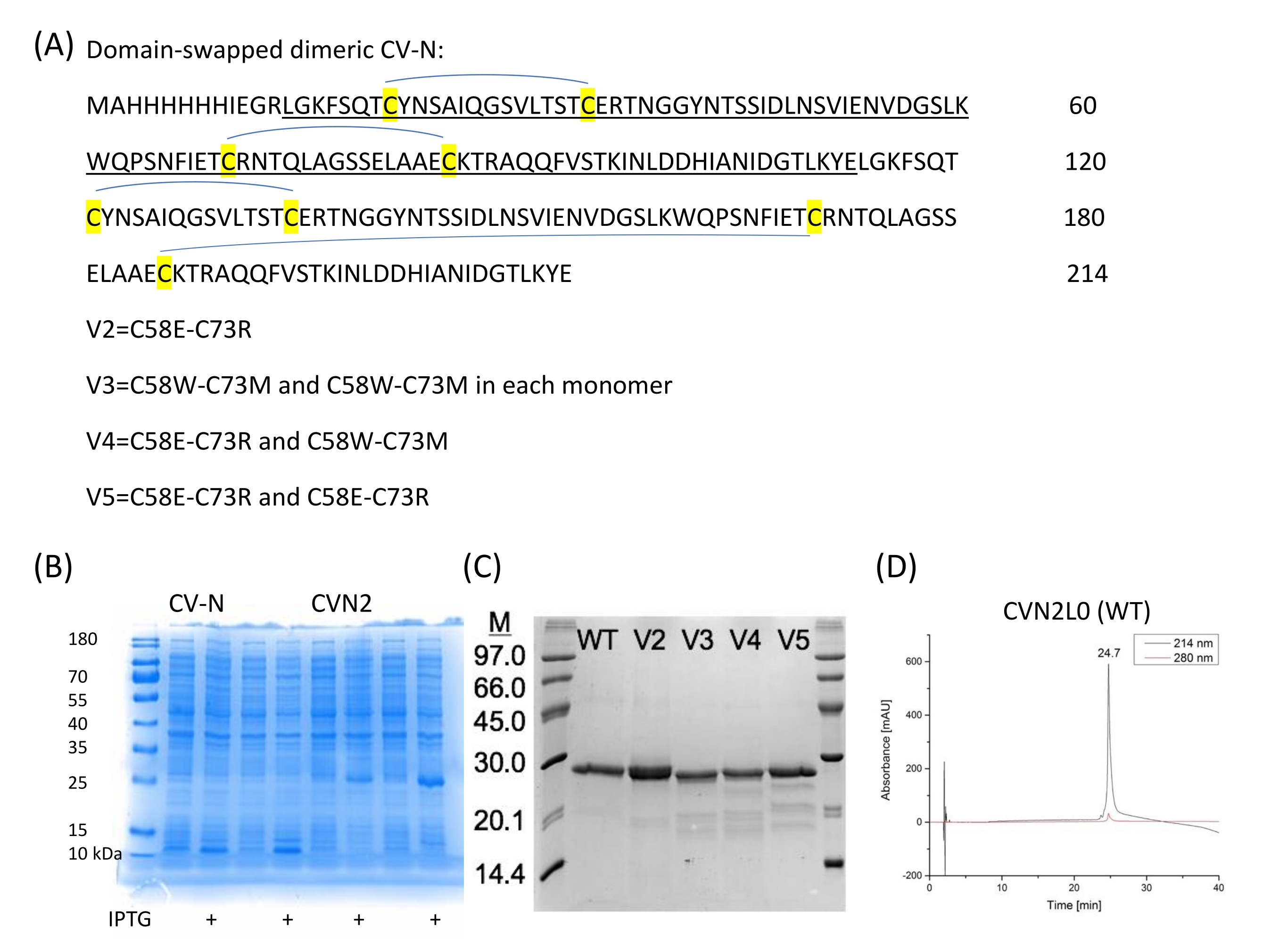
Tandem-gebundenes Dimer CVN2L027 und Bindungsstellenvarianten (V2-V5) werden rekombinant exprimiert, Varianten mit Disulfidbindungsersatz (C58E und C73R) (Abbildung 2A). Auch eine Mutante mit einer Einzelpunktmutation E41A wird hergestellt, da diese Position als intermolekularer Kreuzkontaktrest angesehen wurde. Diese Mutante ist ein weiteres interessantes Molekül für SPR-Bindungsmessungen zwischen den Lektin- und High-Mannose-Oligosacchariden, die Bindungsdomänen entschlüsseln und den Vergleich mit der dimeren Form ermöglichen. Die domänenvertauschte Kristallstruktur von CVN2 zeigt einen flexiblen Linker, der sich zwischen 49 und 54 Resten erstreckt. Die beiden Domänen können sich weiterhin als starre Körper um das Scharnier bewegen und entweder ein Monomer durch intramolekulare Domänenwechselwirkungen entwickeln (Domäne A -Reste 1-39;90-101- mit Domäne B -Reste 40-89) oder ein Dimer durch intermolekularen Domänentausch [Domäne A (des ersten Monomers) mit Domäne B (des zweiten) und Domäne B (des ersten Monomers) mit Domäne A (der zweiten Kopie)]. Es gibt keine engen Wechselwirkungen zwischen den A- und B-Domänen der beiden Protomer, mit Ausnahme von Glu4128. Das Gen für CV-N kann unter Verwendung einer repetitiven PCR-Methode mit 40-mer-synthetisierten Oligos29 entwickelt und dann in die NdeI- und BamHI-Stellen von pET11a subkloniert werden, um sie in elektrokompetente Zellen umzuwandeln (Elektroporation), wie von Keeffe, J.R.27 beschrieben. Das Protein, das zur Erreichung der jeweiligen Kristallstruktur (PDB ID 3S3Y) verwendet wird, enthält eine N-terminale 6-Histidin-Reinigungsmarke, gefolgt von einer Faktor-Xa-Protease-Spaltstelle. Die ortsgesteuerte Mutagenese wird verwendet, um Punktmutationen vorzunehmen, Codons zu wechseln und einzelne oder mehrere Basen oder Codons für den Aminosäureaustausch einzufügen oder zu löschen. Diese Umwandlungen liefern unschätzbare Einblicke in die Funktion und Struktur von Proteinen. Rekombinant exprimierte und gereinigte CV-N, CVN2 und CVN3 wurden biophysikalisch gut untersucht20,21,27, sind billig herzustellen und werden daher zur Charakterisierung von Bindungsassays an Glykane verwendet, die auf SPR-Sensorchips immobilisiert sind. Der konventionelle Enzyme-linked Immunosorbent Assay (ELISA) bietet eine geringere Reproduzierbarkeit in Bezug auf die Immobilisierungstechnik von Glykanliganden und wandelt die Echtzeitbindung verschiedener Bindungsstellenvarianten, die für SPR gezeigt wird, in Endpunktassays um.
Die Bindungsaffinitätsvariante CVN2L0-V2 (eine intakte Falte homodimeren CV-N mit einer Disulfidbrückensubstitution10) wird mit einem His-Tag in Escherichia coli (E. coli) exprimiert, über die Ni-NTA-Säule unter Anwendung der Affinitätschromatographie gereinigt und auf Bindung an HA (H3N2), monomannosyliertes HA-Peptid und dimannosyliertes HA-Peptid unter Verwendung von SPR getestet. Chemisch mannosylierte Peptide oder HA- und S-Proteine sind alle Liganden und Amin, die an die hydrophile Chipoberfläche gekoppelt sind über reaktive Ester oder Biotin-Streptavidin-Protein-Engineering. Das gleiche Verfahren der sequentiellen Durchläufe wird auf diese Liganden angewendet, wobei verschiedene Verdünnungen von CV-N und Varianten von CV-N (und CVN2) injiziert werden, um kinetische Informationen für die molekularen Interaktionsanalysen zu erhalten, wie untenbeschrieben 30. RBD-immobilisierter SPR-Sensorchip wird für Bindungsstudien an CV-N- zu S-Peptiden verwendet, und Affinitäten werden mit der SARS-CoV-2-Bindung mit dem menschlichen ACE2 verglichen.
Protokoll
Für die vorliegende Studie wurde anstelle von CV-N ein CVN-kleines Ubiquitin-like Modifier (SUMO) Fusionsprotein in enzymgebundenen Immunassays verwendet und eignet sich für zellbasierte Assays. Rekombinantes HA-H3-Protein des Influenza-A-Virus in voller Länge wird kommerziell gewonnen (siehe Materialtabelle) oder in HEK293-Zelllinien von Säugetieren und Baculovirus-infizierten Insektenzellen gemäß Standardprotokollen12 exprimiert. Wuhan-1-Spike-Protein wird in HEK293-Zellen von Säugetieren exprimiert. Die Synthese von monomannosyliertem Peptid (MM) und dimannosyliertem Peptid (DM) ermöglicht den Nachweis homogener Liganden zu CVN2 und monomannosyliertem niedermolekularem10.
1. CV-N-Konstrukte erstellen
- Für jede der CVN2-Varianten und das CVN2L0-Protein (PDB ID 3S3Y) erhalten Sie das Genkonstrukt mit einer N-terminalen pelB-Leadersequenz und einem His-Tag in pET27b(+)-Vektor aus kommerziellen Quellen (siehe Materialtabelle, Zusatzdatei 1).
- Erhalten Sie CVN2L0 und seine Varianten (V2, V3, V4 und V5; Abbildung 2A,C) im Hintergrund eines CVN2L0-Template-Gens, das aus zwei verschiedenen DNA-Sequenzen für jede CV-N-Wiederholung besteht.
- Die lyophilisierte Plasmid-DNA wird in sterilem entionisiertem destilliertem Wasser (ddH2O) auf eine Endkonzentration von 100 ng/μL gelöst.
2. Herstellung von LB-Agarplatten mit Plasmid-DNA-transformierten Zellen
- Das Kulturmedium LB-Lennox wird durch Auflösen von 10 g/L Pepton, 5 g/L Hefeextrakt und 5 g/L NaCl inddH2O(siehe Materialtabelle) hergestellt und der pH-Wert auf 7,4 eingestellt. Die Umwandlung in kompetente E. coli BL21 (DE3) für jede Variante (V2-V5) erfolgt nach einem chemischen Verfahren nach einem zuvor veröffentlichten Bericht10.
- Teilen Sie die Lösung (900 μL und 100 μL), übertragen Sie 100 μL auf LB-Agarplatten (50 μg/ml Kanamycin) und verwenden Sie vorsichtig einen sterilen Zellspreizer. Die Agarplatten über Nacht bei 37 °C anbrüten.
3. Klonen
- Subklontierung des Gens für CV-N in die NdeI- und BamHI-Stellen von pET11a (siehe Materialtabelle) zur Umwandlung (Elektroporation) in elektrokompetente Zellen gemäß Referenz27.
4. Ortsbezogene Mutagenese
- Es sollten CVN2L029 und mutierte CVN-E41A im Hintergrund eines CVN2L0-Template-Gens erzeugt werden, das zwei unterschiedene DNA-Sequenzen für jede CV-N-Wiederholungenthält 27.
- Führen Sie Mutationen mit einem ortsgerichteten Mutagenese-Kit (siehe Materialtabelle) und spezifischen mutagenen Primern 5'-gagaaccgtcaacgtttgcgataacagagttcagg-3' und 5'-cctgaactctgttatcgcaaacgttgacggttctc-3' für die Durchführung der PCR31 durch.
- Starten Sie eine Reihe von Probenreaktionen mit mehreren Konzentrationen von doppelsträngiger DNA (dsDNA)-Schablone im Bereich von 5-50 ng (z. B. 5, 10, 20 und 50 ng dsDNA-Vorlage). Halten Sie die Primerkonzentration konstant.
HINWEIS: Die PCR-Mischung und das Thermozyklenprotokoll werden im Allgemeinen wie in der Bedienungsanleitung für das ortsgerichtete Mutagenese-Kit32 beschrieben verwendet.
- Starten Sie eine Reihe von Probenreaktionen mit mehreren Konzentrationen von doppelsträngiger DNA (dsDNA)-Schablone im Bereich von 5-50 ng (z. B. 5, 10, 20 und 50 ng dsDNA-Vorlage). Halten Sie die Primerkonzentration konstant.
- Fügen Sie das Restriktionsenzym Dpn I (1 μL, 10 U/μL, siehe Materialtabelle) unter die Mineralölüberlagerung ein. Gründlich und vorsichtig die Reaktionen mischen, in einer Tisch-Mikrozentrifuge für 1 min herunterdrehen und sofort bei 37 °C für 1 h inkubieren, um die elterliche supercoiled dsDNA zu verdauen.
5. Transformation von Bakterienzellen
- Die XL1-Blue superkompetenten Zellen (siehe Materialtabelle) vorsichtig auf Eis auftauen. Um jede Kontroll- und Probenreaktion zu transformieren, aliquoten Sie die superkompetenten Zellen (50 μL) in ein vorgekühltes Polypropylen-Rundbodenrohr (14 ml).
- Transfer von 1 μL der mit Dpn I behandelten einzelsträngigen DNA (ssDNA) aus jeder Kontroll- und Probenreaktion (mutierte ssDNA) in separate Aliquots der superkompetenten Zellen, die den komplementären Strang synthetisieren. Schwenken Sie die Transformationsreaktionen vorsichtig, um die Reaktionen zu mischen und 30 min auf Eis zu inkubieren.
HINWEIS: Vor dem Transfer der mit Dpn I behandelten DNA in die Transformationsreaktion wird empfohlen, das verbleibende Mineralöl vorsichtig aus der Pipettenspitze zu entfernen. Als optionale Kontrolle muss die Transformationseffizienz der superkompetenten XL1-Blue-Zellen überprüft werden, indem 0,1 ng/μL des pUC18-Kontrollplasmids (1 μL) mit einem Aliquot von 50 μL der superkompetenten Zellen gemischt werden.
- Transfer von 1 μL der mit Dpn I behandelten einzelsträngigen DNA (ssDNA) aus jeder Kontroll- und Probenreaktion (mutierte ssDNA) in separate Aliquots der superkompetenten Zellen, die den komplementären Strang synthetisieren. Schwenken Sie die Transformationsreaktionen vorsichtig, um die Reaktionen zu mischen und 30 min auf Eis zu inkubieren.
- Die Umwandlungsreaktionen werden 45 s lang bei 42 °C mit einem Wärmeimpuls versehen und anschließend 2 min auf Eis gelegt.
HINWEIS: Der angelegte Wärmeimpuls wurde bereits für die genannten Bedingungen in Polypropylen-Rundbodenrohren (14 ml) optimiert. - 0,5 mL NZY+ Brühe (enthält pro Liter: 10 g NZ-Amin (Kaseinhydrolysat), 5 g Hefeextrakt, 5 g NaCl, 12,5 ml 1 M MgCl2, 12,5 mL 1 M MgSO4, 10 mL 2 M Glucose, pH7,5 und auf 42 °C vorgewärmt) zugeben und die Umwandlungsreaktionen bei 37 °C mit Schütteln bei 225-250 U/min für 1 h inkubieren. Plate das korrekte Volumen jeder Transformationsreaktion (5 μL aus Kontrollplasmidtransformation; 250 μL aus Probentransformation) auf LB-Ampicillin-Agarplatten.
HINWEIS: Für die Transformationskontrollen und Mutagenese verteilen Sie Zellen auf LB-Ampicillin-Agarplatten mit 5-Brom-4-chlor-3-indolyl-β-D-galactopyranosid (X-gal, 80 μg/ml) und Isopropyl-1-thio-β-D-galactopyranosid (IPTG, 20 mM) (siehe Materialtabelle). Beimpfen Sie 50 ml der Zellkulturen mit einer einzigen Kolonie transformierter E. coli-Zellen zur Reinigung mutierter Plasmid-DNA für Analysen. Die Mutagenese wird durch DNA-Sequenzierung an einer externen Einrichtung bestätigt.
6. Expression und Proteinreinigung
- Für eine groß angelegte Kultur impfen Sie eine kleine Menge LB (Ampicillin enthaltend) mit einer einzigen Kolonie von der transformierten Platte.
- Unter Verwendung der Übernachtkultur wird die Expressionskultur mit Zusätzen wie 10 mMMgCl2, 10 mMMgSO4 und 20 mM Glucose inokuliert, wobei die Samenkultur auf 1/100 verdünnt wird.
- Zellen mit kräftigem Schütteln bei 37 °C züchten. Die Zellen werden auf ein ABS von 600 nm zwischen 0,4 und 0,6 (Mid-Log-Phase) gezüchtet, bevor die Zellen auf 20 °C abgekühlt werden. Induzieren Sie mit 1 mM IPTG und wachsen Sie über Nacht.
- Dann ernten Sie die Zellen durch Zentrifugieren bei 4.000 x g für 15 min bei 4 °C und verwerfen den Überstand mit einer Pipette.
- Resuspendieren Sie das Zellpellet in phosphatgepufferter Kochsalzlösung (PBS) Puffer und zentrifugieren Sie es bei 4.000 x g für 15 min bei 4 °C. Dann entsorgen Sie den Überstand mit einer Pipette. Das verbleibende Pellet wird in 10 mL Lysepuffer resuspendiert und die Suspension 1 h bei 37 °C inkubiert.
ANMERKUNG: Zusammensetzung des Lysepuffers: (50 mMNaH2PO4, 300 mM NaCl, 2% Triton-X100, 500 ng/ml Lysozym, 1 mM Phenylmethylsulfonylfluorid (PMSF), 1 mM Dithiothreitol, 1 mM MgCl2, pH 8, siehe Materialtabelle).- Das Gemisch zwei Gefrier-Tau-Zyklen (-80 °C) unterziehen. Trennen Sie lösliche und unlösliche Fraktionen durch Zentrifugation bei 4.000 x g für 15 min bei 4 °C und analysieren Sie sie mittels Polyacrylamid-Gelelektrophorese (PAGE), insbesondere Natriumdodecylsulfat (SDS)-SEITE33 (Abbildung 2B,C).
HINWEIS: Zellen können auf verschiedene Arten lysiert werden, z. B. Gefrier-Tauen, Beschallung, Homogenisierung, enzymatische Lyse oder eine Kombination dieser Methoden. Die Reinigung von Einschlusskörpern wird empfohlen, um hohe Proteinausbeuten zu sammeln26.
- Das Gemisch zwei Gefrier-Tau-Zyklen (-80 °C) unterziehen. Trennen Sie lösliche und unlösliche Fraktionen durch Zentrifugation bei 4.000 x g für 15 min bei 4 °C und analysieren Sie sie mittels Polyacrylamid-Gelelektrophorese (PAGE), insbesondere Natriumdodecylsulfat (SDS)-SEITE33 (Abbildung 2B,C).
- Proteine mittels Ni-NTA-Säulenchromatographie reinigen (siehe Materialtabelle). Laden Sie die lösliche Fraktion auf eine regenerierte Ni-NTA-Perlensäule (1 ml/min). Waschen Sie das System mit TBS-Puffer (50 mM Tris, 150 mM Natriumchlorid, pH 7,5), bevor Sie einen Gradienten (0-100% 500 mM Imidazol in TBS über 60 min) beginnen und Fraktionen sammeln (1 ml/min). Dialysieren Sie die gereinigten Proteine zur biochemischen Charakterisierung gegen 100 mM PBS (Abbildung 2C,D).
- Alternativ kann His-Select Ni 2+ Affinitätsgel (siehe Materialtabelle) in 14-ml-Röhrchen verwendet werden, um sein markiertes rekombinant exprimiertes CV-N in Pufferlösungen mit 20 mM Imidazol bzw.250 mM Imidazol zu binden und erneut zu suspendieren. In Charge für mindestens 30 min inkubieren.
HINWEIS: Tragen Sie diese halbgereinigten Proteine auf eine vorverpackte Einwegsäule für den Pufferaustausch und die Reinigung biologischer Proben, z. B. Kohlenhydrate und Proteine, auf, die 1-1,5 ml Eluat aus der immobilisierten Metallaffinitätschromatographie laden können. - Die Proteinlösungen werden mit einem 10 kDa-Sperrfilter in Zentrifugationsröhrchen überführt (siehe Materialtabelle) und durch Zentrifugieren für 10 min bei 4.500 x g und 4 °C konzentriert. Für SPR-Messungen tauschen Sie die Analytlösungen auf 10 mM HEPES, 150 mM Natriumchlorid, 3 mM Ethylendiamintetraessigsäure (EDTA) und 0,05% Tween20, pH 7,4 (HBS-EP (+), siehe Materialtabelle).
- Diesen SPR-Laufpuffer auf einen Verdünnungsfaktor von 1:10 geben und viermal auf das Ausgangsvolumen für 10 min bei 4.500 x g und 4 °C zentrifugieren.
- Bestimmung der Proteinkonzentration bei 280 nm mit einem NanoDrop UV-Vis-Spektralphotometer (siehe Materialtabelle) basierend auf dem berechneten Extinktionskoeffizienten (20.440 M-1 cm-1) für das Hauptprotein CVN2L0 mit einer Größe von 23.474 Da34. Verwenden Sie PBS (100 mM, pH 7,0) oder SPR-Puffer als Blindwert und messen Sie die Proteinkonzentration in drei Verdünnungsschritten (1:1, 1:10 und 1:100).

Abbildung 2: CV-N-Sequenzen und Expression. (A) CVN2 ohne Linker zwischen jeder CV-N-Wiederholung (je 101 Aminosäuren) und vier Disulfidbrücken wird in pET11a-Vektor in E exprimiert. Coli. (B) Ausdrücke zweier unabhängiger Kolonien für CV-N (Monomer) und CVN2 (Dimer). (C) Disulfidbindungsvarianten werden auf SDS-PAGE gereinigt und analysiert. Ein niedermolekularer Marker (6 μL) wird als Referenz verwendet. WT = CVN2L0 mit vier Disulfidbrücken gemäß Buchstabe A. V2 ist eine Variante mit einem Disulfidbindungsersatz durch polare Reste an den Positionen 58 und 73. V3-V5 sind Varianten mit zwei verbleibenden S-S-Bindungen und entweder polaren (C58E-C73R) oder unpolaren (C58W-C73M) substituierenden Aminosäuren oder einer Kombination dieser Restpaarsubstitutionen. (D) HPLC-Chromatogramme von gereinigtem CVN2L0 werden bei einer Flussrate von 1 ml/min mit einem linearen Gradienten von 5% -65% Puffer B in Puffer A über 30 min eluiert. Puffer A ist: 0,1% (v/v) Trifluoressigsäure inddH2O, Puffer B ist: 0,08% (v/v) Trifluoressigsäure in Acetonitril. Das Protein wird auf einer Hochleistungs-Silikagel-Säule 300-5-C4 (150 x 4,6 mm) bei 214 nm und 280 nm analysiert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
7. SPR-Spektroskopie
- Verwenden Sie das Dual-Channel SPR-System (siehe Materialtabelle) mit Laufpuffer HBS-EP(+) und 10 mM Glycin HCl pH 1,5-1,6 als Regenerationspuffer. Schalten Sie das Gerät, den Entgaser, den Autosampler und die Pumpe ein und waschen Sie das gesamte System mit ddH20 für 1 h. Legen Sie den gebrauchsfertigen Laufpuffer in eine separate Flasche.
- Tropfen Sie Emersionsöl auf den Detektor und montieren Sie einen Glassensorchip (siehe Materialtabelle), der mit einem dünnen Goldfilm beschichtet und auf der Oberseite mit Carboxymethyldextran-Hydrogel funktionalisiert ist, direkt auf den Detektor unterhalb der Drei-Port-Durchflusszelle. Korrigieren Sie die Einstellung, indem Sie die Handhabung herunterziehen.
HINWEIS: C19RBDHC30M 200 nm Streptavidin derivatisiertes Carboxymethyldextran Hydrogel mit einer mittleren Dichte des biotinylierten Coronavirus-2 RBD-Proteins für schweres akutes respiratorisches Syndrom ist ein gebrauchsfertiger Sensorchip mit dem vorimmobilisierten Liganden.
8. SPR-Bindungsassay für CV-N-Bindung an HA, S-Protein und RBD
- Immobilisieren Sie die proteinartigen Liganden auf Sensorchips, indem Sie die folgenden Schritte ausführen.
- Öffnen Sie eine Run-Tabelle, indem Sie in der Menüleiste auf Formular und in der integrierten SPRAutoLink-Software auf Table Editor ausführen klicken (siehe Materialtabelle). Wählen und klicken Sie auf BASIC_Immobilization aus der Liste der verfügbaren Ausführungstabellen und folgen Sie den Schritten des experimentellen Verfahrens auf dem Computerbildschirm. Der jeweils verwendete Sample Editor wird in der rechten oberen Ecke angezeigt.
- Klicken Sie auf Sample Set Editor im Abschnitt Formular, um die Reagenzienliste für zwei Racks auszufüllen, die für weitere Analysen im Autosampler platziert sind. Klicken Sie in der Menüleiste auf Autosampler Direct Control als "Tool", um die Racks nach vorne oder nach Hause zu bringen. Wählen Sie 4 °C als Betriebstemperatur.
HINWEIS: Die SPR-Software ermöglicht "SPR Instrument Direct Control" und "Pump Direct Control" durch Auswahl der entsprechenden Werkzeuge, sowie Autosampler-Handhabung, und durch Klicken auf Formular; Auch Run Table Editor, Data-Plot oder Post-Processing können ausgewählt werden, um Datenanalysen durchzuführen. Dateien werden direkt im Standardverzeichnis gespeichert und als scrubber.files aus dem Fenster Nachbearbeitung exportiert.
- Starten Sie die Pumpe, um ddH20 zu infundieren, indem Sie auf Tools und Pump Direct Control klicken und Daten aufzeichnen, indem Sie auf SPR Instrument Direct Control klicken, und jedes Mal Start in den neu erscheinenden Fenstern. Geben Sie die Kupplungsreagenzien (Schritt 8.3) in 300-μL-Fläschchen, legen Sie sie in die Autosampler-Racks und starten Sie die Run-Tabelle, indem Sie auf Run klicken.
HINWEIS: Chipoberflächen werden entweder mit 10 mM Glycinpuffer pH 9,0 konditioniert oder können mit 1 M Natriumchlorid, 0,1 M Natriumboratpuffer pH 9,0 gespült worden sein, um die Carboxyl-derivatisierte Chipoberfläche für EDC / NHS-Aktivierungsmischung30 zu konditionieren. - Für diese einfache Protein-Protein-Interaktion verwenden Sie den CMD500D-Chip (siehe Materialtabelle), um eine Mikrobrechungsindexeinheit (μRIU) = 2500 - 3000 Flusszelle mit immobilisiertem HA und μRIU = 400 Flusszelle mit Spike-Protein zu erzeugen. Bei einer Infusionsflussrate von 15 μL/min wird eine wässrige und gleichmäßige Mischung aus 0,4 M N-ethyl-N'-(dimethylaminopropyl)carbodiimidhydrochlorid (EDC*HCl) und 0,1 M N-Hydroxysuccinimid (NHS) injiziert, indem die folgenden aufeinanderfolgenden Schritte angewendet werden.
- Nachfüllen der Pumpe bei 25.000 μL/min, Baseline-Einstellung für 30 s, Injektion von 90 μL Probenaktivierungslösung (EDC/NHS) über 6 min Kontaktzeit und anschließendes Halten für weitere 5 Minuten.
- Wiederholen Sie diesen Zyklus nach dem Baseline-Lauf für 1 min bei 10 μL/min nur auf der linken Flusszelle (blau), um chemisch synthetisierte Peptide 10, HA und Spike-Protein bei20 μg/ml zu injizieren und zu immobilisieren, und ermöglichen Sie die anschließende Baseline-Einstellung mit ddH2O für 1,5 min, bevor die aktivierte Chipoberfläche mit 1 M Ethanolamin HCl pH 8,5 abgeschreckt wird.
- Schalten Sie die Röhrchen vom Flüssigkeitssammler zum Entgaser von ddH20 in die Flasche mit HBS-EP(+) (ergänzende Abbildung 1).
- Analysieren Sie die SPR-Sensorgramme.
- Um kinetische Studien durchzuführen, verwenden Sie verschiedene Analytkonzentrationen (10-5-10-8 M), mit einem Regenerationsschritt nach jeder Injektion und Blindmessungen nach verschiedenen Analyten. Ändern Sie die Flussrate auf 10 μL/min und beginnen Sie mit den Injektionen für 4 min Kontaktzeit, dann 5 min Baseline-Generierung und zwei Regenerationsschritten von jeweils 2 min mit einem Intervall von 30 s.
- Injizieren Sie die Pufferlösung für Blindmessungen, deren Sensorgramme von Probenläufen subtrahiert werden, um verschiedene Proteinkonzentrationen zu normalisieren.
- Klicken Sie auf Formular, scrollen Sie nach unten und wechseln Sie zu "Nachbearbeitung", indem Sie auf diesen Betriebsmodus klicken. Klicken Sie auf Hinzufügen , um die im Laufe der Zeit im Datenplotformular für jede Flusszelle generierten Bindungskurven auszuwählen, und exportieren Sie das Overlay als Scrubber-Datei (.ovr). Klicken Sie auf Datei, um die Dateispeicheroptionen zu öffnen. Erhalten Sie Antwortkurven, indem Sie linke und rechte Kurven ausrichten und Signale des zweiten Referenzkanals von denen des Ligandenkanals subtrahieren.
HINWEIS: Die Daten werden in "Post-Processing" verarbeitet, indem Sensorgramme rechnerisch definiert werden. Es wird in eine Überlagerung von Sensorgrammen aus linken und rechten Durchflusszellen eingefügt oder als Sensorgramm aus der Differenz beider Kanäle dargestellt. - Reinigen Sie die gesamte Fluidik mit 50-100 mM Glycinpuffer pH 9,5, Wasser und 20% Ethanol vor und nach den Bindungsmessungen, um Spuren von Salz oder Proteinkontamination zu entfernen, oder strenger, mit 0,5% SDS und Glycin.
HINWEIS: Um eine Beschädigung des Instruments zu vermeiden, wird empfohlen, die mechanische Stabilität des Glaschips zu überprüfen, bevor die Chipkartusche wieder in das Instrument eingesetzt wird, wenn die Chips unter Puffer oder bei 100% Feuchtigkeit gelagert wurden.
Ergebnisse
Ein dimeres domänenvertauschtes CVN2L0-Molekül wird in drei separaten SPR-Experimenten auf Bindung an die HA-Top-Region getestet und die Bindungsaffinität wird inKD-Werten dargestellt. Es wird angenommen, dass Domäne B H-Bindungsstellen umfasst, die durch den Ersatz einer Disulfidbindung in ionische Reste beeinflusst werden, und Domäne A bildet L10,18. Einzelinjektionen von CVN2L0 und den Varianten V2 (drei Disulfidbrücken) und V5 (zwei Disulfidb...
Diskussion
Die Bindungsaffinität von CV-N korreliert mit der Anzahl der funktionalen Bindungsstellen [2H auf Domänen B und 2L auf Domänen A, wenn sie als domänenvertauschtes Dimer entwickelt wurden]. Eine Variante mit veränderter Bindungsaffinität (CVN2L0-V2, eine homodimere stabile Falte von CV-N, die einen Disulfidbrücken-Knock-out umfasst) wird in E. coli exprimiert, gereinigt und positiv auf Bindung an HA-Protein (H3N2) unter Verwendung von SPR10 getestet und zeigt eine Konformationsände...
Offenlegungen
Der Autor hat nichts offenzulegen.
Danksagungen
Der Autor dankt Dr. Christian Derntl vom Department für Biotechnologie und Mikrobiologie der TU Wien und der Universitätsklinik für Medizin III, Abteilung für Nephrologie und Dialyse der Medizinischen Universität Wien, insbesondere Dr. Markus Wahrmann für die technische und wissenschaftliche Unterstützung. Die Proteinexpression in Säugetierzellen wurde vom Department für Biotechnologie der Universität für Bodenkultur (BOKU) Wien unterstützt. Die Autorin dankt Dr. Nico Dankbar von XanTec bioanalytics in Düsseldorf für hilfreiche wissenschaftliche Diskussionen zur Durchführung der SPR-Bindungsassays.
Materialien
| Name | Company | Catalog Number | Comments |
| Äkta primeplus | Cytiva | ||
| Amicon tubes | Merck | C7715 | |
| Ampillicin | Sigma-Aldrich | A5354 | |
| Beckmann Coulter Cooler Allegra X-30R centrifuge | Beckman Coulter | B06320 | |
| Cell spreader | Sigma-Aldrich | HS86655 | silver stainless steel, bar L 33 mm |
| Custom DNA Oligos | Sigma-Aldrich | OLIGO | |
| Custom Gensynthesis | GenScript | #1390661 | cloning vector: pET27b(+) |
| Cytiva HBS-EP+ Buffer 10, 4x50mL | Thermo Scientific | 50-105-5354 | |
| Dionex UlitMate 3000 | Thermo Scientific | IQLAAAGABHFAPBMBFB | |
| Dpn I restriction enzyme (10 U/μL) | Fisher Scientific | ER1701 | |
| DTT | Merck | DTT-RO | |
| EDC | Merck | 39391 | |
| EDTA | Merck | E9884 | |
| Eppendorf Safe-Lock Tubes | Eppendorf | 30120086 | |
| Eppendorf Safe-Lock Tubes | Eppendorf | 30120094 | |
| Eppendorf Minispin and MiniSpin Plus personal microcentrifuge | Sigma-Aldrich | Z606235 | |
| Ethanol | Merck | 51976 | |
| Ethanolamine HCl | Merck | E6133 | |
| Falcon 50mL Conical Centrifuge Tubes | Fisher Scientific | 14-432-22 | |
| Falcon 14 mL Round Bottom Polystyrene Test Tube, with Snap Cap, Sterile, 25/Pack | Corning | 352057 | |
| Glucose | Merck | G8270 | |
| Glycine HCl | Merck | 55097 | |
| HA H3 protein | Abcam | ab69751 | |
| HEPES | Merck | H3375 | |
| His-select Ni2+ | Merck | H0537 | |
| Imidazole | Merck | I2399 | |
| IPTG | Merck | I6758 | |
| Kanamycin A | Sigma-Aldrich | K1377 | |
| Kromasil 300-5-C4 | Nouryon | ||
| LB agar | Merck | 52062 | |
| LB agar | Merck | 19344 | |
| LB Lennox | Merck | L3022 | |
| Lysozyme | Merck | 10837059001 | |
| Magnesium chloride | Merck | M8266 | |
| Magnesium sulfate | Merck | M7506 | |
| NaH2P04 | Merck | S0751 | |
| NanoDrop UV-Vis2000c spectrophotometer | Thermo Scientific | ND2000CLAPTOP | |
| NaOH | Merck | S5881 | |
| NHS | Merck | 130672 | |
| NZ amine (casein hydrolysate) | Merck | C0626 | |
| PBS | Merck | 806552 | |
| PD MidiTrap G-10 | Sigma-Aldrich | GE28-9180-11 | |
| Peptone | Merck | 70171 | |
| pET11a | Merck Millipore (Novagen) | 69436 | |
| PMSF | Merck | PMSF-RO | |
| QIAprep Spin Miniprep Kit (1000) | Qiagen | 27106X4 | |
| Reichert Software Package Autolink1-1-9 | Reichert | ||
| Reichert SPR SR7500DC Dual Channel System | Reichert | ||
| Scrubber2-2012-09-04 for data analysis | Reichert | ||
| SDS | Merck | 11667289001 | |
| Site-directed mutagenesis kit incl pUC18 control plasmid | Stratagene | #200518 | |
| Sodim chloride | Merck | S9888 | |
| Sodium acetate.Trihydrate | Merck | 236500 | |
| SPR sensor chip C19RBDHC30M | XanTec bioanalytics | SCR C19RBDHC30M | |
| SPR sensor chip CMD500D | XanTec bioanalytics | SCR CMD500D | |
| Sterilin Standard 90mm Petri Dishes | Thermo Scientific | 101R20 | |
| TBS | Merck | T5912 | 10x, solution |
| Triton-X100 | Merck | T8787 | |
| Tryptone | Merck | 93657 | |
| Tween20 | Merck | P1379 | |
| Vortex-Genie 2 Mixer | Merck | Z258423 | |
| X-gal | Merck | XGAL-RO | |
| XL1-Blue Supercompetent Cells | Stratagene | #200236 | |
| Yeast extract | Merck | Y1625 |
Referenzen
- Perez-Caballero, D., et al. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell. 139 (3), 499-511 (2009).
- Waheed, A. A., Gitzen, A., Swiderski, M., Freed, E. O. High-mannose but not complex-type glycosylation of tetherin is required for restriction of HIV-1 release. Viruses. 10 (1), 26 (2018).
- Wilson, I. A., et al. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature. 289 (5796), 366-373 (1981).
- Otterstrom, J. J., et al. Relating influenza virus membrane fusion kinetics to stoichiometry of neutralizing antibodies at the single-particle level. Proceedings of the National Academy of Sciences of the United States of America. 111 (48), 5143-5148 (2014).
- Kaletsky, R. L., Francica, J. R., Agrawal-Gamse, C., Bates, P. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proceedings of the National Academy of Sciences of the United States of America. 106 (8), 2886-2891 (2009).
- Tokarev, A., Skasko, M., Fitzpatrick, K., Guatelli, J. Antiviral activity of the interferon-induced cellular protein BST-2/tetherin. AIDS Research and Human Retroviruses. 25 (12), 1197-1210 (2009).
- Gnirss, K., et al. Tetherin sensitivity of influenza A viruses is strain specific: Role of hemagglutinin and neuraminidase. Journal of Virology. 89 (18), 9178-9188 (2015).
- Fleury, D., et al. A complex of influenza hemagglutinin with a neutralizing antibody that binds outside the virus receptor binding site. Nature Structural Biology. 6 (6), 530-534 (1999).
- Salunke, S. B., et al. Iron(III) chloride as an efficient catalyst for stereoselective synthesis of glycosyl azides and a cocatalyst with Cu(0) for the subsequent click chemistry. Chemical Communication. (Camb). 47 (37), 10440-10442 (2011).
- Schilling, P. E., et al. Mannosylated hemagglutinin peptides bind cyanovirin-N independent of disulfide-bonds in complementary binding sites. RSC Advances. 10 (19), 11079-11087 (2020).
- Fleury, D., Wharton, S. A., Skehel, J. J., Knossow, M., Bizebard, T. Antigen distortion allows influenza virus to escape neutralization. Nature Structural Biology. 5 (2), 119-123 (1998).
- Maier, I., Schiestl, R. H., Kontaxis, G. Cyanovirin-N binds viral envelope proteins at the low-affinity carbohydrate binding site without direct virus neutralization ability. Molecules. 26 (12), 3621 (2021).
- Ahmed, A. J., Keremane, S. R., Vielmetter, J., Bjorkman, P. J. Structural characterization of GASDALIE Fc bound to the activating Fc receptor FcγRIIIa. Journal of Structural Biology. 194 (1), 78-89 (2016).
- Boyd, R. Discovery of cyanovirin-N, a novel human immunodeficiency virus-inactivating protein that binds viral surface envelope glycoprotein gp120: potential applications to microbicide development. Antimicrobial Agents and Chemotherapy. 41 (7), 1521-1530 (1997).
- Bolmstedt, A. J., O'Keefe, B. R., Shenoy, S. R., McMahon, J. B., Boyd, M. R. Cyanovirin-N defines a new class of antiviral agent targeting N-linked, high-mannose glycans in an oligosaccharide-specific manner. Molecular Pharmacology. 59 (5), 949-954 (2001).
- O'Keefe, B. R., et al. Potent anti-influenza activity of cyanovirin-N and interactions with viral hemagglutinin. Antimicrobial Agents and Chemotherapy. 47 (8), 2518-2525 (2003).
- Shenoy, S. R., et al. Multisite and multivalent binding between cyanovirin-N and branched oligomannosides: calorimetric and NMR characterization. Chemical Biology. 9 (10), 1109-1118 (2002).
- Bewley, C. A., Kiyonaka, S., Hamachi, I. Site-specific discrimination by cyanovirin-N for alpha-linked trisaccharides comprising the three arms of Man(8) and Man(9). Journal of Molecular Biology. 322 (4), 881-889 (2002).
- Barrientos, L. G., Matei, E., Lasala, F., Delgado, R., Gronenborn, A. M. Dissecting carbohydrate-Cyanovirin-N binding by structure-guided mutagenesis: functional implications for viral entry inhibition. Protein Engineering Design & Selection. 19 (12), 525-535 (2006).
- Bewley, C. A., Otero-Quintero, S. The potent anti-HIV protein cyanovirin-N contains two novel carbohydrate binding sites that selectively bind to Man(8) D1D3 and Man(9) with nanomolar affinity: implications for binding to the HIV envelope protein gp120. Journal of the American Chemical Society. 123 (17), 3892-3902 (2001).
- Bewley, C. A. Solution structure of a cyanovirin-N:Man alpha 1-2Man alpha complex: structural basis for high-affinity carbohydrate-mediated binding to gp120. Structure. 9 (10), 931-940 (2001).
- Jensen, S. M. R., et al. Differential inhibitory effects of Cyanovirin-N, Griffithsin, and Scytovirin on entry mediated by envelopes of gammaretroviruses and deltaretroviruses. Journal of Virology. 88 (4), 2327-2332 (2014).
- Lan, J., et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 581 (7807), 215-220 (2020).
- Lingwood, D., et al. Structural and genetic basis for development of broadly neutralizing influenza antibodies. Nature. 489 (7417), 566-570 (2012).
- Ekiert, D. C., et al. Antibody recognition of a highly conserved influenza virus epitope. Science. 324 (5924), 246-251 (2009).
- Pinto, D., et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature. 583 (7815), 290-295 (2020).
- Keeffe, J. R., et al. Designed oligomers of cyanovirin-N show enhanced HIV neutralization. Proceedings of the National Academy of Sciences of the United States of America. 108 (34), 14079-14084 (2011).
- Yang, F., et al. Crystal structure of cyanovirin-N, a potent HIV-inactivating protein, shows unexpected domain swapping. Journal of Molecular Biology. 288 (3), 403-412 (1999).
- Stemmer, W. P., Crameri, A., Ha, K. D., Brennan, T. M., Heyneker, H. L. Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides. Gene. 164 (1), 49-53 (1995).
- Fischer, M. J. E., Mol, N., Fischer, M. Amine coupling through EDC/NHS: A practical approach. Surface Plasmon Resonance. Methods in Molecular Biology (Methods and Protocols). 627, (2010).
- Novoradovsky, A., et al. Computational principles of primer design for site directed mutagenesis. Technical Proceedings of 2005 NSTI Nanotechnology Conference and Trade Show. , 532-535 (2005).
- . QuikChange Site-Directed MutagenesisKit, User Manual Available from: https://users.drew.edu/jliu3/Docs/Stratagene%20Quikchange%20mutagenesis.pdf#:~:text=The%20QuikChange%20sitedirected%20mutagenesis%20kit%20is%20used%20to (2005)
- Laemmli, U. K. Cleavage of structural proteins during the assembly of the head bacteriophage T4. Nature. 227 (5259), 680-685 (1970).
- . Expasy.org Available from: https://web.expasy.org/cgi-bin/protparam/protparam (2022)
- Karlsson, R. Real-time competitive kinetic analysis of interactions between low-molecular-weight ligands in solution and surface-immobilized receptors. Analytical Biochemistry. 221 (1), 142-151 (1994).
- Schuck, P., Zhao, H. The role of mass transport limitation and surface heterogeneity in the biophysical characterization of macromolecular binding processes by SPR biosensing. Methods Molecular Biology. 627, 15-54 (2020).
- Barnes, O., et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature. 588 (7839), 682-687 (2020).
- . Using reichert Surface Plasmon Resonance (SPR) for Antiviral Development, Application Note 28 Available from: https://www.reichertspr.com/clientuploads/directory/application_notes/Application_Note_28__Using_Reichert_Surface_Plasmon_Resonance_for_Antiviral_Testing.pdf (2022)
- Sundberg, E. J., Andersen, P. S., Gorshkova, I. I., Schuck, P., Schuck, P. Surface plasmon resonance biosensing in the study of ternary systems of interacting proteins. Protein Interactions: Biophysical Approaches for the Study of Complex Reversible Systems. 5, 97-141 (2007).
- . Method Development Notes Available from: https://www.reichertspr.com/applications/method-development-notes/ (2022)
- Angulo, J., Enríquez-Navas, P. M., Nieto, P. M. Ligand-receptor binding affinities from saturation transfer difference (STD)-NMR spectroscopy: the binding Isotherm of STD initial growth rates. Chemistry. 16 (26), 7803-7812 (2010).
- Goldflam, M., Tarragó, T., Gairí, M., Giralt, E. NMR studies of protein-ligand interactions. Methods in Molecular Biology. 831, 233-259 (2012).
- Kumar, S., Maurya, V. K., Prasad, A. K., Bhatt, M. L. B., Saxena, S. K. Structural, glycosylation and antigenic variation between 2019 novel coronavirus (2019-nCoV) and SARS coronavirus (SARS-CoV). Virusdisease. 31 (1), 13-21 (2020).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten