
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Ingénierie d’agents antiviraux par résonance plasmonique de surface
Dans cet article
Résumé
Le présent protocole décrit de nouveaux outils pour les tests de liaison SPR afin d’examiner la liaison CV-N à l’HA, à la glycoprotéine S, aux glycanes de type hybride apparentés et aux oligosaccharides à haute teneur en mannose. SPR est utilisé pour déterminer le KD pour lier le CV-N dimérique ou monomère à ces glycanes.
Résumé
La résonance plasmonique de surface (SPR) est utilisée pour mesurer la liaison de l’hémagglutinine (HA) au dimère de cyanovirine-N (CV-N) interverti et pour surveiller les interactions entre les peptides mannosylés et le site de liaison à haute affinité de CV-N. Il a été rapporté que les pics d’enveloppe virale gp120, HA et la glycoprotéine Ebola (GP) 1,2 se lient aux sites de liaison à haute et à faible affinité sur le CVN2 dimérique. Le peptide HA dimannosylé est également lié aux deux sites de liaison de faible affinité à une molécule modifiée de CVN2, qui porte un site de haute affinité pour le ligand respectif et mutée pour remplacer une liaison disulfure stabilisatrice dans la poche de liaison aux glucides, confirmant ainsi la liaison multivalente. La liaison de l’HA est démontrée à un site de liaison à haute affinité du pseudo-anticorps CVN2 à une constante de dissociation (KD) de 275 nM qui neutralise davantage le virus de l’immunodéficience humaine de type 1 (VIH-1) par oligomérisation. La corrélation du nombre de ponts disulfure dans CVN2 permuté de domaine, qui sont diminués de 4 à 2 en substituant les cystines en paires de résidus polaires d’acide glutamique et d’arginine, entraîne une affinité de liaison réduite à l’HA. Parmi les interactions les plus fortes, Ebola GP1,2 est lié par CVN2 avec deux sites de liaison de haute affinité dans la gamme nanomolaire inférieure utilisant le glycane d’enveloppe sans domaine transmembranaire. Dans la présente étude, la liaison de la glycoprotéine de pointe (S) monomère multispécifique au coronavirus 2 du syndrome respiratoire aigu sévère (SRAS-CoV-2) est mesurée à K D = 18,6 μM par rapport à la nanomolaire KD à ces autres pointes virales, et via son domaine de liaison au récepteur dans la gamme molaire moyenne μ.
Introduction
L’activité antivirale associée à la tetherine est induite par l’interféron-α, et elle comprend des attaches à base de protéines, qui conduisent à la rétention de virions entièrement formés sur les surfaces cellulaires infectées1. La nécessité de la glycosylation de l’attache dans l’inhibition de la libération du virus demeure incertaine, ce qui implique l’importance des profils de glycosylation sur les glycanes exprimés par recombinaison pour les études in vitro 1,2, qui dépend de la conformation (dans le cas du virus de la grippe) de l’hémagglutinine HA 3,4 de la grippe exprimée en surface . Il a été noté que la modification de l’oligosaccharide attaché à la glycosylation liée à l’azote est suffisante pour la restriction médiée par l’attache de la libération 2 du VIH de type1, tandis que la dimérisation joue un rôle essentiel dans la prévention de la libération du virus, impliquant ainsi le domaine transmembranaire ou l’ancrage du glycosyl-phosphatidyl-inositol (GPI) pour attacher les virions en herbe5 . Des caractéristiques uniques sont décrites pour l’attachement humain et murin afin de bloquer plusieurs virus, rétrovirus et filovirus enveloppés. BST-2/tetherin est une protéine antivirale inductible par l’interféron de l’immunité innée1,6, agissant avec une activité antivirale à large spectre et est antagonisée par les glycoprotéines d’enveloppe5 pour translocaliser l’attache ou perturber la structure de la tetherin 6. Par exemple, la glycoprotéine HA et la neuraminidase de l’enveloppe exprimée en surface sur le virus de la grippe A sont bien connues pour l’antagonisme de la tetherine d’une manière spécifique à la souche7, facilitant la reconnaissance des sites de liaison aux récepteurs de l’hôte8. Les anticorps ciblant les glycanes sont étudiés dans la stœchiométrie de leurs interactions avec les boucliers glycanes à personnalisation rapide sur l’HA, ce qui entraîne une affinité de liaison avec les sous-types 4 H3N2 et H1N1 de la grippeA.
Pour élucider les mécanismes de liaison entre les agents antiviraux et les pics d’enveloppe virale, c’est-à-dire les ligands glucidiques, et les méthodes immunologiques et spectroscopiques complémentaires, les fractions mono-, di- et tri-mannose sont synthétisées chimiquement. Les peptides mannosylés sont créés par glycosylation azido de glycosyl {bêta}-peracétates en 1,2-trans glycosyl azoturetransformation 9, imitant la N-acétyl glucosamine et les oligosaccharides à haute teneur en mannose à la surface de virus potentiellement mortels. Les bioisostères triazolés sont utilisés pour imiter les liaisons qui forment le résidu mannosylé du peptide HA10 et faciliter les interactions spécifiques au site avec les dérivés antiviraux CV-N autour de la deuxième tache de glycosylation liée à l’azote sur le domaine de la tête HA (sommet HA avec 4 glycanes N54, N97, N181, N301)8,11,12 . Les interactions entre l’acide glutamique (Glu) et l’arginine (Arg) et le dipôle hélicoïdal qui en résulte ont manifesté une bonne stabilité des peptides et des protéines modèles, mais sont visualisées à l’aide de SPR. Si on compare à la reconnaissance d’un seul site de glycosylation synthétisé chimiquement sur HA10 en inhibant directement la liaison du récepteur sur les fractions glycanes, une affinité plus élevée d’une structure Fc mutée à quatre sites avec son récepteur est démontrée pour provoquer des fonctions effectrices in vivo, révélant que la composition non apparentée des glycanes liés à l’azote attachés au mutant Fc doit être déterminée mécaniquement13.
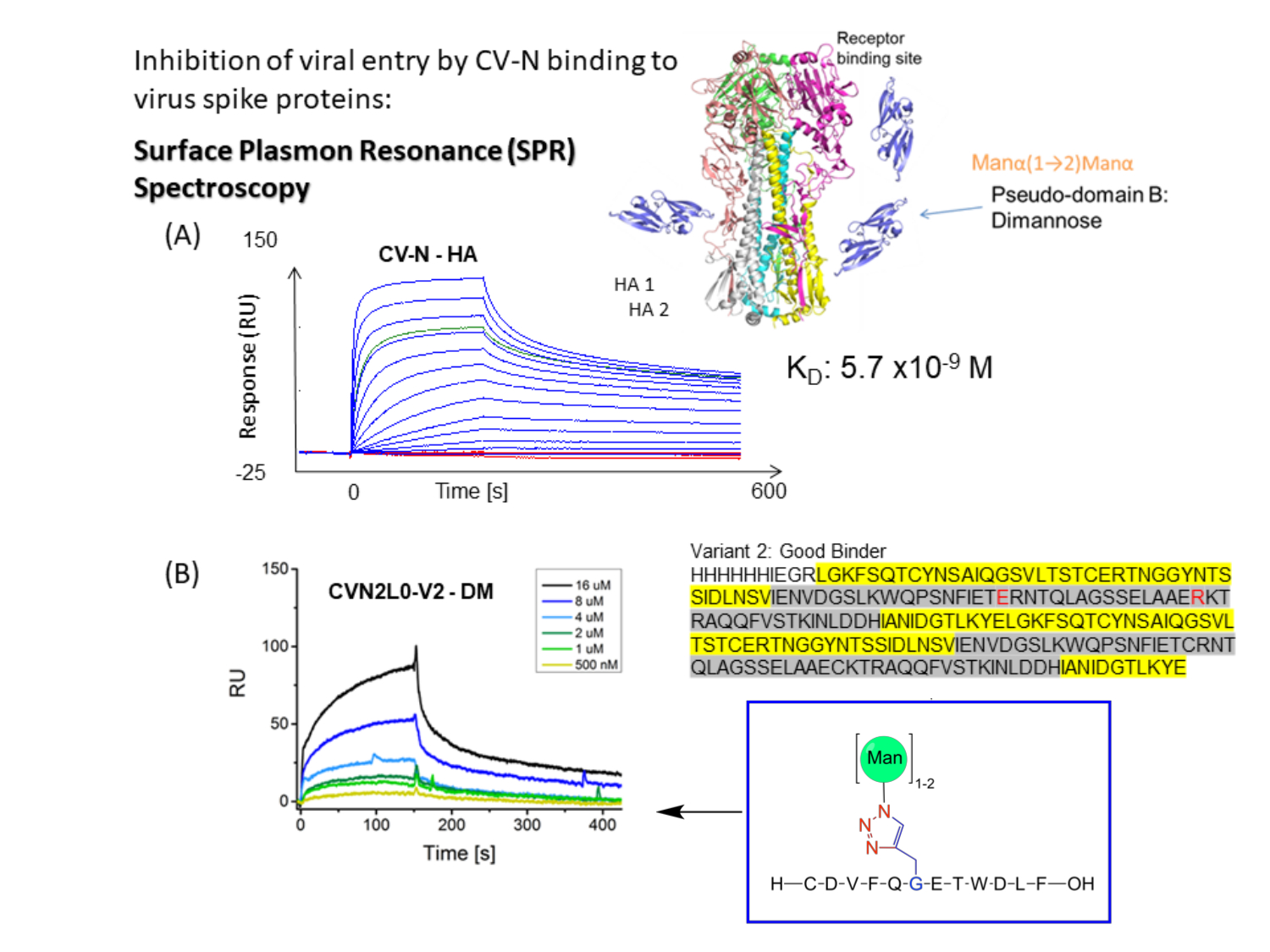
CV-N affiche une activité antivirale contre le VIH 14,15, la grippe16 et le virus Ebola, qui est médiée par la liaison nanomolaire aux modifications oligosaccharidiques à haute teneur en mannose sur les protéines de pointe d’enveloppe12,17,18,19. Il est déterminé que l’HA de la grippe se liant à un site de liaison aux glucides (H) de haute affinité dans le CV-N ou à deux Hs dans le CVN2 dimérique lié par covalence présente des constantes de dissociation à l’équilibre (K D) = 5,7 nM (figure 1A) et KD = 2,7 nM, respectivement. CV-N et CVN2 abritent tous deux un ou deux autres sites de liaison aux glucides de faible affinité (L)s 12,17,20,21. Ebola GP1,2 se lie à 2H de CVN2 avec des affinités dans la gamme nanomolaire inférieure (KD = 26 nM). La liaison CV-N Wt à Ebola GP1,2 et HA présente des affinités de K D = 34 nM à KD = 5,7 nM (A/New York/55/04)12. Les lectines, telles que CV-N, qui ciblent spécifiquement les glycanes à haut niveau de mannose sur les enveloppes virales, inhibent davantage la réplication du virus de l’hépatite C, du SRAS-CoV, de l’herpèsvirus, du virus Marburg et du virus de la rougeole22.
La petite molécule CV-N a été étudiée en profondeur pendant plus de 20 ans car elle se fonctionnalise pour se lier à un large éventail de virus afin d’inhiber l’entrée virale16,18. Les analyses structurales et les tests d’affinité de liaison indiquent la réticulation de deux L dans un dimère CVN2 permuté par domaine par liaison bivalente dans la gamme micromolaire pour améliorer l’avidité des glycoprotéines de l’enveloppe virale10,19. La liaison sélective de Manα1-2Manα sur les bras Man(8) D1D3 et Man(9) comprend deux sites de liaison d’affinités différentes situés sur des protomères protéiques opposés20, atteignant ainsi des affinités de liaison nanomolaire (Figure 1B). Ainsi, CVN2 est considéré comme un pseudo-anticorps concernant son application pour lier les épitopes sur le VIH gp120, similaire aux anticorps neutralisant le virus17. Ici, l’auteur s’intéresse à l’étude de la liaison potentielle de CVN2 au pic SARS-CoV-2 via son domaine de liaison au récepteur (RBD). Les courbes de liaison de l’enzyme de conversion de l’angiotensine humaine (ECA)-2 immobilisée avec le RBD du SRAS-CoV-2 donnent KD = 4,7 nM pour cette interaction de liaison biologiquement pertinente23.
En revanche, certaines classes d’immunoglobulines reconnaissent des schémas protéiques structuraux spécifiques et cohérents, qui confèrent un substrat pour la maturation d’affinité dans les régions HA ancrées dans la membrane24. CV-N montre une activité très puissante dans presque tous les virus grippaux A et B16, et c’est un agent antiviral largement neutralisant. Nos connaissances sont incomplètes sur la localisation des épitopes ciblés sur la tige de HA1 et HA2 qui impliquent éventuellement des structures épitopiques pour le ciblage des glycanes par des anticorps hautement neutralisants et par rapport à la liaison à la lectine25.

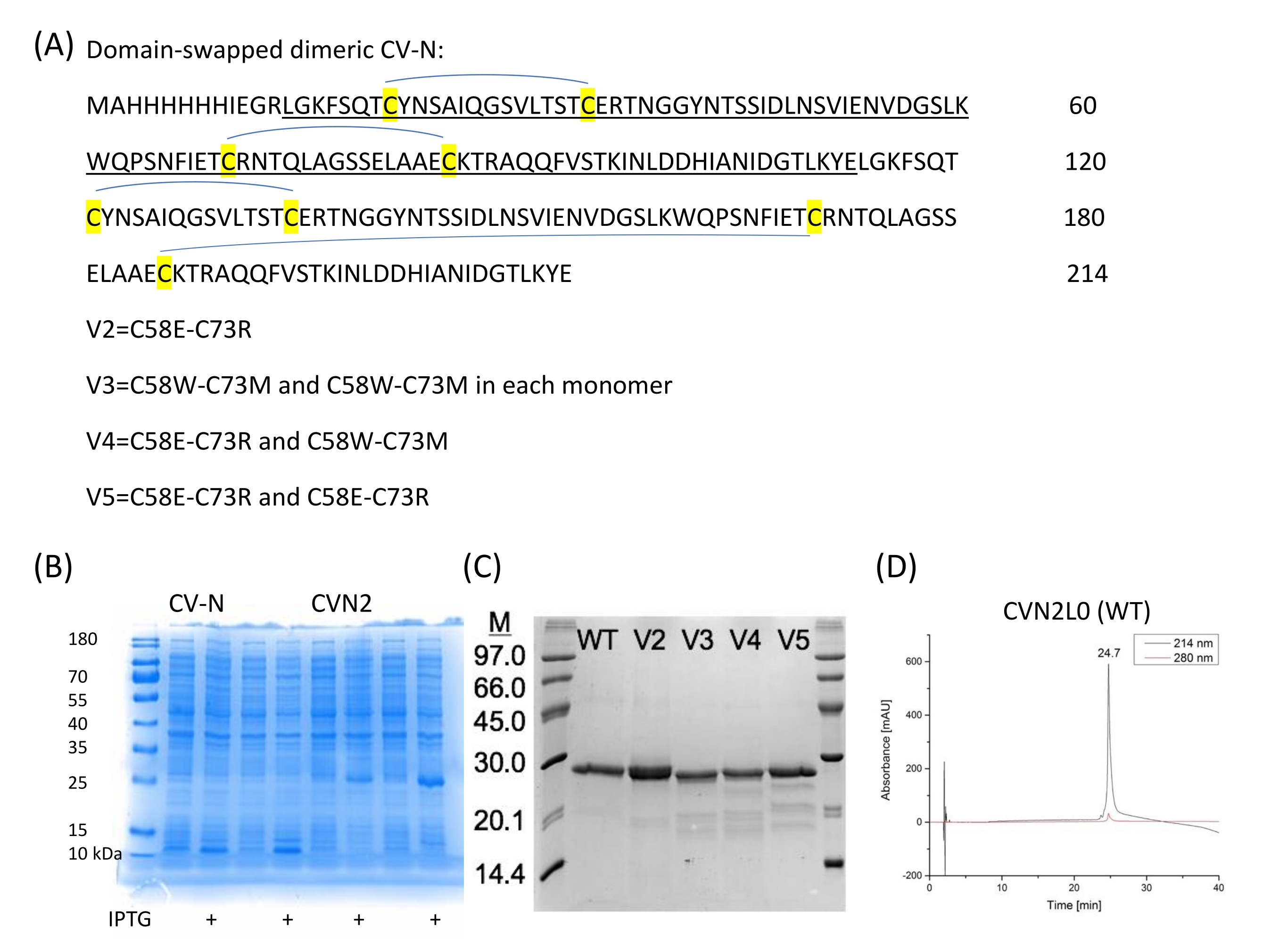
Figure 1 : Représentation schématique du test de liaison SPR pour les pics CV-N aux pics d’enveloppe virale. (A) Dosage SPR pour la liaison CV-N au ligand: protéine pleine longueur HA (90 kDa). Ensemble de données cinétiques (5120, 2560, 1280, 640, 320, 160, 80, 40, 20, 10, 5, 2,5, 0 nM) montrant une liaison en temps réel à double référence à l’HA de la grippe A/New-York/55/04 (H3N2). (B) Liaison de la variante V2 de la variante CVN2L0 au ligand DM immobilisé dans une plage de concentration comprise entre 500 nM et 16 μM. Séquence: Les résidus L sont surlignés en jaune. Les résidus H sont surlignés en gris. E58 et R73 remplacent les cystéines dans la protéine de type sauvage et font de V2 un repliement protéique stable avec trois liaisons disulfure au lieu de quatre Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Alors que le bouclier glycane sur la partie supérieure HA distale de la membrane induit une liaison de haute affinité à CV-N 12, la liaison CVN2 à HA adjacente à un pont disulfure de la partie supérieure HA a également été observée à ses sites de faible affinité10,12. Diverses interactions polaires et sites d’interaction sont identifiés dans la liaison aux glucides par CV-N. Ces interactions sont vérifiées en générant des variantes knock-out dans le site de liaison pour corréler les affinités de liaison à la glycosylation in silico prédite12. Ainsi, le projet vise à comparer des peptides HA chimiquement mannosylés précédemment testés en affinité et spécificité de liaison avec de courtes séquences peptidiques provenant de pics 2019-nCoV liés au SRAS et du SARS-CoV-2, naturellement modifiés par un petit nombre de sites de glycosylation liés à l’N différents et glycosylation liée à O. En utilisant la cryo-microscopie électronique et les tests de liaison, Pinto et ses collègues rapportent un anticorps monoclonal, S309, qui reconnaît potentiellement un épitope sur la protéine de pointe du SRAS-CoV-2 contenant un glycane conservé dans le sous-genre Sarbecovirus, sans entrer en compétition avec la fixation du récepteur26. Le protocole de cette étude décrit comment la conception, l’expression et la caractérisation des variantes CV-N sont importantes pour étudier comment CV-N et CVN2 se lient aux protéines glycosylées et aux peptides mannosylés synthétiques à l’aide de la technologie SPR10,12.
Les variantes de dimères liés en tandem CVN2L027 et de site de liaison (V2-V5) sont exprimées de manière recombinante et les variantes sont avec des remplacements de liaison disulfure (C58E et C73R) (Figure 2A). En outre, un mutant avec une mutation ponctuelle unique E41A est préparé parce que cette position a été vue comme un résidu de contact intermoléculaire croisé. Ce mutant est une autre molécule intéressante pour les mesures de liaison SPR entre la lectine et les oligosaccharides à haut mannose, déchiffrant les domaines de liaison et permettant la comparaison avec la forme dimérique. La structure cristalline permutée de domaine de CVN2 montre un linker flexible, qui s’étend entre 49 et 54 résidus. Les deux domaines peuvent continuer à se déplacer autour de la charnière en tant que corps rigides, développant soit un monomère par des interactions de domaine intramoléculaire (domaine A -résidus 1-39;90-101- avec le domaine B -résidus 40-89) soit un dimère par échange de domaine intermoléculaire [domaine A (du premier monomère) avec le domaine B (du second), et domaine B (du premier monomère) avec le domaine A (de la deuxième copie)]. Il n’y a pas d’interactions étroites entre les domaines A et B des deux protomères, à l’exception de Glu4128. Le gène CV-N peut être développé à l’aide d’une méthode de PCR répétitive avec des oligos synthétisés 40-mer29 et est ensuite sous-cloné dans les sites NdeI et BamHI de pET11a pour la transformation (électroporation) en cellules électrocompétentes comme décrit par Keeffe, J.R.27. La protéine, qui est utilisée pour obtenir la structure cristalline respective (PDB ID 3S3Y), comprend une étiquette de purification N-terminale 6-histidine suivie d’un site de clivage de la protéase du facteur Xa. La mutagénèse dirigée est utilisée pour effectuer des mutations ponctuelles, changer de codon, et insérer ou supprimer des bases ou des codons simples ou multiples pour l’échange d’acides aminés. Ces transformations fournissent des informations inestimables sur la fonction et la structure des protéines. Les CV-N, CVN2 et CVN3 exprimés et purifiés par recombinaison ont été biophysiquement bien étudiés20,21,27, sont peu coûteux à produire et sont donc utilisés pour caractériser les tests de liaison aux glycanes immobilisés sur les puces de capteur SPR. Le test immuno-enzymatique conventionnel (ELISA) fournit moins de reproductibilité en ce qui concerne la technique d’immobilisation des ligands glycanes et transforme la liaison en temps réel de diverses variantes de site de liaison, qui est montrée pour SPR, en tests finaux.
La variante d’affinité de liaison CVN2L0-V2 (un pli intact de CV-N homodimérique avec une substitution de pont disulfure10) est exprimée avec un His-tag chez Escherichia coli (E. coli), purifiée sur colonne Ni-NTA en appliquant une chromatographie d’affinité et testée pour la liaison à HA (H3N2), peptide HA monomannosylé et peptide HA dimannosylé à l’aide de SPR. Les peptides chimiquement mannosylés, ou protéines HA et S, sont tous des ligands et des amines couplés à la surface de la puce hydrophile via des esters réactifs ou l’ingénierie des protéines biotine-streptavidine. La même procédure d’essais séquentiels est appliquée à ces ligands, en injectant diverses dilutions de CV-N et de variantes de CV-N (et CVN2) pour obtenir des informations cinétiques pour les analyses d’interaction moléculaire décrites ci-dessous30. La puce de capteur SPR immobilisée par RBD est utilisée pour les études de liaison sur les peptides CV-N à S, et les affinités sont comparées à la liaison du SARS-CoV-2 avec l’ACE2 humain.
Protocole
Pour la présente étude, une protéine de fusion SUMO (modificateur de type ubiquitine) à petite ubiquitine CVN a été utilisée dans des tests immuno-enzymatiques au lieu de CV-N et convient aux tests cellulaires. La protéine HA H3 recombinante du virus de la grippe A pleine longueur est obtenue commercialement (voir le tableau des matières) ou exprimée dans des lignées cellulaires HEK293 de mammifères et des cellules d’insectes infectées par le baculovirus selon les protocoles standard12. La protéine de pointe Wuhan-1 est exprimée dans les cellules HEK293 de mammifères. La synthèse de peptide monomannosylé (MM) et de peptide dimannosylé (DM) permet la détection de ligands homogènes à CVN2 et monomannosylée à petite molécule10.
1. Création de constructions CV-N
- Pour chacune des variantes CVN2 et de la protéine CVN2L0 (PDB ID 3S3Y), obtenir la construction du gène avec une séquence de tête pelB N-terminale et un marqueur His dans le vecteur pET27b(+) à partir de sources commerciales (voir le tableau des matériaux, dossier supplémentaire 1).
- Procurez-vous CVN2L0 et ses variantes (V2, V3, V4 et V5; Figure 2A,C) à l’arrière-plan d’un gène modèle CVN2L0 constitué de deux séquences d’ADN distinctes pour chaque répétition CV-N.
- Dissoudre l’ADN plasmidique lyophilisé dans de l’eau distillée désionisée stérile (ddH2O) jusqu’à une concentration finale de 100 ng/μL.
2. Préparation de plaques LB-agar avec des cellules transformées en ADN plasmidique
- Préparer le milieu de culture LB-Lennox en dissolvant 10 g/L de peptone, 5 g/L d’extrait de levure et 5 g/L de NaCl dans duddH2O(voir le tableau des matières) et ajuster le pH à 7,4. Effectuer la transformation en E. coli compétent BL21 (DE3) pour chaque variante (V2-V5) par méthode chimique suivant un rapport publié précédemment10.
- Diviser la solution (900 μL et 100 μL), transférer 100 μL sur des plaques de LB-agar (50 μg/mL de kanamycine) et utiliser doucement un épandeur de cellules stériles. Incuber les gélose pendant la nuit à 37 °C.
3. Clonage
- Sous-cloner le gène CV-N dans les sites NdeI et BamHI de pET11a (voir Tableau des matériaux) pour la transformation (électroporation) en cellules électrocompétentes suivant la référence27.
4. Mutagénèse dirigée
- Générer CVN2L029 et CVN-E41A mutant en arrière-plan d’un gène modèle CVN2L0 contenant deux séquences d’ADN distinguées pour chaque répétition CV-N27.
- Faire des mutations à l’aide d’une trousse de mutagénèse dirigée (voir le tableau des matériaux) et d’amorces mutagènes spécifiques 5'-gagaaccgtcaacgtttgcgataacagagttcagg-3' et 5'-cctgaactctgttatccaaacgttgacggttctc-3' pour exécuter la PCR31.
- Commencez une série de réactions d’échantillons en utilisant plusieurs concentrations d’ADN double brin (ADNds) allant de 5 à 50 ng (par exemple, 5, 10, 20 et 50 ng de modèle d’ADNds). Maintenez la concentration de l’apprêt constante.
REMARQUE : Le mélange PCR et le protocole de cyclage thermique sont généralement utilisés comme décrit dans le manuel d’instructions du kit de mutagénèse dirigée32.
- Commencez une série de réactions d’échantillons en utilisant plusieurs concentrations d’ADN double brin (ADNds) allant de 5 à 50 ng (par exemple, 5, 10, 20 et 50 ng de modèle d’ADNds). Maintenez la concentration de l’apprêt constante.
- Ajouter l’enzyme de restriction Dpn I (1 μL, 10 U/μL, voir le tableau des matériaux) sous la couche d’huile minérale. Bien mélanger délicatement les réactions, faire tourner vers le bas dans une microcentrifugeuse de table pendant 1 min, et incuber immédiatement à 37 °C pendant 1 h pour digérer l’ADNds superenroulé parental.
5. Transformation des cellules bactériennes
- Décongeler doucement les cellules supercompétentes XL1-Blue (voir le tableau des matériaux) sur la glace. Pour transformer chaque réaction de contrôle et d’échantillonnage, aliquote les cellules supercompétentes (50 μL) dans un tube à fond rond en polypropylène prérefroidi (14 mL).
- Transférer 1 μL de l’ADN simple brin traité par Dpn I (ADNss) de chaque réaction de contrôle et d’échantillon (ADNss muté) pour séparer les aliquotes des cellules supercompétentes, qui synthétisent le brin complémentaire. Faire tourbillonner soigneusement les réactions de transformation pour mélanger et incuber les réactions sur la glace pendant 30 min.
REMARQUE: Avant de transférer l’ADN traité par Dpn I à la réaction de transformation, il est recommandé de retirer soigneusement toute huile minérale restante de l’embout de la pipette. En tant que contrôle facultatif, l’efficacité de transformation des cellules supercompétentes XL1-Blue doit être vérifiée en mélangeant 0,1 ng/μL du plasmide témoin pUC18 (1 μL) avec une partie aliquote de 50 μL des cellules supercompétentes.
- Transférer 1 μL de l’ADN simple brin traité par Dpn I (ADNss) de chaque réaction de contrôle et d’échantillon (ADNss muté) pour séparer les aliquotes des cellules supercompétentes, qui synthétisent le brin complémentaire. Faire tourbillonner soigneusement les réactions de transformation pour mélanger et incuber les réactions sur la glace pendant 30 min.
- Appliquer une impulsion de chaleur aux réactions de transformation à 42 °C pendant 45 s, puis placer les réactions sur de la glace pendant 2 min.
REMARQUE: L’impulsion thermique appliquée a déjà été optimisée pour les conditions mentionnées dans les tubes à fond rond en polypropylène (14 mL). - Ajouter 0,5 mL de bouillon NZY+ (contenant par litre : 10 g d’amine néo-zélandaise (hydrolysat de caséine), 5 g d’extrait de levure, 5 g de NaCl, 12,5 mL de 1 M MgCl 2, 12,5 mL de 1 M MgSO4, 10 mL de glucose2 M, pH 7,5, et préchauffé à 42 °C) et incuber les réactions de transformation à 37 °C avec agitation à 225-250 rpm pendant 1 h. Plaquer le volume correct de chaque réaction de transformation (5 μL de transformation plasmidique témoin; 250 μL de transformation d’échantillon) sur des plaques de gélose LB-ampicilline.
REMARQUE : Pour les témoins de transformation et la mutagénèse, étaler les cellules sur des plaques de gélose LB-ampicilline contenant du 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal, 80 μg/mL) et de l’isopropyl-1-thio-β-D-galactopyranoside (IPTG, 20 mM) (voir le tableau des matériaux). Inoculer 50 mL de cultures cellulaires avec une seule colonie d’E transformé. cellules coli pour purifier l’ADN plasmidique muté pour les analyses. La mutagenèse est confirmée par séquençage de l’ADN dans une installation externe.
6. Expression et purification des protéines
- Pour une culture à grande échelle, inoculer une petite quantité de LB (contenant de l’ampicilline) avec une seule colonie de la plaque transformée.
- À l’aide de la culture de nuit, inoculer la culture d’expression avec des additifs, tels que 10 mM de MgCl2, 10 mM de MgSO4 et 20 mM de glucose, en diluant la culture de graines à 1/100.
- Cultiver des cellules en agitant vigoureusement à 37 °C. Cultiver les cellules à un Abs 600 nm entre 0,4-0,6 (phase mi-logarithmique) avant de refroidir les cellules à 20 °C. Induire avec 1 mM IPTG et grandir pendant la nuit.
- Ensuite, récoltez les cellules par centrifugation à 4 000 x g pendant 15 min à 4 °C et jetez le surnageant à l’aide d’une pipette.
- Resuspendre la pastille cellulaire dans un tampon de solution saline tamponnée au phosphate (PBS) et recentrifuger à 4 000 x g pendant 15 min à 4 °C. Ensuite, jetez le surnageant avec une pipette. Remettez en suspension la pastille restante dans 10 mL de tampon de lyse et incuber la suspension pendant 1 h à 37 °C.
NOTE: Composition du tampon de lyse: (50 mM de NaH 2 PO4, 300 mM de NaCl, 2% de Triton-X100, 500 ng / mL de lysozyme, 1 mM de fluorure de phénylméthylsulfonyle (PMSF), 1 mM de dithiothréitol, 1 mM de MgCl2, pH 8, voir Tableau des matériaux).- Soumettre le mélange à deux cycles de congélation-dégel (-80 °C). Séparer les fractions solubles et insolubles par centrifugation à 4 000 x g pendant 15 min à 4 °C et les analyser par électrophorèse sur gel de polyacrylamide (PAGE), en particulier le dodécylsulfate de sodium (SDS)-PAGE33 (Figure 2B,C).
REMARQUE: Les cellules peuvent être lysées de plusieurs façons, telles que le gel-décongélation, la sonication, l’homogénéisation, la lyse enzymatique ou une combinaison de ces méthodes. La purification des corps d’inclusion est recommandée pour la collecte de rendements protéiques élevés26.
- Soumettre le mélange à deux cycles de congélation-dégel (-80 °C). Séparer les fractions solubles et insolubles par centrifugation à 4 000 x g pendant 15 min à 4 °C et les analyser par électrophorèse sur gel de polyacrylamide (PAGE), en particulier le dodécylsulfate de sodium (SDS)-PAGE33 (Figure 2B,C).
- Purifier les protéines par chromatographie sur colonne Ni-NTA (voir le tableau des matériaux). Charger la fraction soluble sur une colonne de billes de Ni-NTA régénérée (1 mL/min). Laver le système avec un tampon TBS (50 mM de Tris, 150 mM de chlorure de sodium, pH 7,5) avant de commencer un gradient (0-100% 500 mM d’imidazole dans TBS sur 60 min) et de recueillir les fractions (1 mL/min). Dialyser les protéines purifiées pour la caractérisation biochimique contre 100 mM de PBS (Figure 2C,D).
- Vous pouvez également utiliser le gel d’affinité His-Select Ni 2+ (voir le tableau des matériaux) dans des tubes de 14 mL pour lier et remettre en suspension son CV-N exprimé par recombinaison dans des solutions tampons contenant 20 mM d’imidazole et250 mM d’imidazole, respectivement. Incuber par lots pendant au moins 30 min.
REMARQUE : Appliquez ces protéines semi-purifiées sur une colonne préemballée à usage unique pour l’échange tampon et le nettoyage d’échantillons biologiques, par exemple les glucides et les protéines, qui peuvent charger de 1 à 1,5 mL d’éluat provenant de la chromatographie d’affinité métallique immobilisée. - Transférer les solutions protéiques dans des tubes de centrifugation munis d’un filtre de coupure de 10 kDa (voir tableau des matières) et les concentrer par centrifugation pendant 10 min à 4 500 x g et 4 °C. Pour les mesures SPR, échanger les solutions d’analyte avec 10 mM d’HEPES, 150 mM de chlorure de sodium, 3 mM d’acide éthylènediaminetétraacétique (EDTA) et 0,05 % Tween20, pH 7,4 (HBS-EP(+), voir le tableau des matériaux).
- Ajouter ce tampon de fonctionnement SPR à un facteur de dilution de 1:10 et centrifuger quatre fois au volume initial pendant 10 min à 4 500 x g et 4 °C.
- Déterminer la concentration de protéines à 280 nm à l’aide d’un spectrophotomètre UV-Vis NanoDrop (voir le tableau des matériaux) en fonction du coefficient d’extinction calculé (20 440 M-1 cm-1) pour la protéine principale CVN2L0 montrant une taille de 23 474 Da34. Utilisez le PBS (100 mM, pH 7,0) ou le tampon SPR comme blanc et mesurez la concentration de protéines à trois étapes de dilution (1:1, 1:10 et 1:100).

Figure 2 : Séquences et expression CV-N. (A) CVN2 sans liaison entre chaque répétition CV-N (101 acides aminés chacune) et quatre ponts disulfure est exprimée en vecteur pET11a dans E. coli. (B) Expressions de deux colonies indépendantes pour CV-N (monomère) et CVN2 (dimère). (C) Les variantes de liaison disulfure sont purifiées et analysées sur SDS-PAGE. Un marqueur de faible poids moléculaire (6 μL) est utilisé comme référence. WT = CVN2L0 portant quatre ponts disulfures comme indiqué en (A). V2 est une variante avec un remplacement de liaison disulfure par des résidus polaires aux positions 58 et 73. V3-V5 sont des variantes avec deux liaisons S-S restantes et des acides aminés de substitution polaires (C58E-C73R) ou non polaires (C58W-C73M) ou une combinaison de ces substitutions de paires de résidus. (D) Les chromatogrammes CLHP de CVN2L0 purifié sont élués à un débit de 1 mL/min avec un gradient linéaire de 5 % à 65 % du tampon B dans le tampon A sur 30 min. Le tampon A est : 0,1 % (v/v) d’acide trifluoroacétique dans le ddH2O, le tampon Best : 0,08 % (v/v) d’acide trifluoroacétique dans l’acétonitrile. La protéine est analysée sur une colonne de gel de silice haute performance 300-5-C4 (150 x 4,6 mm) à 214 nm et 280 nm. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
7. Spectroscopie SPR
- Utiliser le système SPR à double canal (voir le tableau des matériaux) avec tampon HBS-EP(+) et glycine HCl pH 1,5-1,6 à 10 mM comme tampon de régénération. Allumez l’instrument, le dégazeur, l’échantillonneur automatique, pompez et lavez l’ensemble du système avec ddH20 pendant 1 h. Placez le tampon de fonctionnement prêt à l’emploi dans une bouteille séparée.
- Déposez de l’huile d’émersion sur le détecteur et montez une puce de capteur en verre (voir Tableau des matériaux) recouverte d’un mince film d’or et fonctionnalisée sur la face supérieure avec de l’hydrogel de carboxyméthyldextrane directement sur le détecteur sous la cellule d’écoulement à trois orifices. Corrigez le paramètre en abaissant la manipulation.
REMARQUE: C19RBDHC30M 200 nm streptavidine dérivatized carboxymethyldextran hydrogel avec une densité moyenne de protéine RBD biotinylée du coronavirus du syndrome respiratoire aigu sévère 2, est une puce de capteur prête à l’emploi avec le ligand pré-immobilisé.
8. Test de liaison SPR pour la liaison CV-N à HA, protéine S et RBD
- Immobiliser les ligands protéiques sur les puces du capteur en suivant les étapes ci-dessous.
- Ouvrez une table d’exécution en cliquant sur Formulaire dans la barre de menu et sur Exécuter l’éditeur de table dans le logiciel SPRAutoLink intégré (voir Table des matériaux). Choisissez et cliquez sur BASIC_Immobilization dans la liste des tables d’exécution disponibles et suivez les étapes de la procédure expérimentale sur l’écran de l’ordinateur. L’éditeur d’échantillons utilisé est affiché dans le coin supérieur droit.
- Cliquez sur Sample Set Editor dans la section Form pour remplir la liste des réactifs pour deux racks placés dans l’échantillonneur automatique pour des analyses plus approfondies. Cliquez sur Autosampler Direct Control en tant qu'« outil » dans la barre de menus pour faire avancer ou ramener les racks à la maison. Choisissez 4 °C comme température de fonctionnement.
REMARQUE: Le logiciel SPR permet « SPR Instrument Direct Control » et « Pump Direct Control » via la sélection des outils correspondants, ainsi que la manipulation automatique de l’échantillonneur, et en cliquant sur Formulaire; Exécuter l’éditeur de table, le diagramme de données ou le post-traitement peuvent également être choisis pour effectuer l’analyse des données. Les fichiers sont directement enregistrés dans le répertoire par défaut et exportés sous le nom scrubber.files à partir de la fenêtre Post-traitement.
- Démarrez la pompe pour infuser ddH20 en cliquant sur Outils et contrôle direct de la pompe et enregistrez les données en cliquant sur SPR Instrument Direct Control, et chaque fois Start dans les fenêtres nouvellement apparues. Placez les réactifs de couplage (étape 8.3) dans des flacons de 300 μL, placez-les dans les racks d’échantillonneur automatique et démarrez la table d’exécution en cliquant sur Exécuter.
REMARQUE: Les surfaces des puces sont soit conditionnées avec un tampon glycine de 10 mM pH 9,0, soit rincées avec du chlorure de sodium de 1 M, un tampon de borate de sodium de 0,1 M pH 9,0 pour conditionner la surface de la puce dérivatisée par carboxyle pour le mélange d’activation EDC / NHS30. - Pour cette interaction protéine-protéine simple, utilisez la puce CMD500D (voir Tableau des matériaux) pour générer une unité d’indice de micro-réfraction (μRIU) = cellule d’écoulement 2500 - 3000 avec HA immobilisé et μRIU = cellule d’écoulement 400 avec protéine de pointe. À un débit de perfusion de 15 μL/min, injecter un mélange aqueux et égal de chlorhydrate de 0,4 M de N-éthyl-N'-(diméthylaminopropyl) carbodiimide (EDC*HCl) et de 0,1 M N-hydroxysuccinimide (NHS) en appliquant les étapes séquentielles suivantes.
- Recharger la pompe de remplissage à 25 000 μL/min, effectuer un réglage de base pendant 30 s, injecter 90 μL de solution d’activation d’échantillon (EDC/NHS) sur un temps de contact de 6 minutes, puis maintenir pendant 5 minutes supplémentaires.
- Répéter ce cycle après l’application pendant 1 min à 10 μL/min uniquement sur la cellule d’écoulement gauche (bleue) pour injecter et immobiliser les peptides10, HA et la protéine de pointe synthétisés chimiquement à 20 μg/mL, et laisser procéder à l’ajustement ultérieur de la ligne de base avec ddH2O pendant 1,5 min avant de tremper la surface de la puce activée avec 1 M éthanolamine HCl pH 8,5.
- Basculer les tubes de l’échantillonneur de liquide vers le dégazeur de ddH20 dans la bouteille avec HBS-EP(+) (Figure supplémentaire 1).
- Analyser les capteurs SPR.
- Pour effectuer des études cinétiques, utiliser différentes concentrations d’analytes (10-5-10-8 M), avec une étape de régénération après chaque injection et des mesures à blanc après différents analytes. Réglez le débit à 10 μL/min et commencez les injections pendant 4 min de temps de contact, puis 5 min de génération de base et deux étapes de régénération de 2 min chacune avec un intervalle de 30 s.
- Injecter la solution tampon pour les mesures à blanc dont les capteurs sont soustraits des prélèvements d’échantillons pour normaliser différentes concentrations de protéines.
- Cliquez sur Formulaire, faites défiler vers le bas et passez à « Post-Processing » en cliquant sur ce mode de fonctionnement. Cliquez sur Ajouter pour sélectionner les courbes de liaison générées au fil du temps dans le formulaire de tracé de données pour chaque cellule de flux et exporter la superposition sous forme de fichier de nettoyage (.ovr). Cliquez sur Fichier pour ouvrir les options d’enregistrement du fichier. Obtenir des courbes de réponse en alignant les courbes gauche et droite et en soustrayant les signaux du second canal de référence de ceux du canal du ligand.
REMARQUE: Les données sont exploitées en « Post-Processing » en définissant des capteurs par calcul. Il est placé dans une superposition de capteurs à partir de cellules de flux gauche et droite, ou représenté comme des capteurs de la différence des deux canaux. - Nettoyez l’ensemble des fluidiques avec un tampon glycine de 50 à 100 mM pH 9,5, de l’eau et de l’éthanol à 20% avant et après les mesures de liaison pour éliminer les traces de sel ou toute contamination protéique, ou plus stricte, avec 0,5% de SDS et de glycine.
REMARQUE: Pour éviter d’endommager l’instrument, il est recommandé de vérifier la stabilité mécanique de la puce de verre avant de réinsérer la cartouche de puce dans l’instrument si les puces ont été stockées sous tampon ou à 100% d’humidité.
Résultats
Une molécule CVN2L0 dimérique échangée de domaine est testée pour se lier à la région supérieure HA dans trois expériences SPR distinctes et l’affinité de liaison est présentée en valeurs KD. Le domaine B est supposé comprendre les sites de liaison H, qui sont affectés par le remplacement d’une liaison disulfure en résidus ioniques, et le domaine A forme L10,18. Les injections simples de CVN2L0 et les variantes V2 (trois ponts disulf...
Discussion
L’affinité de liaison de CV-N est corrélée avec le nombre de sites de liaison fonctionnels [2H sur les domaines B et 2L sur le(s) domaine(s) A lorsqu’ils sont conçus comme dimères permutés de domaine]. Une variante avec une affinité de liaison altérée (CVN2L0-V2, un pli stable homodimérique de CV-N comprenant un pont disulfure) est exprimée dans E. coli, purifiée et testée positivement pour la liaison à la protéine HA (H3N2) en utilisant SPR10, et montre un changement c...
Déclarations de divulgation
L’auteur n’a rien à divulguer.
Remerciements
L’auteur remercie le Dr Christian Derntl du Département de biotechnologie et de microbiologie de la TU Wien et du Département de médecine III, Division de néphrologie et de dialyse de l’Université de médecine de Vienne, en particulier le Dr Markus Wahrmann pour son soutien technique et scientifique. L’expression des protéines dans les cellules de mammifères a été soutenue par le Département de biotechnologie de l’Université des ressources naturelles et des sciences de la vie (BOKU) de Vienne. L’auteur tient à exprimer sa profonde gratitude au Dr Nico Dankbar de XanTec bioanalytics à Düsseldorf, en Allemagne, pour ses discussions scientifiques utiles sur la réalisation des tests de liaison SPR.
matériels
| Name | Company | Catalog Number | Comments |
| Äkta primeplus | Cytiva | ||
| Amicon tubes | Merck | C7715 | |
| Ampillicin | Sigma-Aldrich | A5354 | |
| Beckmann Coulter Cooler Allegra X-30R centrifuge | Beckman Coulter | B06320 | |
| Cell spreader | Sigma-Aldrich | HS86655 | silver stainless steel, bar L 33 mm |
| Custom DNA Oligos | Sigma-Aldrich | OLIGO | |
| Custom Gensynthesis | GenScript | #1390661 | cloning vector: pET27b(+) |
| Cytiva HBS-EP+ Buffer 10, 4x50mL | Thermo Scientific | 50-105-5354 | |
| Dionex UlitMate 3000 | Thermo Scientific | IQLAAAGABHFAPBMBFB | |
| Dpn I restriction enzyme (10 U/μL) | Fisher Scientific | ER1701 | |
| DTT | Merck | DTT-RO | |
| EDC | Merck | 39391 | |
| EDTA | Merck | E9884 | |
| Eppendorf Safe-Lock Tubes | Eppendorf | 30120086 | |
| Eppendorf Safe-Lock Tubes | Eppendorf | 30120094 | |
| Eppendorf Minispin and MiniSpin Plus personal microcentrifuge | Sigma-Aldrich | Z606235 | |
| Ethanol | Merck | 51976 | |
| Ethanolamine HCl | Merck | E6133 | |
| Falcon 50mL Conical Centrifuge Tubes | Fisher Scientific | 14-432-22 | |
| Falcon 14 mL Round Bottom Polystyrene Test Tube, with Snap Cap, Sterile, 25/Pack | Corning | 352057 | |
| Glucose | Merck | G8270 | |
| Glycine HCl | Merck | 55097 | |
| HA H3 protein | Abcam | ab69751 | |
| HEPES | Merck | H3375 | |
| His-select Ni2+ | Merck | H0537 | |
| Imidazole | Merck | I2399 | |
| IPTG | Merck | I6758 | |
| Kanamycin A | Sigma-Aldrich | K1377 | |
| Kromasil 300-5-C4 | Nouryon | ||
| LB agar | Merck | 52062 | |
| LB agar | Merck | 19344 | |
| LB Lennox | Merck | L3022 | |
| Lysozyme | Merck | 10837059001 | |
| Magnesium chloride | Merck | M8266 | |
| Magnesium sulfate | Merck | M7506 | |
| NaH2P04 | Merck | S0751 | |
| NanoDrop UV-Vis2000c spectrophotometer | Thermo Scientific | ND2000CLAPTOP | |
| NaOH | Merck | S5881 | |
| NHS | Merck | 130672 | |
| NZ amine (casein hydrolysate) | Merck | C0626 | |
| PBS | Merck | 806552 | |
| PD MidiTrap G-10 | Sigma-Aldrich | GE28-9180-11 | |
| Peptone | Merck | 70171 | |
| pET11a | Merck Millipore (Novagen) | 69436 | |
| PMSF | Merck | PMSF-RO | |
| QIAprep Spin Miniprep Kit (1000) | Qiagen | 27106X4 | |
| Reichert Software Package Autolink1-1-9 | Reichert | ||
| Reichert SPR SR7500DC Dual Channel System | Reichert | ||
| Scrubber2-2012-09-04 for data analysis | Reichert | ||
| SDS | Merck | 11667289001 | |
| Site-directed mutagenesis kit incl pUC18 control plasmid | Stratagene | #200518 | |
| Sodim chloride | Merck | S9888 | |
| Sodium acetate.Trihydrate | Merck | 236500 | |
| SPR sensor chip C19RBDHC30M | XanTec bioanalytics | SCR C19RBDHC30M | |
| SPR sensor chip CMD500D | XanTec bioanalytics | SCR CMD500D | |
| Sterilin Standard 90mm Petri Dishes | Thermo Scientific | 101R20 | |
| TBS | Merck | T5912 | 10x, solution |
| Triton-X100 | Merck | T8787 | |
| Tryptone | Merck | 93657 | |
| Tween20 | Merck | P1379 | |
| Vortex-Genie 2 Mixer | Merck | Z258423 | |
| X-gal | Merck | XGAL-RO | |
| XL1-Blue Supercompetent Cells | Stratagene | #200236 | |
| Yeast extract | Merck | Y1625 |
Références
- Perez-Caballero, D., et al. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell. 139 (3), 499-511 (2009).
- Waheed, A. A., Gitzen, A., Swiderski, M., Freed, E. O. High-mannose but not complex-type glycosylation of tetherin is required for restriction of HIV-1 release. Viruses. 10 (1), 26 (2018).
- Wilson, I. A., et al. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature. 289 (5796), 366-373 (1981).
- Otterstrom, J. J., et al. Relating influenza virus membrane fusion kinetics to stoichiometry of neutralizing antibodies at the single-particle level. Proceedings of the National Academy of Sciences of the United States of America. 111 (48), 5143-5148 (2014).
- Kaletsky, R. L., Francica, J. R., Agrawal-Gamse, C., Bates, P. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proceedings of the National Academy of Sciences of the United States of America. 106 (8), 2886-2891 (2009).
- Tokarev, A., Skasko, M., Fitzpatrick, K., Guatelli, J. Antiviral activity of the interferon-induced cellular protein BST-2/tetherin. AIDS Research and Human Retroviruses. 25 (12), 1197-1210 (2009).
- Gnirss, K., et al. Tetherin sensitivity of influenza A viruses is strain specific: Role of hemagglutinin and neuraminidase. Journal of Virology. 89 (18), 9178-9188 (2015).
- Fleury, D., et al. A complex of influenza hemagglutinin with a neutralizing antibody that binds outside the virus receptor binding site. Nature Structural Biology. 6 (6), 530-534 (1999).
- Salunke, S. B., et al. Iron(III) chloride as an efficient catalyst for stereoselective synthesis of glycosyl azides and a cocatalyst with Cu(0) for the subsequent click chemistry. Chemical Communication. (Camb). 47 (37), 10440-10442 (2011).
- Schilling, P. E., et al. Mannosylated hemagglutinin peptides bind cyanovirin-N independent of disulfide-bonds in complementary binding sites. RSC Advances. 10 (19), 11079-11087 (2020).
- Fleury, D., Wharton, S. A., Skehel, J. J., Knossow, M., Bizebard, T. Antigen distortion allows influenza virus to escape neutralization. Nature Structural Biology. 5 (2), 119-123 (1998).
- Maier, I., Schiestl, R. H., Kontaxis, G. Cyanovirin-N binds viral envelope proteins at the low-affinity carbohydrate binding site without direct virus neutralization ability. Molecules. 26 (12), 3621 (2021).
- Ahmed, A. J., Keremane, S. R., Vielmetter, J., Bjorkman, P. J. Structural characterization of GASDALIE Fc bound to the activating Fc receptor FcγRIIIa. Journal of Structural Biology. 194 (1), 78-89 (2016).
- Boyd, R. Discovery of cyanovirin-N, a novel human immunodeficiency virus-inactivating protein that binds viral surface envelope glycoprotein gp120: potential applications to microbicide development. Antimicrobial Agents and Chemotherapy. 41 (7), 1521-1530 (1997).
- Bolmstedt, A. J., O'Keefe, B. R., Shenoy, S. R., McMahon, J. B., Boyd, M. R. Cyanovirin-N defines a new class of antiviral agent targeting N-linked, high-mannose glycans in an oligosaccharide-specific manner. Molecular Pharmacology. 59 (5), 949-954 (2001).
- O'Keefe, B. R., et al. Potent anti-influenza activity of cyanovirin-N and interactions with viral hemagglutinin. Antimicrobial Agents and Chemotherapy. 47 (8), 2518-2525 (2003).
- Shenoy, S. R., et al. Multisite and multivalent binding between cyanovirin-N and branched oligomannosides: calorimetric and NMR characterization. Chemical Biology. 9 (10), 1109-1118 (2002).
- Bewley, C. A., Kiyonaka, S., Hamachi, I. Site-specific discrimination by cyanovirin-N for alpha-linked trisaccharides comprising the three arms of Man(8) and Man(9). Journal of Molecular Biology. 322 (4), 881-889 (2002).
- Barrientos, L. G., Matei, E., Lasala, F., Delgado, R., Gronenborn, A. M. Dissecting carbohydrate-Cyanovirin-N binding by structure-guided mutagenesis: functional implications for viral entry inhibition. Protein Engineering Design & Selection. 19 (12), 525-535 (2006).
- Bewley, C. A., Otero-Quintero, S. The potent anti-HIV protein cyanovirin-N contains two novel carbohydrate binding sites that selectively bind to Man(8) D1D3 and Man(9) with nanomolar affinity: implications for binding to the HIV envelope protein gp120. Journal of the American Chemical Society. 123 (17), 3892-3902 (2001).
- Bewley, C. A. Solution structure of a cyanovirin-N:Man alpha 1-2Man alpha complex: structural basis for high-affinity carbohydrate-mediated binding to gp120. Structure. 9 (10), 931-940 (2001).
- Jensen, S. M. R., et al. Differential inhibitory effects of Cyanovirin-N, Griffithsin, and Scytovirin on entry mediated by envelopes of gammaretroviruses and deltaretroviruses. Journal of Virology. 88 (4), 2327-2332 (2014).
- Lan, J., et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 581 (7807), 215-220 (2020).
- Lingwood, D., et al. Structural and genetic basis for development of broadly neutralizing influenza antibodies. Nature. 489 (7417), 566-570 (2012).
- Ekiert, D. C., et al. Antibody recognition of a highly conserved influenza virus epitope. Science. 324 (5924), 246-251 (2009).
- Pinto, D., et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature. 583 (7815), 290-295 (2020).
- Keeffe, J. R., et al. Designed oligomers of cyanovirin-N show enhanced HIV neutralization. Proceedings of the National Academy of Sciences of the United States of America. 108 (34), 14079-14084 (2011).
- Yang, F., et al. Crystal structure of cyanovirin-N, a potent HIV-inactivating protein, shows unexpected domain swapping. Journal of Molecular Biology. 288 (3), 403-412 (1999).
- Stemmer, W. P., Crameri, A., Ha, K. D., Brennan, T. M., Heyneker, H. L. Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides. Gene. 164 (1), 49-53 (1995).
- Fischer, M. J. E., Mol, N., Fischer, M. Amine coupling through EDC/NHS: A practical approach. Surface Plasmon Resonance. Methods in Molecular Biology (Methods and Protocols). 627, (2010).
- Novoradovsky, A., et al. Computational principles of primer design for site directed mutagenesis. Technical Proceedings of 2005 NSTI Nanotechnology Conference and Trade Show. , 532-535 (2005).
- . QuikChange Site-Directed MutagenesisKit, User Manual Available from: https://users.drew.edu/jliu3/Docs/Stratagene%20Quikchange%20mutagenesis.pdf#:~:text=The%20QuikChange%20sitedirected%20mutagenesis%20kit%20is%20used%20to (2005)
- Laemmli, U. K. Cleavage of structural proteins during the assembly of the head bacteriophage T4. Nature. 227 (5259), 680-685 (1970).
- . Expasy.org Available from: https://web.expasy.org/cgi-bin/protparam/protparam (2022)
- Karlsson, R. Real-time competitive kinetic analysis of interactions between low-molecular-weight ligands in solution and surface-immobilized receptors. Analytical Biochemistry. 221 (1), 142-151 (1994).
- Schuck, P., Zhao, H. The role of mass transport limitation and surface heterogeneity in the biophysical characterization of macromolecular binding processes by SPR biosensing. Methods Molecular Biology. 627, 15-54 (2020).
- Barnes, O., et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature. 588 (7839), 682-687 (2020).
- . Using reichert Surface Plasmon Resonance (SPR) for Antiviral Development, Application Note 28 Available from: https://www.reichertspr.com/clientuploads/directory/application_notes/Application_Note_28__Using_Reichert_Surface_Plasmon_Resonance_for_Antiviral_Testing.pdf (2022)
- Sundberg, E. J., Andersen, P. S., Gorshkova, I. I., Schuck, P., Schuck, P. Surface plasmon resonance biosensing in the study of ternary systems of interacting proteins. Protein Interactions: Biophysical Approaches for the Study of Complex Reversible Systems. 5, 97-141 (2007).
- . Method Development Notes Available from: https://www.reichertspr.com/applications/method-development-notes/ (2022)
- Angulo, J., Enríquez-Navas, P. M., Nieto, P. M. Ligand-receptor binding affinities from saturation transfer difference (STD)-NMR spectroscopy: the binding Isotherm of STD initial growth rates. Chemistry. 16 (26), 7803-7812 (2010).
- Goldflam, M., Tarragó, T., Gairí, M., Giralt, E. NMR studies of protein-ligand interactions. Methods in Molecular Biology. 831, 233-259 (2012).
- Kumar, S., Maurya, V. K., Prasad, A. K., Bhatt, M. L. B., Saxena, S. K. Structural, glycosylation and antigenic variation between 2019 novel coronavirus (2019-nCoV) and SARS coronavirus (SARS-CoV). Virusdisease. 31 (1), 13-21 (2020).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.