
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Präparation und strukturelle Bewertung von Epithelzell-Monoschichten in einer physiologisch dimensionierten mikrofluidischen Kulturanlage
In diesem Artikel
Zusammenfassung
Das vorgestellte Protokoll beschreibt die Entwicklung und den Einsatz einer Phalloidin-basierten filamentösen Aktin-Färbetechnik mit konfokaler Laser-Scanning-Mikroskopie (CLSM) zur Visualisierung der adhärenten Zellschichtstruktur in mikrofluidischen dynamischen Kulturkanälen und traditionellen statischen Fix-Well-Kulturkammern. Dieser Ansatz hilft bei der Bewertung der Zellschichtkonfluenz, der Monolagenbildung und der Gleichmäßigkeit der Schichtdicke.
Zusammenfassung
In-vitro-mikrofluidische Experimente bergen ein großes Potenzial, viele Einblicke in die mikrophysiologischen Phänomene zu gewinnen, die bei Erkrankungen wie dem akuten Atemnotsyndrom (ARDS) und der beatmungsinduzierten Lungenschädigung (VILI) auftreten. Studien in mikrofluidischen Kanälen mit physiologisch relevanten Dimensionen für die terminalen Bronchiolen der menschlichen Lunge stehen jedoch derzeit vor mehreren Herausforderungen, insbesondere aufgrund von Schwierigkeiten bei der Festlegung geeigneter Zellkulturbedingungen, einschließlich Medienflussraten, innerhalb einer bestimmten Kulturumgebung. Das vorgestellte Protokoll beschreibt einen bildbasierten Ansatz zur Bewertung der Struktur von NCI-H441 humanen Lungenepithelzellen, die in einem sauerstoffundurchlässigen mikrofluidischen Kanal kultiviert wurden, dessen Dimensionen physiologisch relevant für die terminalen Bronchiolen der menschlichen Lunge sind. Mittels Phalloidin-basierter filamentöser Aktin-Färbung werden die zytoskelettalen Strukturen der Zellen mittels konfokaler Laser-Scanning-Mikroskopie aufgedeckt, was die Visualisierung sowohl einzelner als auch geschichteter Zellen ermöglicht. Die anschließende Quantifizierung bestimmt, ob die verwendeten Zellkulturbedingungen einheitliche Monoschichten erzeugen, die für weitere Experimente geeignet sind. Das Protokoll beschreibt Zellkultur- und Schichtbewertungsmethoden in mikrofluidischen Kanälen und traditionellen Fixed-Well-Umgebungen. Dazu gehören Kanalkonstruktion, Zellkultur und erforderliche Bedingungen, Fixierung, Permeabilisierung und Färbung, konfokale mikroskopische Bildgebung, Bildverarbeitung und Datenanalyse.
Einleitung
Das akute Atemnotsyndrom (ARDS) ist eine akute Erkrankung, die durch eine Verletzung des Lungenparenchyms verursacht wird und zu einem Lungenödem der Alveolen, einem unzureichenden Gasaustausch und einer anschließenden Hypoxämie führt1. Dadurch wird ein Zyklus aus entzündungsfördernder Zytokinfreisetzung, Rekrutierung neutrophiler Granulozyten, Freisetzung toxischer Mediatoren und Gewebeschädigung in Gang gesetzt, die wiederum eine weitere Entzündungsreaktion hervorruft2. Darüber hinaus kann pulmonales Surfactant, das die Atemwege stabilisiert und Schäden durch wiederholte Rekrutierung/Derekrutierung (F/E) verhindert, durch die während des ARDS ablaufenden chemischen Prozesse inaktiviert oder anderweitig funktionsgestört werden, was zu weiterer Belastung und Verletzung des umgebenden Parenchyms führt3. Wenn eine ausreichende Schädigung vorliegt, kann eine mechanische Beatmung erforderlich sein, um eine ausreichende systemische Sauerstoffversorgung zu gewährleisten4. Die mechanische Beatmung bringt jedoch ihre eigenen Herausforderungen und Traumata mit sich, einschließlich der Möglichkeit einer beatmungsinduzierten Lungenschädigung (VILI), die als Verletzung des Lungenparenchyms gekennzeichnet ist, die durch die mechanischen Belastungen verursacht wird, die während der Überblähung (Volutrauma) und/oder der F/E der Luft-Flüssigkeits-Grenzfläche im flüssigkeitsverschlossenen Atemweg (Atelectrauma) auftreten5. Der Druckgradient, den Epithelzellen, die einer Luft-Flüssigkeits-Grenzfläche (wie in einer flüssigkeitsverschlossenen Bronchiole) ausgesetzt sind, im Atelectrauma-Modell erfahren, kann zu einer durch Permeabilität verursachten obstruktiven Reaktion (POOR) führen, die zu einem POOR-get-POORer-positiven Verletzungszyklus führt 6,7,8.
In-vitro-Experimente können Einblicke in diese Phänomene im Mikromaßstab liefern, aber aktuelle Studien in mikrofluidischen Kanalumgebungen mit physiologisch relevanten Dimensionen stehen vor mehreren Herausforderungen9. Zum einen stellt die Optimierung der Zellkulturbedingungen eine erhebliche Eintrittsbarriere für die Zellkulturforschung in mikrofluidischen Umgebungen dar, da es eine enge Schnittstelle gibt, innerhalb derer Medienflussparameter, Kulturdauer und andere Kulturbedingungen eine optimale Zellschichtbildung ermöglichen. Dazu gehören auch die Diffusionsbeschränkungen, die sich aus der Sauerstoffundurchlässigkeit des mikrofluidischen Kulturkanalgehäuses ergeben. Dies erfordert eine sorgfältige Berücksichtigung der Strömungsparameter des Mediums, da niedrige Durchflussraten den Zellen Sauerstoff entziehen können, insbesondere denen, die am weitesten vom Einlass entfernt sind. Andererseits können hohe Flussraten Zellen aus dem Kulturkanal verdrängen oder zu einer unsachgemäßen oder ungleichmäßigen Schichtentwicklung führen. Diffusionsbeschränkungen können durch die Verwendung sauerstoffdurchlässiger Materialien wie Polydimethylsiloxan (PDMS) in einer Luft-Flüssigkeits-Grenzfläche (ALI)-Kulturapparatur behoben werden. Viele herkömmliche mikrofluidische Kulturkanäle, wie z. B. die des ECIS-Systems (Electric Cell-Substrat Impedance Sensing), sind jedoch aufgrund der Beschaffenheit des hergestellten Gehäuses10 von Natur aus sauerstoffundurchlässig. Dieses Protokoll zielt darauf ab, eine Technik zur Analyse von Zellschichten bereitzustellen, die in einem sauerstoffundurchlässigen Gehäuse kultiviert wurden.
Beim Vergleich der Lebensfähigkeit von Kulturbedingungen sind Beobachtungen spezifischer Schichteigenschaften, wie z. B. das Vorhandensein einer Monoschicht, die Oberflächentopologie, die Konfluenz und die Gleichmäßigkeit der Schichtdicke, erforderlich, um festzustellen, ob die Zellschicht, die durch eine bestimmte Gruppe von Kulturbedingungen erzeugt wird, die gewünschten Spezifikationen erfüllt und tatsächlich für das Versuchsdesign relevant ist. Eine begrenzte Bewertung kann mit Methoden wie ECIS durchgeführt werden, bei denen Messungen des elektrischen Potentials (Spannung) verwendet werden, das durch den Widerstand gegen hochfrequenten Wechselstrom (AC) (Impedanz) erzeugt wird, der von elektrisch isolierenden Membranen von Zellen erzeugt wird, die auf Goldelektroden innerhalb des Durchflussarrays kultiviert wurden. Durch die Modulation der Frequenz des auf Zellen angelegten AC können spezifische frequenzabhängige zelluläre Eigenschaften der Zellen und Zellschichten, wie z. B. Oberflächenadhärenzstärke, Tight-Junction-Bildung und Zellproliferation oder -konfluenz, gezielt anvisiert und untersucht werden11. Diese indirekten Formen der Messung sind jedoch zu Beginn eines Experiments etwas schwierig zu interpretieren und quantifizieren möglicherweise nicht alle relevanten Aspekte der Zellschicht. Die einfache Beobachtung der Zellschicht unter einem Phasenkontrastmikroskop kann die Natur bestimmter Eigenschaften wie der Konfluenz aufdecken; Viele relevante Merkmale, wie z. B. das Vorhandensein einer Monoschicht und die Gleichmäßigkeit der Schichtdicke, erfordern jedoch eine dreidimensionale (3D) Auswertung, die mit Hellfeld-, Phasenkontrast- oder fluoreszenzmikroskopischer Bildgebung nicht möglich ist12.
Das Ziel dieser Studie war es, eine filamentöse Aktin-Färbetechnik zu entwickeln, die eine bildgebende Verifizierung einer Monoschicht und die Bewertung der Zellschichtgleichmäßigkeit mittels konfokaler Laser-Scanning-Mikroskopie (CLSM) ermöglicht. Filamentöses Aktin (F-Aktin) wurde als geeignetes Ziel für das Fluorophor-Konjugat angesehen, was zum Teil auf die Art und Weise zurückzuführen ist, wie F-Aktin eng der Zellmembran folgt und eine visuelle Annäherung an das gesamte Zellvolumen ermöglicht13. Ein weiterer wichtiger Vorteil des Targetings von F-Aktin ist die Art und Weise, wie die Färbung von F-Aktin Störungen oder Veränderungen des Zytoskeletts, die durch die Belastungen der Zellen verursacht werden, visuell aufklärt. Die Vernetzungsfixierung mit methanolfreiem Formaldehyd wurde verwendet, um die Morphologie der Zellen und der Zellschicht zu erhalten, da dehydrierende Fixiermittel wie Methanol dazu neigen, Zellen abzuflachen, die Zellschicht stark zu verzerren und ihre Eigenschaften zu verändern14,15.
Um zu bestimmen, ob die Schichtbewertungstechnik in der Lage ist, diese Herausforderungen zu mindern, wurden die Zellen in traditionellen Acht-Well-Kulturkammern sowie in mikrofluidischen Kanälen kultiviert, um die Unterschiede in den erzeugten Zellschichten zu bewerten. Für Fixkulturwellen wurden Deckglaseinheiten mit acht Wells verwendet. Für die mikrofluidische Kultur wurden Flow-Arrays (Kanallänge 50 mm, Breite 5 mm, Tiefe 0,6 mm) optimiert, um immortalisierte humane Lungenepithelzellen (NCI-H441) in einer Umgebung zu kultivieren, deren Abmessungen physiologisch relevant für die terminalen Bronchiolen in der Atemzone der menschlichen Lunge sind16. Obwohl dieses Protokoll unter Berücksichtigung der Kulturumgebung von ECIS-Flow-Arrays entwickelt wurde, kann es für jede sauerstoffundurchlässige dynamische Kulturumgebung gelten, für die eine Bewertung der Eigenschaften der kultivierten Zellschicht oder der Kulturbedingungen erforderlich ist.
Protokoll
Für die vorliegende Studie wurde die humane epitheliale Lungenzelllinie NCI-H441 verwendet (siehe Materialtabelle).
1. Zellkultur im mikrofluidischen Kanal
- Stellen Sie den mikrofluidischen Kanal her und führen Sie die Vorbehandlung gemäß den folgenden Schritten durch.
- Besorgen Sie sich ein einkanaliges Durchfluss-Array (siehe Materialtabelle) und trennen Sie den oberen Teil von der Polycarbonat-Grundplatte.
- Besorgen Sie sich ein rechteckiges Deckglas #1.5 (Dicke 0.17 mm) mit Abmessungen von 60 mm x 22 mm. Reinigen Sie die Oberflächen des Deckglases in einem Ultraschallbad und behandeln Sie eine Seite 5 Minuten lang mit einer 0,1 mg/ml-Lösung Poly-D-Lysin bei Raumtemperatur, bevor Sie sie 30 Minuten lang bei 60 °C trocknen.
- Bringen Sie einen 0,13 mm dicken doppelseitigen Klebstoff (siehe Materialtabelle), der auf die Abmessungen der Oberseite des Strömungsarrays und des Strömungskanals (50 mm Länge, 5 mm Breite) zugeschnitten ist, auf die Oberseite des Strömungsarrays auf, wobei darauf zu achten ist, dass die Kanalausschnitte17 genau ausgerichtet sind.
- Befestigen Sie einen 0,1 mm dicken Mylar-Abstandshalter (siehe Materialtabelle), lasergeschnitten, um die Abmessungen der Oberseite des Flow-Arrays und des Flow-Kanals aufzunehmen, auf den Klebestreifen und achten Sie dabei auf eine exakte Ausrichtung der Kanalausschnitte.
- Wiederholen Sie die Schritte 1.1.3 und 1.1.4, bis die gewünschte Kanalhöhe erreicht ist (z. B. bei einer Kanalhöhe von 0,6 mm zwei Abstandshalter und drei Klebestreifen verwenden).
- Befestigen Sie ein rechteckiges Deckglas auf dem untersten Klebestreifen mit der Poly-D-Lysin-behandelten Seite, die dem Klebstoff zugewandt ist. Üben Sie nach Abschluss der Montage, wie in Abbildung 1 gezeigt, festen und gleichmäßigen Druck auf die Ober- und Unterseite der Konstruktion aus und halten Sie sie 1 Minute lang.
Anmerkungen: Die Konstruktion des Kanalgehäuses, einschließlich Deckglas, Klebstoffen, Abstandshaltern und Durchfluss-Array-Oberseite, ist jetzt abgeschlossen. - Spülen Sie den Kanal mit einer Spritze mit deionisiertem Wasser aus und prüfen Sie gleichzeitig, ob er undicht ist.
- Sterilisieren Sie das Kanalgehäuse in einem UV-Sterilisator für 30 min18.
- Behandeln Sie den Kanal steril mit 2,0 μg/ml humanem Fibronektin (siehe Materialtabelle) in phosphatgepufferter Kochsalzlösung (PBS) und inkubieren Sie ihn mindestens 30 min bei 37 °C19.
- Führen Sie die Zellkultur im mikrofluidischen Kanal durch, indem Sie die folgenden Schritte ausführen.
- In einer sterilen Laminar-Flow-Haube wird eine gleichmäßige Suspension von NCI-H441-Zellen in RPMI 1640-Medium mit 10 % fetalem Kälberserum (FBS) (siehe Materialtabelle) übertragen, um zwei mikrofluidische Kanäle mit jeweils Zellen mit einer Oberflächendichte von 150.000 Zellen/cmzu besiedeln2.
- Verwenden Sie für Kanäle mit den Maßen 50 mm x 5 mm x 0,6 mm 0,25 ml einer 2,5 x 10Suspension mit 6 Zellen/ml, um jeden Kanal sowie einen Teil der Anschlüsse zu füllen. Überprüfen Sie mit einem Hellfeldmikroskop, ob die Zellen gleichmäßig innerhalb der Kanäle verteilt sind.
- Die beiden Kanäle werden 24 h bzw. 48 h lang bei 37 °C mit 5 % CO2 unter Verwendung einer programmierbaren Spritzenpumpe (siehe Materialtabelle) kultiviert, wobei verbrauchte Medien aus dem Kanal und frische Medien aus einem sterilen Medienreservoir entnommen werden, das am Kanaleinlass befestigt ist und aus einer aufgeschnittenen 20-ml-Spritze besteht, die aus einer mit Paraffinfolie bedeckten 20-ml-Spritze besteht.
- Nach einer Wartezeit von 10 Minuten nach der Aussaat der Zelle wird frisches Medium aus dem Reservoir mit einer variablen Durchflussrate, die bei 0,2 μl/min beginnt und auf 10 μl/min für 4 h ansteigt, durch den Kanal gepumpt, wobei diese Rate danach20 Stunden beibehalten wird.
HINWEIS: Diese variable Flussrate bietet Kulturbedingungen, die es den Zellen ermöglichen, (1) sich gravitativ an der Kulturoberfläche abzusetzen, (2) an der Kulturoberfläche zu haften und (3) eine konfluente Monoschicht zu bilden.
- Nach einer Wartezeit von 10 Minuten nach der Aussaat der Zelle wird frisches Medium aus dem Reservoir mit einer variablen Durchflussrate, die bei 0,2 μl/min beginnt und auf 10 μl/min für 4 h ansteigt, durch den Kanal gepumpt, wobei diese Rate danach20 Stunden beibehalten wird.
- In einer sterilen Laminar-Flow-Haube wird eine gleichmäßige Suspension von NCI-H441-Zellen in RPMI 1640-Medium mit 10 % fetalem Kälberserum (FBS) (siehe Materialtabelle) übertragen, um zwei mikrofluidische Kanäle mit jeweils Zellen mit einer Oberflächendichte von 150.000 Zellen/cmzu besiedeln2.
- Führen Sie die Zellfixierung innerhalb der mikrofluidischen Kanäle mit Formaldehydlösung durch.
VORSICHT: Formaldehyd ist giftig und muss in einem geeigneten chemischen Abzug21 gehandhabt werden.- In einem chemischen Abzug werden Formaldehydlösungen hergestellt, indem 4 % Formaldehyd in PBS (methanolfrei) verwendet werden (siehe Materialtabelle), um zwei 4-ml-Portionen herzustellen, wobei die erste auf eine Konzentration von 1 % Formaldehyd und die zweite auf 2 % Formaldehyd verdünnt wird, wobei die phosphatgepufferte Kochsalzlösung von Dulbecco (DPBS; mit Ca 2+ und Mg2+) als Verdünnungsmittel verwendet wird. Übertragen Sie Formaldehydlösungen in separate 5-ml-Spritzen und etikettieren Sie sie entsprechend. 20 ml DPBS in eine separate 20-ml-Spritze aufziehen.
- Entfernen Sie mikrofluidische Kanäle aus dem Kulturapparat und legen Sie sie in den chemischen Abzug.
- Montieren Sie das Fixier- und Färbegerät.
- Befestigen Sie ein 10 cm langes Segment des Transferschlauchs über einen männlichen Luer-Lock-to-Schlauchtüllenadapter (siehe Materialtabelle) an der seitlichen Öffnung eines Dreiwege-Absperrhahns und verbinden Sie dann den Absperrhahn mit der Einlassöffnung des Durchflussarrays.
- Befestigen Sie als Nächstes ein weiteres 10-cm-Segment des Transferschlauchs mit demselben Schlauchtüllenadapter an der Auslassöffnung des Durchflussarrays.
- Befestigen Sie schließlich die freien Enden beider Transferröhrchen in einem für Chemikalien und biologische Gefahren geeigneten Abfallbehälter, z. B. einem beschrifteten leeren konischen 50-ml-Zentrifugenröhrchen.
- Drehen Sie den Absperrhahn, um die Einlassöffnung des Durchflussbereichs zu blockieren, und spülen Sie die Abflussleitung mit DPBS. Drehen Sie dann den Absperrhahn, um die Abfallleitung zu verstopfen, und waschen Sie die Zellen langsam mit 2 ml DPBS. Wiederholen Sie den Spülschritt jedes Mal mit der neuen Lösung. Eine neue Lösung (oder Konzentration der Lösung) wird in den Kanal eingeführt.
- Schieben Sie langsam 2 ml 1%ige Fixierlösung durch den Kanal und lassen Sie sie dann 5 Minuteneinwirken 22.
- Schieben Sie langsam 2 ml 2%ige Fixierlösung durch den Kanal und lassen Sie sie dann 15 Minuten einwirken.
- Waschen Sie die Zellen, indem Sie langsam 2 ml frisches DPBS in drei separaten Instanzen (jeweils 5 Minuten) in den Kanal einführen.
- Führen Sie die Schritte 1.3.3 bis 1.3.7 für beide mikrofluidischen Kanäle parallel aus.
- Färben, permeabilizieren und fügen Sie den Zellen im mikrofluidischen Kanal Einbettmedien hinzu.
- Bereiten Sie eine 0,1%ige Saponinlösung vor, indem Sie 1 mg Saponin (siehe Materialtabelle) pro ml DPBS hinzufügen, um 4 ml Lösung herzustellen, und vorsichtig wirbeln, um23 zu mischen. 8 ml DPBS in eine 20-ml-Spritze aufziehen.
- In der 0,1%igen Saponinlösung werden ein F-Aktin-färbendes Phalloidin-Reagenz und ein Zellkern-färbendes Hoechst-Reagenz (siehe Materialtabelle) in zwei Tropfen (0,1 ml) jedes Reagenzes pro ml Saponinlösung gegeben. Halten Sie die vorbereitete Färbe-/Permeabilisierungslösung lichtfern, indem Sie sie mit Alufolie24 abdecken.
- Spülen Sie die Linie mit einer kleinen Menge Färbe-/Permeabilisierungslösung (wie in Schritt 1.3.4 beschrieben), geben Sie dann 2 ml der Lösung in den mikrofluidischen Kanal und decken Sie den Kanal mit Aluminiumfolie ab, bevor Sie ihn 30 Minuten lang bei Raumtemperatur ruhen lassen.
- Spülen Sie die Färbe-/Permeabilierlösung zweimal mit 2 ml DPBS für 5 Minuten pro Spülung aus.
- Um eine bessere Bildqualität zu erzielen, fügen Sie ein geeignetes Einbettmedium (mit einem Brechungsindex, der eng mit dem Öl des Mikroskopobjektivs und dem Deckglas übereinstimmt, siehe Materialtabelle) in den Kanal ein.
- Führen Sie unter Verwendung einer Mikropipette eine minimale Menge eines weich fixierten Antifade-Eindeckmittels in jeden Anschluss des mikrofluidischen Kanals ein, wobei sichergestellt wird, dass die Bodenfläche vollständig bedeckt ist und keine Blasen im gewünschten Bildgebungsbereich25 eingeschlossen werden. Versiegeln Sie die Enden des Kanals und überprüfen Sie die Integrität der Zellschicht durch Beobachtung unter einem Hellfeldmikroskop.
- Führen Sie die Schritte 1.4.3 bis 1.4.5 für beide Mikrofluidikkanäle parallel aus.
HINWEIS: Bildzellen so schnell wie möglich nach dem Färben ab, um maximale Bildqualität zu erzielen. Wenn Photobleichung auftritt oder eine Langzeitlagerung gewünscht wird, können andere Einbettmedien mit lichtbeständigen oder probenerhaltenden Eigenschaften verwendet werden. Beachten Sie, dass harthärtende Einbettmedien die 3D-Struktur der Zellen und damit auch die Zellschicht verzerren. Aus diesem Grund sind weich abbindende Eindeckmedien26 vorzuziehen.
- Bilden Sie Zellen im mikrofluidischen Kanal ab, indem Sie die folgenden Schritte ausführen.
- Passen Sie die Einstellungen des konfokalen Mikroskops (siehe Materialtabelle) an, einschließlich Laserleistung, Verstärkung, Offset und Scanparameter wie Scangeschwindigkeit, Scanbereich, Scanformat, Auflösung und Lochblendendurchmesser27.
- Testen Sie die Position der Bildgebung, indem Sie sowohl Referenzscans als auch Z-Stapel erstellen, bis die gewünschten Bildparameter und -bedingungen erfüllt sind. Einwahlparameter bei Objektiven mit sequentiell höherer Vergrößerung bis zum Erreichen des 40-fachen Ölimmersionsobjektivs28.
- Konstruieren Sie unter Verwendung der Flow-Array-Grundplatte als Referenz Z-Stapel an fünf Stellen, an der zukünftigen Position der ersten Elektrode auf der Einlassseite, auf halbem Weg zwischen der Mitte und der vorherigen Position, in der Mitte, auf halbem Weg zwischen der Mitte und der Position der letzten Elektrode (auf der Auslassseite) und an der letzten Elektrode, wie in Abbildung 2 dargestellt.
- Führen Sie Bildverarbeitung und Datenanalyse durch.
- Exportieren Sie XZ- und YZ-Querschnitte mit dem Softwarepaket für konfokale Mikroskope (siehe Materialtabelle).
- Messen Sie unter Verwendung einer Bildverarbeitungssoftware (siehe Materialtabelle) mit Kantenerkennungsfunktionen (Schwellenwert 15,0) die gesamte Bildfläche in Pixeln mit dem Zauberstab-Werkzeug außerhalb des Bildes, dann den Bereich ohne die gesamte Querschnittsfläche der Zellenebene in Pixeln, indem Sie das Zauberstab-Werkzeug in dem Teil des Bildes außerhalb der Zellenschicht29 verwenden.
- Subtrahieren Sie mit einer Datenverarbeitungssoftware (siehe Materialtabelle) den Pixelwert der Außenfläche vom Pixelwert der Gesamtfläche, um den Pixelwert für die Querschnittsfläche zu ermitteln.
- Konvertieren Sie die Pixelwerte der Querschnittsfläche in μm2-Werte , indem Sie die Pixelwerte mit dem Quadrat des μm/Pixel-Werts multiplizieren, wie in der Mikroskopsoftware für das jeweilige Bild angegeben (0,31 μm/Pixel für Z-Stapel, die in 1024p-Auflösung ohne Zoom auf einem 40-fachen Ölimmersionsobjektiv aufgenommen wurden).
- Berechnen Sie Mittelwerte und Standardabweichungen von Daten und Diagrammergebnissen.

Abbildung 1: Explosionszeichnung der mikrofluidischen Kanalkonstruktion. Das obere Element ist der obere Teil des Flow-Arrays, dünne graue Elemente sind Klebestreifen, dünne blaue Elemente sind Mylar-Abstandshalter und das untere Element ist das rechteckige Deckglas. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
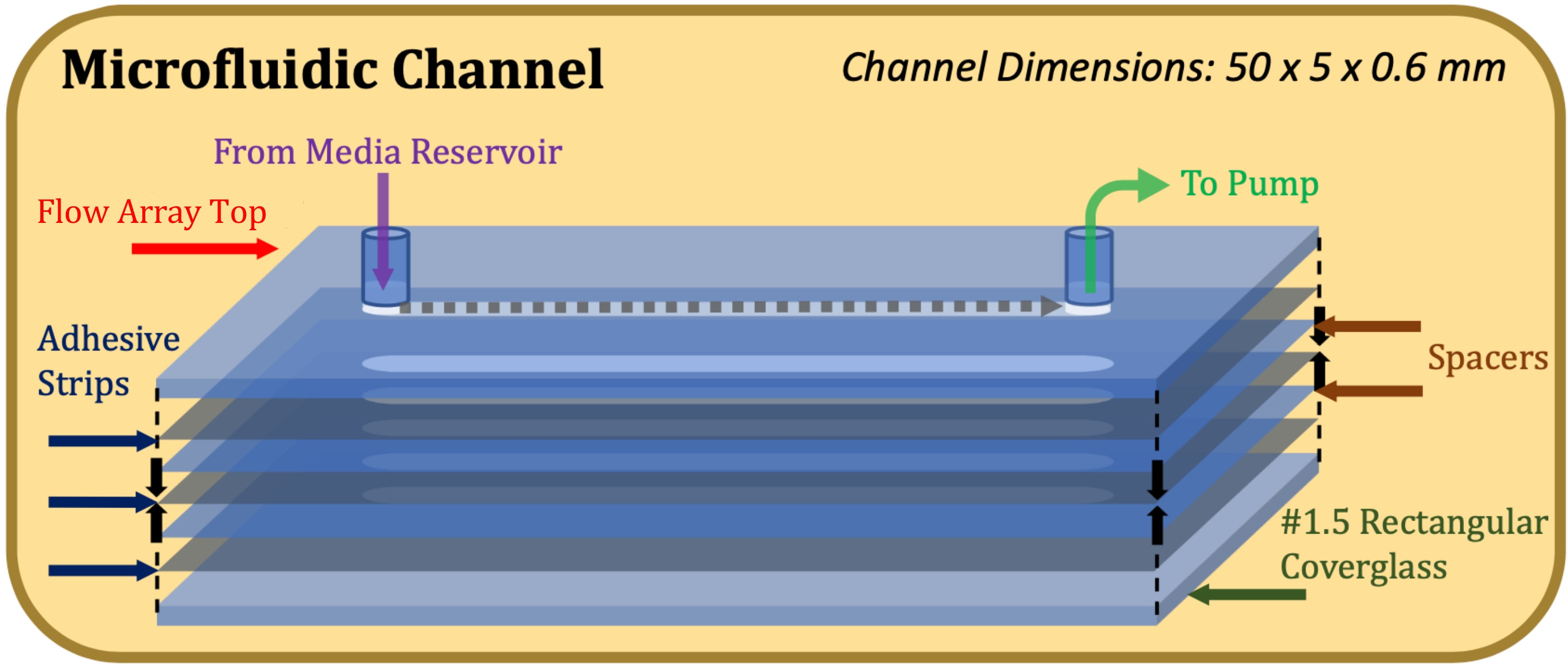{kind=link}

Abbildung 2: Fünf Bildgebungspositionen entlang des konsistent schichtproduzierenden Bereichs des mikrofluidischen Kulturkanals. Die Bildgebungspositionen sind wie folgt: Einlassseite, in der Nähe der Stelle, an der sich die erste Elektrode auf dem intakten Durchflussarray befinden würde; auf halbem Weg zwischen der Lage auf der Einlassseite und der Mitte des Kanals; Mitte des Kanals; auf halbem Weg zwischen der Mitte und der auslassseitigen Position und der Auslassseite, in der Nähe der Stelle, an der sich die letzte Elektrode auf dem intakten Durchflussarray befinden würde. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
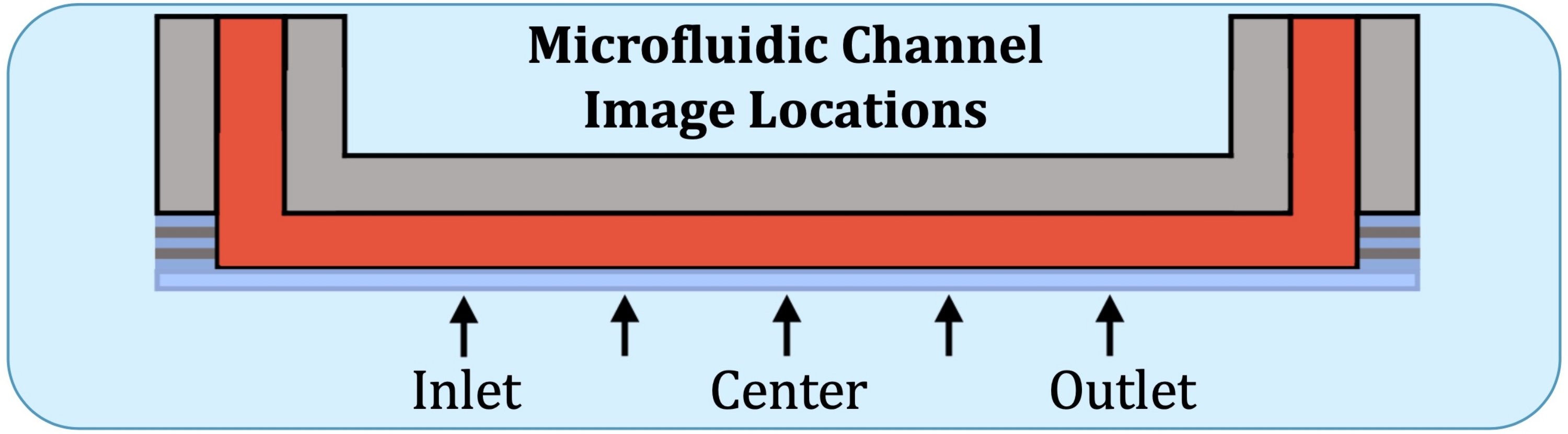{kind=link}
2. Zellkultur im Acht-Well-Deckglas
- Führen Sie eine Vorbehandlung des Deckglases mit acht Wells durch.
- Besorgen Sie sich ein steriles Deckglas mit acht Wells, das mit einem #1.5-Deckglas und einer Oberflächenbehandlung hergestellt wurde, die die Zellhaftung erhöht (Abbildung 3, siehe Materialtabelle).
- Die Oberfläche von Kulturvertiefungen wird steril mit 2,0 μg/ml humanem Fibronektin in PBS behandelt und mindestens 30 min bei 37 °C inkubiert19.
- Führen Sie die Zellkultur im gekammerten Deckglas durch, indem Sie die folgenden Schritte ausführen.
- In einer sterilen Laminar-Flow-Haube werden 0,5-ml-Portionen von Lösungen von NCI-H441-Zellen übertragen, die gleichmäßig in RPMI 1640-Medium mit 10 % FBS bei Volumendichten von 81.000, 162.000 und 324.000 Zellen/ml suspendiert sind, um die Kulturvertiefungen mit Oberflächendichten von 45.000, 90.000 bzw. 180.000 Zellen/cm2 zu besiedeln. Überprüfen Sie mit einem Hellfeldmikroskop, ob die Zellen gleichmäßig in den Vertiefungen verteilt sind.
- Kulturzellen für 24 h, 48 h und 96 h bei 37 °C mit 5 % CO2 und täglichem Austausch der Medien.
- Führen Sie die Formaldehydfixierung im Deckglas mit acht Kammern durch.
VORSICHT: Formaldehyd ist giftig und muss in einem geeigneten chemischen Abzug21 gehandhabt werden.- In einem chemischen Abzug werden Formaldehydlösungen hergestellt, indem zwei Portionen von 4 % Formaldehyd in PBS (methanolfrei) hergestellt werden, wobei die erste auf eine Konzentration von 1 % Formaldehyd und die andere auf 2 % Formaldehyd verdünnt wird, wobei die phosphatgepufferte Kochsalzlösung von Dulbecco (DPBS; mit Ca 2+ und Mg2+) als Verdünnungsmittel verwendet wird.
- Entfernen Sie das Deckglas der Acht-Well-Kulturkammer aus dem Inkubator und setzen Sie es in den Chemikalienabzug ein.
- Waschen Sie die Zellen vorsichtig mit 0,5 ml DPBS mit einer Mikropipette, indem Sie langsam Flüssigkeit entlang des oberen Teils der Ecke jeder Vertiefung einführen.
- Entfernen Sie die vorhandene Flüssigkeit in jeder Vertiefung, indem Sie sie mit einer Mikropipette langsam aus der Ecke der Vertiefungen herausziehen. Unter Verwendung der Methode der Flüssigkeitseinführung (Schritt 2.3.3) werden 0,5 ml 1%ige Fixierlösung in jede Vertiefung gegeben und 5 min22 Minuten ruhen gelassen.
- Entfernen Sie die vorhandene Flüssigkeit in jeder Vertiefung mit der in Schritt 2.3.4 beschriebenen Methode zur Flüssigkeitsextraktion. Unter Anwendung der in Schritt 2.3.3 beschriebenen Methode zur Flüssigkeitseinführung werden 0,5 ml 2%ige Fixierlösung in jede Vertiefung gegeben und 15 Minuten ruhen gelassen.
- Unter Verwendung der Flüssigkeitszufuhr- und Extraktionsmethoden (Schritte 2.3.3 und 2.3.4) werden die Zellen durch Einbringen und Entfernen von 0,5 ml frischem DPBS in jeder Vertiefung in drei getrennten Instanzen für jeweils 5 min gewaschen.
- Führen Sie das Färben, Permeabilisieren und das Einbinden von Medien in das Deckglas mit acht Wells durch.
- Bereiten Sie eine 0,1%ige Saponinlösung vor, indem Sie 1 mg Saponin pro ml DPBS hinzufügen und vorsichtig wirbeln, um23 zu mischen.
- In die 0,1%ige Saponinlösung werden je zwei Tropfen (0,1 ml) eines F-Aktin-färbenden Phalloidin-Reagenzes und eines Nukleus-färbenden Hoechst-Reagenzes pro ml Saponinlösung gegeben. Halten Sie die vorbereitete Lösung lichtfern, indem Sie sie mit Alufolie24 abdecken.
- Geben Sie 0,2 ml Färbe-/Permeabilisierungslösung in jede Vertiefung und decken Sie das gekammerte Deckglas mit Aluminiumfolie ab, bevor Sie es 30 Minuten lang bei Raumtemperatur ruhen lassen.
- Färbe-/Permeabilisierungslösung zweimal mit 0,5 ml DPBS ausspülen.
- Um eine bessere Bildqualität zu erzielen, fügen Sie ein geeignetes Eindeckmedium (mit einem Brechungsindex, der dem Öl des Mikroskopobjektivs und dem Deckglas genau entspricht) in die Vertiefungen hinzu.
- Geben Sie mit einer Mikropipette eine minimale Menge eines weich abbindenden Eindeckmittels in jede Vertiefung, wobei darauf zu achten ist, dass die Bodenfläche vollständig bedeckt ist und keine Blasen im gewünschten Bildgebungsbereich25 eingeschlossen werden. Überprüfen Sie die Integrität der Zellschicht durch Beobachtung unter einem Hellfeldmikroskop.
HINWEIS: Bildzellen so schnell wie möglich nach dem Färben ab, um maximale Bildqualität zu erzielen. Wenn Photobleichung auftritt oder eine Langzeitlagerung gewünscht wird, können andere Einbettmedien mit lichtbeständigen oder probenerhaltenden Eigenschaften verwendet werden. Es ist zu beachten, dass harthärtende Einbettmedien die 3D-Struktur der Zellen und damit auch der Zellschicht verzerren, so dass weich aushärtende Eindeckmedien vorzuziehen sind26.
- Geben Sie mit einer Mikropipette eine minimale Menge eines weich abbindenden Eindeckmittels in jede Vertiefung, wobei darauf zu achten ist, dass die Bodenfläche vollständig bedeckt ist und keine Blasen im gewünschten Bildgebungsbereich25 eingeschlossen werden. Überprüfen Sie die Integrität der Zellschicht durch Beobachtung unter einem Hellfeldmikroskop.
- Führen Sie die Bildgebung im Deckglas mit acht Wells durch.
- Passen Sie die Einstellungen des konfokalen Mikroskops an, einschließlich Laserleistung, Verstärkung, Offset und Scanparameter wie Scangeschwindigkeit, Scanbereich, Scanformat, Auflösung und Lochblendendurchmesser27.
- Testen Sie die Position der Bildgebung, indem Sie sowohl Referenzscans als auch Z-Stapel erstellen, bis die gewünschten Bildparameter und -bedingungen erfüllt sind. Einwahlparameter bei Objektiven mit sequentiell höherer Vergrößerung bis zum Erreichen des 40-fachen Ölimmersionsobjektivs28.
- Konstruieren Sie Z-Stapel aus drei zufälligen Positionen in jeder Übereinstimmung zwischen Seedingdichte und Kulturdauer.
- Führen Sie Bildverarbeitung und Datenanalyse durch.
- Exportieren Sie XZ- und YZ-Querschnitte mit dem Softwarepaket für konfokale Mikroskope.
- Messen Sie unter Verwendung einer Bildverarbeitungssoftware mit Kantenerkennungsfunktionen (Schwellenwert 15,0) den gesamten Bildbereich in Pixeln mit dem Zauberstab-Werkzeug außerhalb des Bildes und dann den Bereich ohne die gesamte Querschnittsfläche der Zellenebene in Pixeln, indem Sie das Zauberstab-Werkzeug in dem Teil des Bildes außerhalb der Zellenebene29 verwenden.
- Subtrahieren Sie mit einer Datenverarbeitungssoftware den Pixelwert für den Außenbereich vom Pixelwert der Gesamtfläche, um den Pixelwert für die Querschnittsfläche zu ermitteln.
- Konvertieren Sie die Pixelwerte der Querschnittsfläche in μm2-Werte , indem Sie die Pixelwerte mit dem Quadrat des μm/Pixel-Werts multiplizieren, wie in der Mikroskopsoftware für das jeweilige Bild angegeben (0,31 μm/Pixel für Z-Stapel, die in 1024p-Auflösung ohne Zoom auf einem 40-fachen Ölimmersionsobjektiv aufgenommen wurden).
- Berechnen Sie Mittelwerte und Standardabweichungen und stellen Sie die Ergebnisse grafisch dar.

Abbildung 3: Diagramm des Deckglases mit acht Wells, das für das Fixed-Well-Kultur-, Färbe- und Bildgebungsexperiment verwendet wurde, um die Auswirkungen der anfänglichen Zellaussaatdichte und der Kulturdauer auf die Bildung von Zellschichten zu vergleichen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
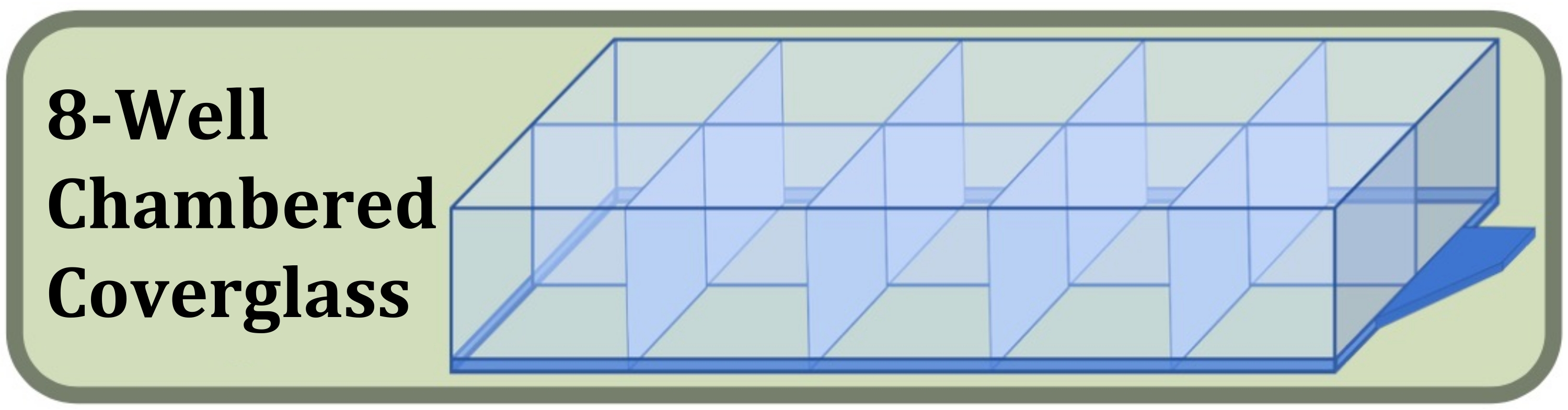{kind=link}
Ergebnisse
Die vorgestellte Methode ermöglicht die Visualisierung von Epithelzellschichten, die in mikrofluidischen Kulturkanälen kultiviert wurden, und nutzt eine Demonstration in traditionellen Fixed-Well-Zellkulturumgebungen als Validierung. Die aufgenommenen Bilder liegen in einem Spektrum von Qualität, Signalintensität und zellulärer Zielspezifität vor. Erfolgreiche Bilder weisen einen hohen Kontrast auf, der eine Bildanalyse und Quantifizierung der Daten für die anschließende statistische Auswertung ermöglicht. Erfol...
Diskussion
Das vorgestellte Protokoll beschreibt die Kultur, Vernetzungsfixierung, Färbung, Permeabilisierung und konfokale mikroskopische Visualisierung von NCI-H441 menschlichen Lungenepithelzellen in der dynamischen Umgebung eines einkanaligen mikrofluidischen Flussarrays sowie in der statischen Umgebung eines traditionellen Acht-Well-Deckglases. Bei jedem mikrofluidischen Zellkulturprotokoll sind die Fließbedingungen der Zellkulturmedien von größter Bedeutung, da die hohe Fließgeschwindigkeit das Potenzial hat, die Zellen ...
Offenlegungen
Die Autoren erklären keine Interessenkonflikte.
Danksagungen
Die Autoren danken Alan Shepardson für die Entwicklung des Schnittmusters für den 3M-Klebstoff und die Mylar-Folie, die bei der Konstruktion von Mikrofluidikkanälen verwendet werden, sowie für die Prüfung der Durchflussrate des Zellkulturmediums und der Programmierung der Spritzenpumpe. Die Finanzierung erfolgte durch NIH R01 HL0142702, NSF CBET 1706801 und den Newcomb-Tulane College Dean's Grant.
Materialien
| Name | Company | Catalog Number | Comments |
| A1R HD25 Confocal Microscope System | Nikon | A1R HD25 | https://www.microscope.healthcare.nikon. com/products/confocal-microscopes/a1hd25-a1rhd25/specifications |
| ActinGreen 488 ReadyProbes Reagent (AlexaFluor 488 phalloidin) | Invitrogen | R37110 | https://www.thermofisher.com/order/catalog/product/R37110 |
| Adhesive Transfer Tape Double Linered | 3M | 468MP | https://gizmodorks.com/3m-468mp-adhesive-transfer-tape-sheet-5-pack/ |
| Air-Tite HSW Soft-Ject Disposable Syringes | Air-Tite RL5 | 14-817-53 | https://www.fishersci.com/shop/products/air-tite-hsw-soft-ject-disposable-syringes-6/1481753#?keyword=syringe%20leur%20locking%205ml |
| BAISDY 4 mil (0.1 mm) Thick Mylar Sheet | BAISDY | AS022 | https://www.amazon.ca/Stencil-Perfect-Silhouette-Machines-BAISDY/dp/B07RJJ9BNC |
| Branson Ultrasonics M Series Ultrasonic Cleaning Bath | Branson Ultrasonics | 15-336-100 | https://www.fishersci.com/shop/products/m-series-ultrasonic-cleaning-bath/15336100 |
| Corning Fibronectin, Human | Fisher Scientific | CB-40008 | https://www.fishersci.com/shop/products/corning-fibronectin-human-3/CB40008?keyword=true |
| DPBS, calcium, magnesium | Gibco | 14040133 | https://www.thermofisher.com/order/catalog/product/14040133?SID=srch-srp-14040133 |
| ECIS Cultureware Disposable Electrode Arrays 8 x 10 ECIS Flow Array | Applied BioPhysics | 1F8x10E PC | https://www.biophysics.com/cultureware.php#1F8x10E |
| Enterprise Technology Solutions UV Sterilizer Cabinet, White | Enterprise Technology Solutions | 50-211-1163 | https://www.fishersci.com/shop/products/uv-sterilizer-cabinet-white/502111163 |
| Fetal Bovine Serum (FBS) | Gibco | 26140079 | https://www.thermofisher.com/order/catalog/product/26140079 |
| Finnpipette F2 Variable Volume Pipettes | Thermo Scientific | 4642090 | https://www.thermofisher.com/order/catalog/product/4642090 |
| Fisherbrand 50mL Easy Reader Plastic Centrifuge Tubes | Fisher Scientific | 06-443-21 | https://www.fishersci.com/shop/products/fisherbrand-higher-speed-easy-reader-plastic-centrifuge-tubes-8/p-193269 |
| Fisherbrand Cover Glasses: Rectangles (#1.5) | Fisher Scientific | 12-544-GP | https://www.fishersci.com/shop/products/cover-glasses-rectangles-promo-22/12544GP#coverglass |
| Fisherbrand Sterile Syringes for Single Use | Fisher Scientific | 14-955-458 | https://www.fishersci.com/shop/products/sterile-syringes-single-use-12/14955458 |
| Gibco RPMI 1640 Medium | Gibco | 11875093 | https://www.thermofisher.com/order/catalog/product/11875093 |
| Image-iT Fixative Solution (4% formaldehyde, methanol-free) | Invitrogen | FB002 | https://www.thermofisher.com/order/catalog/product/FB002 |
| ImageJ Fiji | ImageJ | ImageJ Fiji | https://imagej.net/downloads |
| Immersion Oil F 30 cc | Nikon | MXA22168 | https://www.microscope.healthcare.nikon. com/products/accessories/immersion-oil/specifications |
| Large-Capacity Reach-In CO2 Incubator, 821 L, Polished Stainless Steel | Thermo Scientific | 3950 | https://www.thermofisher.com/order/catalog/product/3950 |
| Laxco LMC-3000 Series Brightfield Compound Microscope System | Laxco | LMC3BF1 | https://www.fishersci.com/shop/products/lmc-3000-series-brightfield-compound-microscope-system-8/LMC3BF1 |
| Masterflex Fitting, Nylon, Straight, Male Luer Lock to Hose Barb Adapters, 1/16" ID; 25/PK | Masterflex | ZY-45505-31 | https://www.masterflex.com/i/masterflex-fitting-nylon-straight-male-luer-lock-to-hose-barb-adapters-1-16-id-25-pk/4550531?PubID=ZY&persist=true&ip=no& gclid=Cj0KCQiA3rKQBhCNARIsAC UEW_Zb5yXy1em6bGs0a9KFOk5k pdlkHCvAEslHumdqcnlwSN0MdR0 udmwaAuDHEALw_wcB |
| Microsoft Excel | Microsoft | 0016 | https://www.microsoft.com/en-us/download/details.aspx?id=56547 |
| National Target All-Plastic Disposable Syringes | Thermo Scientific | 03-377-24 | https://www.fishersci.com/shop/products/national-target-all-plastic-disposable-syringes/0337724#tab8 |
| NCI-H441 Human Epithelial Lung Cells | American Type Culture Collection (ATCC) | HTB-174 | https://www.atcc.org/products/htb-174 |
| NE-1600 Six Channel Programmable Syringe Pump | New Era Pump Systems | NE-1600 | https://www.syringepump.com/NE-16001800.php |
| NIS Elements AR | Nikon | NIS Elements AR | https://www.microscope.healthcare.nikon. com/products/software/nis-elements/nis-elements-advanced-research |
| NucBlue Live ReadyProbes Reagent (Hoechst 33342) | Invitrogen | R37605 | https://www.thermofisher.com/order/catalog/product/R37605?SID=srch-srp-R37605 |
| Nunc Lab-Tek Chambered Coverglass | Thermo Scientific | 155411 | https://www.thermofisher.com/order/catalog/product/155361 |
| Parafilm M Wrapping Film | Fisher Scientific | S37441 | https://www.fishersci.com/shop/products/parafilm-m-wrapping-film-3/S37441 |
| PendoTech 3-Way Stopcock, Polysulfone, Male/Female Luer Inlet x Female Luer Branch | PendoTech | ZY-19406-49 | https://www.masterflex.com/i/pendotech-3-way-stopcock-polysulfone-male-female-luer-inlet-x-female-luer-branch/1940649 |
| Phosphate Buffered Solution (PBS), pH 7.4 | Gibco | 10010023 | https://www.thermofisher.com/order/catalog/product/10010023 |
| Poly-D-Lysine | Gibco | A3890401 | https://www.thermofisher.com/order/catalog/product/A3890401#/A3890401 |
| Reynolds Aluminum Wrap Foil | Reynolds | 458742928317 | https://www.amazon.com/Reynolds-Wrap-Aluminum-Foil-Square/dp/B00UNT0Y2M |
| Saponin | Millipore Sigma (Sigma Aldrich) | 47036 | https://www.sigmaaldrich.com/US/en/product/sigma/47036 |
| SlowFade Glass Soft-set Antifade Mountant | Invitrogen | S36917-5X2ML | https://www.thermofisher.com/order/catalog/product/S36917-5X2ML |
| Thermo Scientific 1300 Series Class II, Type A2 Biological Safety Cabinet Package | Thermo Scientific | 13-100-752PM | https://www.fishersci.com/shop/products/1300-series-class-ii-type-a2-biological-safety-cabinet-package-promo/p-9049003#?keyword=biosafety%20hood |
| Tygon Transfer Tubing, BioPharm Platinum-Cured Silicone, 1/16" ID x 1/8" OD; 50 Ft | Cole-Parmer | EW-95702-01 | https://www.coleparmer.com/i/tygon-transfer-tubing-biopharm-platinum-cured-silicone-1-16-id-x-1-8-od-50-ft/9570201?searchterm=95702-01 |
Referenzen
- Matthay, M. A., et al. Acute respiratory distress syndrome. Nature Reviews Disease Primers. 5, 18 (2019).
- Rawal, G., Yadav, S., Kumar, R. Acute respiratory distress syndrome: An update and Review. Journal of Translational Internal Medicine. 6 (2), 74-77 (2018).
- Bilek, A. M., Dee, K. C., Gaver, D. P. Mechanisms of surface-tension-induced epithelial cell damage in a model of pulmonary airway reopening. Journal of Applied Physiology. 94 (2), 770-783 (2003).
- Modrykamien, A. M., Gupta, P. The acute respiratory distress syndrome. Baylor University Medical Center Proceedings. 28 (2), 163-171 (2017).
- Jacob, A. -. M., Gaver, D. P. Atelectrauma disrupts pulmonary epithelial barrier integrity and alters the distribution of tight junction proteins ZO-1 and Claudin 4. Journal of Applied Physiology. 113 (9), 1377-1387 (2012).
- Kay, S. S., Bilek, A. M., Dee, K. C., Gaver, D. P. Pressure gradient, not exposure duration, determines the extent of epithelial cell damage in a model of pulmonary airway reopening. Journal of Applied Physiology. 97 (1), 269-276 (2004).
- Jacob, A. M., Gaver, D. P. An investigation of the influence of cell topography on epithelial mechanical stresses during pulmonary airway reopening. Physics of Fluids. 17 (3), 031502 (1994).
- Gaver, D. P., et al. The POOR get POORer: A hypothesis for the pathogenesis of ventilator-induced lung injury. American Journal of Respiratory and Critical Care Medicine. 202 (8), 1081-1087 (2020).
- Jain, P., et al. Reconstruction of ultra-thin alveolar-capillary basement membrane mimics. Advanced Biology. 5 (8), 2000427 (2021).
- Byrne, M. B., Leslie, M. T., Gaskins, H. R., Kenis, P. J. A. Methods to study the tumor microenvironment under controlled oxygen conditions. Trends in Biotechnology. 32 (11), 556-563 (2014).
- Szulcek, R., Bogaard, H. J., van Nieuw Amerongen, G. P. Electric cell-substrate impedance sensing for the quantification of endothelial proliferation, barrier function, and motility. Journal of Visualized Experiments. (85), e51300 (2014).
- Jaccard, N., et al. Automated method for the rapid and precise estimation of adherent cell culture characteristics from phase contrast microscopy images. Biotechnology and Bioengineering. 111 (3), 504-517 (2013).
- Hagiyama, M., et al. Modest static pressure suppresses columnar epithelial cell growth in association with cell shape and cytoskeletal modifications. Frontiers in Physiology. 8, 00997 (2017).
- Srinivasan, M., Sedmak, D., Jewell, S. Effect of fixatives and tissue processing on the content and integrity of Nucleic Acids. The American Journal of Pathology. 161 (6), 1961-1971 (2002).
- Zhu, L., Rajendram, M., Huang, K. C. Effects of fixation on bacterial cellular dimensions and integrity. Iscience. 24 (4), 102348 (2021).
- Lust, R. M. . The Pulmonary System. XPharm: The Comprehensive Pharmacology Reference. , 1-6 (2007).
- EpilogueLaser. FusionSeries: Pro & Edge Laser System Manual and Original Instructions. EpilogueLaser. , (2022).
- Chitnis, D. S., Katara, G., Hemvani, N., Chitnis, S., Chitnis, V. Surface disinfection by exposure to germicidal UV light. Indian Journal of Medical Microbiology. 26 (3), 241 (2008).
- Sandell, L., Sakai, D. Mammalian cell culture. Current Protocols Essential Laboratory Techniques. 5 (1), 4 (2011).
- New Era Pump Systems. Multi-Phaser Programmable Syringe Pump: NE-1000 Series User Manual. New Era Pump Systems. , (2014).
- Thermo Fisher Scientific. Safety Data Sheet: Image-iT Fixative Solution (4% formaldehyde, methanol-free). Thermo Fisher Scientific. , (2018).
- Thavarajah, R., Mudimbaimannar, V. K., Rao, U. K., Ranganathan, K., Elizabeth, J. Chemical and physical basics of routine formaldehyde fixation. Journal of Oral and Maxillofacial Pathology. 16 (3), 400-405 (2012).
- Jamur, M. C., Oliver, C. Permeabilization of cell membranes. Immunocytochemical Methods and Protocols. 588, 63-66 (2009).
- Thermo Fisher Scientific. ActinGreen 488 ReadyProbes Reagent Protocol. Thermo Fisher Scientific. , (2022).
- Slowfade Glass soft-set Antifade Mountant. Thermo Fisher Scientific Available from: https://www.thermofisher.com/order/catalog/product/S36917-5X2ML?SID=srch-hj-S36917-5X2ML (2022)
- Ravikumar, S., Surekha, R., Thavarajah, R. Mounting media: An overview. Journal of Dr. NTR University of Health Sciences. 3 (5), 1-8 (2014).
- Shihan, M. H., Novo, S. G., Le Marchand, S. J., Wang, Y., Duncan, M. K. A simple method for quantitating confocal fluorescent images. Biochemistry and Biophysics Reports. 25, 100916 (2021).
- North, A. J. Seeing is believing? A beginners' guide to practical pitfalls in image acquisition. Journal of Cell Biology. 172 (1), 9-18 (2006).
- Ferriera, F., Rasband, W. ImageJ User Guide. National Institutes of Health. , (2012).
- Halldorsson, S., Lucumi, E., Gómez-Sjöberg, R., Fleming, R. M. T. Advantages and challenges of microfluidic cell culture in polydimethylsiloxane devices. Biosensors and Bioelectronics. 63, 218-231 (2015).
- Smith, H. S., Riggs, J. L., Mosesson, M. W. Production of fibronectin by human epithelial cells in culture. American Association for Cancer Research. 39 (10), 4138-4144 (1979).
- Sieck, G. C., Mantilla, C. B., Prakash, Y. S. Volume measurements in confocal microscopy. Methods in Enzymology. 307, 296-315 (1999).
- Heijink, I. H., et al. Characterisation of cell adhesion in airway epithelial cell types using electric cell-substrate impedance sensing. European Respiratory Journal. 35 (4), 894-903 (2009).
- Zhang, X., Wang, W., Li, F., Voiculescu, I. Stretchable impedance sensor for mammalian cell proliferation measurements. Lab on a Chip. 17 (12), 2054-2066 (2017).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten