
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Préparation et évaluation structurale de monocouches de cellules épithéliales dans un dispositif de culture microfluidique de taille physiologique
Dans cet article
Résumé
Le protocole présenté décrit le développement et l’utilisation d’une technique de coloration filamenteuse à base de phalloïdine avec microscopie confocale à balayage laser (CLSM) pour visualiser la structure de la couche cellulaire adhérente dans les canaux de culture dynamique microfluidique et les chambres de culture statique traditionnelles à puits fixes. Cette approche aide à évaluer la confluence de la couche cellulaire, la formation de monocouches et l’uniformité de l’épaisseur de la couche.
Résumé
L’expérimentation microfluidique in vitro recèle un grand potentiel pour révéler de nombreuses informations sur les phénomènes microphysiologiques qui se produisent dans des conditions telles que le syndrome de détresse respiratoire aiguë (SDRA) et les lésions pulmonaires induites par un ventilateur (VILI). Cependant, les études sur les canaux microfluidiques dont les dimensions sont physiologiquement pertinentes pour les bronchioles terminales du poumon humain font actuellement face à plusieurs défis, en particulier en raison des difficultés à établir des conditions de culture cellulaire appropriées, y compris les débits de milieu, dans un environnement de culture donné. Le protocole présenté décrit une approche basée sur l’image pour évaluer la structure des cellules épithéliales pulmonaires humaines NCI-H441 cultivées dans un canal microfluidique imperméable à l’oxygène avec des dimensions physiologiquement pertinentes pour les bronchioles terminales du poumon humain. En utilisant la coloration filamenteuse d’actine à base de phalloïdine, les structures cytosquelettiques des cellules sont révélées par microscopie confocale à balayage laser, permettant la visualisation de cellules individuelles et en couches. La quantification ultérieure détermine si les conditions de culture cellulaire utilisées produisent des monocouches uniformes adaptées à une expérimentation ultérieure. Le protocole décrit les méthodes de culture cellulaire et d’évaluation des couches dans les canaux microfluidiques et les environnements traditionnels de puits fixes. Cela comprend la construction de canaux, la culture cellulaire et les conditions requises, la fixation, la perméabilisation et la coloration, l’imagerie microscopique confocale, le traitement d’images et l’analyse de données.
Introduction
Le syndrome de détresse respiratoire aiguë (SDRA) est une affection aiguë résultant de l’agression et de la propagation d’une lésion dans le parenchyme pulmonaire, entraînant un œdème pulmonaire des alvéoles, un échange gazeux insuffisant et une hypoxémiesubséquente 1. Cela déclenche un cycle de libération de cytokines pro-inflammatoires, de recrutement de neutrophiles, de libération de médiateurs toxiques et de lésions tissulaires, qui lui-même entraîne une réponse inflammatoiresupplémentaire 2. De plus, le surfactant pulmonaire, qui stabilise les voies respiratoires et prévient les dommages causés par le recrutement/dérecrutement répétitif (R/D), peut être inactivé ou autrement rendu dysfonctionnel par les processus chimiques se produisant pendant le SDRA, entraînant un stress supplémentaire et des lésions au parenchyme environnant3. Si des dommages suffisants sont subis, une ventilation mécanique peut être nécessaire pour assurer une oxygénation systémique adéquate4. Cependant, la ventilation mécanique impose ses propres défis et traumatismes, y compris la possibilité d’une lésion pulmonaire induite par la ventilation (VILI), caractérisée comme une lésion du parenchyme pulmonaire causée par les contraintes mécaniques imposées lors du surgonflage (volutraumatisme) et/ou la R/D de l’interface air-liquide dans les voies respiratoires obstruées par le fluide (atelectrauma)5. Le gradient de pression subi par les cellules épithéliales exposées à une interface air-liquide (comme dans un bronchiole obstrué par un fluide) dans le modèle d’atelectrauma peut entraîner une réponse obstructive d’origine perméabilité (POOR), conduisant à un cercle vertueux de lésions POOR-get-POORer 6,7,8.
L’expérimentation in vitro peut fournir des informations à micro-échelle sur ces phénomènes, mais les études actuelles dans des environnements de canaux microfluidiques avec des dimensions physiologiquement pertinentes font face à plusieurs défis9. D’une part, l’optimisation des conditions de culture cellulaire constitue une barrière importante à l’entrée pour la recherche en culture cellulaire dans des environnements microfluidiques, car il existe une intersection étroite dans laquelle les paramètres de flux de milieu, la durée de culture et d’autres conditions de culture permettent une formation optimale de la couche cellulaire. Cela inclut les limitations de diffusion imposées par la nature imperméable à l’oxygène de l’enceinte du canal de culture microfluidique. Cela nécessite un examen attentif des paramètres de débit du milieu, car de faibles débits peuvent priver les cellules d’oxygène, en particulier celles les plus éloignées de l’entrée; D’autre part, des débits élevés peuvent pousser les cellules hors du canal de culture ou entraîner un développement inapproprié ou inégal de la couche. Les limitations de diffusion peuvent être abordées en utilisant des matériaux perméables à l’oxygène tels que le polydiméthylsiloxane (PDMS) dans un appareil de culture à interface air-liquide (ALI); cependant, de nombreux canaux de culture microfluidique conventionnels, tels que ceux du système de détection d’impédance cellule électrique et substrat (ECIS), sont intrinsèquement imperméables à l’oxygène, compte tenu de la nature de l’enceinte fabriquée10. Ce protocole vise à fournir une technique d’analyse des couches cellulaires cultivées dans une enceinte imperméable à l’oxygène.
Lors de la comparaison de la viabilité des conditions de culture, des observations des caractéristiques spécifiques de la couche, telles que la présence d’une monocouche, la topologie de surface, la confluence et l’uniformité de l’épaisseur de la couche, sont nécessaires pour déterminer si la couche cellulaire produite par un ensemble particulier de conditions de culture répond aux spécifications souhaitées et est effectivement pertinente pour la conception expérimentale. Une évaluation limitée peut être effectuée par des méthodes telles que ECIS, qui utilise des mesures du potentiel électrique (tension) créé par la résistance au courant alternatif (AC) à haute fréquence (impédance) imposée par les membranes électriquement isolantes de cellules cultivées sur des électrodes en or dans le réseau d’écoulement. En modulant la fréquence de l’AC appliquée aux cellules, les propriétés cellulaires spécifiques dépendantes de la fréquence des cellules et des couches cellulaires, telles que la force d’adhérence de surface, la formation de jonctions serrées et la prolifération ou la confluence cellulaire, peuvent être ciblées et examinées11. Cependant, ces formes indirectes de mesures sont quelque peu difficiles à interpréter au début d’une expérience et peuvent ne pas quantifier tous les aspects pertinents de la couche cellulaire. Le simple fait d’observer la couche cellulaire au microscope à contraste de phase peut révéler la nature de certaines qualités telles que la confluence; cependant, de nombreuses caractéristiques pertinentes telles que la présence d’une monocouche et l’uniformité de l’épaisseur de la couche nécessitent une évaluation tridimensionnelle (3D) qui n’est pas possible avec l’imagerie microscopique en fond clair, en contraste de phase ou en fluorescence12.
L’objectif de cette étude était de développer une technique de coloration à l’actine filamenteuse pour permettre la vérification basée sur l’imagerie d’une monocouche et l’évaluation de l’uniformité de la couche cellulaire à l’aide de la microscopie confocale à balayage laser (CLSM). L’actine filamenteuse (F-actine) a été jugée une cible appropriée pour le conjugué fluorophore, en partie à cause de la façon dont la F-actine suit étroitement la membrane cellulaire, permettant une approximation visuelle de l’ensemble du volume cellulaire13. Un autre avantage important du ciblage de la F-actine est la manière dont la coloration de la F-actine élucide visuellement les perturbations ou altérations du cytosquelette imposées par les contraintes et les tensions subies par les cellules. La fixation par réticulation avec du formaldéhyde sans méthanol a été utilisée pour préserver la morphologie des cellules et de la couche cellulaire, car les fixateurs déshydratants tels que le méthanol ont tendance à aplatir les cellules, déformant grossièrement la couche cellulaire et altérant ses propriétés14,15.
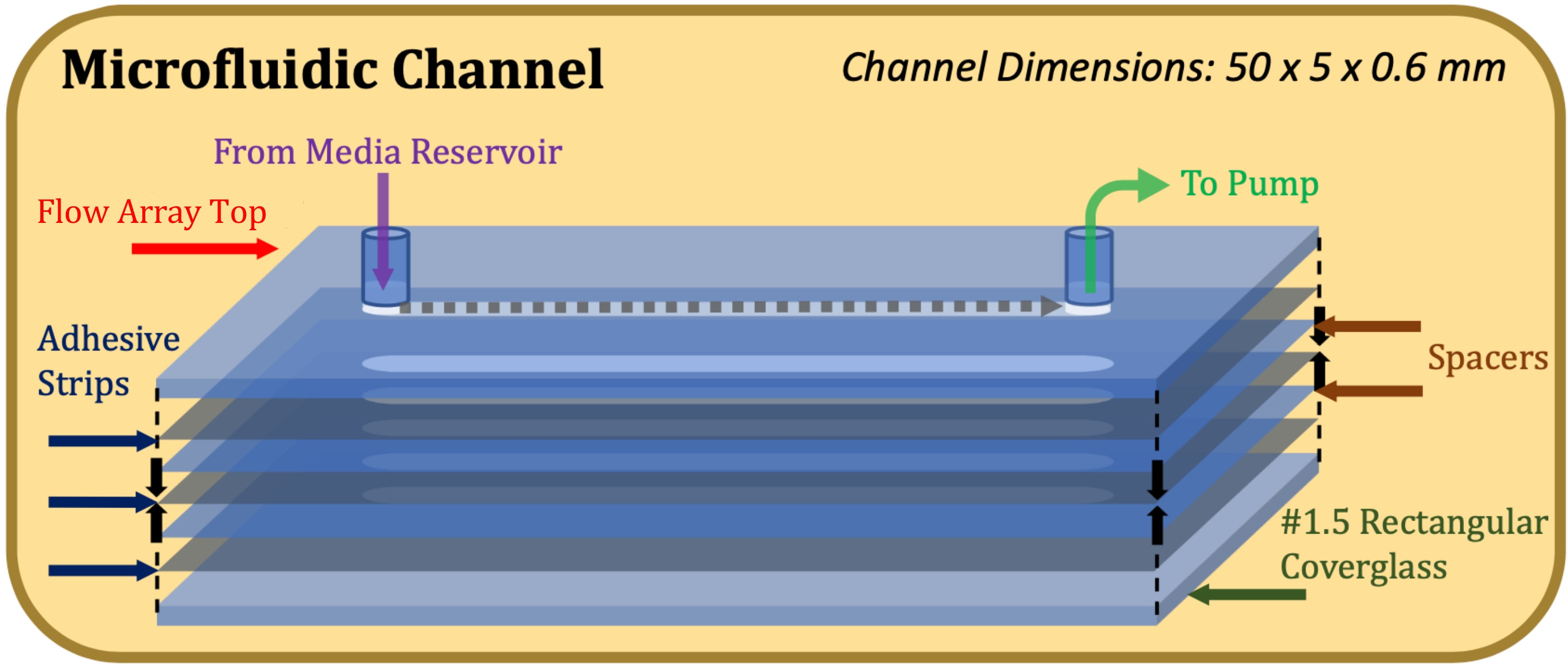
Pour déterminer la capacité de la technique d’évaluation des couches à atténuer ces défis, les cellules ont été cultivées dans des chambres de culture traditionnelles à huit puits ainsi que dans des canaux microfluidiques pour évaluer les différences, le cas échéant, dans les couches cellulaires produites. Pour les puits de culture fixes, des unités de verre de couverture chambrées à huit puits ont été utilisées. Pour la culture microfluidique, les réseaux d’écoulement (longueur du canal 50 mm, largeur 5 mm, profondeur 0,6 mm) ont été optimisés pour la culture de cellules épithéliales pulmonaires humaines immortalisées (NCI-H441) dans un environnement dont les dimensions sont physiologiquement pertinentes pour les bronchioles terminales présentes dans la zone respiratoire du poumon humain16. Bien que ce protocole ait été développé en tenant compte de l’environnement de culture des réseaux d’écoulement ECIS, il peut s’appliquer à tout environnement de culture dynamique imperméable à l’oxygène pour lequel l’évaluation des caractéristiques de la couche cellulaire cultivée ou des conditions de culture est nécessaire.
Protocole
La lignée cellulaire pulmonaire épithéliale humaine NCI-H441 a été utilisée pour la présente étude (voir le tableau des matériaux).
1. Culture cellulaire dans le canal microfluidique
- Fabriquez le canal microfluidique et effectuez le prétraitement en suivant les étapes ci-dessous.
- Procurez-vous un réseau d’écoulement à canal unique (voir le tableau des matériaux) et séparez la partie supérieure de la plaque de base en polycarbonate.
- Obtenez un verre rectangulaire #1.5 (épaisseur 0,17 mm) avec des dimensions de 60 mm x 22 mm. Nettoyez les surfaces du verre de couverture dans un bain à ultrasons et traitez un côté avec une solution de 0,1 mg/mL de Poly-D-Lysine à température ambiante pendant 5 min avant de sécher à 60 °C pendant 30 min.
- Fixer un adhésif double face de 0,13 mm d’épaisseur (voir tableau des matériaux), découpé au laser pour s’adapter aux dimensions du sommet du réseau d’écoulement et du canal d’écoulement (50 mm de longueur, 5 mm de largeur), au sommet du réseau d’écoulement, en prenant soin d’aligner précisément les découpes du canal17.
- Apposer une entretoise mylar de 0,1 mm d’épaisseur (voir tableau des matériaux), découpée au laser pour s’adapter aux dimensions du sommet du réseau d’écoulement et du canal d’écoulement, sur la bande adhésive, en prenant soin d’aligner précisément les découpes du canal.
- Répétez les étapes 1.1.3 et 1.1.4 jusqu’à ce que la hauteur de canal souhaitée soit atteinte (par exemple, pour une hauteur de canal de 0,6 mm, utilisez deux entretoises et trois bandes adhésives).
- Fixez un verre rectangulaire sur la bande adhésive la plus basse avec le côté traité à la poly-D-lysine face à l’adhésif. Une fois l’assemblage terminé, comme indiqué à la figure 1, appliquer une pression ferme et égale sur le haut et le bas de la construction et maintenir pendant 1 min.
REMARQUE : La construction de l’enceinte du canal, y compris la vitre, les adhésifs, les entretoises et le dessus du réseau d’écoulement, est maintenant terminée. - Rincer le canal avec de l’eau désionisée à l’aide d’une seringue, en vérifiant simultanément les fuites.
- Stériliser l’enceinte du canal dans un stérilisateur ultraviolet (UV) pendant 30 min18.
- À l’aide d’une technique stérile, traiter le canal avec 2,0 μg/mL de fibronectine humaine (voir le tableau des matières) dans une solution saline tamponnée au phosphate (PBS) et incuber pendant au moins 30 minutes à 37 °C19.
- Effectuer une culture cellulaire dans le canal microfluidique en suivant les étapes ci-dessous.
- Dans une hotte laminaire stérile, utiliser une micropipette pour transférer une suspension uniforme de cellules NCI-H441 dans un milieu RPMI 1640 avec 10 % de sérum bovin fœtal (FBS) (voir le tableau des matières) pour ensemencer deux canaux microfluidiques, chacun avec des cellules à une densité de surface de 150 000 cellules/cm2.
- Pour les canaux de 50 mm x 5 mm x 0,6 mm, utilisez 0,25 mL d’une suspension de 2,5 x 106 cellules/ml pour remplir chaque canal, ainsi qu’une partie des orifices. Vérifiez que les cellules ont été réparties uniformément dans les canaux à l’aide d’un microscope à fond clair.
- Culture des deux canaux pendant 24 h et 48 h respectivement à 37 °C avec 5 % de CO2 à l’aide d’une pompe à seringue programmable (voir le tableau des matériaux), en aspirant les fluides usés du canal et les milieux frais dans le canal à partir d’un réservoir de média stérile fixé à l’entrée du canal composé d’une seringue de 20 mL à ouverture coupée recouverte d’un film de paraffine.
- Après une période d’attente de 10 minutes suivant l’ensemencement cellulaire, introduire et pomper le fluide frais du réservoir à travers le canal à un débit variable commençant à 0,2 μL/min et augmentant jusqu’à 10 μL/min pendant 4 h, en maintenant à ce rythme par la suite20.
NOTE: Ce débit variable fournit des conditions de culture qui permettent aux cellules (1) de se déposer gravitationnellement à la surface de culture, (2) d’adhérer à la surface de culture et (3) de former une monocouche confluente.
- Après une période d’attente de 10 minutes suivant l’ensemencement cellulaire, introduire et pomper le fluide frais du réservoir à travers le canal à un débit variable commençant à 0,2 μL/min et augmentant jusqu’à 10 μL/min pendant 4 h, en maintenant à ce rythme par la suite20.
- Dans une hotte laminaire stérile, utiliser une micropipette pour transférer une suspension uniforme de cellules NCI-H441 dans un milieu RPMI 1640 avec 10 % de sérum bovin fœtal (FBS) (voir le tableau des matières) pour ensemencer deux canaux microfluidiques, chacun avec des cellules à une densité de surface de 150 000 cellules/cm2.
- Effectuer la fixation cellulaire à l’intérieur des canaux microfluidiques à l’aide d’une solution de formaldéhyde.
ATTENTION : Le formaldéhyde est toxique et doit être manipulé dans une hotte chimiqueappropriée 21.- Dans une hotte chimique, préparer des solutions de formaldéhyde en utilisant 4 % de formaldéhyde dans du PBS (sans méthanol) (voir le tableau des matières) pour créer deux portions de 4 mL, en diluant la première à une concentration de 1 % de formaldéhyde et la seconde à 2 % de formaldéhyde en utilisant la solution saline tamponnée au phosphate de Dulbecco (DPBS; avec Ca 2+ et Mg2+) comme diluant. Transférer les solutions de formaldéhyde dans des seringues séparées de 5 ml et étiqueter en conséquence. Prélever 20 mL de DPBS dans une seringue séparée de 20 mL.
- Retirez les canaux microfluidiques de l’appareil de culture et placez-les dans la hotte chimique.
- Assembler l’appareil de fixation et de coloration.
- Fixez un segment de tube de transfert de 10 cm à l’orifice latéral d’un robinet d’arrêt à trois voies au moyen d’un adaptateur de barbe de verrouillage Luer mâle à tuyau (voir le tableau des matériaux), puis connectez le robinet d’arrêt à l’orifice d’entrée du réseau d’écoulement.
- Ensuite, fixez un autre segment de tube de transfert de 10 cm à l’orifice de sortie du réseau d’écoulement à l’aide du même type d’adaptateur barbe de tuyau.
- Enfin, fixez les extrémités libres des deux tubes de transfert dans un contenant de déchets chimiques et de risques biologiques, comme un tube à centrifuger conique vide étiqueté de 50 mL.
- Tournez le robinet d’arrêt pour bloquer l’orifice d’entrée du réseau de débit et rincez la conduite de déchets avec DPBS. Ensuite, tournez le robinet d’arrêt pour bloquer la ligne de déchets et lavez lentement les cellules avec 2 ml de DPBS. Répétez l’étape de rinçage à l’aide de la nouvelle solution à chaque fois. Une nouvelle solution (ou concentration de solution) est introduite dans le canal.
- Pousser lentement 2 ml de solution fixatrice à 1% dans le canal, puis laisser reposer pendant 5 min22.
- Pousser lentement 2 mL de solution fixatrice à 2% à travers le canal, puis laisser reposer pendant 15 min.
- Lavez les cellules en introduisant lentement 2 ml de DPBS frais dans le canal en trois instances distinctes (5 min chacune).
- Complétez les étapes 1.3.3-1.3.7 pour les deux canaux microfluidiques en parallèle.
- Colorer, perméabiliser et ajouter un support de montage aux cellules du canal microfluidique.
- Préparer une solution de saponine à 0,1 % en ajoutant 1 mg de saponine (voir le tableau des matières) par mL de DPBS pour produire 4 mL de solution et un léger vortex pour mélanger23. Aspirer 8 mL de DPBS dans une seringue de 20 mL.
- Ajouter un réactif de phalloïdine colorant la F-actine et un réactif Hoechst colorant le noyau (voir le tableau des matières) à la solution de saponine à 0,1 %, à raison de deux gouttes (0,1 mL) de chaque réactif par mL de solution de saponine. Conserver la solution de coloration/perméabilisation préparée à l’abri de la lumière en la recouvrant de papier d’aluminium24.
- Rincer la ligne avec une petite quantité de solution de coloration/perméabilisation (comme décrit à l’étape 1.3.4), puis introduire 2 mL de solution dans le canal microfluidique et recouvrir le canal de papier d’aluminium avant de le laisser reposer à température ambiante pendant 30 minutes.
- Rincer deux fois la solution de coloration/perméabilisation avec 2 mL de DPBS pendant 5 minutes par rinçage.
- Pour une meilleure qualité d’image, ajoutez un support de montage approprié (avec un indice de réfraction correspondant étroitement à l’huile de l’objectif du microscope et au verre de couverture, voir Tableau des matériaux) dans le canal.
- À l’aide d’une micropipette, introduire une quantité minimale d’un support antifade souple à chaque orifice du canal microfluidique, en veillant à ce que la surface inférieure soit complètement recouverte et qu’aucune bulle ne soit piégée dans la zone d’imagerie souhaitée25. Scellez les extrémités du canal et vérifiez l’intégrité de la couche cellulaire en observant au microscope à fond clair.
- Complétez les étapes 1.4.3-1.4.5 pour les deux canaux microfluidiques en parallèle.
REMARQUE: Les cellules d’image dès que possible après la coloration pour une qualité d’image maximale. Si le photoblanchiment se produit ou si un stockage à long terme est souhaité, d’autres supports de montage ayant des propriétés antidécoloration ou de préservation des échantillons peuvent être utilisés. Notez que le support de montage durcissant déformera la structure 3D des cellules et, par extension, la couche de cellule; Pour cette raison, un support de montage souple est préférable26.
- Cellules d’image dans le canal microfluidique en suivant les étapes ci-dessous.
- Ajustez les paramètres du microscope confocal (voir Tableau des matériaux), y compris la puissance laser, le gain, le décalage et les paramètres de balayage tels que la vitesse de numérisation, la zone de numérisation, le format de numérisation, la résolution et le diamètre du trou d’épingle27.
- Testez l’emplacement de l’imagerie en prenant des scans de référence ainsi que des piles Z jusqu’à ce que les paramètres et conditions d’image souhaités soient satisfaits. Paramètres d’appel à des objectifs de grossissement séquentiellement plus élevés jusqu’à ce que l’objectif d’immersion dans l’huile de 40x soit atteint et optimisé28.
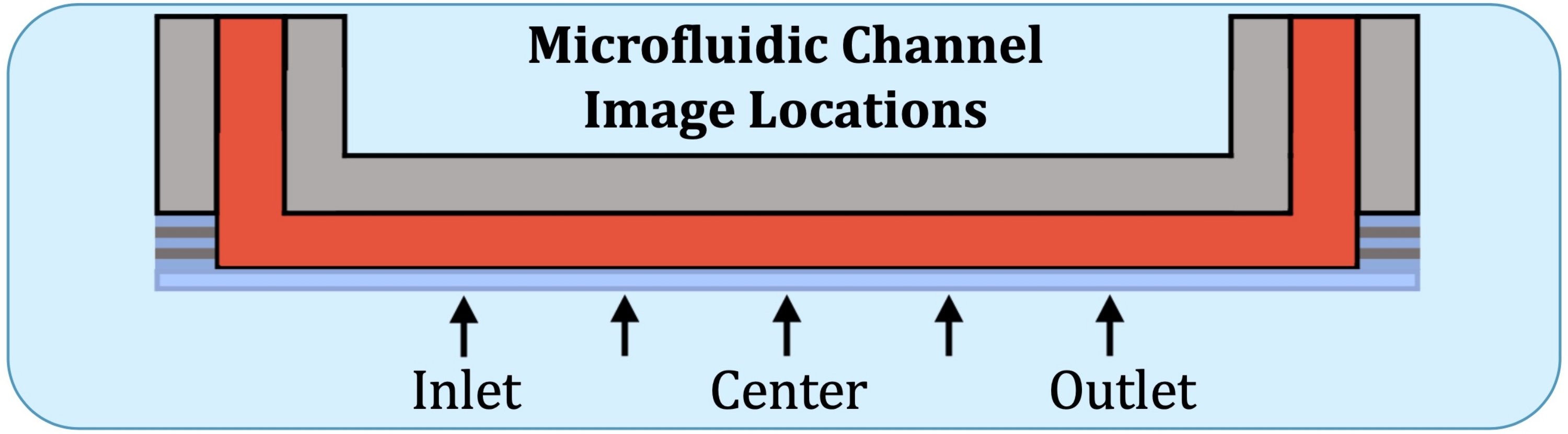
- En utilisant la plaque de base du réseau d’écoulement comme référence, construire des piles en Z à cinq endroits, à l’emplacement potentiel de la première électrode du côté de l’entrée, à mi-chemin entre le centre et l’emplacement précédent, le centre, à mi-chemin entre le centre et l’emplacement de la dernière électrode (du côté de la sortie), et à la dernière électrode, comme l’indique la figure 2.
- Effectuer le traitement d’images et l’analyse de données.
- Exportez les sections transversales XZ et YZ à l’aide du progiciel du microscope confocal (voir Tableau des matériaux).
- À l’aide d’un logiciel de traitement d’image (voir Tableau des matériaux) doté de capacités de détection des bords (valeur de seuil 15,0), mesurez la surface totale de l’image en pixels à l’aide de l’outil Baguette magique en dehors de l’image, puis la zone excluant la section transversale totale de la couche de cellule en pixels à l’aide de l’outil Baguette magique dans la partie de l’image située à l’extérieur de la couche de cellule29.
- À l’aide d’un logiciel de traitement des données (voir le tableau des matériaux), soustrayez la valeur en pixels de la zone extérieure de la valeur en pixels de la surface totale pour trouver la valeur en pixels de la section transversale.
- Convertissez les valeurs de pixels de section transversale en valeurs μm2 en multipliant les valeurs de pixels par le carré de la valeur μm/pixel, comme indiqué dans le logiciel de microscope pour l’image particulière (0,31 μm/pixel pour les piles Z prises en résolution 1024p sans zoom sur un objectif à immersion dans l’huile 40x).
- Calculer les moyennes et les écarts-types des résultats des données et des graphiques.

Figure 1 : Schéma en vue éclatée de la construction du canal microfluidique. L’élément supérieur est la partie supérieure du réseau d’écoulement, les éléments gris minces sont des bandes adhésives, les éléments bleus minces sont des entretoises en mylar et l’élément inférieur est le verre rectangulaire. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2 : Cinq emplacements d’imagerie le long de la région de production constante des couches du canal de culture microfluidique. Les emplacements d’imagerie sont les suivants : côté entrée, près de l’endroit où la première électrode se trouverait sur le réseau d’écoulement intact; à mi-chemin entre l’emplacement du côté de l’entrée et le centre du chenal; centre du canal; À mi-chemin entre le centre et l’emplacement côté sortie, et côté sortie, près de l’endroit où la dernière électrode serait sur le réseau d’écoulement intact. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
2. Culture cellulaire dans le verre de couverture chambré à huit puits
- Effectuer le prétraitement du verre de couverture chambré à huit puits.
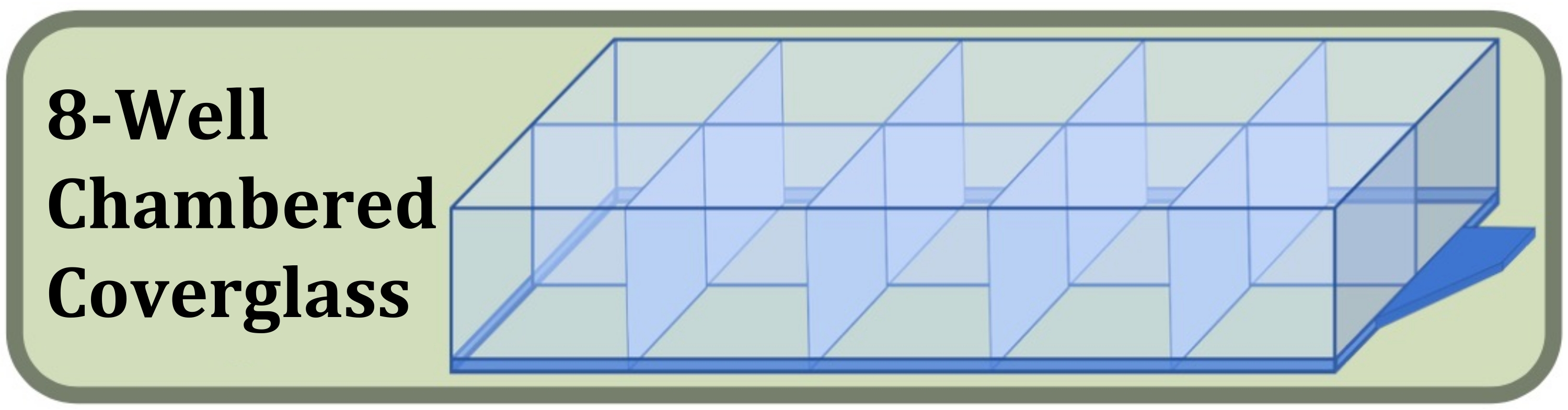
- Procurez-vous un verre de couverture stérile chambré à huit puits fabriqué avec un verre de couverture #1.5 et un traitement de surface augmentant l’adhérence cellulaire (figure 3, voir le tableau des matériaux).
- À l’aide d’une technique stérile, traiter la surface des puits de culture avec 2,0 μg/mL de fibronectine humaine dans du PBS et incuber pendant au moins 30 minutes à 37 °C19.
- Effectuez la culture cellulaire dans le verre de couverture chambré en suivant les étapes ci-dessous.
- Dans une hotte laminaire stérile, transférer des portions de 0,5 mL de solutions de cellules NCI-H441 uniformément suspendues dans un milieu RPMI 1640 avec 10% FBS à des densités volumétriques de 81 000, 162 000 et 324 000 cellules / ml pour ensemencer les puits de culture à des densités de surface de 45 000, 90 000 et 180 000 cellules / cm2, respectivement. Vérifiez que les cellules ont été réparties uniformément dans les puits à l’aide d’un microscope à fond clair.
- Cellules de culture pendant 24 h, 48 h et 96 h à 37 °C avec 5 % de CO2, en remplaçant le milieu quotidiennement.
- Effectuer la fixation du formaldéhyde dans le verre de couverture chambré à huit puits.
ATTENTION : Le formaldéhyde est toxique et doit être manipulé dans une hotte chimiqueappropriée 21.- Dans une hotte chimique, préparer des solutions de formaldéhyde en faisant deux portions de formaldéhyde à 4% dans du PBS (sans méthanol), en diluant la première à une concentration de 1% de formaldéhyde et l’autre à 2% de formaldéhyde en utilisant la solution saline tamponnée au phosphate de Dulbecco (DPBS; avec Ca 2+ et Mg2+) comme diluant.
- Retirez le verre de protection chambré de culture à huit puits de l’incubateur et placez-le dans la hotte chimique.
- Lavez doucement les cellules avec 0,5 mL de DPBS à l’aide d’une micropipette en introduisant lentement du liquide le long de la partie supérieure du coin de chaque puits.
- Retirez le liquide existant dans chaque puits en l’extrayant lentement du coin des puits à l’aide d’une micropipette. À l’aide de la méthode d’introduction de liquide (étape 2.3.3), introduire 0,5 mL de solution fixatrice à 1 % dans chaque puits et laisser reposer pendant 5 min22.
- Éliminer le liquide existant dans chaque puits en utilisant la méthode d’extraction liquide mentionnée à l’étape 2.3.4. En utilisant la méthode d’introduction de liquide mentionnée à l’étape 2.3.3, introduire 0,5 mL de solution fixatrice à 2 % dans chaque puits et laisser reposer pendant 15 minutes.
- À l’aide des méthodes d’introduction et d’extraction du liquide (étapes 2.3.3 et 2.3.4), laver les cellules en introduisant et en retirant 0,5 mL de DPBS frais dans chaque puits en trois instances distinctes pendant 5 minutes chacune.
- Effectuez la coloration, la perméabilisation et l’ajout de supports de montage dans le verre de couverture chambré à huit puits.
- Préparer la solution de saponine à 0,1 % en ajoutant 1 mg de saponine par mL de DPBS et en mélangeant doucement23 vortex.
- À la solution de saponine à 0,1 %, ajouter deux gouttes (0,1 mL) chacune d’un réactif de phalloïdine colorant la F-actine et d’un réactif Hoechst colorant le noyau par mL de solution de saponine. Conserver la solution préparée à l’abri de la lumière en la recouvrant d’une feuille d’aluminium24.
- Introduire 0,2 mL de solution de coloration/perméabilisation dans chaque puits et recouvrir le verre chambré de papier d’aluminium avant de le laisser reposer à température ambiante pendant 30 minutes.
- Rincer deux fois la solution de coloration/perméabilisation avec 0,5 mL de DPBS.
- Pour une meilleure qualité d’image, ajoutez un support de montage approprié (avec un indice de réfraction correspondant étroitement à l’huile de l’objectif du microscope et au verre de couverture) dans les puits.
- À l’aide d’une micropipette, introduire une quantité minimale d’un support anti-décoloration souple dans chaque puits, en veillant à ce que la surface inférieure soit complètement recouverte et qu’aucune bulle ne soit piégée dans la zone d’imageriesouhaitée 25. Vérifier l’intégrité de la couche cellulaire en observant au microscope à fond clair.
REMARQUE: Les cellules d’image dès que possible après la coloration pour une qualité d’image maximale. Si le photoblanchiment se produit ou si un stockage à long terme est souhaité, d’autres supports de montage ayant des propriétés antidécoloration ou de préservation des échantillons peuvent être utilisés. Notez que les supports de montage durcis déformeront la structure 3D des cellules et, par extension, la couche de cellule, de sorte que le support de montage à réglage souple est préférable26.
- À l’aide d’une micropipette, introduire une quantité minimale d’un support anti-décoloration souple dans chaque puits, en veillant à ce que la surface inférieure soit complètement recouverte et qu’aucune bulle ne soit piégée dans la zone d’imageriesouhaitée 25. Vérifier l’intégrité de la couche cellulaire en observant au microscope à fond clair.
- Effectuez des images dans la vitre à huit puits chambrés.
- Ajustez les paramètres du microscope confocal, y compris la puissance laser, le gain, le décalage et les paramètres de balayage tels que la vitesse de numérisation, la zone de numérisation, le format de numérisation, la résolution et le diamètre du sténopé27.
- Testez l’emplacement de l’imagerie en prenant des scans de référence ainsi que des piles Z jusqu’à ce que les paramètres et conditions d’image souhaités soient satisfaits. Paramètres d’appel à des objectifs de grossissement séquentiellement plus élevés jusqu’à ce que l’objectif d’immersion dans l’huile de 40x soit atteint et optimisé28.
- Construisez des piles Z de trois emplacements aléatoires dans chaque correspondance densité de semis/durée de culture.
- Effectuer le traitement d’images et l’analyse de données.
- Exportez les sections efficaces XZ et YZ à l’aide du logiciel de microscope confocal.
- À l’aide d’un logiciel de traitement d’image doté de capacités de détection des bords (valeur de seuil 15,0), mesurez la surface totale de l’image en pixels à l’aide de l’outil Baguette magique en dehors de l’image, puis la zone excluant la section transversale totale de la couche de cellule en pixels à l’aide de l’outil Baguette magique dans la partie de l’image située à l’extérieur de la couche de cellule29.
- À l’aide d’un logiciel de traitement des données, soustrayez la valeur en pixels de la zone extérieure de la valeur totale en pixels de surface pour trouver la valeur de pixel de la section transversale.
- Convertissez les valeurs de pixels de la section transversale en valeurs μm2 en multipliant les valeurs de pixels par le carré de la valeur μm/pixel indiquée dans le logiciel de microscope pour l’image particulière (0,31 μm/pixel pour les piles Z prises en résolution 1024p sans zoom sur un objectif à immersion dans l’huile 40x).
- Calculer les moyennes et les écarts-types et les résultats graphiques.

Figure 3 : Schéma du verre de couverture chambré à huit puits utilisé pour l’expérience de culture, de coloration et d’imagerie à puits fixes comparant les effets de la densité initiale d’ensemencement cellulaire et de la durée de culture sur la formation des couches cellulaires. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Résultats
La méthode présentée permet la visualisation de couches de cellules épithéliales cultivées dans des canaux de culture microfluidiques et utilise une démonstration dans des environnements traditionnels de culture cellulaire à puits fixes comme validation. Les images acquises existeront sur un spectre de qualité, d’intensité du signal et de spécificité cellulaire de la cible. Les images réussies présenteront un contraste élevé, ce qui permettra l’analyse des images et la quantification des données pour...
Discussion
Le protocole présenté décrit la culture, la fixation par réticulation, la coloration, la perméabilisation et la visualisation microscopique confocale des cellules épithéliales pulmonaires humaines NCI-H441 dans l’environnement dynamique d’un réseau d’écoulement microfluidique à canal unique, ainsi que dans l’environnement statique d’un verre de couverture chambré traditionnel à huit puits. Avec tout protocole de culture cellulaire microfluidique, les conditions d’écoulement du milieu de culture c...
Déclarations de divulgation
Les auteurs ne déclarent aucun conflit d’intérêts.
Remerciements
Les auteurs remercient Alan Shepardson pour avoir conçu le modèle de coupe de la feuille adhésive et mylar 3M utilisée dans la construction de canaux microfluidiques et pour avoir testé le débit du milieu de culture cellulaire et la programmation de la pompe à seringues. Le financement a été fourni par NIH R01 HL0142702, NSF CBET 1706801 et la subvention du doyen du Newcomb-Tulane College.
matériels
| Name | Company | Catalog Number | Comments |
| A1R HD25 Confocal Microscope System | Nikon | A1R HD25 | https://www.microscope.healthcare.nikon. com/products/confocal-microscopes/a1hd25-a1rhd25/specifications |
| ActinGreen 488 ReadyProbes Reagent (AlexaFluor 488 phalloidin) | Invitrogen | R37110 | https://www.thermofisher.com/order/catalog/product/R37110 |
| Adhesive Transfer Tape Double Linered | 3M | 468MP | https://gizmodorks.com/3m-468mp-adhesive-transfer-tape-sheet-5-pack/ |
| Air-Tite HSW Soft-Ject Disposable Syringes | Air-Tite RL5 | 14-817-53 | https://www.fishersci.com/shop/products/air-tite-hsw-soft-ject-disposable-syringes-6/1481753#?keyword=syringe%20leur%20locking%205ml |
| BAISDY 4 mil (0.1 mm) Thick Mylar Sheet | BAISDY | AS022 | https://www.amazon.ca/Stencil-Perfect-Silhouette-Machines-BAISDY/dp/B07RJJ9BNC |
| Branson Ultrasonics M Series Ultrasonic Cleaning Bath | Branson Ultrasonics | 15-336-100 | https://www.fishersci.com/shop/products/m-series-ultrasonic-cleaning-bath/15336100 |
| Corning Fibronectin, Human | Fisher Scientific | CB-40008 | https://www.fishersci.com/shop/products/corning-fibronectin-human-3/CB40008?keyword=true |
| DPBS, calcium, magnesium | Gibco | 14040133 | https://www.thermofisher.com/order/catalog/product/14040133?SID=srch-srp-14040133 |
| ECIS Cultureware Disposable Electrode Arrays 8 x 10 ECIS Flow Array | Applied BioPhysics | 1F8x10E PC | https://www.biophysics.com/cultureware.php#1F8x10E |
| Enterprise Technology Solutions UV Sterilizer Cabinet, White | Enterprise Technology Solutions | 50-211-1163 | https://www.fishersci.com/shop/products/uv-sterilizer-cabinet-white/502111163 |
| Fetal Bovine Serum (FBS) | Gibco | 26140079 | https://www.thermofisher.com/order/catalog/product/26140079 |
| Finnpipette F2 Variable Volume Pipettes | Thermo Scientific | 4642090 | https://www.thermofisher.com/order/catalog/product/4642090 |
| Fisherbrand 50mL Easy Reader Plastic Centrifuge Tubes | Fisher Scientific | 06-443-21 | https://www.fishersci.com/shop/products/fisherbrand-higher-speed-easy-reader-plastic-centrifuge-tubes-8/p-193269 |
| Fisherbrand Cover Glasses: Rectangles (#1.5) | Fisher Scientific | 12-544-GP | https://www.fishersci.com/shop/products/cover-glasses-rectangles-promo-22/12544GP#coverglass |
| Fisherbrand Sterile Syringes for Single Use | Fisher Scientific | 14-955-458 | https://www.fishersci.com/shop/products/sterile-syringes-single-use-12/14955458 |
| Gibco RPMI 1640 Medium | Gibco | 11875093 | https://www.thermofisher.com/order/catalog/product/11875093 |
| Image-iT Fixative Solution (4% formaldehyde, methanol-free) | Invitrogen | FB002 | https://www.thermofisher.com/order/catalog/product/FB002 |
| ImageJ Fiji | ImageJ | ImageJ Fiji | https://imagej.net/downloads |
| Immersion Oil F 30 cc | Nikon | MXA22168 | https://www.microscope.healthcare.nikon. com/products/accessories/immersion-oil/specifications |
| Large-Capacity Reach-In CO2 Incubator, 821 L, Polished Stainless Steel | Thermo Scientific | 3950 | https://www.thermofisher.com/order/catalog/product/3950 |
| Laxco LMC-3000 Series Brightfield Compound Microscope System | Laxco | LMC3BF1 | https://www.fishersci.com/shop/products/lmc-3000-series-brightfield-compound-microscope-system-8/LMC3BF1 |
| Masterflex Fitting, Nylon, Straight, Male Luer Lock to Hose Barb Adapters, 1/16" ID; 25/PK | Masterflex | ZY-45505-31 | https://www.masterflex.com/i/masterflex-fitting-nylon-straight-male-luer-lock-to-hose-barb-adapters-1-16-id-25-pk/4550531?PubID=ZY&persist=true&ip=no& gclid=Cj0KCQiA3rKQBhCNARIsAC UEW_Zb5yXy1em6bGs0a9KFOk5k pdlkHCvAEslHumdqcnlwSN0MdR0 udmwaAuDHEALw_wcB |
| Microsoft Excel | Microsoft | 0016 | https://www.microsoft.com/en-us/download/details.aspx?id=56547 |
| National Target All-Plastic Disposable Syringes | Thermo Scientific | 03-377-24 | https://www.fishersci.com/shop/products/national-target-all-plastic-disposable-syringes/0337724#tab8 |
| NCI-H441 Human Epithelial Lung Cells | American Type Culture Collection (ATCC) | HTB-174 | https://www.atcc.org/products/htb-174 |
| NE-1600 Six Channel Programmable Syringe Pump | New Era Pump Systems | NE-1600 | https://www.syringepump.com/NE-16001800.php |
| NIS Elements AR | Nikon | NIS Elements AR | https://www.microscope.healthcare.nikon. com/products/software/nis-elements/nis-elements-advanced-research |
| NucBlue Live ReadyProbes Reagent (Hoechst 33342) | Invitrogen | R37605 | https://www.thermofisher.com/order/catalog/product/R37605?SID=srch-srp-R37605 |
| Nunc Lab-Tek Chambered Coverglass | Thermo Scientific | 155411 | https://www.thermofisher.com/order/catalog/product/155361 |
| Parafilm M Wrapping Film | Fisher Scientific | S37441 | https://www.fishersci.com/shop/products/parafilm-m-wrapping-film-3/S37441 |
| PendoTech 3-Way Stopcock, Polysulfone, Male/Female Luer Inlet x Female Luer Branch | PendoTech | ZY-19406-49 | https://www.masterflex.com/i/pendotech-3-way-stopcock-polysulfone-male-female-luer-inlet-x-female-luer-branch/1940649 |
| Phosphate Buffered Solution (PBS), pH 7.4 | Gibco | 10010023 | https://www.thermofisher.com/order/catalog/product/10010023 |
| Poly-D-Lysine | Gibco | A3890401 | https://www.thermofisher.com/order/catalog/product/A3890401#/A3890401 |
| Reynolds Aluminum Wrap Foil | Reynolds | 458742928317 | https://www.amazon.com/Reynolds-Wrap-Aluminum-Foil-Square/dp/B00UNT0Y2M |
| Saponin | Millipore Sigma (Sigma Aldrich) | 47036 | https://www.sigmaaldrich.com/US/en/product/sigma/47036 |
| SlowFade Glass Soft-set Antifade Mountant | Invitrogen | S36917-5X2ML | https://www.thermofisher.com/order/catalog/product/S36917-5X2ML |
| Thermo Scientific 1300 Series Class II, Type A2 Biological Safety Cabinet Package | Thermo Scientific | 13-100-752PM | https://www.fishersci.com/shop/products/1300-series-class-ii-type-a2-biological-safety-cabinet-package-promo/p-9049003#?keyword=biosafety%20hood |
| Tygon Transfer Tubing, BioPharm Platinum-Cured Silicone, 1/16" ID x 1/8" OD; 50 Ft | Cole-Parmer | EW-95702-01 | https://www.coleparmer.com/i/tygon-transfer-tubing-biopharm-platinum-cured-silicone-1-16-id-x-1-8-od-50-ft/9570201?searchterm=95702-01 |
Références
- Matthay, M. A., et al. Acute respiratory distress syndrome. Nature Reviews Disease Primers. 5, 18 (2019).
- Rawal, G., Yadav, S., Kumar, R. Acute respiratory distress syndrome: An update and Review. Journal of Translational Internal Medicine. 6 (2), 74-77 (2018).
- Bilek, A. M., Dee, K. C., Gaver, D. P. Mechanisms of surface-tension-induced epithelial cell damage in a model of pulmonary airway reopening. Journal of Applied Physiology. 94 (2), 770-783 (2003).
- Modrykamien, A. M., Gupta, P. The acute respiratory distress syndrome. Baylor University Medical Center Proceedings. 28 (2), 163-171 (2017).
- Jacob, A. -. M., Gaver, D. P. Atelectrauma disrupts pulmonary epithelial barrier integrity and alters the distribution of tight junction proteins ZO-1 and Claudin 4. Journal of Applied Physiology. 113 (9), 1377-1387 (2012).
- Kay, S. S., Bilek, A. M., Dee, K. C., Gaver, D. P. Pressure gradient, not exposure duration, determines the extent of epithelial cell damage in a model of pulmonary airway reopening. Journal of Applied Physiology. 97 (1), 269-276 (2004).
- Jacob, A. M., Gaver, D. P. An investigation of the influence of cell topography on epithelial mechanical stresses during pulmonary airway reopening. Physics of Fluids. 17 (3), 031502 (1994).
- Gaver, D. P., et al. The POOR get POORer: A hypothesis for the pathogenesis of ventilator-induced lung injury. American Journal of Respiratory and Critical Care Medicine. 202 (8), 1081-1087 (2020).
- Jain, P., et al. Reconstruction of ultra-thin alveolar-capillary basement membrane mimics. Advanced Biology. 5 (8), 2000427 (2021).
- Byrne, M. B., Leslie, M. T., Gaskins, H. R., Kenis, P. J. A. Methods to study the tumor microenvironment under controlled oxygen conditions. Trends in Biotechnology. 32 (11), 556-563 (2014).
- Szulcek, R., Bogaard, H. J., van Nieuw Amerongen, G. P. Electric cell-substrate impedance sensing for the quantification of endothelial proliferation, barrier function, and motility. Journal of Visualized Experiments. (85), e51300 (2014).
- Jaccard, N., et al. Automated method for the rapid and precise estimation of adherent cell culture characteristics from phase contrast microscopy images. Biotechnology and Bioengineering. 111 (3), 504-517 (2013).
- Hagiyama, M., et al. Modest static pressure suppresses columnar epithelial cell growth in association with cell shape and cytoskeletal modifications. Frontiers in Physiology. 8, 00997 (2017).
- Srinivasan, M., Sedmak, D., Jewell, S. Effect of fixatives and tissue processing on the content and integrity of Nucleic Acids. The American Journal of Pathology. 161 (6), 1961-1971 (2002).
- Zhu, L., Rajendram, M., Huang, K. C. Effects of fixation on bacterial cellular dimensions and integrity. Iscience. 24 (4), 102348 (2021).
- Lust, R. M. . The Pulmonary System. XPharm: The Comprehensive Pharmacology Reference. , 1-6 (2007).
- EpilogueLaser. FusionSeries: Pro & Edge Laser System Manual and Original Instructions. EpilogueLaser. , (2022).
- Chitnis, D. S., Katara, G., Hemvani, N., Chitnis, S., Chitnis, V. Surface disinfection by exposure to germicidal UV light. Indian Journal of Medical Microbiology. 26 (3), 241 (2008).
- Sandell, L., Sakai, D. Mammalian cell culture. Current Protocols Essential Laboratory Techniques. 5 (1), 4 (2011).
- New Era Pump Systems. Multi-Phaser Programmable Syringe Pump: NE-1000 Series User Manual. New Era Pump Systems. , (2014).
- Thermo Fisher Scientific. Safety Data Sheet: Image-iT Fixative Solution (4% formaldehyde, methanol-free). Thermo Fisher Scientific. , (2018).
- Thavarajah, R., Mudimbaimannar, V. K., Rao, U. K., Ranganathan, K., Elizabeth, J. Chemical and physical basics of routine formaldehyde fixation. Journal of Oral and Maxillofacial Pathology. 16 (3), 400-405 (2012).
- Jamur, M. C., Oliver, C. Permeabilization of cell membranes. Immunocytochemical Methods and Protocols. 588, 63-66 (2009).
- Thermo Fisher Scientific. ActinGreen 488 ReadyProbes Reagent Protocol. Thermo Fisher Scientific. , (2022).
- Slowfade Glass soft-set Antifade Mountant. Thermo Fisher Scientific Available from: https://www.thermofisher.com/order/catalog/product/S36917-5X2ML?SID=srch-hj-S36917-5X2ML (2022)
- Ravikumar, S., Surekha, R., Thavarajah, R. Mounting media: An overview. Journal of Dr. NTR University of Health Sciences. 3 (5), 1-8 (2014).
- Shihan, M. H., Novo, S. G., Le Marchand, S. J., Wang, Y., Duncan, M. K. A simple method for quantitating confocal fluorescent images. Biochemistry and Biophysics Reports. 25, 100916 (2021).
- North, A. J. Seeing is believing? A beginners' guide to practical pitfalls in image acquisition. Journal of Cell Biology. 172 (1), 9-18 (2006).
- Ferriera, F., Rasband, W. ImageJ User Guide. National Institutes of Health. , (2012).
- Halldorsson, S., Lucumi, E., Gómez-Sjöberg, R., Fleming, R. M. T. Advantages and challenges of microfluidic cell culture in polydimethylsiloxane devices. Biosensors and Bioelectronics. 63, 218-231 (2015).
- Smith, H. S., Riggs, J. L., Mosesson, M. W. Production of fibronectin by human epithelial cells in culture. American Association for Cancer Research. 39 (10), 4138-4144 (1979).
- Sieck, G. C., Mantilla, C. B., Prakash, Y. S. Volume measurements in confocal microscopy. Methods in Enzymology. 307, 296-315 (1999).
- Heijink, I. H., et al. Characterisation of cell adhesion in airway epithelial cell types using electric cell-substrate impedance sensing. European Respiratory Journal. 35 (4), 894-903 (2009).
- Zhang, X., Wang, W., Li, F., Voiculescu, I. Stretchable impedance sensor for mammalian cell proliferation measurements. Lab on a Chip. 17 (12), 2054-2066 (2017).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.