
Method Article
Isolierung von Zellkernen aus schockgefrorenem Lebergewebe für Einzelzell-Multiomics
In diesem Artikel
Zusammenfassung
Hier stellen wir ein Protokoll zur Isolierung von Kernen aus schockgefrorenem, archiviertem Lebergewebe für Einzelkern-RNA-seq, ATAC-seq und Gelenk-Multiomics (RNA-seq und ATAC-seq) vor.
Zusammenfassung
Die Leber ist ein komplexes und heterogenes Gewebe, das für die Durchführung vieler kritischer physiologischer Funktionen verantwortlich ist, wie unter anderem die Aufrechterhaltung der Energiehomöostase und den Stoffwechsel von Xenobiotika. Diese Aufgaben werden durch enge Koordination zwischen Leberparenchym- und Nicht-Parenchymzellen durchgeführt. Darüber hinaus sind verschiedene metabolische Aktivitäten auf bestimmte Bereiche des Leberläppchens beschränkt - ein Phänomen, das Leberzonation genannt wird. Jüngste Fortschritte bei Einzelzellsequenzierungstechnologien haben es Forschern ermöglicht, die Gewebeheterogenität mit Einzelzellauflösung zu untersuchen. In vielen komplexen Geweben, einschließlich der Leber, können harte enzymatische und/oder mechanische Dissoziationsprotokolle die Lebensfähigkeit oder die Qualität der Einzelzellsuspensionen negativ beeinflussen, die zur umfassenden Charakterisierung dieses Organs bei Gesundheit und Krankheit erforderlich sind.
Dieser Artikel beschreibt ein robustes und reproduzierbares Protokoll zur Isolierung von Kernen aus gefrorenem, archiviertem Lebergewebe. Diese Methode liefert qualitativ hochwertige Kerne, die mit nachgeschalteten Einzelzell-Omics-Ansätzen kompatibel sind, einschließlich Einzelkern-RNA-seq, Assay für Transposase-zugängliches Chromatin mit Hochdurchsatzsequenzierung (ATAC-seq) sowie multimodaler Omics (gemeinsame RNA-seq und ATAC-seq). Diese Methode wurde erfolgreich zur Isolierung von Kernen aus gesunden und kranken gefrorenen Leberproben von Menschen, Mäusen und nichtmenschlichen Primaten eingesetzt. Dieser Ansatz ermöglicht die unvoreingenommene Isolierung aller wichtigen Zelltypen in der Leber und bietet daher eine robuste Methodik zur Untersuchung der Leber bei Einzelzellauflösung.
Einleitung
Die Einzelzellgenomik entwickelt sich schnell zu einer wesentlichen Methodik zur Untersuchung der Leberfunktion und zur Bewertung der Auswirkungen zellulärer Heterogenität auf Gesundheit und Krankheitszustände1. Die rasante Entwicklung von "Multiomics" für die gleichzeitige Messung verschiedener Informationsschichten und den parallelen Ausbau robuster Computational Pipelines ebnet den Weg für die Entdeckung bisher unbekannter Zelltypen und Subtypen in der normalen und kranken Leber2.
Die Möglichkeit, Biobanken und archivierte gefrorene Proben zu erforschen, hat die Möglichkeiten, die Rolle nicht-parenchymalerZellen erneut zu untersuchen und zu entdecken 3,4,5 und die Rolle polyploider Hepatozyten während des Alterns und bei chronischen Krankheiten zu untersuchen, erheblich erhöht 6,7,8,9 . Daher beschreibt dieser Artikel ein robustes und reproduzierbares Einzelkern-Isolationsprotokoll für schockgefrorene (FF) archivierte Lebern, das mit der nachgeschalteten Einzelkern-RNA-Sequenzierung und ATAC-Sequenzierung sowie mit multimodalen Omics (gemeinsame RNA-seq und ATAC-seq) kompatibel ist (Abbildung 1).
Dieser Workflow ermöglicht die Untersuchung des Transkriptoms und der Chromatinzugänglichkeit aller Zelltypen in der Leber, unabhängig von der Zellgröße oder Zerbrechlichkeit, in enzymatischen Dissoziationsprotokollen. Es kann mit kleinen Gewebeschnitten (15-30 mg oder 5-10 mm3) von wertvollen menschlichen Proben oder transgenen Mäusen durchgeführt werden. Die Bestimmung der hohen Reinheit der Kernisolierung umfasst die Quantifizierung und Messung der Kerngröße, die mit erhöhter Zellgröße und Seneszenz korrelieren könnte 10,11, und diese Reinheit ist relevant für die Analyse sowohl der Hepatozytenploidie12 als auch der zellgrößenabhängigen Transkriptionsmechanismen 11,13,14,15 . Darüber hinaus behalten Kerne, die aus gefrorenen Lebern isoliert wurden, wertvolle Informationen über die Leberzonierung. Der Workflow und die Gewebeentnahme ermöglichen die Validierung von Einzelzell-Genomik-Daten oder weitere komplementäre Analysen, wie Immunhistochemie oder räumliche Transkriptomik aus demselben Gewebe und demselben Individuum. Daher kann dieser Ansatz auf mehrere Lebererkrankungen angewendet werden und Organismen systematisch und zuverlässig modellieren.
Protokoll
Alle Tierversuche wurden in Übereinstimmung mit dem deutschen Tierschutzgesetz und den Vorschriften der Regierung von Oberbayern durchgeführt. Die Tierhaltung wurde nach §11 Tierschutzgesetz genehmigt und gemäß Richtlinie 2010/63/EU durchgeführt.
1. Gewebevorbereitung
- Opfern Sie eine 3 Monate alte bis 22 Monate alte männliche C57BL / 6J-Maus durch zervikale Dislokation. Legen Sie das Tier auf ein Sezierbrett, sichern Sie die Extremitäten mit Stiften und desinfizieren Sie den Bauch mit 70% Ethanol.
- Führen Sie eine Nekropsie durch, wie von Treuting et al.16 empfohlen.
- Öffnen Sie den Bauch bis zum Brustkorb, visualisieren Sie die Leber und verwenden Sie eine Pinzette, um die Leber vorsichtig zu entfernen, ohne die Läppchen zu durchbohren.
- Extrahieren Sie die intakte Leber, indem Sie das Zwerchfell mit einer Pinzette halten und das Bindegewebe mit einer Schere entfernen.
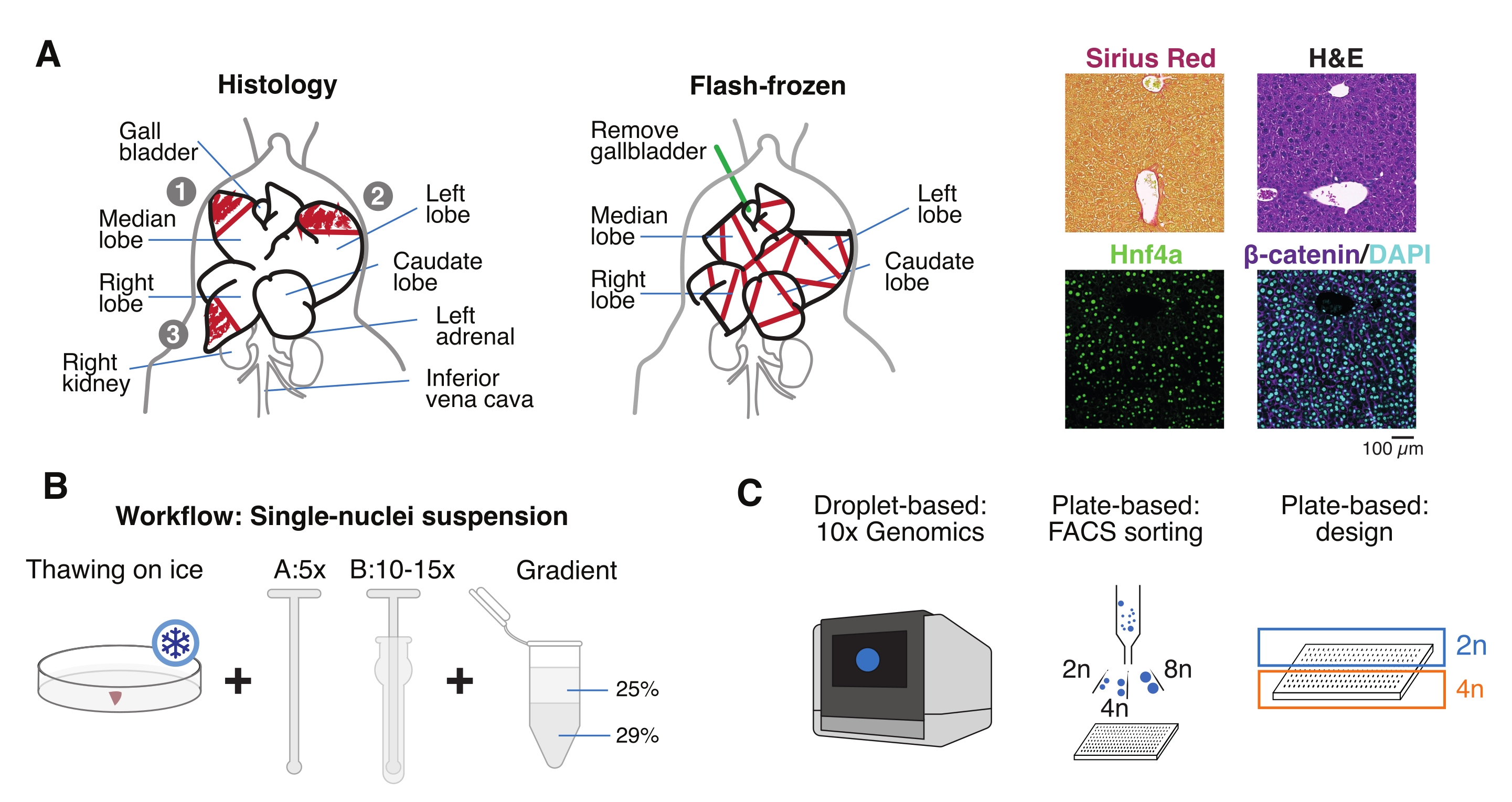
- Waschen Sie das Organ in kalter phosphatgepufferter Kochsalzlösung (PBS), tupfen Sie es auf einem sauberen Papiertuch trocken und schneiden Sie die Leberläppchen für verschiedene Zwecke in mehrere Stücke: FF für Einzelkernisolierung und Multiomik; paraformaldehydfixiert (10% PFA) zur Paraffineinbettung; und/oder eingebettet in die optimale Schnitttemperatur (OCT) für weitere histologische Analysen (Abbildung 1A).
- Aliquot die Leberstücke in Kryozellen oder 5 ml Schraubverschlussröhrchen und sofort in flüssigem Stickstoff schockgefrieren. Lagern Sie die gefrorenen Leberproben bei −80 °C für nachgeschaltete Einzelkern-Multiomics-Experimente (Abbildung 1B, C).
HINWEIS: Das kryokonservierte Gewebe kann sicher bei −80 °C für mehrere Jahre gelagert werden und sollte vor der Verwendung immer auf Trockeneis bei −80 °C transportiert werden.
2. Isolierung der Kerne
- Tischreinigung und Vorbereitung der Puffer und Verbrauchsmaterialien
- Reinigen Sie die Arbeitstischplatten und Pipetten mit 70% Ethanol und RNase-Dekontaminationslösung oder verwenden Sie spezielle RNase-freie Tischplatten und Materialien.
- Kühlen Sie sowohl den Schwenklöffel als auch die Zentrifugen mit festem Winkel, 1,5 ml/2 ml Röhrchen und Multi-Well-Platten auf 4 °C, wie unten beschrieben.
- Kühlen Sie die RNase-freie Einwegpinzette für die Handhabung von Gewebe, ein steriles Einwegskalpell und eine Petrischale auf Trockeneis (−60 °C) vor.
- Dounce Glashomogenisator und Stößel auf Eis (4 °C) vorkühlen. Legen Sie jeden Stößel (A und B) in ein 5-ml-Röhrchen, um direkten Kontakt mit dem Eis und eine mögliche RNase-Kontamination zu vermeiden.
- Bereiten Sie eine Verdünnungslösung von Iodixanolmedium (IDM) (Tabelle 1A) her und verwenden Sie sie, um 50% und 29% Verdünnungen des 60% igen Iodixanol-Stammdichtegradientenmediums herzustellen (Tabelle 1B bzw. Tabelle 1C).
- Alle Tuben vorkühlen. Bereiten Sie für jede zu verarbeitende Probe die folgenden Röhrchen vor:
- Bereiten Sie drei 1,5-ml-Low-DNA-Binderöhrchen vor (eines für das gefilterte Gewebehomogenat, ein zweites mit 250 μL einer 50%igen Verdünnung von Iodixanollösung und ein drittes für die saubere Kernsuspension).
- Bereiten Sie ein 2-ml-Rundbodenröhrchen mit 500 μL einer 29%igen Verdünnung von Jodixanollösung für die Dichtegradiententrennung vor.
- Bereiten Sie ein 15-ml-konisches Röhrchen für das Kernisolierungsmedium-2 (NIM-2) und den Homogenisierungspuffer (HB) vor.
- Bereiten Sie das Kernisolierungsmedium-1 (NIM-1) (Tabelle 1D) vor und verwenden Sie es, um das NIM-2 (Tabelle 1E) und anschließend das HB (Tabelle 1F) herzustellen. Fügen Sie beide RNAse Inhibitoren kurz vor der Verwendung hinzu, wie unten im Protokoll beschrieben.
- Bereiten Sie den Kernspeicherpuffer (NSB) wie in Tabelle 1G beschrieben vor. Fügen Sie kurz vor der Anwendung einen rekombinanten RNASE-Inhibitor hinzu. Fügen Sie proteinbasierten RNAse Inhibitor in die NSB ein, bevor Sie sie für die FACS-Sortierung verwenden (optional).
- Bereiten Sie ein 500-ml-Becherglas mit sterilem Wasser vor, um die Dounce-Homogenisatoren und Stößel nach der Gewebehomogenisierung für die optimale Reinigung und Wartung der Homogenisatoren einzuweichen.
- Gewebehomogenisierung
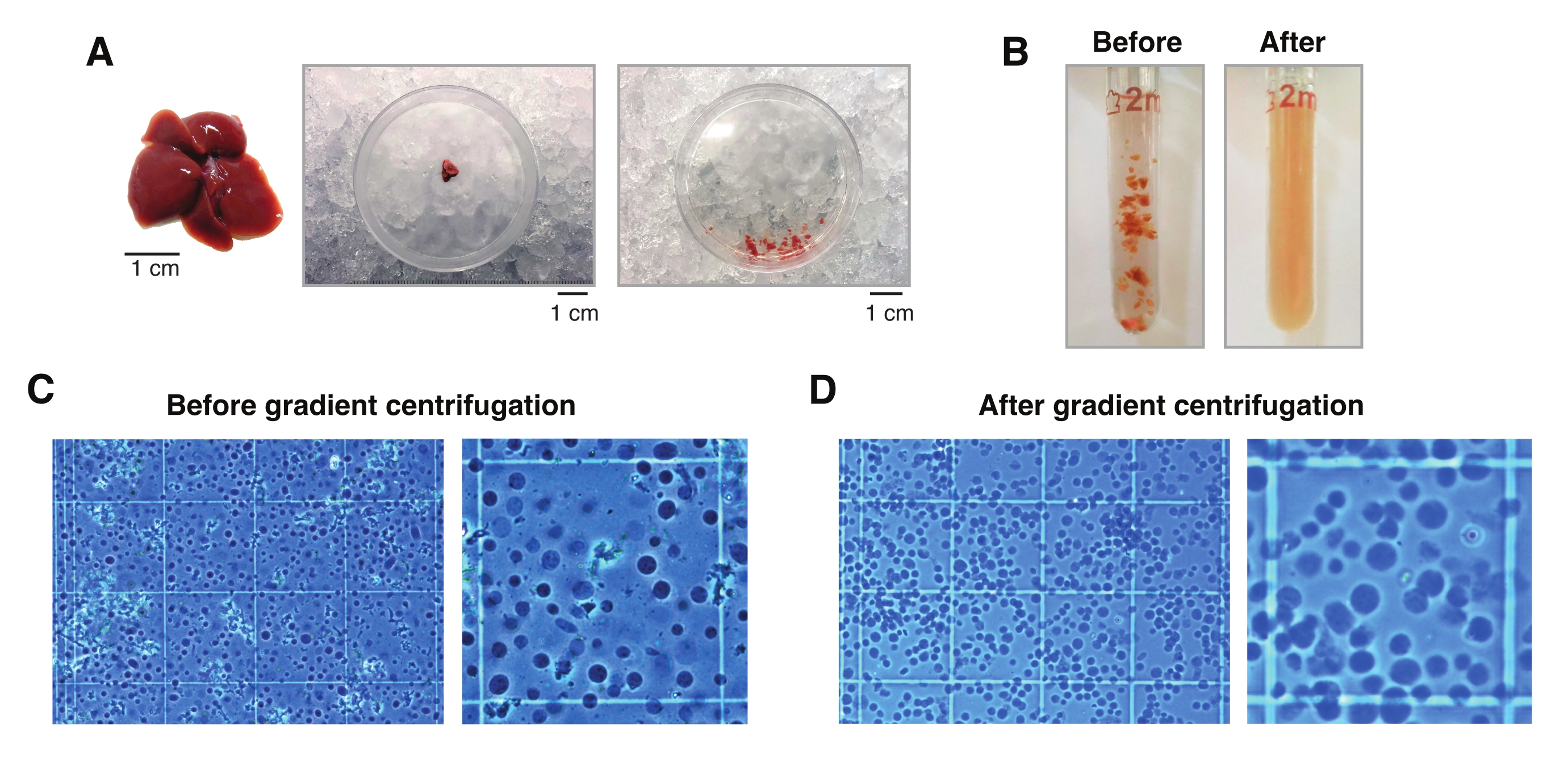
- Schneiden Sie ein 20-30 mg (oder 5 mm3) Gewebestück mit einem vorgekühlten Skalpell in die Petrischale auf Trockeneis. Anschließend die Petrischale sofort auf Nasseis (4 °C) umfüllen. Fügen Sie 1 ml HB hinzu und zerkleinern Sie das Gewebe mit dem kalten Skalpell so weit wie möglich, damit es leicht mit einer 1 ml breiten Öffnungsspitze abgesaugt werden kann.
HINWEIS: Verwenden Sie immer breite Öffnungsspitzen für alle Gewebe-/Kerntransfers. Alternativ können 1-ml-Spitzen mit einem sterilen Skalpell auf einer sterilen Kunststoffabdeckung geschnitten werden, um breite Öffnungen zu erzeugen (Abbildung 2A). - Sammeln Sie die Gewebesuspension, und geben Sie sie in einen vorgekühlten 2-ml-Glas-Dounce-Homogenisator (Abbildung 2B).
- Waschen Sie die Petrischale mit zusätzlichen 0,5-1 ml HB und sammeln Sie alle restlichen Gewebestücke, während Sie alles auf Eis halten.
- Langsam und vorsichtig fünf Schläge mit losem Stößel A auf Eis machen. Vermeiden Sie es, Blasen zu erzeugen, indem Sie Drehbewegungen des Stößels verwenden, wenn Sie ihn auf und ab ziehen. Stellen Sie sicher, dass sich der Stößel bei jedem Schlag vorsichtig von oben nach unten des Homogenisators bewegt.
- Führen Sie anschließend 10-15 langsame Schläge mit engem Stößel B auf Eis durch. Vermeiden Sie es, Blasen zu erzeugen.
HINWEIS: Es wird empfohlen, die Kerne nach 10 Schlägen mit Stößel B visuell unter dem Mikroskop zu untersuchen, um festzustellen, ob weitere Schläge erforderlich sind. Verwenden Sie dazu eine Trypanblau-Färbung (1:1-Verhältnis von Trypan zu Probe) und ein manuelles Hämozytometer (z. B. mischen Sie 10 μL Trypanblau mit 10 μL Kernsuspension und verwenden Sie 10 μL der Mischung zur Untersuchung unter dem Mikroskop; Abbildung 2C). - Filtern Sie das Homogenat durch ein 50-μm-Zellsieb, während Sie es in ein vorgekühltes 1,5-ml-Röhrchen überführen. Verwenden Sie mehr als einen Filter und/oder Röhrchen für Homogenate, die einen hohen Anteil an Bindegewebsklumpen enthalten.
- Spülen Sie den Homogenisator und die verwendeten Filter mit zusätzlichen 0,5-1 ml HB aus, um das gesamte Gewebehomogenat gründlich zu sammeln. Fahren Sie mit der Zentrifugation des Dichtegradienten fort.
- Schneiden Sie ein 20-30 mg (oder 5 mm3) Gewebestück mit einem vorgekühlten Skalpell in die Petrischale auf Trockeneis. Anschließend die Petrischale sofort auf Nasseis (4 °C) umfüllen. Fügen Sie 1 ml HB hinzu und zerkleinern Sie das Gewebe mit dem kalten Skalpell so weit wie möglich, damit es leicht mit einer 1 ml breiten Öffnungsspitze abgesaugt werden kann.
- Dichtegradientenzentrifugation
- Zentrifugieren Sie das gefilterte Homogenat in einer vorgekühlten Zentrifuge mit festem Winkel bei 1.000 × g für 8 min bei 4 °C.
- Während sich die Probe dreht, bereiten Sie ein 1,5-ml-Röhrchen mit 250 μL 50%iger Iodixanol-Verdünnung und ein 2-ml-Röhrchen mit 500 μL 29%iger Iodixanol-Verdünnung vor. Beide Röhrchen auf Eis halten.
- Nach der Zentrifugation saugen Sie den Überstand mit einer Vakuumpumpe ab, ohne das Pellet zu stören.
HINWEIS: Die Verwendung von manuellem Pipettieren beeinträchtigt die Qualität der endgültigen Kernsuspension. - Mit der 1-ml-Pipettenspitze mit breiter Öffnung 250 μL HB in das Pellet geben und sehr langsam resuspendieren.
- 250 μL der Kernsuspension werden in ein vorgekühltes 1,5-ml-Röhrchen mit 250 μL 50%iger Jodixanol-Verdünnung überführt und vorsichtig, aber gründlich gemischt, um eine 25%ige Iodixanol/Kern-Suspension zu erzeugen.
- 500 μL der 25%igen Iodixanol/Kern-Suspension in ein vorgekühltes 2-ml-Röhrchen mit 500 μL 29%iger Iodixanol-Verdünnung überführen.
HINWEIS: Die 500 μL der 25%igen Iodixanol/Kern-Suspension sollten vorsichtig auf die 500 μL 29%ige Iodixanol-Lösung abgeschieden werden, so dass die 25%/29%-Iodixanol-Gemische eine klare Phasentrennung aufweisen. Verwenden Sie die Seite der Rohrwand mit der Pipettenspitze, die in einem Winkel von 45° positioniert ist, um diese Farbverlaufsschnittstelle zu erstellen. Von da an muss das Rohr sanft gehandhabt werden, um dieses Gefälle nicht zu stören. - Zentrifugieren Sie das Röhrchen in einer vorgekühlten Schaufelzentrifuge bei 12.500 g für 20 min bei ausgeschalteter Bremse.
- Kurz bevor der Zentrifugationsschritt abgeschlossen ist, fügen Sie die RNAse Inhibitoren dem NSB-Puffer hinzu, wenn Sie zu den scRNA-seq-Pipelines übergehen (siehe Tabelle 1G).
- Nach der Zentrifugation saugen Sie den Überstand mit einer Vakuumpumpe ab, ohne das Pellet zu stören.
HINWEIS: Die Verwendung von manuellem Pipettieren beeinträchtigt die Qualität der endgültigen Kernsuspension. - Resuspendieren Sie das Pellet vorsichtig mit der 1-ml-Pipettenspitze mit breiter Öffnung in 100-300 μl NSB und geben Sie die Kernsuspension in ein sauberes, vorgekühltes 1,5-ml-Röhrchen.
- Zählen Sie die Kerne mit Trypanblau-Lösung (Verhältnis 1:1 von Trypan zu Probe) und einem manuellen Hämozytometer (z. B. mischen Sie 10 μL Trypanblau mit 10 μL Kernsuspension und verwenden Sie 10 μL der Mischung zum Zählen; Abbildung 2D).
- Verwenden Sie die erhaltene Kernsuspension sofort für den Einzelkern-Genomik-Assay.
HINWEIS: Die Kernsuspension in NSB kann gekühlt bei 4 °C für bis zu 1 Woche zur weiteren Analyse durch Durchfluss- und/oder bildgebende Durchflusszytometrie gelagert werden, jedoch nicht für snRNA-seq oder snATAC-seq.
3. Kernsortierung für Hepatozyten-Ploidie-Profiling oder gut basierte Sequenzierungsansätze
- Für die durchflusszytometrische Zellsortierung filtrieren Sie die Kernsuspension durch einen 50-μm-Filter in ein vorgekühltes 5-ml-FACS-Röhrchen.
- Verwenden Sie einen Durchflusszytometrie-Sortierer mit einer 100-μm-Düse. Legen Sie das FACS-Röhrchen in den Sortierer ein, und zeigen Sie eine Vorschau der Probe an.
- Richten Sie die Gating-Strategie für die Kernsortierung ein, beginnend mit einem Streugatter, indem Sie den Vorwärtsstreubereich gegen den Seitenstreubereich (FSC-A/SSC-A) aufzeichnen, gefolgt von Hoechst-Höhe versus Hoechst-Bereich (Kerngatter) und dann Hoechst-Breite versus Hoechst-Fläche (Singlet-Gatter). Visualisieren Sie das Ploidieprofil der Kerne im Hoechst-Area-Histogramm.
HINWEIS: Für eine bessere Spitzenauflösung visualisieren Sie den Hoechst-Kanal (450/50) auf der linearen Skala (Abbildung 3A). - Für die Sortierung in 96-/384-Well-Platten stellen Sie die Tröpfchenverzögerung ein und optimieren Sie die Plattenausrichtung mit der kolorimetrischen Methode mit Benzidin-Substrat-Meerrettichperoxidase (TMB-HRP), wie zuvor beschrieben6.
- Stellen Sie sowohl die Probenkühlung als auch den Plattenhalter auf 4 °C ein, wobei die Probendrehung bei 300 U/min eingeschaltet ist.
- Sortieren Sie die einzelnen Kerne bei einer Probenkonzentration von ~1 x 105 Kernen/ml und mit der Flussrate bei 200-500 Ereignissen/s.
4. Sichtprüfung und Quantifizierung von Kernparametern durch bildgebende Zytometrie (optional)
- Laden Sie ein 0,5-ml-Röhrchen mit 50 μL der Kernsuspension in einer Konzentration von 2 × 107 Kernen/ml.
- Richten Sie ein All-Events-Gate basierend auf dem Seitenverhältnis im Vergleich zum Hoechst-Fluoreszenzkanal ein, um das Ploidieprofil der Kerne zu visualisieren (Abbildung 3B).
- Erfassen Sie die Probe mit einer 40-fachen Vergrößerung mit Hellfeld (BF) und dem Hoechst-Fluoreszenzkanal.
- Inspektion und Quantifizierung von 2n- und 4n-Kernen mit der Hellfeldmessung und der Hoechst-Fluoreszenzintensität mit bildgebender Zytometrie-Software (Abbildung 3C).
5. Aufbau und Sequenzierung von Einzelkern-RNA-seq, ATAC-seq oder Multiombibliotheken
- Für tröpfchenbasierte snRNA-seq-Ansätze laden Sie die gereinigte Kernsuspension direkt in die mikrofluidische Vorrichtung für automatisierte parallele Partitionierung und molekulares Barcoding17.
- Nachdem der mikrofluidische Lauf in der Einzelzellpartitionierungsvorrichtung abgeschlossen ist, sammeln Sie die gelperlenverkapselten Kerne, inkubieren und reinigen Sie sie, wie zuvor in den Richtlinien des Herstellers17 beschrieben.
- Führen Sie 11 Polymerase-Kettenreaktionszyklen (PCR) für die cDNA-Präamplifikation mit dem folgenden Programm durch: 3 min bei 98 °C (15 s bei 98 °C, 20 s bei 63 °C und 1 min bei 72 °C) x 11, 1 min bei 72 °C und halten bei 4 °C. Fahren Sie mit einer Endreparatur und einem A-Tailing-Schritt und einer Adapterligatur fort, wie vom Hersteller angegeben17. Für den anschließenden endgültigen Aufbau der Genexpressionsbibliothek führen Sie 10 PCR-Zyklen mit dem folgenden Programm durch: 45 s bei 98 °C (20 s bei 98 °C, 30 s bei 54 °C, 20 s bei 72 °C) x 10, 1 min bei 72 °C und bei 4 °C halten.
- Sequenzieren Sie die erhaltenen Bibliotheken auf eine Lesetiefe von ~20.000-50.000 mittleren Lesevorgängen pro Kern.
- Für die gemeinsame Multiomics (RNA + ATAC) Tröpfchen-basierte Sequenzierung inkubieren Sie die Leberkerne in Lysepuffer für 5 min und markieren Sie sie dann für 1 h, wie zuvor beschrieben18.
- Laden Sie die markierten Kerne direkt in ein mikrofluidisches Gerät für eine automatisierte parallele Partitionierung und molekulares Barcoding.
- Nachdem der mikrofluidische Lauf abgeschlossen ist, sammeln Sie die gelperlenverkapselten Kerne, inkubieren und reinigen Sie, wie vom Hersteller18 beschrieben.
- Führen Sie sechs PCR-Zyklen für den cDNA-Präamplifikationsschritt mit dem folgenden Programm durch: 5 min bei 72 °C, 3 min bei 98 °C (20 s bei 98 °C, 30 s bei 63 °C, 1 min bei 72 °C) x 6, 1 min bei 72 °C und bei 4 °C halten.
- Nehmen Sie 35 μL der voramplifizierten Probe und führen Sie eine cDNA-Amplifikation wie folgt durch: 3 min bei 98 °C (15 s bei 98 °C, 20 s bei 63 °C, 1 min bei 72 °C) x 6, 1 min bei 72 °C und bei 4 °C halten. Fahren Sie mit der Endreparatur und dem A-Tailing-Schritt und der Adapterligatur fort, wie vom Hersteller angegeben18 . Für die anschließende PCR zur endgültigen Probenindizierung werden 15 PCR-Zyklen wie folgt durchgeführt: 45 s bei 98 °C (20 s bei 98 °C, 30 s bei 54 °C, 20 s bei 72 °C) x 15, 1 min bei 72 °C und bei 4 °C halten.
- Verwenden Sie für den Aufbau von ATAC-Bibliotheken 40 μL und amplifizieren Sie für sechs PCR-Zyklen für die Probenindizierung mit dem folgenden Programm: 45 s bei 98 °C (20 s bei 98 °C, 30 s bei 67 °C, 20 s bei 72 °C) x 6, 1 min bei 72 °C und bei 4 °C halten.
- Sequenzieren Sie die erhaltenen Multiom-Genexpressionsbibliotheken auf eine minimale Lesetiefe von 20.000 Lesepaaren pro Kern und die Multiom-ATAC-Bibliotheken auf eine minimale Lesetiefe von 25.000 Lesepaaren pro Zelle, wie vom Hersteller empfohlen18.
Ergebnisse
Dieser Workflow für die Einzelkernisolierung aus gefrorenen Leberproben ist auf Single-Nucleus-Multiomics zugeschnitten und beruht auf drei Hauptschritten, die wie folgt zusammengefasst werden können: i) Probenentnahme für die parallele Analyse der zellulären Heterogenität und Gewebearchitektur, ii) Einzelkern-Suspension und iii) Einzelkern-Multiomics (Abbildung 1 ). Die extrahierten Lebern werden von euthanasierten Mäusen seziert und zur histologischen Inspektion in Stücke geschnitten, um entweder Paraffin-Einbettung, Kryosektion oder beides zu erhalten. Andere geschnittene Teile werden sofort in flüssigem Stickstoff schockgefroren, um sie für die nachgeschaltete Einzelkernisolierung für Multiomics-Analysen zu isolieren. Dieses System der Gewebesammlung ermöglicht es dem Benutzer, die Einzelkern-Omics-Daten an Gewebeschnitten desselben Individuums weiter zu validieren und dabei den Datensatz bei Bedarf mit räumlicher Transkriptomik oder immunhistochemischen Analysen zu ergänzen.
Die mikroskopische Untersuchung der aus gefrorenen Lebern extrahierten Kerne mit der hier beschriebenen Methode zeigt, dass der Schritt der Dichtegradientenzentrifugation die Entfernung unerwünschter Zell- und Gewebetrümmer erheblich erleichtert (Abbildung 2C,D). Darüber hinaus bewahrt diese Methodik alle Ebenen der Ploidie, die durch zytometrische Analysen validiert und quantifiziert werden können (Abbildung 3).
Um die Leistung dieses Protokolls weiter zu validieren, führten wir tropfenbasierte snRNA-seq an unsortierten und FACS-sortierten Kernen durch und analysierten die Daten nach der Seurat-Pipeline. Kurz gesagt, die extrahierten Einzelkerne wurden für tröpfchenbasierte snRNA-seq präpariert, wie zuvor beschrieben19. Für die snRNA-seq von 2n- und 4n-Kernen oder höheren Ploidie-Niveaus wurden 11 Zyklen für den cDNA-Amplifikationsschritt und 10 Zyklen für die endgültige Konstruktion der Genexpressionsbibliothek verwendet. Die resultierenden Bibliotheken wurden auf eine Lesetiefe von ~25.000-39.000 mittleren Lesevorgängen pro Kern sequenziert. Die erhaltenen Einzelkern-Messwerte wurden dem GRCm39/mm39-Mausgenom zugeordnet. Beim Ausführen der Vorverarbeitungspipeline wurde der Befehl -- include-intron hinzugefügt, um den Einschluss und die Quantifizierung der in den Kernen vorhandenen ungespleißten Boten-RNA (mRNA) zu ermitteln. Der im Aligner integrierte EmptyDrops-Algorithmus filterte die leeren Tröpfchen heraus und entfernte sie.
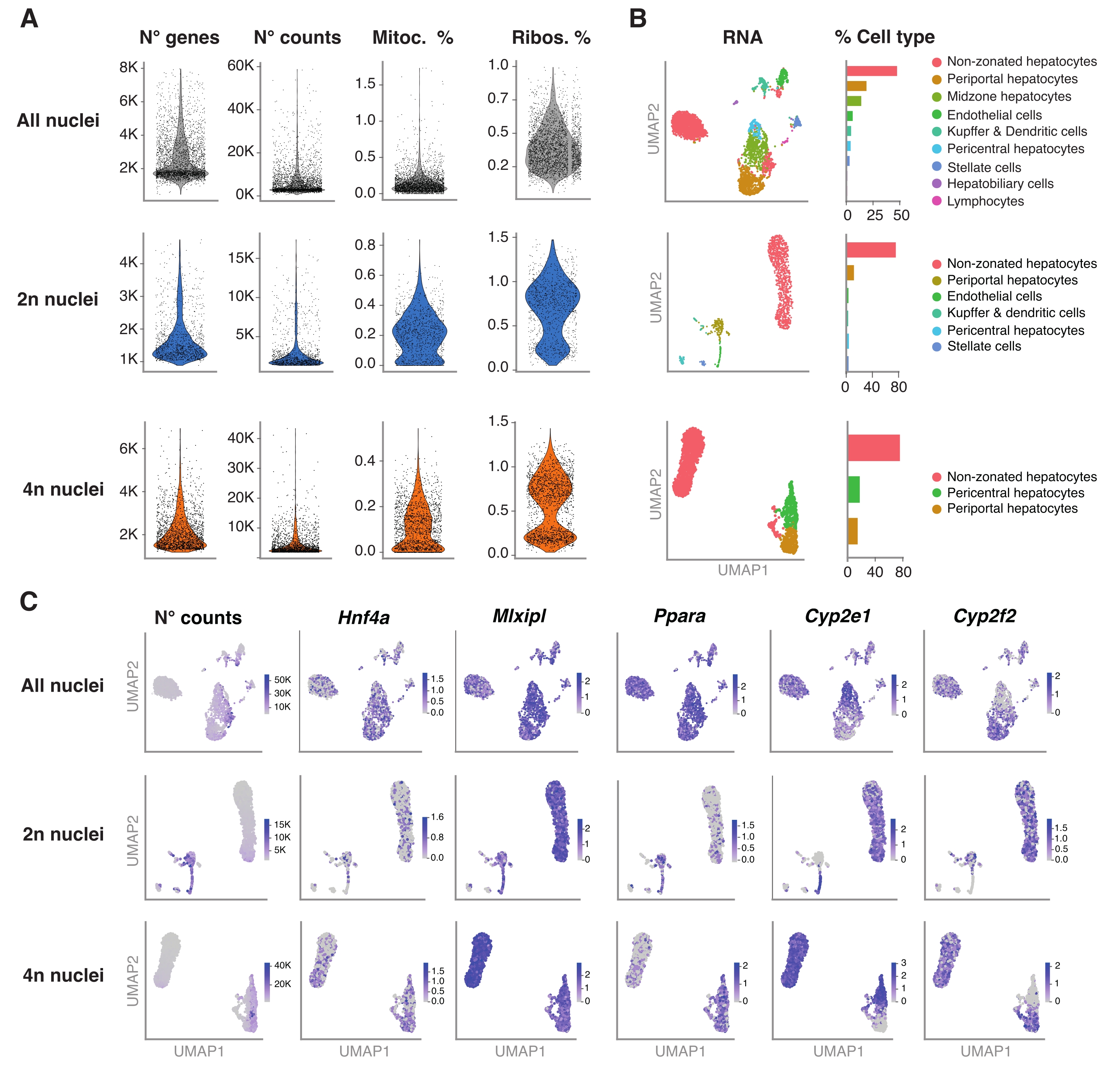
Das R-Paket Seurat (Version 4.1.1) wurde verwendet, um Qualitätskontrollmetriken (QC) unter Verwendung der Unique Molecular Identifier (UMI)-Zählmatrix zu berechnen, wie sie von der snRNA-seq-Analysepipeline ausgegeben wird. Zählungen mit weniger als 100 Merkmalen (Genen) und weniger als 10 Zellen wurden entfernt. Die Kerne wurden nach identifizierten QC-Schwellenwerten gefiltert: minimale Anzahl von Genen = 200 und maximale Anzahl von Genen = 8.000, mitochondriale Fraktion <1% und ribosomale Fraktion <2%. Die Top 3.000 hochvariablen Gene (HVGs) wurden für die Hauptkomponentenanalyse (PCA) verwendet, wie sie in Seurat implementiert wurde. Graph-basiertes Clustering wurde nach Eingabe der Top 15 PC-Dimensionen durchgeführt, die sich aus der PCA-Analyse ergaben. Um die Zellen zu clustern, haben wir die Modularitätsoptimierungstechnik (Louvain-Algorithmus) mit einem Auflösungsparameter auf 0,5 angewendet. Um diesen Datensatz zu visualisieren und zu untersuchen, haben wir eine nichtlineare dimensionale Reduktion durchgeführt, nämlich Uniform Manifold Approximation and Projection (UMAP). Die Identität jedes Clusters wurde auf der Grundlage von Vorkenntnissen der Markergene 6,20,21 zugeordnet.
Abbildung 4A zeigt qualitativ hochwertige Metriken aus den Daten, die mit dieser Methode der Kernextraktion gewonnen wurden. UMAP stellt die Anzahl der Zählungen über alle Kerne und die wichtigsten Zelltypen dar, die nur mit dem Kerntranskriptom und der relativ flachen Sequenzierung (~25.000-40.000 mittlere Lesevorgänge pro Zelle) sicher identifiziert werden können (Abbildung 4B). Dieser Ansatz ermöglicht die Untersuchung leberspezifischer Transkriptionsfaktoren wie Hnf4a, Mlxipl und Ppara sowie nachgeschalteter Zielgene, die am Metabolismus von Xenobiotika beteiligt sind (d.h. Cyp2e1 und Cyp2f2) (Abbildung 4C). Bemerkenswert ist, dass die extrahierten Kerne wichtige Informationen über die Leberzonierung enthielten, wie das komplementäre Muster von Hallmark-Genen wie dem perizentralen Cyp2e1 und dem periportalen Cyp2f2 zeigt (Abbildung 4C).
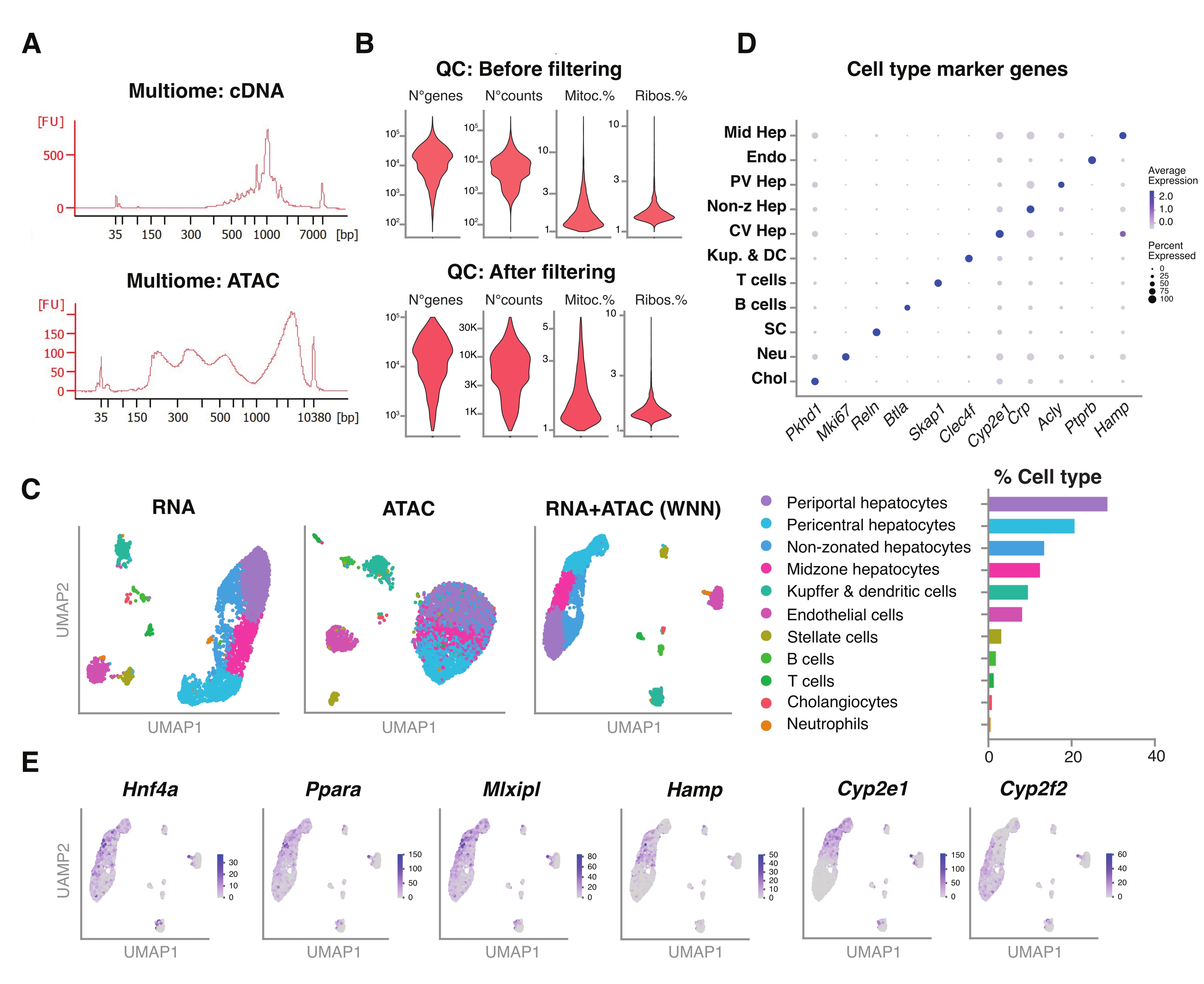
Wir untersuchten ferner die Kompatibilität der unsortierten Kerne, die mit dieser Methode extrahiert wurden, mit neueren Multiomics-Assays für die gleichzeitige Profilierung der epigenomischen Landschaft (ATAC) und der Genexpression (RNA) in denselben Einzelkernen18. Wir haben die Inkubationszeit der Kernlyse auf 5 min vor der Transposition optimiert. Die Sequenzierungsbibliotheken wurden durch die Durchführung von sechs PCR-Zyklen für die cDNA-Präamplifikation erstellt. Aus der voramplifizierten Probe wurden 35 μL entnommen und durch 15 PCR-Zyklen für die Probenindizierung weiter amplifiziert, um die Genexpressionsbibliothek aufzubauen. Ein repräsentatives Elektropherogramm der cDNA-Spur ist in Abbildung 5A (oben) dargestellt. Für den Aufbau der ATAC-Bibliotheken wurden 40 μL voramplifizierte Probe verwendet und für weitere sechs PCR-Zyklen zur Probenindizierung durchgeführt, wobei das repräsentative Elektropherogramm der DNA-Spur in Abbildung 5A (unten) gezeigt ist. Die resultierende Genexpressionsbibliothek (RNA) wurde bis zu einer Tiefe von 44.600 Lesevorgängen pro Kern und die ATAC-Bibliothek bis zu einer Tiefe von 43.500 Lesevorgängen pro Kern sequenziert (Abbildung 5B).
Ähnlich wie das oben beschriebene, tröpfchenbasierte Sequenzierungsprotokoll mit einer Modalität wurden Lesemapping, Ausrichtung, Entfernung leerer Tropfen und Fragmentzählung nach Standardrichtlinien durchgeführt, wie zuvor beschrieben, unter Verwendung des GRCm39/mm39-Referenzgenoms18. Unter Verwendung der Seurat- und Signac-Pakete22 führten wir eine "Weighted Nearest Neighbor" (WNN)-Analyse für mehrere Messungen beider Modalitäten (RNA + ATAC) durch (Abbildung 5C), die es uns ermöglichte, sowohl die Haupt- als auch die Nebenleberzelltypen ohne prominente Verzerrungen aufgrund von Zellgröße oder nuklearer Fragilität zu identifizieren und zu kommentieren (Abbildung 5D). Die Pipeline, wie vom Satija-Labor22,23 veröffentlicht, umfasst Standard-QC-Schritte - Vorverarbeitung und Dimensionsreduktion - an beiden Assays unabhängig voneinander. Um eine gute Darstellung der gewichteten Kombination der RNA-seq- und ATAC-seq-Modalitäten zu erhalten, wurde der WNN-Graph aufgetragen und für die UMAP-Visualisierung, Clustering und Annotation basierend auf zuvor identifizierten Markergenen 6,20,21 verwendet. Ähnlich wie bei den Assays mit einer einzigen Modalität entdeckten wir auch vorgeschaltete Transkriptionsregulatoren (Hnf4a, Ppara, Mlxipl) und charakteristische Gene der Leberzonierung (Hamp, Cyp2e1 und Cyp2f2) (Abbildung 5E).

Abbildung 1: Experimenteller Überblick, Workflow und genomische Einzelzellanwendungen. (A) Illustrative Darstellung der Gewebeentnahme für die Histologie (links, drei Abschnitte sind für die Paraffineinbettung und/oder Kryosektion ausgewählt), die Flash-Frozen Tissue Collection für die Einzelzellgenomik (Mitte) und repräsentative Immunhistochemie und Immunfluoreszenzanalysen (rechts); Maßstabsbalken = 100 μm. (B) Kritische Schritte für hochwertige Einzelkernsuspensionen. (C) Die Kernsuspensionen können auf einen 10-fachen Chromchip geladen oder für FACS-Sortierung und plattenbasierte Ansätze verwendet werden. Abkürzungen: H&E = Hämatoxylin und Eosin; DAPI = 4',6-Diamidino-2-phenylindol; FACS = fluoreszenzaktivierte Zellsortierung. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 2: Leberdissektion und Dounce-Glasgewebehomogenisierung. (A) Repräsentative Mausleber von einer 3 Monate alten C57BL6/J-Maus (links); Der Leberabschnitt, der für die Einzelkernisolierungen vor (Mitte) und nach dem Zerkleinern des Gewebes mit dem Skalpell (rechts) verwendet wird. Maßstabsbalken = 1 cm. (B) Illustrative Bilder der 2 mL Dounce-Glashomogenisierung vor Schlägen mit "lockerem" Stößel A (links) und nach "straffem" Stößel B (rechts). Überwachung der Gewebehomogenisierung mit einem Hämozytometer (C) vor der Gradientenzentrifugation und (D) nach der Gradientenzentrifugation. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 3: Fluoreszenzaktivierte Zellsortierung und bildgebende Durchflusszytometrie zur Charakterisierung von Hochdurchsatzkernen . (A) Gating-Strategie für die Einkern-FACS-Sortierung in eine Platte zur Abfrage verschiedener Ploidiegrade. Streugatter basierend auf dem vorderen Streubereich im Vergleich zum seitlichen Streubereich, der so eingestellt ist, dass Trümmer ausgeschlossen werden; Kerngatter basierend auf Hoechst-Höhe versus Hoechst-Area mit mehreren Kernpopulationen; Singlets-Gate basierend auf Hoechst-Width versus Hoechst-A-Set für Dublettendiskriminierung; Das Hoechst-A-Histogramm ermöglicht die Visualisierung des Kernploidieprofils. (B) Repräsentative bildgebende zytometrische Quantifizierung aller Ereignisse (links) und einzelner (rechts) Ereignisse mit diploiden und tetraploiden Kernen. (C) Hellfeld- und Hoechst-Bilder von 2n- und 4n-Kernen und deren Quantifizierung mittels bildgebender Zytometrie. Abkürzungen: FACS = fluoreszenzaktivierte Zellsortierung; FSC-A = Vorwärtsstreufläche; SSC-A = Seitenstreufläche; Hoechst-H = Hoechst-Höhe; Hoechst-A = Hoechst-Area; Hoechst-W = Hoechst-Breite. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 4: Tiefencharakterisierung der Hepatozytenploidie mit snRNA-seq . (A) Geigendiagramme, die die Anzahl der Gene, die Anzahl, den Prozentsatz der mitochondrialen Gene und den Prozentsatz der nachgewiesenen ribosomalen Gene zeigen. (B) UMAP, das die mit snRNA-seq nachgewiesenen Zelltypen demonstriert (links), wobei die Quantifizierung in Prozent der Zellkerne ausgedrückt wird (rechts). (C) UMAP veranschaulicht die Anzahl der Zählungen und zeigt die Expression von Hepatozyten-spezifischen Genen an. Alle Kerne (obere Reihe), 2n Kerne (mittlere Reihe) und 4n Kerne (untere Reihe). Abkürzungen: snRNA-seq = Single-nucleus RNA-seq; UMAP = Uniform Manifold Approximation and Projection; Mitoc. = mitochondriale Gene; Ribos. = ribosomale Gene. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 5: Qualitätskontrolle und Analyse von Multiomics (gemeinsame RNA-seq und ATAC-seq) aus gefrorenen, archivierten jungen Lebern . (A) Repräsentative automatische elektrophoretische Spuren, die nach der Multiom-Pipeline erhalten wurden und das Molekulargewichtsprodukt nach cDNA-Synthese und ATAC zeigen. (B) Geigendiagramm, das die Anzahl der Zählungen für ATAC-seq, RNA-seq, den Prozentsatz der mitochondrialen Gene und den Prozentsatz der ribosomalen Gene vor und nach der Filterung zeigt. (C) UMAP zeigt die Genexpression aus RNA-seq (links), ATAC-seq (Mitte) und gemeinsamen Modalitäten RNA-seq und ATAC-seq (rechts). (D) Verschiedene Zelltypen sind in verschiedenen Farben annotiert, mit der Expression von indizierten Hepatozyten-spezifischen Genen. (E) Merkmalsdiagramm, das die clusterspezifische Expression der angegebenen Gene in den angegebenen Zelltypen zeigt. Abkürzungen: ATAC-seq = Assay for transposase-accessible chromatin with high-throughput sequencing; Mid Hep = Mid-zonale Hepatozyten; Endo = Endothelzellen; PV Hep = periportale Hepatozyten; Non-z Hep = nicht zonierte Hepatozyten; CV Hep = perizentrale Hepatozyten; Kup & DC = Kupffer & dendritische Zellen; SC = Sternzellen, Neu = Neutrophile; Chol = Cholangiozyten; Mitoc. = mitochondriale Gene; Ribos. = ribosomale Gene. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
| Reagenz | Lager | 10 ml | 15 ml | 50 ml | ||
| (a ) Iodixanol Medium (IDM) | ||||||
| 250 mM Saccharose | 1M | 12,5 ml | ||||
| 150 mM KCl | 2M | 3,75 ml | ||||
| 30 mM MgCl2 | 1M | 1,5 ml | ||||
| 60 mM Tris Puffer pH 8,0 | 1M | 3 ml | ||||
| Reinstes RNase-freies Wasser | 29,25 ml | |||||
| b) 50 % IDM | ||||||
| Joodixanol | 60% | 12,5 ml | ||||
| IDM | 2,5 ml | |||||
| (C) 29% IDM | ||||||
| Joodixanol | 60% | 7,25 ml | ||||
| IDM | 7,75 ml | |||||
| (D ) Nuclei Isolation Medium-1 (NIM-1) | ||||||
| 250 mM Saccharose | 1M | 12,5 ml | ||||
| 25 mM KCl | 2M | 0,625 ml | ||||
| 5 mM MgCl2 | 1M | 0,25 ml | ||||
| 10 mM Tris Puffer pH 8,0 | 1M | 0,5 ml | ||||
| Reinstes Rnase-freies Wasser | 36,125 ml | |||||
| (E ) Nuclei Isolation Medium-2 (NIM-2) | ||||||
| NIM-1-Puffer | 9,99 ml | |||||
| Dithiothreitol (DTT) | 1 mM | 0,01 ml | ||||
| Protease-Inhibitor-Tablette (EDTA-frei) | 1 | 1 Tablette | ||||
| (f ) Homogenisierungspuffer (HB) | ||||||
| NIM-2-Puffer | 9.697 ml | |||||
| Rekombinanter RNase-Inhibitor | 40 U/μL | 0,1 ml | ||||
| Proteinbasierter RNAse Inhibitor (SUPERase•IN) | 20 U/μL | 0,1 ml | ||||
| 0,1% Triton-X | 10% | 0,1 ml | ||||
| 3 μg/ml Hoechst 33342 | 10 mg/ml | 0,003 ml | ||||
| g ) Kernspeicherpuffer (NSB) | ||||||
| 166,5 mM Saccharose | 1M | 1.665 ml | ||||
| 5 mM MgCl2 | 1M | 0,05 ml | ||||
| 10 mM Tris pH 8,0 | 1M | 0,1 ml | ||||
| Rekombinanter RNase-Inhibitor | 40 U/μL | 0,1 ml | ||||
| Proteinbasierter RNase-Inhibitor (SUPERase•IN) * | 20 U/μL | 0,1 ml | ||||
| Reinstes RNase-freies Wasser | 8.085 mL | |||||
| * Optional (nur für FACS-Sortierung) | ||||||
Tabelle 1: Lösungsrezepte. (A) Herstellung von Jodixanolmedium (IDM); (B) 50%ige Verdünnung von Jodixanollösung; (C) 29%ige Verdünnung der Jodixanollösung. (D) Herstellung von Kernisolierungsmedium-1 (NIM-1). (E) Herstellung von Kernisolierungsmedium-2 (NIM-2). (F) Herstellung eines Homogenisierungspuffers (HB). (G) Herstellung von Kernspeicherpuffer (NSB).
Diskussion
Die Analyse der zellulären Zusammensetzung der Leber durch Einzelzell- oder Einzelkern-RNA-seq liefert ein tieferes Verständnis der Entwicklung und Progression von Lebererkrankungen 3,4,5,24. Die Einzelzellisolierung aus Lebern ist zeitaufwendig und erfordert Protokolle, die eine harte mechanische oder enzymatische Dissoziation beinhalten25,26,27. Es ist allgemein anerkannt, dass jedes Gewebe eine systematische Bewertung erfordert, um das optimale Gewebedissoziationsprotokoll zu bestimmen, sowie eine geeignete Speichermethode zur Erfassung fragiler Zelltypen oder Kerne28. Abhängig von der Gewebeverfügbarkeit, der interessierenden Erkrankung, dem Entwicklungsstadium oder dem Modellorganismus könnte die Herstellung einer Einzelkernsuspension für die nachgeschaltete Verarbeitung eine geeignetere Methode sein als die Verwendung von Einzelzellsuspensionen. Wichtig ist, dass in der Leber scRNA-seq und snRNA-seq eine hohe Korrelation zwischen nukleärer und zytoplasmatischer mRNA gezeigt haben, was darauf hindeutet, dass beide Ansätze komplementäreInformationen 2,3,4,6,29 enthalten.
Dieses Papier bietet eine standardisierte, robuste und reproduzierbare Einzelkernisolierung aus gefrorenen, archivierten Leberproben von Mäusen und anderen Arten, einschließlich Menschen und Makaken. Diese Methode kann für Wildtyp-Mäuse verwendet werden, die mit Chow und einer fettreichen Diät (HFD) gefüttert werden, und für Mausmodelle der Leberfibrose, die sowohl gut plattenbasierte als auch tröpfchenbasierte Einkern-genomische Ansätze verwenden6. Diese Methode basiert auf dem Protokoll, das ursprünglich von Krishnaswami et al.30 für Hirngewebe beschrieben wurde, mit zusätzlichen Modifikationen, die auf schockgefrorene Leber zugeschnitten sind. Eine optimale Homogenisierung befreit die meisten Kerne aus dem Gewebe, ohne die Integrität der Kernmembran negativ zu beeinflussen. Eine Überdosierung kann jedoch die zerbrechlichen Kerne schädigen und ihre Gesamtqualität verringern. Junge und/oder fettige Lebern benötigen normalerweise nur 5 Schläge mit Stößel A und 10 Schläge mit Stößel B, während alte und/oder fibrotische Lebern 15 Schläge mit Stößel B benötigen, aber nicht mehr. Es wird daher nicht empfohlen, mehr Schläge als die hier angegebenen Zahlen auszuführen. Eine Überdosierung kann die Qualität der Einzelkernsuspension negativ beeinflussen und die Menge an Umgebungs-RNA erhöhen. In der Folge kann dies dazu führen, dass während der nachgelagerten Datenanalysen zusätzliche computergestützte Filterschritte durchgeführt werden müssen.
Das hier vorgestellte Protokoll ist vielseitig und kann an unterschiedliche Leberzustände bei jungen (3 Monate) und alten (24 Monate) Mäusen angepasst werden. Da wir festgestellt haben, dass ein größerer Abschnitt der Leber für altes, HFD- und fibrotisches Gewebe notwendig ist, kann die Größe des für die Verarbeitung verfügbaren Gewebes für einige Benutzer mit kleineren Mengen an biologischem Ausgangsmaterial eine Einschränkung darstellen. Die Gradientenreinigung wird jedoch dringend für die sofortige Probenverarbeitung mit tröpfchenbasierten genomischen Assays empfohlen. Wenn die Kerne für gut basierte Assays in 96-/384-Well-Platten sortiert werden müssen, kann auf eine Gradientenreinigung verzichtet werden. Wir empfehlen den Benutzern, die Gradientenreinigung durchzuführen, wenn genügend Gewebeproben vorhanden sind, um die empfohlene Kernkonzentration für die FACS-Sortierung zu erhalten (dh ~ 1 × 105 Kerne / ml).
Die Leber ist durch die polyploide Natur der Hepatozytengekennzeichnet 9, aber die Rolle der Hepatozytenploidie in der normalen Physiologie und Krankheit ist noch nicht klar. Es gibt eine wachsende Zahl von Beweisen, die darauf hindeuten, dass Ploidie genomische Variabilität bietet31, und es ist bekannt, dass die Ploidie mitdem Alter von 32,33 Jahren zunimmt. Die Anreicherung von einkernigen tetraploiden Hepatozyten ist jedoch auch klinisch mit einer schlechten Prognose beim humanen hepatozellulären Karzinom (HCC) verbunden34. In ähnlicher Weise sind Veränderungen der Hepatozytenploidie mit altersbedingten chronischen Lebererkrankungen wie der nichtalkoholischen Fettlebererkrankung (NAFLD) verbunden35,36,37. Ploidie ist die Bedingung, mehr als zwei Kopien des Genoms zu besitzen, die durch Färbung des Genominhalts mit einem DNA-Farbstoff wie Hoechst38 erforscht werden können. Hoechst-Farbstoff, der dem HB vor der Extraktion zugesetzt wird, markiert alle Kerne während des Isolierungsprotokolls. Dies ermöglicht die Unterscheidung zwischen diploiden und polyploiden Kernen anhand ihres DNA-Gehalts, wenn sie von einem UV- (350 nm) oder violetten (450 nm) Laser auf einem Durchflusszytometriegerät angeregt werden. Mit der vorgestellten Gating-Strategie können 2n, 4n, 8n und höhere Hepatozytenploidie in gefrorenen, archivierten Lebern untersucht werden, um die Rolle der zellulären Heterogenität in der Gewebefunktion besser zu verstehen1 (Abbildung 3A). Darüber hinaus kann die Kernmorphologie, einschließlich der Größe und des Volumens, mittels bildgebender Durchflusszytometrie quantifiziert werden, um Veränderungen der Kerngröße mit Änderungen der Gesamtzahl der Zählungen oder der Anzahl der Gene in Abhängigkeit vom Ploidieniveau zu korrelieren (Abbildung 3B, C).
Die multimodale Omics-Messung bietet die Möglichkeit, mehrere Schichten genomischer Organisation gleichzeitig zu untersuchen. Der gemeinsame RNA + ATAC-Multiomics-Ansatz ermöglicht die Untersuchung von vorgeschalteten Regulatoren und nachgeschalteten metabolischen Genen und bietet einen umfassenden Ansatz zur Untersuchung von Transkriptionsnetzwerken und der Chromatinarchitektur, die mit der Leberfunktion bei Einzelzellauflösung verbunden ist. Darüber hinaus leistet Single-Cell-Multiomics mit den Fortschritten bei Berechnungsmethoden, die Datenspärlichkeit und die Reduzierung der Sequenzierungskosten berücksichtigen können, Pionierarbeit bei der Bewertung mehrerer Modalitäten aus derselben Zelle. Dieses Einzelkern-Isolationsprotokoll ist kompatibel mit der individuellen und gemeinsamen Bewertung von Expressions- und Chromatindatensätzen. Wir haben Standardpipelines verwendet, die von Stuart et al.23 (Signac-Paket) festgelegt wurden, um die Qualität der Daten zu veranschaulichen, während mehrere verfügbare und alternative Berechnungsmethoden leicht für nachgelagerte Analysen übernommen werden können23,39,40,41.
Insgesamt ermöglicht Single-Nucleus Multiomics die Untersuchung von bioarchiviertem, FF-Maus-, menschlichem und nicht-menschlichem Primatenlebergewebe unter Verwendung einer sehr kleinen Menge an Ausgangsprobenmaterial durch Implementierung des hier vorgestellten Kernextraktionsprotokolls. Dieses unschätzbare Werkzeug wird Leberbiologen in die Lage versetzen, sowohl die Genexpression als auch die Chromatinzugänglichkeit im Zusammenhang mit verschiedenen Leberpathologien zu hinterfragen. Darüber hinaus könnten verschiedene Ebenen der Hepatozytenploidie und die daraus resultierende Anpassung der Genexpression in Abhängigkeit von ihrer Position im Leberläppchen ihre Rolle bei Leberpathologien aufzeigen. Daher erwarten wir, dass die Untersuchung der zellulären Heterogenität neue Möglichkeiten für die Entwicklung der Präzisionsmedizin und gezielte Interventionen gegen Krankheiten wie HCC und NAFLD bieten wird.
Offenlegungen
Die Autoren erklären, dass sie keine Interessenkonflikte haben.
Danksagungen
Unterstützt wurde diese Forschung vom Helmholtz Pioneer Campus (M.S., K.Y., C.P.M.-J.) und dem Institut für Computational Biology (C. T.-L.). Diese Forschung wurde auch von AMED unter der Fördernummer JP20jm0610035 (C.P.M.-J.) unterstützt. Wir danken der Unterstützung von Core Genomics am HMGU (I. de la Rosa) und Bioinformatik (T. Walzthoeni), insbesondere Xavier Pastor für die Schulung und Anleitung. Wir danken A. Feuchtinger, U. Buchholz, J. Bushe und allen anderen Mitarbeitern der HMGU Pathology and Tissue Analytic Core Facility für ihre technische und wissenschaftliche Unterstützung sowie J. Zorn, R. Erdelen, D. Würzinger, Mitarbeitern von E-Streifen, sowie der Core Facility Animal Services für ihre kontinuierliche wissenschaftliche Unterstützung und Diskussion. Wir danken der Core Facility Cell Analysis bei TranslaTUM (R. Mishra) und Luminex, A DiaSorin Company (P. Rein). Wir danken Dr. I Deligiannis für seine technische Unterstützung. Dr. M. Hartman, Dr. A. Schröder und Frau A. Barden (Helmholtz Pioneer Campus) waren grundlegend für ihre rechtliche, betriebswirtschaftliche und administrative Unterstützung.
Materialien
| Name | Company | Catalog Number | Comments |
| 10% Tween 20 - 5 mL | Bio-Rad | 1662404 | |
| 2100 Bioanalyzer Instrument | Agilent | G2939BA | |
| Adhesive PCR film | Thermo Fisher Scientific | AB0558 | |
| Bioanalyzer High Sensitivity DNA Analysis | Agilent | 5067-4626 | |
| C1000 Touch Thermal Cycler | Bio-Rad | 1851196 | |
| Cell Sorter | For fluoresence-activated cell sorting (FACS); e.g. BD FACSAria Cell Sorter. | ||
| Centrifuge 5430R | Eppendorf | 5428000619 | Use chilled at 4 °C |
| Chromium Controller & Next GEM Accessory Kit | 10X Genomics | 1000204 | |
| Chromium Next GEM Chip G Single Cell Kit | 10X Genomics | 1000127 | |
| Chromium Next GEM Chip J Single cell kit, 16 reactions | 10X Genomics | 1000230 | |
| Chromium Next GEM Single Cell 3' Kit v3.1, 4 reactions | 10X Genomics | 1000269 | |
| Chromium Next GEM Single Cell Multiome ATAC + Gene Expression Reagent Bundle, 4 reactions | 10X Genomics | 1000285 | |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Sigma Aldrich | 11873580001 | |
| Dithiothreitol (DTT) 1 M solution | Sigma Aldrich | 646563 | Make 1 mM stock solution and use at 1 µM final concentration. |
| DNA AWAY Surface Decontaminant | Thermo Fisher Scientific | 10223471 | Wipe surfaces and pipettes before start of experiment |
| DNA LoBind Tubes, 1.5 mL | Thermo Fisher Scientific | 16628742 | |
| DNA LoBind Tubes, 2.0 mL | Thermo Fisher Scientific | 16638742 | |
| Elution Buffer (EB) - 250 mL | Qiagen | 19086 | |
| Eppendorf ThermoMixer C | Thermo Fisher Scientific | 13527550 | |
| Eppendorf ThermoMixer C Accessory: Smartblock | Thermo Fisher Scientific | 13518470 | |
| Ethanol, Absolute (200 Proof), Molecular Biology Grade - 100 mL | Thermo Fisher Scientific | 10517694 | |
| Filters 50 µm, sterile | SYSMEX PARTEC - CELLTRICS | 04-004-2327 | Adjust filter diameter according with tissue and nuclei size |
| Glycerin (Glycerol), 50% (v/v) - 1 L | Ricca Chemical Company | 3290-32 | |
| Hard-Shell, 384-Well PCR Plates, thin wall, skirted, clear/white | Bio-Rad | HSP3905 | |
| Herenz Heinz ABS Forceps | Thermo Fisher Scientific | 1131884 | |
| Hoechst 33342, Trihydrochloride, Trihydrate, 10 mg/mL Solution in Water | Invitrogen | H3570 | Light-sensitive |
| Imaging Flow Cytometer | For imaging flow cytometry analysis, e.g. Luminex Amnis ImageStream | ||
| Invitrogen TE Buffer - 100 mL | Thermo Fisher Scientific | 11568846 | |
| KCl (2 M), RNase-free, 100 mL | Thermo Fisher Scientific | AM9640G | |
| MgCl2 (1 M), 100 mL | Thermo Fisher Scientific | AM9530G | |
| MicroAmp 8-Cap Strip, clear-300 strips | Thermo Fisher Scientific | 10209104 | |
| MicroAmp 8-Tube Strip, 0.2 mL-125 strips | Thermo Fisher Scientific | 10733087 | |
| Mörser 2 mL DOUNCE | Wagner & Munz GmbH | 9651632 | RNase zap and rinse with MillQ before use |
| MyFuge 12 Mini MicroCentrifuge C1012 | Benchmark Scientific | C1012 | Or any other strip and tube mini centrifuge |
| Neubauer Hemocytometer | OMNILAB LABORZENTRUM | 5435293 | Visualize and count nuclei under microscope |
| Nuclease-Free Water (not DEPC-Treated) | Thermo Fisher Scientific | AM9937 | |
| OptiPrep Density Gradient Medium | Sigma Aldrich | D1556 | |
| Pipette tips RT LTS 1000 µL, Wide-O | Mettler Toledo | 30389218 | |
| Pistill "A" 2 mL | Wagner & Munz GmbH | 9651621 | RNase zap and rinse with MillQ before use |
| Pistill "B" 2 mL | Wagner & Munz GmbH | 9651627 | RNase zap and rinse with MillQ before use |
| Polypropylene 15 mL Centrifuge Tube | Thermo Fisher Scientific | 10579691 | |
| Polystyrene Petri dish, 60 mm x 15 mm | Thermo Fisher Scientific | 10634141 | |
| Polystyrene Round-Bottom 5 mL FACS Tubes | Thermo Fisher Scientific | 10100151 | |
| Protector RNase inhibitor - 2,000 U | Sigma Aldrich | 3335399001 | Keep in -20 °C until use |
| Protein-based RNase Inhibitor SUPERase•In (20 U/μL) | Thermo Fisher Scientific | AM2696 | Keep in -20 °C until use |
| Recombinant RNase Inhibitor | Clontech Takara | 2313B | Keep in -20 °C until use |
| RNAse free microfuge tubes - 0.5 mL | Thermo Fisher Scientific | AM12450 | |
| RNaseZap RNase Decontamination Solution | Thermo Fisher Scientific | AM9780 | Wipe surfaces and pipettes before start of experiment |
| SPRIselect - 60 mL | Beckman Coulter | B23318 | Aliquot and store in 4 °C |
| Sucrose, 500 g | Sigma Aldrich | S0389-500G | Make a 1 M stock solution |
| Swann-Morton Sterile Disposable Stainless Steel Scalpels | Thermo Fisher Scientific | 11798343 | |
| Tris-HCI (1M), pH 8.0 | Invitrogen | 15568025 | |
| Triton X-100, 98%, 100 mL | Thermo Fisher Scientific | 10671652 | Make 10% stock solution. Keep at 4 °C and protect from light. |
| Trypan Blue Solution, 0.4% | Thermo Fisher Scientific | 11538886 | |
| Vortex- Mixer | VWR | 444-1372 | Or any other type of vortex |
Referenzen
- Kamies, R., Martinez-Jimenez, C. P. Advances of single-cell genomics and epigenomics in human disease: where are we now. Mamm Genome. 31 (5-6), 170-180 (2020).
- Ramachandran, P., Matchett, K. P., Dobie, R., Wilson-Kanamori, J. R., Henderson, N. C. Single-cell technologies in hepatology: New insights into liver biology and disease pathogenesis. Nature Reviews Gastroenterology & Hepatology. 17 (8), 457-472 (2020).
- Ramachandran, P., et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. 575 (7783), 512-518 (2019).
- Andrews, T. S., et al. Single-cell, single-nucleus, and spatial RNA sequencing of the human liver identifies cholangiocyte and mesenchymal heterogeneity. Hepatology Communications. 6 (4), 821-840 (2022).
- Xiong, X., et al. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Molecular Cell. 75 (3), 644-660 (2019).
- Richter, M. L., et al. Single-nucleus RNA-seq2 reveals functional crosstalk between liver zonation and ploidy. Nature Communications. 12 (1), 4264 (2021).
- Matsumoto, T., Wakefield, L., Tarlow, B. D., Grompe, M. In vivo lineage tracing of polyploid hepatocytes reveals extensive proliferation during liver regeneration. Cell Stem Cell. 26 (1), 34-47 (2020).
- Chen, F., et al. Broad distribution of hepatocyte proliferation in liver homeostasis and regeneration. Cell Stem Cell. 26 (1), 27-33 (2020).
- Donne, R., Saroul-Ainama, M., Cordier, P., Celton-Morizur, S., Desdouets, C. Polyploidy in liver development, homeostasis and disease. Nature Reviews. Gastroenterology & Hepatology. 17 (7), 391-405 (2020).
- Lengefeld, J., et al. Cell size is a determinant of stem cell potential during aging. Science Advances. 7 (46), 0271 (2021).
- Lanz, M. C., et al. Increasing cell size remodels the proteome and promotes senescence. Mol Cell. 82 (17), 3255-3269 (2022).
- Kim, J. Y., et al. PIDDosome-SCAP crosstalk controls high-fructose-diet-dependent transition from simple steatosis to steatohepatitis. Cell Metabolism. 34 (10), 1548-1560 (2022).
- Padovan-Merhar, O., et al. Single mammalian cells compensate for differences in cellular volume and DNA copy number through independent global transcriptional mechanisms. Molecular Cell. 58 (2), 339-352 (2015).
- Miettinen, T. P., et al. Identification of transcriptional and metabolic programs related to mammalian cell size. Current Biology. 24 (6), 598-608 (2014).
- Vargas-Garcia, C. A., Ghusinga, K. R., Singh, A. Cell size control and gene expression homeostasis in single-cells. Current Opinion in Systems Biology. 8, 109-116 (2018).
- Knoblaugh, S. E., Randolph-Habecker, J. . Necropsy and histology. In Comparative Anatomy and Histology: A Mouse, Rat, and Human Atlas (Second Edition). , (2018).
- Chromium Next GEM Single Cell 3 Reagent Kits v3.1 User Guide. Document number CG000204 (Rev D). 10x Genomics Available from: https://www.10xgenomics.com/support/single-cell-gene-expression/documentation/steps/library-prep/chromium-single-cell-3-reagent-kits-user-guide-v-3-1-chemistry (2019)
- Chromium Next GEM Single Cell Multiome ATAC + Gene Expression User Guide. Document number CG000338 (Rev D). 10x Genomics Available from: https://www.10xgenomics.com/support/single-cell-multiome-atac-plus-gene-expression/documentation/steps/library-prep/chromium-next-gem-single-cell-multiome-atac-plus-gene-expression-reagent-kits-user-guide (2021)
- MacParland, S. A., et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nature Communications. 9 (1), 4383 (2018).
- Martinez-Jimenez, C. P., Kyrmizi, I., Cardot, P., Gonzalez, F. J., Talianidis, I. Hepatocyte nuclear factor 4alpha coordinates a transcription factor network regulating hepatic fatty acid metabolism. Molecular and Cell Biology. 30 (3), 565-577 (2010).
- Schmidt, D., et al. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science. 328 (5981), 1036-1040 (2010).
- Hao, Y., et al. Integrated analysis of multimodal single-cell data. Cell. 184 (13), 3573-3587 (2021).
- Stuart, T., Srivastava, A., Madad, S., Lareau, C. A., Satija, R. Single-cell chromatin state analysis with Signac. Nature Methods. 18 (11), 1333-1341 (2021).
- Sampaziotis, F., et al. Cholangiocyte organoids can repair bile ducts after transplantation in the human liver. Science. 371 (6531), 839-846 (2021).
- Gomez-Lechon, M. J., Donato, M. T., Castell, J. V., Jover, R. Human hepatocytes as a tool for studying toxicity and drug metabolism. Current Drug Metabolism. 4 (4), 292-312 (2003).
- Gomez-Lechon, M. J., Donato, M. T., Castell, J. V., Jover, R. Human hepatocytes in primary culture: The choice to investigate drug metabolism in man. Current Drug Metabolism. 5 (5), 443-462 (2004).
- vanden Brink, S. C., et al. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nature Methods. 14 (10), 935-936 (2017).
- Denisenko, E., et al. Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biology. 21 (1), 130 (2020).
- Nault, R., Fader, K. A., Bhattacharya, S., Zacharewski, T. R. Single-nuclei RNA sequencing assessment of the hepatic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Cellular and Molecular Gastroenterology and Hepatology. 11 (1), 147-159 (2021).
- Krishnaswami, S. R., et al. Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons. Nature Protocols. 11 (3), 499-524 (2016).
- Duncan, A. W., et al. Aneuploidy as a mechanism for stress-induced liver adaptation. Journal of Clinical Investigation. 122 (9), 3307-3315 (2012).
- Kreutz, C., et al. Hepatocyte ploidy is a diversity factor for liver homeostasis. Frontiers in Physiology. 8, 862 (2017).
- Hunt, N. J., Kang, S. W. S., Lockwood, G. P., Le Couteur, D. G., Cogger, V. C. Hallmarks of aging in the liver. Computational and Structural Biotechnology Journal. 17, 1151-1161 (2019).
- Bou-Nader, M., et al. Polyploidy spectrum: a new marker in HCC classification. Gut. 69 (2), 355-364 (2019).
- Gentric, G., et al. Oxidative stress promotes pathologic polyploidization in nonalcoholic fatty liver disease. Journal of Clinical Investigation. 125 (3), 981-992 (2015).
- Schwartz-Arad, D., Zajicek, G., Bartfeld, E. The streaming liver IV: DNA content of the hepatocyte increases with its age. Liver. 9 (2), 93-99 (1989).
- Kudryavtsev, B. N., Kudryavtseva, M. V., Sakuta, G. A., Stein, G. I. Human hepatocyte polyploidization kinetics in the course of life cycle. Virchows Archiv. B, Cell Pathology Including Molecular Pathology. 64 (6), 387-393 (1993).
- Duncan, A. W., et al. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature. 467 (7316), 707-710 (2010).
- Granja, J. M., et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nature Genetics. 53 (3), 403-411 (2021).
- Bredikhin, D., Kats, I., Stegle, O. MUON: Multimodal omics analysis framework. Genome Biology. 23 (1), 42 (2022).
- Velten, B., et al. Identifying temporal and spatial patterns of variation from multimodal data using MEFISTO. Nature Methods. 19 (2), 179-186 (2022).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten