
Method Article
Isolamento di nuclei da tessuto epatico congelato per multiomiche monocellulari
In questo articolo
Riepilogo
Qui, presentiamo un protocollo per isolare i nuclei da tessuti epatici congelati e archiviati per RNA-seq a nucleo singolo, ATAC-seq e multiomici articolari (RNA-seq e ATAC-seq).
Abstract
Il fegato è un tessuto complesso ed eterogeneo responsabile dello svolgimento di molte funzioni fisiologiche critiche, come il mantenimento dell'omeostasi energetica e il metabolismo degli xenobiotici, tra gli altri. Questi compiti vengono eseguiti attraverso uno stretto coordinamento tra cellule parenchimali epatiche e non parenchimali. Inoltre, varie attività metaboliche sono limitate ad aree specifiche del lobulo epatico, un fenomeno chiamato zonazione epatica. I recenti progressi nelle tecnologie di sequenziamento a singola cellula hanno permesso ai ricercatori di studiare l'eterogeneità dei tessuti a una risoluzione a singola cellula. In molti tessuti complessi, incluso il fegato, duri protocolli di dissociazione enzimatica e / o meccanica possono influenzare negativamente la vitalità o la qualità delle sospensioni unicellulari necessarie per caratterizzare in modo completo questo organo in salute e malattia.
Questo documento descrive un protocollo robusto e riproducibile per isolare i nuclei da tessuti epatici congelati e archiviati. Questo metodo produce nuclei di alta qualità compatibili con gli approcci omici a valle a singola cellula, tra cui RNA-seq a singolo nucleo, test per cromatina accessibile alla trasposasi con sequenziamento ad alta produttività (ATAC-seq), nonché omiche multimodali (RNA-seq congiunto e ATAC-seq). Questo metodo è stato utilizzato con successo per l'isolamento di nuclei da campioni di fegato congelato di primati umani sani e malati. Questo approccio consente l'isolamento imparziale di tutti i principali tipi di cellule nel fegato e, quindi, offre una solida metodologia per studiare il fegato alla risoluzione di una singola cellula.
Introduzione
La genomica monocellulare sta rapidamente diventando una metodologia essenziale per studiare la funzionalità epatica e valutare l'impatto dell'eterogeneità cellulare nelle condizioni di salute e malattia1. Il rapido sviluppo della "multiomica" per la misurazione simultanea di diversi strati di informazioni e l'espansione parallela di robuste pipeline computazionali sta aprendo la strada alla scoperta di tipi e sottotipi di cellule precedentemente sconosciuti nel fegato normale e malato2.
La possibilità di esplorare biobanche e campioni congelati archiviati ha aumentato significativamente le opportunità di rivisitare e scoprire il ruolo delle cellule non parenchimali 3,4,5 e indagare il ruolo degli epatociti poliploidi durante l'invecchiamento e nelle malattie croniche 6,7,8,9 . Pertanto, questo articolo descrive un protocollo di isolamento a singolo nucleo robusto e riproducibile per fegati archiviati congelati (FF) che è compatibile con il sequenziamento dell'RNA a singolo nucleo a valle e il sequenziamento ATAC, nonché con omiche multimodali (RNA-seq congiunto e ATAC-seq) (Figura 1).
Questo flusso di lavoro consente lo studio del trascrittoma e dell'accessibilità della cromatina di tutti i tipi di cellule nel fegato, indipendentemente dalle dimensioni o dalla fragilità della cellula, nei protocolli di dissociazione enzimatica. Può essere eseguita con piccole sezioni di tessuto (15-30 mg o 5-10 mm3) da preziosi campioni umani o topi transgenici. La determinazione dell'elevata purezza dell'isolamento dei nuclei include la quantificazione e la misurazione della dimensione nucleare, che potrebbe essere correlata con l'aumento delle dimensioni cellulari e della senescenza 10,11, e questa purezza è rilevante per l'analisi sia della ploidia degli epatociti 12 che dei meccanismi trascrizionali dipendenti dalla dimensione cellulare11,13,14,15 . Inoltre, i nuclei isolati dai fegati congelati conservano preziose informazioni sulla zonazione del fegato. Il flusso di lavoro e la raccolta dei tessuti consentono la convalida di dati genomici a singola cellula o ulteriori analisi complementari, come l'immunoistochimica o la trascrittomica spaziale dallo stesso tessuto e dallo stesso individuo. Pertanto, questo approccio può essere applicato a più condizioni di malattia epatica e organismi modello in modo sistematico e affidabile.
Protocollo
Tutti gli esperimenti sugli animali sono stati condotti in conformità con la legislazione tedesca sul benessere degli animali e le normative del governo dell'Alta Baviera. La stabulazione degli animali è stata approvata ai sensi del § 11 della legge tedesca sul benessere degli animali ed eseguita in conformità con la direttiva 2010/63 / UE.
1. Preparazione dei tessuti
- Sacrificare un topo maschio C57BL / 6J di 3 mesi a 22 mesi per lussazione cervicale. Posare l'animale su una tavola da dissezione, fissare le estremità con spilli e disinfettare l'addome con etanolo al 70%.
- Eseguire la necroscopia come raccomandato da Treuting et al.16.
- Apri l'addome fino alla gabbia toracica, visualizza il fegato e usa una pinza per rimuovere con cura il fegato senza perforare i lobuli.
- Estrarre il fegato intatto tenendo il diaframma con una pinza e rimuovendo il tessuto di collegamento con le forbici.
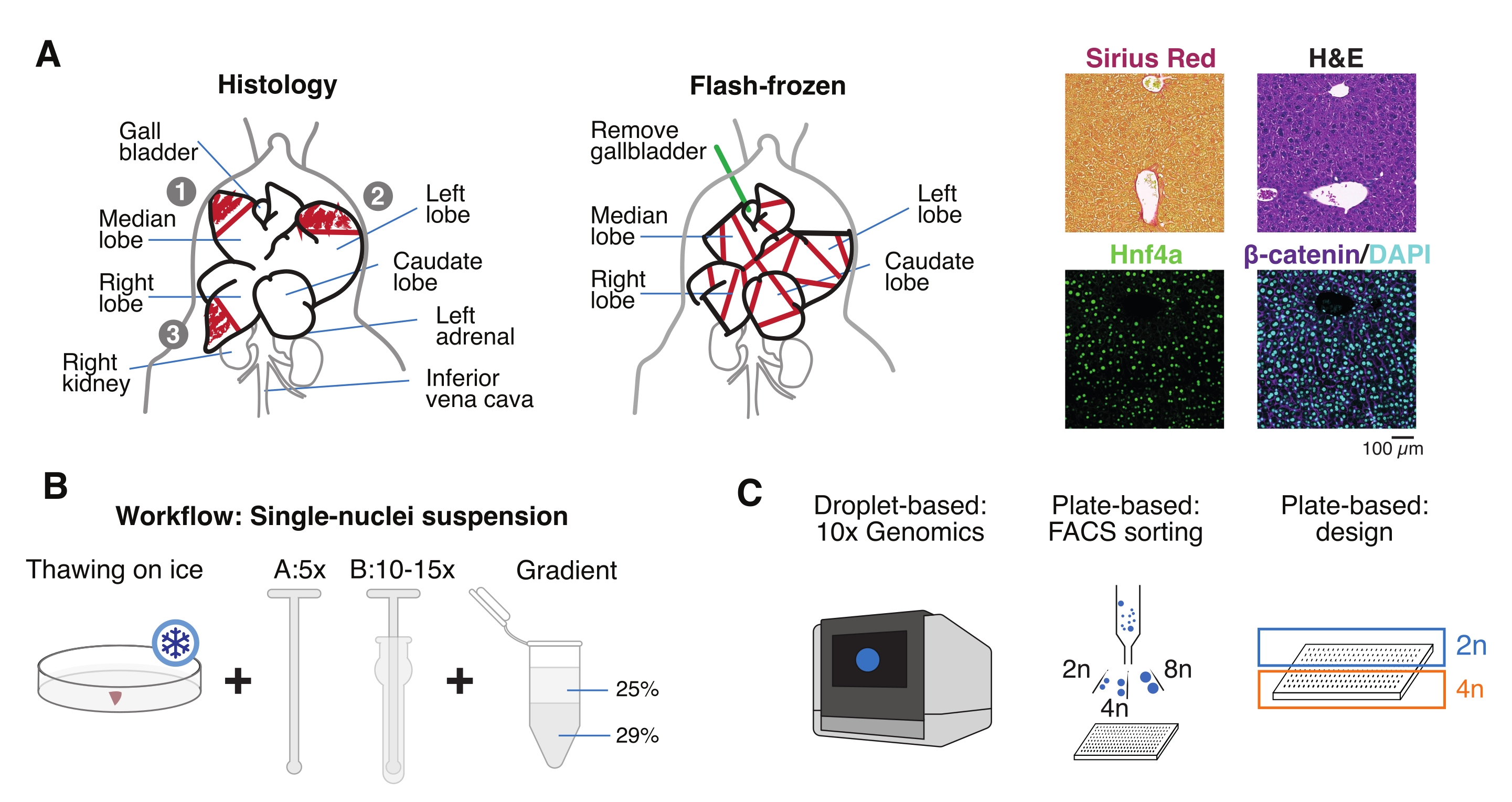
- Lavare l'organo in soluzione salina tamponata con fosfato freddo (PBS), tamponarlo su un tovagliolo di carta pulito e tagliare i lobuli del fegato in più pezzi per scopi diversi: FF per isolamento di un singolo nucleo e multiomica; paraformaldeide (10% PFA) per l'incorporazione di paraffina; e/o incorporato nel composto della temperatura di taglio ottimale (OCT) per ulteriori analisi istologiche (Figura 1A).
- Aliquot i pezzi di fegato in crioviali o tubi con tappo a vite da 5 ml e congelare immediatamente in azoto liquido. Conservare i campioni di fegato congelati a -80 °C per esperimenti multiomici a nucleo singolo a valle (Figura 1B, C).
NOTA: Il tessuto crioconservato può essere conservato in modo sicuro a -80 °C per diversi anni e deve essere sempre trasportato su ghiaccio secco a -80 °C prima dell'uso.
2. Isolamento dei nuclei
- Pulizia da banco e preparazione dei tamponi e dei materiali di consumo
- Pulire i piani di lavoro e le pipette con una soluzione di decontaminazione di etanolo e RNasi al 70% oppure utilizzare piani di lavoro e materiali dedicati privi di RNasi.
- Preraffreddare sia la benna oscillante che le centrifughe ad angolo fisso, i tubi da 1,5 ml/2 ml e le piastre multipozzetto a 4 °C, come descritto di seguito.
- Preraffreddare le pinzette monouso prive di RNasi utilizzate per la manipolazione dei tessuti, un bisturi sterile monouso e una capsula di Petri su ghiaccio secco (-60 °C).
- Preraffreddare l'omogeneizzatore di vetro Dounce e i pestelli sul ghiaccio (4 °C). Posizionare ogni pestello (A e B) in un tubo da 5 ml per evitare il contatto diretto con il ghiaccio e la potenziale contaminazione da RNasi.
- Preparare una soluzione diluente di mezzo di iodixanolo (IDM) (Tabella 1A) e utilizzarla per effettuare diluizioni al 50% e al 29% del mezzo con gradiente di densità dello stock di iodixanolo al 60% (Tabella 1B e Tabella 1C, rispettivamente).
- Preraffreddare tutti i tubi. Per ogni campione che verrà elaborato, preparare le seguenti provette:
- Preparare tre provette a basso legame del DNA da 1,5 ml (una per l'omogenato tissutale filtrato, una seconda contenente 250 μL di una diluizione al 50% della soluzione di iodixanolo e una terza per la sospensione di nuclei puliti).
- Preparare un tubo a fondo tondo da 2 mL contenente 500 μL di una diluizione al 29% della soluzione di iodixanolo per la separazione del gradiente di densità.
- Preparare un tubo conico da 15 mL per il mezzo di isolamento dei nuclei-2 (NIM-2) e il tampone di omogeneizzazione (HB).
- Preparare il nucleo di isolamento medium-1 (NIM-1) (Tabella 1D), e usarlo per fare il NIM-2 (Tabella 1E) e, successivamente, l'HB (Tabella 1F). Aggiungere entrambi gli inibitori della RNAsi appena prima dell'uso, come descritto di seguito nel protocollo.
- Preparare il buffer di stoccaggio dei nuclei (NSB) come descritto nella tabella 1G. Aggiungere l'inibitore della RNAsi ricombinante appena prima dell'uso. Aggiungere l'inibitore della RNAsi basato su proteine nell'NSB prima dell'uso per l'ordinamento FACS (opzionale).
- Preparare un becher da 500 mL con acqua sterile per immergere gli omogeneizzatori e i pestelli Dounce dopo l'omogeneizzazione dei tessuti per la pulizia e il mantenimento ottimali degli omogeneizzatori.
- Omogeneizzazione dei tessuti
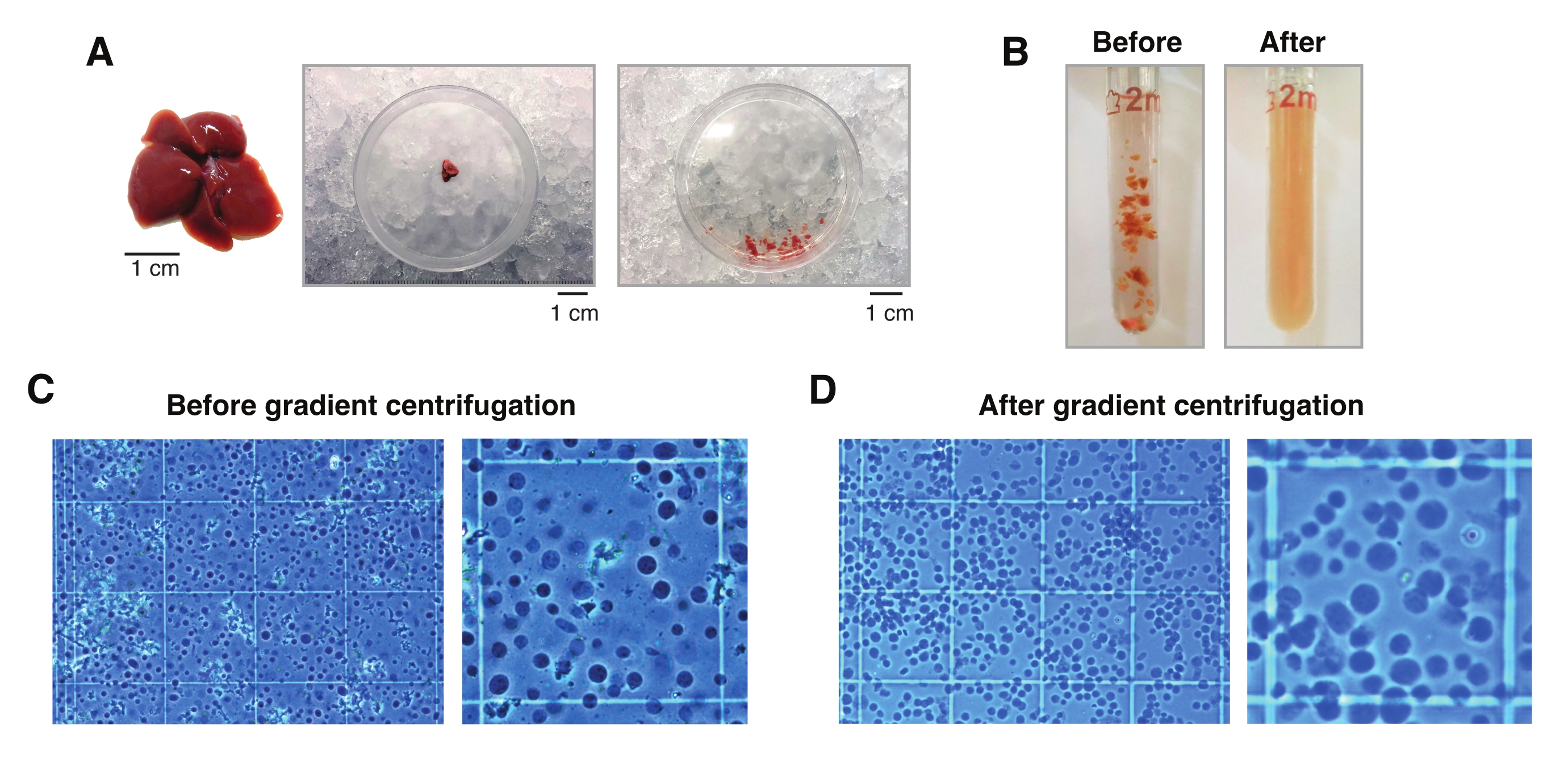
- Tagliare un pezzo di tessuto da 20-30 mg (o 5 mm3) con un bisturi prerefrigerato all'interno della piastra di Petri su ghiaccio secco. Quindi, trasferire immediatamente la capsula di Petri sul ghiaccio umido (4 °C). Aggiungere 1 mL di HB e, usando il bisturi freddo, tritare il tessuto il più possibile per consentirne la facile aspirazione con una punta a orifizio largo da 1 ml.
NOTA: Utilizzare sempre punte a orifizio largo per tutti i trasferimenti di tessuti/nuclei. In alternativa, le punte da 1 mL possono essere tagliate con un bisturi sterile su una copertura di plastica sterile per generare ampi orifizi (Figura 2A). - Raccogliere la sospensione di tessuto e trasferirla in un omogeneizzatore Dounce in vetro da 2 mL prerefrigerato (Figura 2B).
- Lavare la capsula di Petri con altri 0,5-1 ml di HB e raccogliere tutti i pezzi di tessuto rimanenti mantenendo tutto sul ghiaccio.
- Lentamente e con attenzione fai cinque colpi con il pestello sciolto A sul ghiaccio. Evita di creare bolle usando movimenti di torsione del pestello quando lo tiri su e giù. Assicurati che il pestello si muova con attenzione dall'alto verso il basso dell'omogeneizzatore ad ogni colpo.
- Successivamente, eseguire 10-15 colpi lenti con pestello stretto B sul ghiaccio. Evita di creare bolle.
NOTA: Si consiglia di ispezionare visivamente i nuclei al microscopio dopo 10 colpi con pestello B per accertare se sono necessari più colpi. Farlo usando la colorazione blu triponica (rapporto 1:1 tra tripano e campione) e un emocitometro manuale (ad esempio, mescolare 10 μL di blu tripano con 10 μL di sospensione di nuclei e utilizzare 10 μL della miscela per l'esame al microscopio; Figura 2C). - Filtrare l'omogenato attraverso un filtro cellulare da 50 μm trasferendolo in un tubo prerefrigerato da 1,5 ml. Utilizzare più di un filtro e/o tubo per omogenati contenenti una quantità elevata di grumi di tessuto connettivo.
- Risciacquare l'omogeneizzatore e i filtri utilizzati con ulteriori 0,5-1 ml di HB per raccogliere accuratamente tutto l'omogenato tissutale. Procedere alla centrifugazione del gradiente di densità.
- Tagliare un pezzo di tessuto da 20-30 mg (o 5 mm3) con un bisturi prerefrigerato all'interno della piastra di Petri su ghiaccio secco. Quindi, trasferire immediatamente la capsula di Petri sul ghiaccio umido (4 °C). Aggiungere 1 mL di HB e, usando il bisturi freddo, tritare il tessuto il più possibile per consentirne la facile aspirazione con una punta a orifizio largo da 1 ml.
- Centrifugazione a gradiente di densità
- Centrifugare l'omogenato filtrato in una centrifuga prerefrigerata ad angolo fisso a 1.000 × g per 8 minuti a 4 °C.
- Mentre il campione gira, preparare una provetta da 1,5 mL contenente 250 μL di diluizione di iodixanolo al 50% e una provetta da 2 mL contenente 500 μL di diluizione di iodixanolo al 29%. Tenere entrambi i tubi sul ghiaccio.
- Dopo la centrifugazione, aspirare il surnatante senza disturbare il pellet utilizzando una pompa per vuoto.
NOTA: l'utilizzo del pipettaggio manuale compromette la qualità della sospensione finale dei nuclei. - Aggiungere 250 μL di HB al pellet utilizzando la punta della pipetta a orifizio largo da 1 mL e risospendere molto lentamente.
- Trasferire 250 μL della sospensione di nuclei in un tubo prerefrigerato da 1,5 mL contenente 250 μL di diluizione di iodixanolo al 50% e mescolare delicatamente ma accuratamente per ottenere una sospensione di iodixanolo / nuclei al 25%.
- Trasferire 500 μL della sospensione di iodixanolo/nuclei al 25% in una provetta prerefrigerata da 2 mL contenente 500 μL di diluizione di iodixanolo al 29%.
NOTA: I 500 μL della sospensione di iodixanolo/nuclei al 25% devono essere depositati delicatamente sopra i 500 μL di soluzione di iodixanolo al 29% in modo che le miscele di iodixanolo al 25%/29% mostrino una chiara separazione di fase. Utilizzare il lato della parete del tubo con la punta della pipetta posizionata con un angolo di 45° per creare questa interfaccia sfumata. Da quel momento in poi, il tubo deve essere maneggiato delicatamente in modo da non disturbare questa pendenza. - Centrifugare il tubo in una centrifuga a secchio oscillante prerefrigerata a 12.500 g per 20 minuti con il freno impostato su OFF.
- Poco prima che la fase di centrifugazione sia completata, aggiungere gli inibitori dell'RNAsi al tampone NSB quando si procede verso le pipeline scRNA-seq (vedere Tabella 1G).
- Dopo la centrifugazione, aspirare il surnatante senza disturbare il pellet utilizzando una pompa per vuoto.
NOTA: l'utilizzo del pipettaggio manuale compromette la qualità della sospensione finale dei nuclei. - Utilizzando la punta della pipetta a orifizio largo da 1 ml, risospendere delicatamente il pellet in 100-300 μL di NSB e trasferire la sospensione dei nuclei in un tubo pulito e prerefrigerato da 1,5 ml.
- Contare i nuclei utilizzando la soluzione di tripano blu (rapporto 1:1 tra tripano e campione) e un emocitometro manuale (ad esempio, mescolare 10 μL di blu di tripano con 10 μL di sospensione di nuclei e utilizzare 10 μL della miscela per il conteggio; Figura 2D).
- Utilizzare immediatamente la sospensione di nuclei ottenuta per il test di genomica a nucleo singolo.
NOTA: La sospensione di nuclei in NSB può essere conservata refrigerata a 4 °C per un massimo di 1 settimana per ulteriori analisi mediante citometria a flusso e/o imaging a flusso, ma non per snRNA-seq o snATAC-seq.
3. Ordinamento dei nuclei per il profilo della ploidia degli epatociti o approcci di sequenziamento ben basati
- Per la selezione cellulare basata sulla citometria a flusso, filtrare la sospensione dei nuclei attraverso un filtro da 50 μm in una provetta FACS da 5 mL prerefrigerata.
- Utilizzare una selezionatrice per citometria a flusso dotata di un ugello da 100 μm. Caricare il tubo FACS sullo smistatore e visualizzare in anteprima il campione.
- Impostare la strategia di gating per l'ordinamento dei nuclei, iniziando con un cancello di dispersione tracciando l'area di dispersione diretta rispetto all'area di dispersione laterale (FSC-A / SSC-A), seguita da Hoechst-Height contro Hoechst-Area (nuclei gate), e quindi Hoechst-Width contro Hoechst-Area (porta singoletti). Visualizzare il profilo di ploidia dei nuclei sull'istogramma dell'area di Hoechst.
NOTA: per una migliore risoluzione dei picchi, visualizzare il canale di Hoechst (450/50) sulla scala lineare (Figura 3A). - Per lo smistamento in piastre da 96/384 pozzetti, impostare il ritardo delle gocce e ottimizzare l'allineamento delle lastre utilizzando il metodo colorimetrico con substrato benzidina-perossidasi di rafano (TMB-HRP), come descritto in precedenza6.
- Impostare sia il raffreddamento del campione che il supporto della piastra per raffreddare a 4 °C, con la rotazione del campione attivata a 300 giri/min.
- Ordinare i singoli nuclei ad una concentrazione del campione di ~1 x 105 nuclei/mL e con la portata a 200-500 eventi/s.
4. Ispezione visiva e quantificazione dei parametri nucleari mediante citometria per immagini (opzionale)
- Caricare una provetta da 0,5 mL contenente 50 μL di nuclei sospesi ad una concentrazione di 2 × 107 nuclei/mL.
- Impostare un gate di tutti gli eventi basato sulle proporzioni rispetto al canale di fluorescenza di Hoechst per visualizzare il profilo di ploidia dei nuclei (Figura 3B).
- Acquisire il campione con un ingrandimento di 40x utilizzando il campo luminoso (BF) e il canale di fluorescenza di Hoechst.
- Ispezionare e quantificare i nuclei 2n e 4n utilizzando la misurazione del campo luminoso e l'intensità della fluorescenza di Hoechst con il software di citometria per immagini (Figura 3C).
5. Costruzione e sequenziamento di librerie di RNA a singolo nucleo, ATAC-seq o multiomi
- Per gli approcci snRNA-seq basati su goccioline, caricare la sospensione di nuclei purificati direttamente nel dispositivo microfluidico per il partizionamento parallelo automatizzato e il codice a barre molecolare17.
- Dopo aver completato la corsa microfluidica nel dispositivo di partizionamento a cella singola, raccogliere i nuclei incapsulati in gel incapsulati, incubarli e pulirli come descritto in precedenza nelle linee guida del produttore17.
- Eseguire 11 cicli di reazione a catena della polimerasi (PCR) per la preamplificazione del cDNA utilizzando il seguente programma: 3 min a 98 °C, (15 s a 98 °C, 20 s a 63 °C e 1 min a 72 °C) x 11, 1 min a 72 °C e mantenere a 4 °C. Continuare con una riparazione finale e una fase di coda A e la legatura dell'adattatore, come indicato dal produttore17. Per la successiva costruzione finale della libreria di espressione genica, eseguire 10 cicli di PCR utilizzando il seguente programma: 45 s a 98 °C, (20 s a 98 °C, 30 s a 54 °C, 20 s a 72 °C) x 10, 1 min a 72 °C e mantenere a 4 °C.
- Sequenziare le librerie ottenute a una profondità di lettura di ~20.000-50.000 letture medie per nucleo.
- Per il sequenziamento basato su goccioline multiomiche articolari (RNA + ATAC), incubare i nuclei epatici in tampone di lisi per 5 minuti, quindi etichettarli per 1 ora, come descritto in precedenza18.
- Caricare i nuclei etichettati direttamente in un dispositivo microfluidico per il partizionamento parallelo automatizzato e il codice a barre molecolare.
- Al termine della corsa microfluidica, raccogliere i nuclei incapsulati in gel con perline, incubarli e pulirli come descritto dal produttore18.
- Eseguire sei cicli di PCR per la fase di preamplificazione del cDNA utilizzando il seguente programma: 5 min a 72 °C, 3 min a 98 °C, (20 s a 98 °C, 30 s a 63 °C, 1 min a 72 °C) x 6, 1 min a 72 °C e tenere a 4 °C.
- Prelevare 35 μL del campione preamplificato ed eseguire un'amplificazione del cDNA come segue: 3 min a 98 °C, (15 s a 98 °C, 20 s a 63 °C, 1 min a 72 °C) x 6, 1 min a 72 °C e tenere a 4 °C. Continuare con la riparazione finale e la fase di coda ad A e la legatura dell'adattatore, come indicato dal produttore18 . Per la successiva PCR di indicizzazione del campione finale, eseguire 15 cicli di PCR come segue: 45 s a 98 °C, (20 s a 98 °C, 30 s a 54 °C, 20 s a 72 °C) x 15, 1 min a 72 °C e tenere a 4 °C.
- Per la costruzione di librerie ATAC, utilizzare 40 μL e amplificare per sei cicli PCR per l'indicizzazione dei campioni utilizzando il seguente programma: 45 s a 98 °C, (20 s a 98 °C, 30 s a 67 °C, 20 s a 72 °C) x 6, 1 min a 72 °C e mantenere a 4 °C.
- Sequenziare le librerie di espressione genica multiome ottenute a una profondità di lettura minima di 20.000 coppie di lettura per nucleo e le librerie ATAC multiome a una profondità di lettura minima di 25.000 coppie di lettura per cella, come raccomandato dal produttore18.
Risultati
Questo flusso di lavoro per l'isolamento di un singolo nucleo da campioni di fegato congelati è personalizzato per la multiomica a singolo nucleo e si basa su tre fasi principali, che possono essere riassunte come i) raccolta di campioni per l'analisi parallela dell'eterogeneità cellulare e dell'architettura tissutale, ii) sospensione a nucleo singolo e iii) multiomica a nucleo singolo (Figura 1 ). I fegati estratti vengono sezionati da topi eutanizzati e tagliati a pezzi per l'ispezione istologica per l'incorporazione di paraffina, la criosezione o entrambi. Altri pezzi tagliati vengono immediatamente congelati in azoto liquido per l'isolamento a valle a singolo nucleo per analisi multiomiche. Questo sistema di raccolta tissutale consente all'utente di convalidare ulteriormente i dati omici a singolo nucleo su sezioni di tessuto dello stesso individuo, integrando così il set di dati con trascrittomica spaziale o con analisi immunoistochimiche, se necessario.
L'esame microscopico dei nuclei estratti da fegati congelati con il metodo qui descritto mostra che la fase di centrifugazione del gradiente di densità facilita notevolmente la rimozione di detriti cellulari e tissutali indesiderati (Figura 2C,D). Inoltre, questa metodologia preserva tutti i livelli di ploidia, che possono essere convalidati e quantificati mediante analisi citometriche (Figura 3).
Per convalidare ulteriormente le prestazioni di questo protocollo, abbiamo condotto snRNA-seq basato su goccioline su nuclei non ordinati e FACS e analizzato i dati seguendo la pipeline Seurat. In breve, i singoli nuclei estratti sono stati preparati per snRNA-seq a base di goccioline come descritto in precedenza19. Per lo snRNA-seq dei nuclei 2n e 4n o livelli superiori di ploidia, sono stati utilizzati 11 cicli per la fase di amplificazione del cDNA e 10 cicli per la costruzione finale della libreria di espressione genica. Le librerie risultanti sono state sequenziate a una profondità di lettura di ~ 25.000-39.000 letture medie per nucleo. Le letture dei singoli nuclei ottenute sono state mappate sul genoma del topo GRCm39/mm39. Durante l'esecuzione della pipeline di pre-elaborazione, è stato aggiunto il comando -- include-intron per accertare l'inclusione e la quantificazione dell'RNA messaggero unspliced (mRNA) presente nei nuclei. L'algoritmo EmptyDrops incorporato all'interno dell'allineatore ha filtrato e rimosso le goccioline vuote.
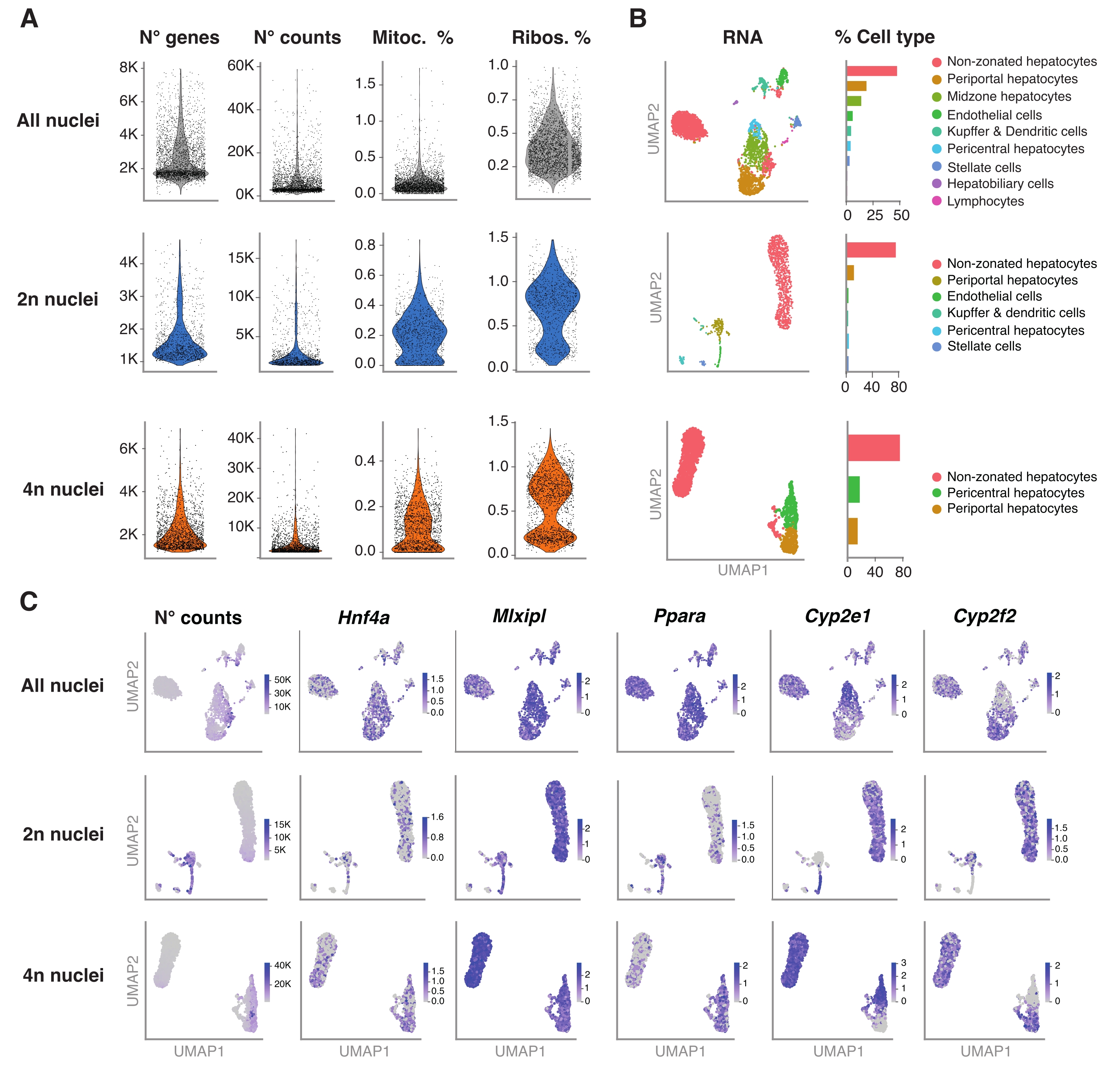
Il pacchetto R Seurat (versione 4.1.1) è stato utilizzato per calcolare le metriche di controllo qualità (QC) utilizzando la matrice di conteggio degli identificatori molecolari univoci (UMI) come prodotto dalla pipeline di analisi snRNA-seq. Sono stati rimossi conteggi con meno di 100 caratteristiche (geni) e meno di 10 cellule. I nuclei sono stati filtrati secondo soglie QC identificate: numero minimo di geni = 200 e numero massimo di geni = 8.000, frazione mitocondriale <1% e frazione ribosomiale <2%. I primi 3.000 geni altamente variabili (HVG) sono stati utilizzati per l'analisi delle componenti principali (PCA), come implementato in Seurat. Il clustering basato su grafici è stato eseguito dopo aver inserito le prime 15 dimensioni del PC risultanti dall'analisi PCA. Per raggruppare le celle, abbiamo applicato la tecnica di ottimizzazione della modularità (algoritmo di Lovanio) con un parametro di risoluzione impostato su 0,5. Per visualizzare ed esplorare questo set di dati, abbiamo eseguito la riduzione dimensionale non lineare, ovvero l'approssimazione e la proiezione uniforme delle varietà (UMAP). L'identità di ciascun cluster è stata assegnata sulla base della conoscenza precedente dei geni marcatori 6,20,21.
La figura 4A mostra metriche di alta qualità dai dati ottenuti utilizzando questo metodo di estrazione dei nuclei. UMAP rappresenta il numero di conteggi in tutti i nuclei e i principali tipi di cellule che possono essere identificati con sicurezza solo con il trascrittoma nucleare e il sequenziamento relativamente superficiale (~ 25.000-40.000 letture medie per cellula) (Figura 4B). Questo approccio consente lo studio di fattori di trascrizione specifici del fegato come Hnf4a, Mlxipl e Ppara, nonché di geni bersaglio a valle coinvolti nel metabolismo degli xenobiotici (cioè Cyp2e1 e Cyp2f2) (Figura 4C). Da notare che i nuclei estratti hanno conservato informazioni critiche sulla zonazione epatica, come mostrato dal modello complementare di geni caratteristici come il Cyp2e1 pericentrale e il periportale Cyp2f2 (Figura 4C).
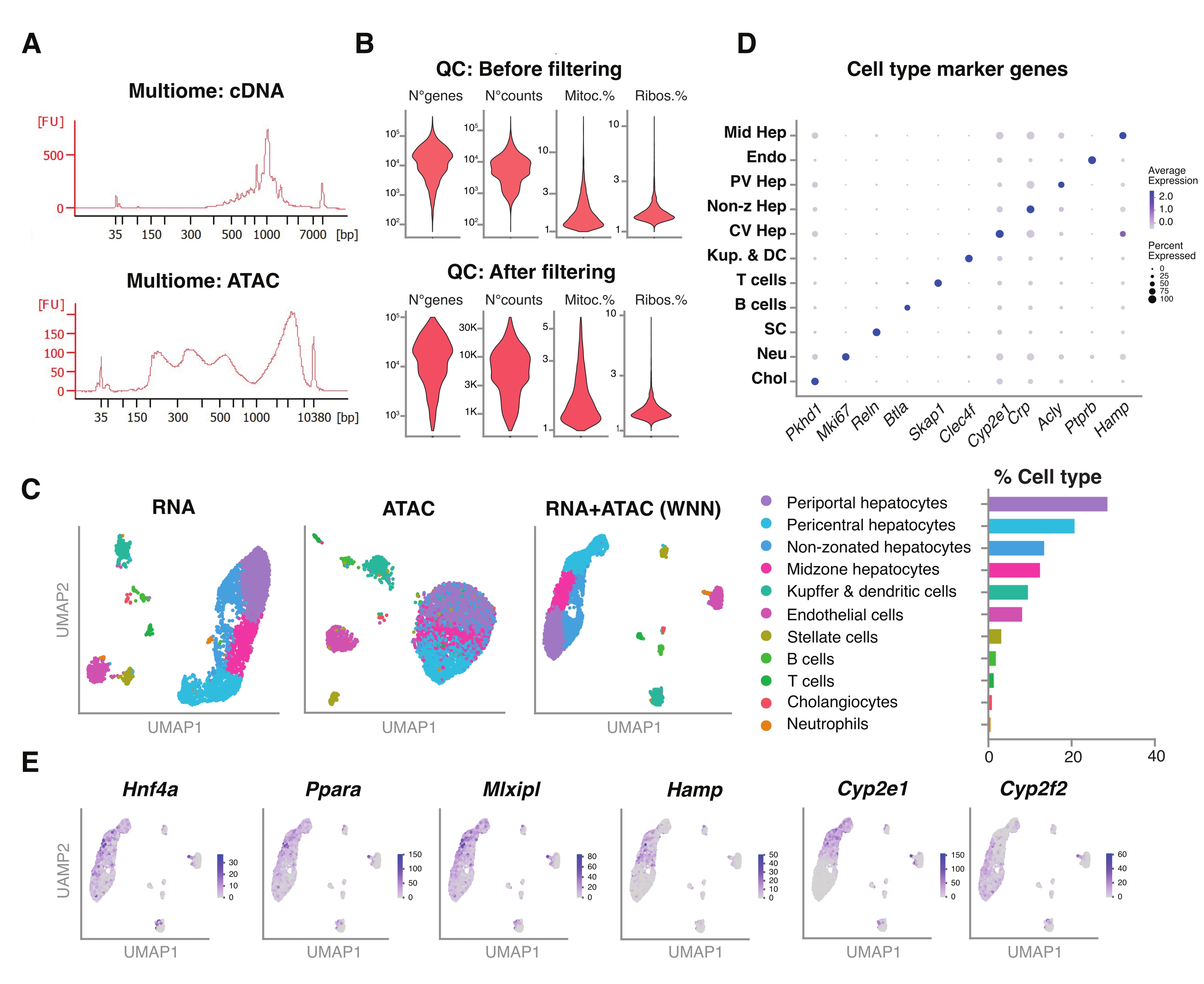
Abbiamo ulteriormente valutato la compatibilità dei nuclei non ordinati estratti utilizzando questo metodo con i più recenti saggi multiomici per la profilazione simultanea del paesaggio epigenomico (ATAC) e dell'espressione genica (RNA) negli stessi nuclei singoli18. Abbiamo ottimizzato il tempo di incubazione della lisi dei nuclei a 5 minuti prima della trasposizione. Le librerie di sequenziamento sono state costruite eseguendo sei cicli di PCR per la preamplificazione del cDNA. Dal campione preamplificato, sono stati prelevati 35 μL e ulteriormente amplificati da 15 cicli di PCR per l'indicizzazione del campione per costruire la libreria di espressione genica. Un elettroferogramma rappresentativo della traccia di cDNA è mostrato nella Figura 5A (in alto). Per la costruzione delle librerie ATAC, sono stati utilizzati 40 μL di campione preamplificato ed eseguiti per ulteriori sei cicli di PCR per l'indicizzazione del campione, con l'elettroferogramma rappresentativo della traccia di DNA mostrato in Figura 5A (in basso). La libreria di espressione genica risultante (RNA) è stata sequenziata a una profondità di 44.600 letture per nucleo e la libreria ATAC a una profondità di 43.500 letture per nucleo (Figura 5B).
Analogamente al protocollo di sequenziamento basato su goccioline a modalità singola sopra descritto, la mappatura di lettura, l'allineamento, la rimozione delle gocce vuote e il conteggio dei frammenti sono stati eseguiti seguendo le linee guida standard, come descritto in precedenza, utilizzando il genoma di riferimento GRCm39 / mm3918. Utilizzando i pacchetti Seurat e Signac22, abbiamo eseguito un'analisi "weighted nearest neighbor" (WNN) per misurazioni multiple di entrambe le modalità (RNA + ATAC) (Figura 5C), che ci ha permesso di identificare e annotare sia i tipi di cellule epatiche maggiori che minori senza pregiudizi prominenti dovuti alle dimensioni delle cellule o alla fragilità nucleare (Figura 5D). La pipeline, come pubblicato dal laboratorio Satija22,23, include fasi standard di controllo qualità - pre-elaborazione e riduzione dimensionale - eseguite su entrambi i saggi in modo indipendente. Per avere una buona rappresentazione della combinazione ponderata delle modalità RNA-seq e ATAC-seq, il grafico WNN è stato tracciato e utilizzato per la visualizzazione, il clustering e l'annotazione UMAP sulla base dei geni marcatori precedentemente identificati 6,20,21. Analogamente ai test che utilizzano la modalità singola, abbiamo anche rilevato regolatori trascrizionali a monte (Hnf4a, Ppara, Mlxipl) e geni caratteristici della zonazione epatica (Hamp, Cyp2e1 e Cyp2f2) (Figura 5E).

Figura 1: Panoramica sperimentale, flusso di lavoro e applicazioni genomiche a singola cellula. (A) Rappresentazione illustrativa del campionamento tissutale per istologia (a sinistra, tre sezioni sono selezionate per l'incorporazione di paraffina e/o criosezione), raccolta di tessuti congelati flash-per genomica monocellulare (al centro) e analisi rappresentative di immunoistochimica e immunofluorescenza (a destra); barra di scala = 100 μm. (B) Passaggi critici per sospensioni mononucleo-di alta qualità. (C) Le sospensioni dei nuclei possono essere caricate su un chip di cromo 10x o utilizzate per la selezione FACS e gli approcci basati su piastre. Abbreviazioni: H&E = ematossilina ed eosina; DAPI = 4',6-diamidino-2-fenilindolo; FACS = selezione cellulare attivata dalla fluorescenza. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 2: Dissezione epatica e omogeneizzazione dei tessuti Dounce-glass. (A) fegato murino rappresentativo di un topo C57BL6/J di 3 mesi (a sinistra); la sezione epatica utilizzata per gli isolamenti del singolo nucleo prima (al centro) e dopo aver tritato il tessuto con il bisturi (a destra). Barre della scala = 1 cm. (B) Immagini illustrative di 2 mL di omogeneizzazione del vetro di Dounce prima dei colpi con pestello "sciolto" A (sinistra) e dopo pestello "stretto" B (destra). Monitoraggio dell'omogeneizzazione tissutale mediante emocitometro (C) prima della centrifugazione a gradiente e (D) dopo la centrifugazione a gradiente. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 3: Selezione cellulare attivata da fluorescenza e citometria a flusso di imaging per la caratterizzazione di nuclei ad alto rendimento . (A) Strategia di gating per lo smistamento FACS a singolo nucleo in una piastra per l'interrogazione di diversi livelli di ploidia. Scatter gate basato sull'area di dispersione anteriore rispetto all'area di dispersione laterale impostata per escludere i detriti; porta dei nuclei basata su Hoechst-Height versus Hoechst-Area che incorpora più popolazioni di nuclei; canottiere basate su Hoechst-Width contro Hoechst-A set per la discriminazione del doppietto; l'istogramma Hoechst-A permette la visualizzazione del profilo ploidico dei nuclei. (B) Quantificazione rappresentativa della citometria per immagini di tutti gli eventi (a sinistra) e singoli (a destra), mostrando nuclei diploidi e tetraploidi. (C) Immagini in campo chiaro e Hoechst di nuclei 2n e 4n e loro quantificazione mediante citometria per immagini. Abbreviazioni: FACS = selezione cellulare attivata da fluorescenza; FSC-A = area di dispersione diretta; SSC-A = area di dispersione laterale; Hoechst-H = Hoechst-Altezza; Hoechst-A = Area Hoechst; Hoechst-W = Hoechst-Width. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 4: Caratterizzazione approfondita della ploidia degli epatociti con snRNA-seq . (A) Grafici di violino che mostrano il numero di geni, la contea, la percentuale di geni mitocondriali e la percentuale di geni ribosomiali rilevati. (B) UMAP che mostra i tipi cellulari rilevati utilizzando snRNA-seq (a sinistra), con la quantificazione espressa in percentuale di nuclei (a destra). (C) UMAP illustra il numero di conteggi e indica l'espressione di geni specifici degli epatociti. Tutti i nuclei (riga superiore), 2n nuclei (fila centrale) e 4n nuclei (riga inferiore). Abbreviazioni: snRNA-seq = RNA-seq a nucleo singolo; UMAP = approssimazione e proiezione uniforme della varietà; mitoc. = geni mitocondriali; Ribo. = geni ribosomiali. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 5: Controllo di qualità e analisi di multiomiche (RNA-seq congiunto e ATAC-seq) da giovani fegati congelati e archiviati. (A) Tracce elettroforetiche automatiche rappresentative ottenute dopo la pipeline multiomica che mostrano il prodotto a peso molecolare dopo sintesi di cDNA e ATAC. (B) Diagramma di violino che mostra il numero di conteggi per ATAC-seq, RNA-seq, la percentuale di geni mitocondriali e la percentuale di geni ribosomiali prima e dopo il filtraggio. (C) UMAP mostra l'espressione genica da RNA-seq (a sinistra), ATAC-seq (al centro) e le modalità articolari - RNA-seq e ATAC-seq (a destra). (D) Diversi tipi di cellule sono annotati in diversi colori, con l'espressione di geni specifici degli epatociti indicati. (E) Grafico delle caratteristiche che mostra l'espressione cluster-specifica dei geni indicati nei tipi cellulari indicati. Abbreviazioni: ATAC-seq = saggio per cromatina accessibile alla trasposasi con sequenziamento ad alta produttività; Hep medio = epatociti medio-zonali; Endo = cellule endoteliali; PV Hep = epatociti periportali; Hep non-z = epatociti non zonati; CV Hep = epatociti pericentrali; Kup & DC = Kupffer e cellule dendritiche; SC = cellule stellate, Neu = neutrofili; Chol = colangiociti; mitoc. = geni mitocondriali; Ribo. = geni ribosomiali. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}
| Reagente | Ceppo | 10 ml | 15 ml | 50 ml | ||
| (A ) Iodixanol Medium (IDM) | ||||||
| 250 mM di saccarosio | 1M | 12,5 ml | ||||
| 150 mM KCl | 2M | 3,75 ml | ||||
| 30 mM MgCl2 | 1M | 1,5 ml | ||||
| 60 mM Tris tampone pH 8,0 | 1M | 3 ml | ||||
| Acqua ultrapura priva di RNasi | 29,25 ml | |||||
| b) 50 % IDM | ||||||
| Iodixanolo | 60% | 12,5 ml | ||||
| IDM | 2,5 ml | |||||
| (C) 29% IDM | ||||||
| Iodixanolo | 60% | 7,25 ml | ||||
| IDM | 7,75 ml | |||||
| (D ) Nuclei Isolation Medium-1 (NIM-1) | ||||||
| 250 mM di saccarosio | 1M | 12,5 ml | ||||
| 25 mM KCl | 2M | 0,625 ml | ||||
| 5 mM MgCl2 | 1M | 0,25 ml | ||||
| Tampone Tris da 10 mM pH 8,0 | 1M | 0,5 ml | ||||
| Acqua ultrapura priva di RNASI | 36,125 ml | |||||
| (E ) Nuclei Isolation Medium-2 (NIM-2) | ||||||
| Tampone NIM-1 | 9,99 ml | |||||
| Ditiotreitolo (DTT) | 1 mM | 0,01 ml | ||||
| Compresse di inibitori della proteasi (senza EDTA) | 1 | 1 compressa | ||||
| f ) Tampone di omogeneizzazione (HB) | ||||||
| Buffer NIM-2 | 9,697 ml | |||||
| Inibitore della RNasi ricombinante | 40 U/μL | 0,1 mL | ||||
| Inibitore della RNAsi basato su proteine (SUPERase•IN) | 20 U/μL | 0,1 mL | ||||
| 0,1% Triton-X | 10% | 0,1 mL | ||||
| 3 μg/mL Hoechst 33342 | 10 mg/ml | 0,003 mL | ||||
| g ) Nuclei Storage Buffer (NSB) | ||||||
| 166,5 mM di saccarosio | 1M | 1,665 ml | ||||
| 5 mM MgCl2 | 1M | 0,05 ml | ||||
| 10 mM Tris pH 8,0 | 1M | 0,1 mL | ||||
| Inibitore della RNasi ricombinante | 40 U/μL | 0,1 mL | ||||
| Inibitore della RNasi basato su proteine (SUPERase•IN) * | 20 U/μL | 0,1 mL | ||||
| Acqua ultrapura priva di RNasi | 8,085 ml | |||||
| * Opzionale (solo per lo smistamento FACS) | ||||||
Tabella 1: Ricette di soluzione. (A) Preparazione del mezzo di iodixanolo (IDM); B) diluizione al 50% della soluzione di iodixanolo; C) Diluizione al 29% della soluzione di iodixanolo. (D) Preparazione del mezzo di isolamento dei nuclei-1 (NIM-1). (E) Preparazione del mezzo di isolamento dei nuclei-2 (NIM-2). (F) Preparazione del tampone di omogeneizzazione (HB). (G) Preparazione del buffer di stoccaggio dei nuclei (NSB).
Discussione
La dissezione della composizione cellulare del fegato mediante RNA-seq a singola cellula o a singolo nucleo fornisce una comprensione più profonda dello sviluppo e della progressione della malattia epatica 3,4,5,24. L'isolamento di singole cellule dal fegato richiede molto tempo e richiede protocolli che comportano una dura dissociazione meccanica o enzimatica25,26,27. È ampiamente accettato che ogni tessuto richiede una valutazione sistematica per determinare il protocollo ottimale di dissociazione tissutale, nonché un metodo di conservazione adatto per catturare tipi di cellule o nuclei fragili28. A seconda della disponibilità tissutale, della malattia di interesse, dello stadio di sviluppo o dell'organismo modello, la preparazione di una sospensione a nucleo singolo per l'elaborazione a valle potrebbe essere una metodologia più adatta rispetto all'utilizzo di sospensioni unicellulari. È importante sottolineare che, nel fegato, scRNA-seq e snRNA-seq hanno mostrato un'alta correlazione tra mRNA nucleare e citoplasmatico, suggerendo che entrambi gli approcci presentano informazioni complementari 2,3,4,6,29.
Questo documento fornisce un isolamento standardizzato, robusto e riproducibile di un singolo nucleo da campioni di fegato congelati e archiviati di topi e altre specie, inclusi esseri umani e macachi. Questo metodo può essere utilizzato per topi wild-type alimentati con chow e una dieta ricca di grassi (HFD) e per modelli murini di fibrosi epatica utilizzando approcci genomici a singolo nucleo sia a base di piastre che a base di goccioline6. Questo metodo si basa sul protocollo descritto originariamente per il tessuto cerebrale da Krishnaswami et al.30 con ulteriori modifiche su misura per il fegato congelato. L'omogeneizzazione ottimale libera la maggior parte dei nuclei dal tessuto senza influire negativamente sull'integrità della membrana nucleare. L'overdouncing, tuttavia, può danneggiare i nuclei fragili e diminuire la loro qualità complessiva. I fegati giovani e / o grassi di solito richiedono solo 5 colpi con pestello A e 10 colpi con pestello B, mentre fegati vecchi e / o fibrotici potrebbero richiedere 15 colpi con pestello B ma non di più. Non è raccomandato, quindi, eseguire più tratti oltre i numeri qui indicati. L'overdouncing potrebbe influire negativamente sulla qualità della sospensione a singolo nucleo e aumentare la quantità di RNA ambientale. Successivamente, ciò potrebbe comportare la necessità di eseguire ulteriori passaggi di filtraggio computazionale durante le analisi dei dati a valle.
Il protocollo qui presentato è versatile e può essere adattato a diverse condizioni del fegato in topi giovani (3 mesi) e anziani (24 mesi). Poiché abbiamo scoperto che una sezione più ampia del fegato è necessaria per i tessuti vecchi, HFD e fibrotici, la dimensione del tessuto disponibile per la lavorazione può rappresentare una limitazione per alcuni utenti con minori quantità di materiale biologico iniziale. Tuttavia, la purificazione del gradiente è altamente raccomandata per l'elaborazione immediata dei campioni con saggi genomici basati su goccioline. Se i nuclei devono essere FACS ordinati in piastre da 96/384 pozzetti per saggi ben basati, la purificazione del gradiente può essere omessa. Incoraggiamo gli utenti a eseguire ancora la purificazione del gradiente se c'è abbastanza campione di tessuto per ottenere la concentrazione di nuclei raccomandata per lo smistamento FACS (cioè ~ 1 × 105 nuclei / ml).
Il fegato è caratterizzato dalla natura poliploide degli epatociti9, ma il ruolo della ploidia degli epatociti nella normale fisiologia e malattia non è ancora chiaro. C'è un numero crescente di prove che indicano che la ploidia fornisce variabilità genomica31, ed è ben noto che la ploidia aumenta con l'etàdi 32,33 anni. L'arricchimento degli epatociti tetraploidi mononucleati, tuttavia, è anche clinicamente associato a prognosi infausta nel carcinoma epatocellulare umano (HCC)34. Allo stesso modo, i cambiamenti nei livelli di ploidia degli epatociti sono legati a malattie croniche del fegato correlate all'invecchiamento come la steatosi epatica non alcolica (NAFLD)35,36,37. La ploidia è la condizione di possedere più di due copie del genoma, che può essere esplorata colorando il contenuto del genoma con un colorante del DNA come Hoechst38. Il colorante Hoechst, che viene aggiunto all'HB prima dell'estrazione, etichetta tutti i nuclei durante il protocollo di isolamento. Ciò consente di distinguere tra nuclei diploidi e poliploidi in base al loro contenuto di DNA quando eccitati da un laser UV (350 nm) o viola (450 nm) su uno strumento di citometria a flusso. Con la strategia di gating presentata, 2n, 4n, 8n e livelli più elevati di ploidia degli epatociti possono essere studiati in fegati congelati e archiviati per comprendere meglio il ruolo dell'eterogeneità cellulare nella funzione tissutale1 (Figura 3A). Inoltre, la morfologia nucleare, comprese le dimensioni e il volume, può essere quantificata utilizzando la citometria a flusso di imaging per correlare i cambiamenti nella dimensione del nucleo con i cambiamenti nel numero totale di conteggi o nel numero di geni a seconda del livello di ploidia (Figura 3B, C).
La misurazione dell'omica multimodale offre l'opportunità di studiare contemporaneamente diversi livelli di organizzazione genomica. L'approccio multiomico congiunto RNA + ATAC consente lo studio dei regolatori a monte e dei geni metabolici a valle, fornendo un approccio completo per lo studio delle reti trascrizionali e dell'architettura della cromatina associata alla funzionalità epatica alla risoluzione di una singola cellula. Inoltre, con i progressi nei metodi computazionali che possono spiegare la scarsità dei dati e la riduzione dei costi di sequenziamento, la multiomica a cella singola è pioniera nella valutazione di più modalità dalla stessa cella. Questo protocollo di isolamento a nucleo singolo è compatibile con la valutazione individuale e congiunta dei set di dati di espressione e cromatina. Abbiamo utilizzato pipeline standard stabilite da Stuart et al.23 (pacchetto Signac) per illustrare la qualità dei dati, mentre diversi metodi computazionali disponibili e alternativi possono essere facilmente adottati per le analisi a valle23,39,40,41.
Nel complesso, la multiomica a singolo nucleo consente lo studio di tessuti epatici di primati bio-archiviati, FF, umani e non umani utilizzando una quantità molto piccola di materiale campione di partenza implementando il protocollo di estrazione del nucleo qui presentato. Questo strumento inestimabile consentirà ai biologi del fegato di interrogare sia l'espressione genica che l'accessibilità della cromatina nel contesto di varie patologie epatiche. Inoltre, vari livelli di ploidia degli epatociti e il conseguente aggiustamento dell'espressione genica dipendente dalla loro posizione nel lobulo epatico potrebbero rivelare il loro ruolo nelle patologie epatiche. Pertanto, prevediamo che lo studio dell'eterogeneità cellulare fornirà nuove opportunità per lo sviluppo della medicina di precisione e interventi mirati contro malattie come HCC e NAFLD.
Divulgazioni
Gli autori dichiarano di non avere conflitti di interesse.
Riconoscimenti
Questa ricerca è stata sostenuta dall'Helmholtz Pioneer Campus (M.S., K.Y., C.P.M.-J.) e dall'Institute of Computational Biology (C. T.-L.). Questa ricerca è stata supportata anche da AMED con il numero di sovvenzione JP20jm0610035 (C.P.M.-J.). Ringraziamo il supporto di Core Genomics presso HMGU (I. de la Rosa) e Bioinformatics (T. Walzthoeni), in particolare Xavier Pastor per la formazione e l'orientamento. Ringraziamo A. Feuchtinger, U. Buchholz, J. Bushe e tutti gli altri membri dello staff della struttura principale HMGU Pathology and Tissue Analytic per il loro supporto tecnico e scientifico, nonché J. Zorn, R. Erdelen, D. Würzinger, membri dello staff di E-Streifen, nonché la struttura principale di Laboratory Animal Services per il loro continuo supporto e discussione scientifica. Siamo grati alla Core Facility Cell Analysis di TranslaTUM (R. Mishra) e a Luminex, A DiaSorin Company (P. Rein). Ringraziamo il Dr. I Deligiannis per il suo supporto tecnico. Il Dr. M. Hartman, il Dr. A. Schröder e la Sig.ra A. Barden (Helmholtz Pioneer Campus) sono stati fondamentali per il loro supporto legale, gestionale e amministrativo.
Materiali
| Name | Company | Catalog Number | Comments |
| 10% Tween 20 - 5 mL | Bio-Rad | 1662404 | |
| 2100 Bioanalyzer Instrument | Agilent | G2939BA | |
| Adhesive PCR film | Thermo Fisher Scientific | AB0558 | |
| Bioanalyzer High Sensitivity DNA Analysis | Agilent | 5067-4626 | |
| C1000 Touch Thermal Cycler | Bio-Rad | 1851196 | |
| Cell Sorter | For fluoresence-activated cell sorting (FACS); e.g. BD FACSAria Cell Sorter. | ||
| Centrifuge 5430R | Eppendorf | 5428000619 | Use chilled at 4 °C |
| Chromium Controller & Next GEM Accessory Kit | 10X Genomics | 1000204 | |
| Chromium Next GEM Chip G Single Cell Kit | 10X Genomics | 1000127 | |
| Chromium Next GEM Chip J Single cell kit, 16 reactions | 10X Genomics | 1000230 | |
| Chromium Next GEM Single Cell 3' Kit v3.1, 4 reactions | 10X Genomics | 1000269 | |
| Chromium Next GEM Single Cell Multiome ATAC + Gene Expression Reagent Bundle, 4 reactions | 10X Genomics | 1000285 | |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Sigma Aldrich | 11873580001 | |
| Dithiothreitol (DTT) 1 M solution | Sigma Aldrich | 646563 | Make 1 mM stock solution and use at 1 µM final concentration. |
| DNA AWAY Surface Decontaminant | Thermo Fisher Scientific | 10223471 | Wipe surfaces and pipettes before start of experiment |
| DNA LoBind Tubes, 1.5 mL | Thermo Fisher Scientific | 16628742 | |
| DNA LoBind Tubes, 2.0 mL | Thermo Fisher Scientific | 16638742 | |
| Elution Buffer (EB) - 250 mL | Qiagen | 19086 | |
| Eppendorf ThermoMixer C | Thermo Fisher Scientific | 13527550 | |
| Eppendorf ThermoMixer C Accessory: Smartblock | Thermo Fisher Scientific | 13518470 | |
| Ethanol, Absolute (200 Proof), Molecular Biology Grade - 100 mL | Thermo Fisher Scientific | 10517694 | |
| Filters 50 µm, sterile | SYSMEX PARTEC - CELLTRICS | 04-004-2327 | Adjust filter diameter according with tissue and nuclei size |
| Glycerin (Glycerol), 50% (v/v) - 1 L | Ricca Chemical Company | 3290-32 | |
| Hard-Shell, 384-Well PCR Plates, thin wall, skirted, clear/white | Bio-Rad | HSP3905 | |
| Herenz Heinz ABS Forceps | Thermo Fisher Scientific | 1131884 | |
| Hoechst 33342, Trihydrochloride, Trihydrate, 10 mg/mL Solution in Water | Invitrogen | H3570 | Light-sensitive |
| Imaging Flow Cytometer | For imaging flow cytometry analysis, e.g. Luminex Amnis ImageStream | ||
| Invitrogen TE Buffer - 100 mL | Thermo Fisher Scientific | 11568846 | |
| KCl (2 M), RNase-free, 100 mL | Thermo Fisher Scientific | AM9640G | |
| MgCl2 (1 M), 100 mL | Thermo Fisher Scientific | AM9530G | |
| MicroAmp 8-Cap Strip, clear-300 strips | Thermo Fisher Scientific | 10209104 | |
| MicroAmp 8-Tube Strip, 0.2 mL-125 strips | Thermo Fisher Scientific | 10733087 | |
| Mörser 2 mL DOUNCE | Wagner & Munz GmbH | 9651632 | RNase zap and rinse with MillQ before use |
| MyFuge 12 Mini MicroCentrifuge C1012 | Benchmark Scientific | C1012 | Or any other strip and tube mini centrifuge |
| Neubauer Hemocytometer | OMNILAB LABORZENTRUM | 5435293 | Visualize and count nuclei under microscope |
| Nuclease-Free Water (not DEPC-Treated) | Thermo Fisher Scientific | AM9937 | |
| OptiPrep Density Gradient Medium | Sigma Aldrich | D1556 | |
| Pipette tips RT LTS 1000 µL, Wide-O | Mettler Toledo | 30389218 | |
| Pistill "A" 2 mL | Wagner & Munz GmbH | 9651621 | RNase zap and rinse with MillQ before use |
| Pistill "B" 2 mL | Wagner & Munz GmbH | 9651627 | RNase zap and rinse with MillQ before use |
| Polypropylene 15 mL Centrifuge Tube | Thermo Fisher Scientific | 10579691 | |
| Polystyrene Petri dish, 60 mm x 15 mm | Thermo Fisher Scientific | 10634141 | |
| Polystyrene Round-Bottom 5 mL FACS Tubes | Thermo Fisher Scientific | 10100151 | |
| Protector RNase inhibitor - 2,000 U | Sigma Aldrich | 3335399001 | Keep in -20 °C until use |
| Protein-based RNase Inhibitor SUPERase•In (20 U/μL) | Thermo Fisher Scientific | AM2696 | Keep in -20 °C until use |
| Recombinant RNase Inhibitor | Clontech Takara | 2313B | Keep in -20 °C until use |
| RNAse free microfuge tubes - 0.5 mL | Thermo Fisher Scientific | AM12450 | |
| RNaseZap RNase Decontamination Solution | Thermo Fisher Scientific | AM9780 | Wipe surfaces and pipettes before start of experiment |
| SPRIselect - 60 mL | Beckman Coulter | B23318 | Aliquot and store in 4 °C |
| Sucrose, 500 g | Sigma Aldrich | S0389-500G | Make a 1 M stock solution |
| Swann-Morton Sterile Disposable Stainless Steel Scalpels | Thermo Fisher Scientific | 11798343 | |
| Tris-HCI (1M), pH 8.0 | Invitrogen | 15568025 | |
| Triton X-100, 98%, 100 mL | Thermo Fisher Scientific | 10671652 | Make 10% stock solution. Keep at 4 °C and protect from light. |
| Trypan Blue Solution, 0.4% | Thermo Fisher Scientific | 11538886 | |
| Vortex- Mixer | VWR | 444-1372 | Or any other type of vortex |
Riferimenti
- Kamies, R., Martinez-Jimenez, C. P. Advances of single-cell genomics and epigenomics in human disease: where are we now. Mamm Genome. 31 (5-6), 170-180 (2020).
- Ramachandran, P., Matchett, K. P., Dobie, R., Wilson-Kanamori, J. R., Henderson, N. C. Single-cell technologies in hepatology: New insights into liver biology and disease pathogenesis. Nature Reviews Gastroenterology & Hepatology. 17 (8), 457-472 (2020).
- Ramachandran, P., et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. 575 (7783), 512-518 (2019).
- Andrews, T. S., et al. Single-cell, single-nucleus, and spatial RNA sequencing of the human liver identifies cholangiocyte and mesenchymal heterogeneity. Hepatology Communications. 6 (4), 821-840 (2022).
- Xiong, X., et al. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Molecular Cell. 75 (3), 644-660 (2019).
- Richter, M. L., et al. Single-nucleus RNA-seq2 reveals functional crosstalk between liver zonation and ploidy. Nature Communications. 12 (1), 4264 (2021).
- Matsumoto, T., Wakefield, L., Tarlow, B. D., Grompe, M. In vivo lineage tracing of polyploid hepatocytes reveals extensive proliferation during liver regeneration. Cell Stem Cell. 26 (1), 34-47 (2020).
- Chen, F., et al. Broad distribution of hepatocyte proliferation in liver homeostasis and regeneration. Cell Stem Cell. 26 (1), 27-33 (2020).
- Donne, R., Saroul-Ainama, M., Cordier, P., Celton-Morizur, S., Desdouets, C. Polyploidy in liver development, homeostasis and disease. Nature Reviews. Gastroenterology & Hepatology. 17 (7), 391-405 (2020).
- Lengefeld, J., et al. Cell size is a determinant of stem cell potential during aging. Science Advances. 7 (46), 0271 (2021).
- Lanz, M. C., et al. Increasing cell size remodels the proteome and promotes senescence. Mol Cell. 82 (17), 3255-3269 (2022).
- Kim, J. Y., et al. PIDDosome-SCAP crosstalk controls high-fructose-diet-dependent transition from simple steatosis to steatohepatitis. Cell Metabolism. 34 (10), 1548-1560 (2022).
- Padovan-Merhar, O., et al. Single mammalian cells compensate for differences in cellular volume and DNA copy number through independent global transcriptional mechanisms. Molecular Cell. 58 (2), 339-352 (2015).
- Miettinen, T. P., et al. Identification of transcriptional and metabolic programs related to mammalian cell size. Current Biology. 24 (6), 598-608 (2014).
- Vargas-Garcia, C. A., Ghusinga, K. R., Singh, A. Cell size control and gene expression homeostasis in single-cells. Current Opinion in Systems Biology. 8, 109-116 (2018).
- Knoblaugh, S. E., Randolph-Habecker, J. . Necropsy and histology. In Comparative Anatomy and Histology: A Mouse, Rat, and Human Atlas (Second Edition). , (2018).
- Chromium Next GEM Single Cell 3 Reagent Kits v3.1 User Guide. Document number CG000204 (Rev D). 10x Genomics Available from: https://www.10xgenomics.com/support/single-cell-gene-expression/documentation/steps/library-prep/chromium-single-cell-3-reagent-kits-user-guide-v-3-1-chemistry (2019)
- Chromium Next GEM Single Cell Multiome ATAC + Gene Expression User Guide. Document number CG000338 (Rev D). 10x Genomics Available from: https://www.10xgenomics.com/support/single-cell-multiome-atac-plus-gene-expression/documentation/steps/library-prep/chromium-next-gem-single-cell-multiome-atac-plus-gene-expression-reagent-kits-user-guide (2021)
- MacParland, S. A., et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nature Communications. 9 (1), 4383 (2018).
- Martinez-Jimenez, C. P., Kyrmizi, I., Cardot, P., Gonzalez, F. J., Talianidis, I. Hepatocyte nuclear factor 4alpha coordinates a transcription factor network regulating hepatic fatty acid metabolism. Molecular and Cell Biology. 30 (3), 565-577 (2010).
- Schmidt, D., et al. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science. 328 (5981), 1036-1040 (2010).
- Hao, Y., et al. Integrated analysis of multimodal single-cell data. Cell. 184 (13), 3573-3587 (2021).
- Stuart, T., Srivastava, A., Madad, S., Lareau, C. A., Satija, R. Single-cell chromatin state analysis with Signac. Nature Methods. 18 (11), 1333-1341 (2021).
- Sampaziotis, F., et al. Cholangiocyte organoids can repair bile ducts after transplantation in the human liver. Science. 371 (6531), 839-846 (2021).
- Gomez-Lechon, M. J., Donato, M. T., Castell, J. V., Jover, R. Human hepatocytes as a tool for studying toxicity and drug metabolism. Current Drug Metabolism. 4 (4), 292-312 (2003).
- Gomez-Lechon, M. J., Donato, M. T., Castell, J. V., Jover, R. Human hepatocytes in primary culture: The choice to investigate drug metabolism in man. Current Drug Metabolism. 5 (5), 443-462 (2004).
- vanden Brink, S. C., et al. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nature Methods. 14 (10), 935-936 (2017).
- Denisenko, E., et al. Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biology. 21 (1), 130 (2020).
- Nault, R., Fader, K. A., Bhattacharya, S., Zacharewski, T. R. Single-nuclei RNA sequencing assessment of the hepatic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Cellular and Molecular Gastroenterology and Hepatology. 11 (1), 147-159 (2021).
- Krishnaswami, S. R., et al. Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons. Nature Protocols. 11 (3), 499-524 (2016).
- Duncan, A. W., et al. Aneuploidy as a mechanism for stress-induced liver adaptation. Journal of Clinical Investigation. 122 (9), 3307-3315 (2012).
- Kreutz, C., et al. Hepatocyte ploidy is a diversity factor for liver homeostasis. Frontiers in Physiology. 8, 862 (2017).
- Hunt, N. J., Kang, S. W. S., Lockwood, G. P., Le Couteur, D. G., Cogger, V. C. Hallmarks of aging in the liver. Computational and Structural Biotechnology Journal. 17, 1151-1161 (2019).
- Bou-Nader, M., et al. Polyploidy spectrum: a new marker in HCC classification. Gut. 69 (2), 355-364 (2019).
- Gentric, G., et al. Oxidative stress promotes pathologic polyploidization in nonalcoholic fatty liver disease. Journal of Clinical Investigation. 125 (3), 981-992 (2015).
- Schwartz-Arad, D., Zajicek, G., Bartfeld, E. The streaming liver IV: DNA content of the hepatocyte increases with its age. Liver. 9 (2), 93-99 (1989).
- Kudryavtsev, B. N., Kudryavtseva, M. V., Sakuta, G. A., Stein, G. I. Human hepatocyte polyploidization kinetics in the course of life cycle. Virchows Archiv. B, Cell Pathology Including Molecular Pathology. 64 (6), 387-393 (1993).
- Duncan, A. W., et al. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature. 467 (7316), 707-710 (2010).
- Granja, J. M., et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nature Genetics. 53 (3), 403-411 (2021).
- Bredikhin, D., Kats, I., Stegle, O. MUON: Multimodal omics analysis framework. Genome Biology. 23 (1), 42 (2022).
- Velten, B., et al. Identifying temporal and spatial patterns of variation from multimodal data using MEFISTO. Nature Methods. 19 (2), 179-186 (2022).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati