
Method Article
Isolement de noyaux de tissu hépatique surgelé pour la multiomique unicellulaire
Dans cet article
Résumé
Ici, nous présentons un protocole pour isoler les noyaux des tissus hépatiques archivés surgelés pour le séquençage de l’ARN mononoyau, le séquençage ATAC et la multiomique articulaire (RNA-seq et ATAC-seq).
Résumé
Le foie est un tissu complexe et hétérogène responsable de l’exécution de nombreuses fonctions physiologiques critiques, telles que le maintien de l’homéostasie énergétique et le métabolisme des xénobiotiques, entre autres. Ces tâches sont effectuées grâce à une coordination étroite entre les cellules parenchymateuses hépatiques et non parenchymateuses. De plus, diverses activités métaboliques sont confinées à des zones spécifiques du lobule hépatique, un phénomène appelé zonation hépatique. Les progrès récents dans les technologies de séquençage unicellulaire ont permis aux chercheurs d’étudier l’hétérogénéité tissulaire à une résolution unicellulaire. Dans de nombreux tissus complexes, y compris le foie, des protocoles de dissociation enzymatiques et/ou mécaniques agressifs peuvent affecter négativement la viabilité ou la qualité des suspensions unicellulaires nécessaires pour caractériser complètement cet organe dans la santé et la maladie.
Cet article décrit un protocole robuste et reproductible pour isoler les noyaux des tissus hépatiques archivés congelés. Cette méthode produit des noyaux de haute qualité compatibles avec les approches omiques monocellulaires en aval, y compris le séquençage de l’ARN mononucléaire, le dosage de la chromatine accessible par transposase avec séquençage à haut débit (ATAC-seq), ainsi que les omiques multimodales (joint RNA-seq et ATAC-seq). Cette méthode a été utilisée avec succès pour l’isolement de noyaux provenant d’échantillons de foie congelés d’humains, de souris et de primates non humains sains et malades. Cette approche permet l’isolement impartial de tous les principaux types de cellules dans le foie et, par conséquent, offre une méthodologie robuste pour étudier le foie à la résolution unicellulaire.
Introduction
La génomique unicellulaire devient rapidement une méthodologie essentielle pour étudier la fonction hépatique et évaluer l’impact de l’hétérogénéité cellulaire sur les conditions de santé et de maladie1. Le développement rapide de la « multiomique » pour la mesure simultanée de différentes couches d’information et l’expansion parallèle de pipelines de calcul robustes ouvrent la voie à la découverte de types et de sous-types cellulaires jusqu’alors inconnus dans le foie normal et malade2.
La possibilité d’explorer des biobanques et des échantillons congelés archivés a considérablement augmenté les possibilités de revisiter et de découvrir le rôle des cellules non parenchymateuses 3,4,5 et d’étudier le rôle des hépatocytes polyploïdes au cours du vieillissement et dans les maladies chroniques 6,7,8,9 . Par conséquent, cet article décrit un protocole robuste et reproductible d’isolement d’un seul noyau pour les foies archivés surgelés (FF) qui est compatible avec le séquençage de l’ARN mononoyau en aval et le séquençage ATAC, ainsi qu’avec les omiques multimodales (ARN-seq conjoint et ATAC-seq) (Figure 1).
Ce flux de travail permet d’étudier le transcriptome et l’accessibilité de la chromatine de tous les types de cellules dans le foie, indépendamment de la taille ou de la fragilité de la cellule, dans des protocoles de dissociation enzymatique. Elle peut être réalisée avec de petites coupes de tissus (15-30 mg ou 5-10 mm3) à partir d’échantillons humains précieux ou de souris transgéniques. La détermination de la grande pureté de l’isolement des noyaux comprend la quantification et la mesure de la taille nucléaire, ce qui pourrait être corrélé à une augmentation de la taille des cellules et de la sénescence 10,11, et cette pureté est pertinente pour l’analyse de la ploïdie des hépatocytes 12 et des mécanismes transcriptionnels dépendants de la taille des cellules11,13,14,15 . De plus, les noyaux isolés des foies congelés conservent des informations précieuses sur la zonation du foie. Le flux de travail et la collecte de tissus permettent la validation de données génomiques unicellulaires ou d’autres analyses complémentaires, telles que l’immunohistochimie ou la transcriptomique spatiale du même tissu et du même individu. Par conséquent, cette approche peut être appliquée à de multiples maladies du foie et à des organismes modèles de manière systématique et fiable.
Protocole
Toutes les expérimentations sur les animaux ont été réalisées conformément à la législation allemande sur le bien-être animal et aux règlements du gouvernement de Haute-Bavière. Le logement des animaux a été approuvé conformément au § 11 de la loi allemande sur le bien-être des animaux et réalisé conformément à la directive 2010/63/UE.
1. Préparation des tissus
- Sacrifier une souris C57BL/6J mâle de 3 mois à une souris mâle de 22 mois par luxation cervicale. Posez l’animal sur une planche de dissection, fixez les extrémités avec des épingles et désinfectez l’abdomen avec de l’éthanol à 70%.
- Effectuer une nécropsie comme recommandé par Treuting et coll.16.
- Ouvrez l’abdomen jusqu’à la cage thoracique, visualisez le foie et utilisez une pince pour retirer soigneusement le foie sans percer les lobules.
- Extraire le foie intact en tenant le diaphragme avec une pince et en enlevant le tissu de connexion avec des ciseaux.
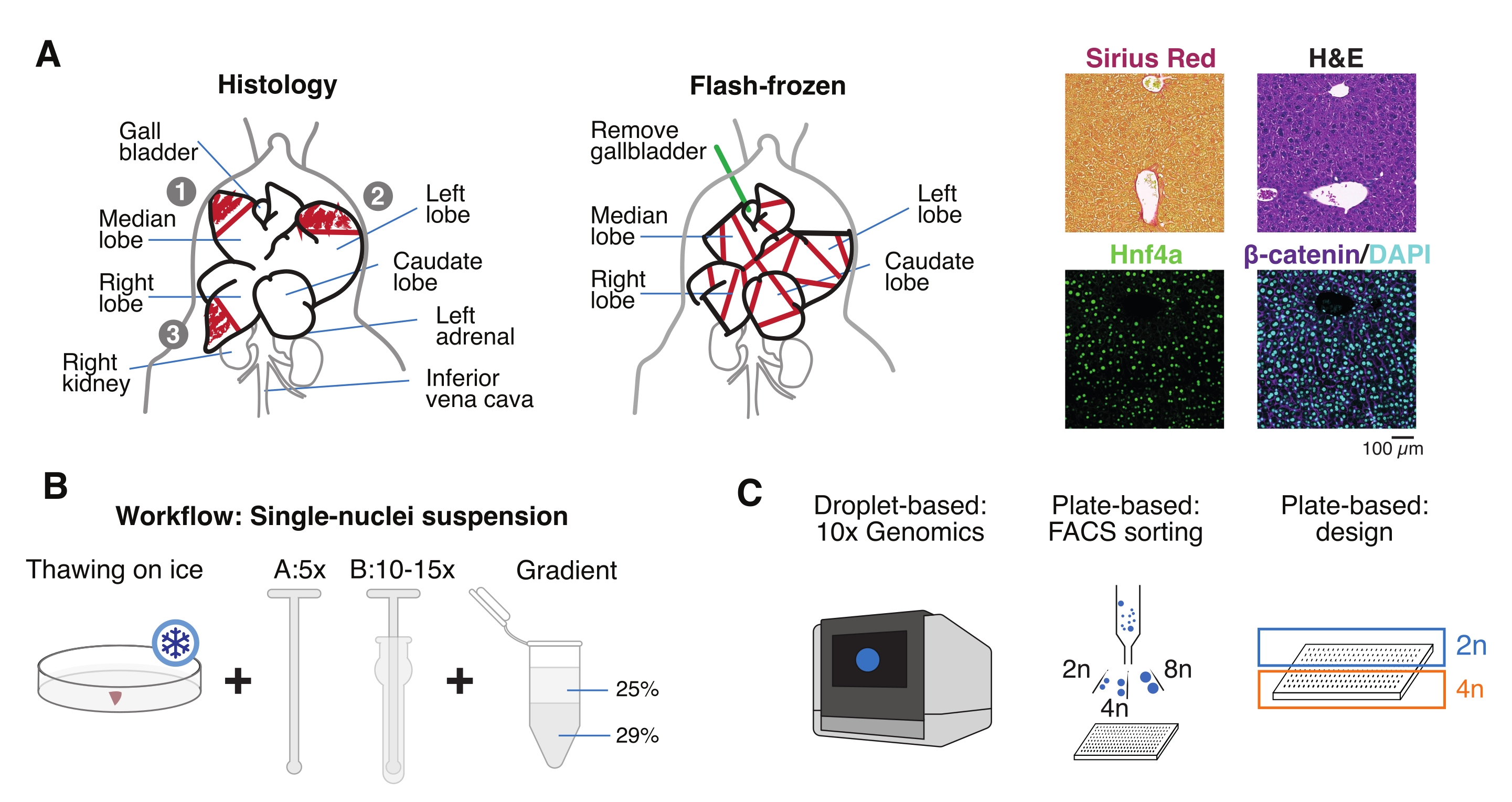
- Lavez l’organe dans une solution saline tamponnée au phosphate froid (PBS), séchez-le sur une serviette en papier propre et coupez les lobules du foie en plusieurs morceaux à des fins différentes: FF pour l’isolement mononucléique et multiomique; fixation au paraformaldéhyde (PFA à 10 %) pour l’incorporation à la paraffine; et/ou incorporé dans un composé à température de coupe optimale (OCT) pour des analyses histologiques plus approfondies (Figure 1A).
- Aliquote des morceaux de foie dans des cryoviales ou des tubes à vis de 5 mL, et immédiatement congeler dans de l’azote liquide. Entreposer les échantillons de foie congelés à −80 °C pour les expériences multiomiques mononoyaux en aval (Figure 1B, C).
REMARQUE: Le tissu cryoconservé peut être conservé en toute sécurité à -80 ° C pendant plusieurs années et doit toujours être transporté sur de la glace sèche à -80 ° C avant utilisation.
2. Isolement des noyaux
- Nettoyage de paillasse et préparation des tampons et consommables
- Nettoyez les paillasses et les pipettes de travail avec une solution de décontamination à 70 % d’éthanol et de RNase, ou utilisez des paillasses et des matériaux dédiés sans RNase.
- Prérefroidir le godet pivotant et les centrifugeuses à angle fixe, les tubes de 1,5 mL/2 mL et les plaques multipuits à 4 °C, comme indiqué ci-dessous.
- Prérefroidissez la pince à épiler sans RNase à usage unique utilisée pour la manipulation des tissus, un scalpel stérile jetable et une boîte de Petri sur de la glace sèche (-60 °C).
- Prérefroidir l’homogénéisateur en verre à boudin et les pilons sur glace (4 °C). Placez chaque pilon (A et B) dans un tube de 5 mL pour éviter tout contact direct avec la glace et toute contamination potentielle par la RNase.
- Préparer une solution diluante de milieu iodixanol (IDM) (tableau 1A) et l’utiliser pour obtenir des dilutions de 50 % et 29 % du milieu de gradient de densité du stock d’iodixanol à 60 % (tableau 1B et tableau 1C, respectivement).
- Prérefroidir tous les tubes. Pour chaque échantillon qui va être traité, préparez les tubes suivants:
- Préparer trois tubes de liaison à faible ADN de 1,5 mL (un pour l’homogénat tissulaire filtré, un deuxième contenant 250 μL d’une solution de dilution à 50% de iodixanol et un troisième pour la suspension de noyaux propres).
- Préparer un tube à fond rond de 2 mL contenant 500 μL d’une solution de dilution à 29 % d’iodixanol pour la séparation du gradient de densité.
- Préparer un tube conique de 15 mL pour le milieu d’isolement des noyaux 2 (NIM-2) et le tampon d’homogénéisation (HB).
- Préparer le milieu d’isolement des noyaux-1 (NIM-1) (tableau 1D) et l’utiliser pour fabriquer le NIM-2 (tableau 1E) et, par la suite, le HB (tableau 1F). Ajouter les deux inhibiteurs de l’ARNase juste avant utilisation, comme décrit ci-dessous dans le protocole.
- Préparez le tampon de stockage des noyaux (NSB) comme décrit dans le tableau 1G. Ajouter l’inhibiteur de l’ARN recombinant juste avant utilisation. Ajouter un inhibiteur de l’ARNase à base de protéines dans le NSB avant de l’utiliser pour le tri FACS (facultatif).
- Préparez un bécher de 500 ml avec de l’eau stérile pour faire tremper les homogénéisateurs et les pilons Dounce après l’homogénéisation des tissus pour le nettoyage et l’entretien optimaux des homogénéisateurs.
- Homogénéisation tissulaire
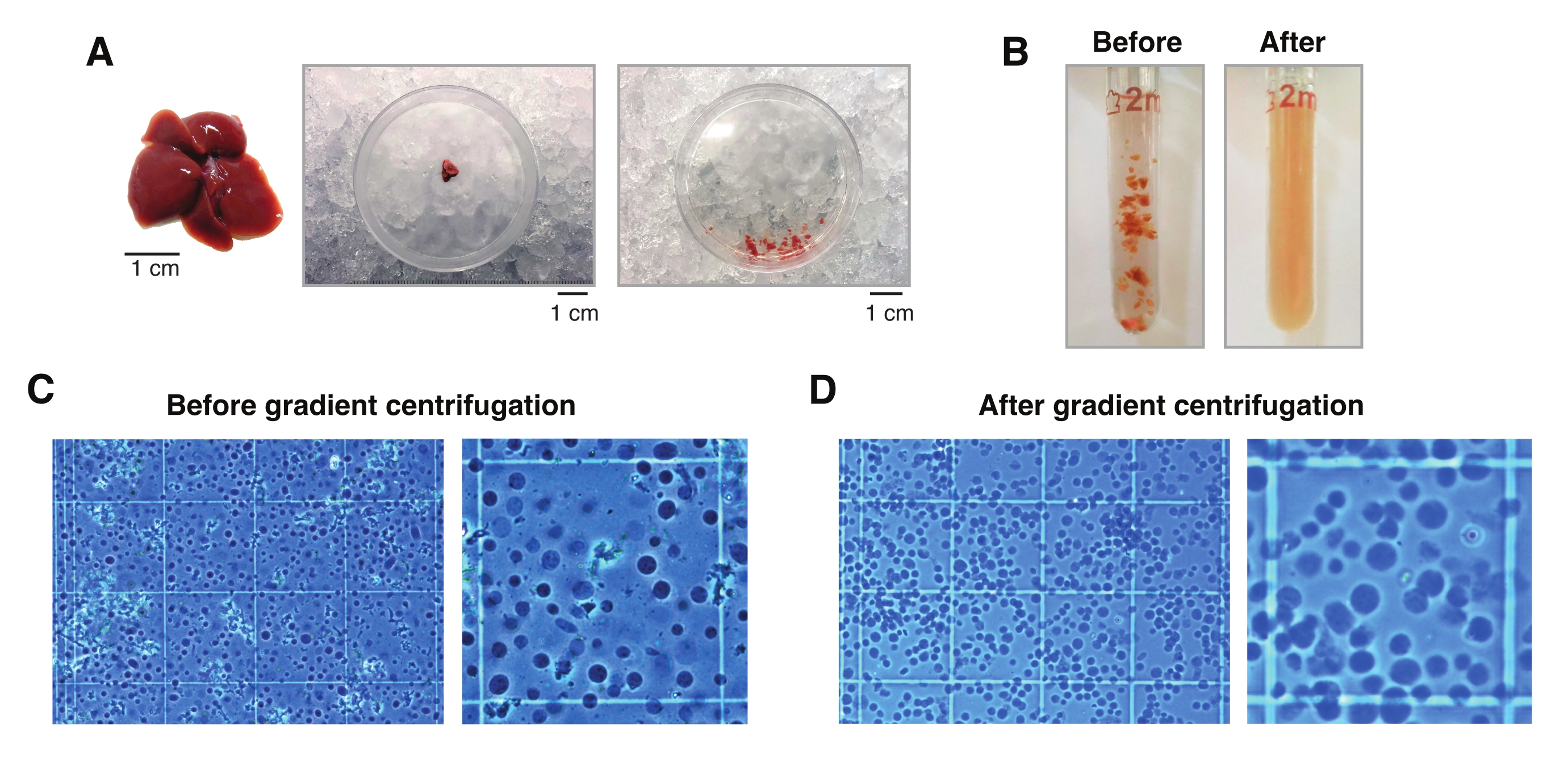
- Couper un morceau de tissu de 20 à 30 mg (ou 5 mm3) avec un scalpel prérefroidi à l’intérieur de la boîte de Petri sur de la glace sèche. Ensuite, transférer immédiatement la boîte de Petri sur de la glace humide (4 °C). Ajouter 1 mL de HB et, à l’aide du scalpel froid, hacher le tissu autant que possible pour lui permettre d’être facilement aspiré avec un embout à orifice large de 1 mL.
REMARQUE: Utilisez toujours des pointes à orifice large pour tous les transferts de tissus / noyaux. Comme alternative, des pointes de 1 mL peuvent être coupées avec un scalpel stérile sur un couvercle en plastique stérile pour générer de larges orifices (Figure 2A). - Prélever la suspension tissulaire et la transférer dans un homogénéisateur Dounce en verre prérefroidi de 2 ml (figure 2B).
- Lavez la boîte de Petri avec 0,5 à 1 ml de HB supplémentaire et recueillez tous les morceaux de tissu restants tout en gardant le tout sur la glace.
- Faites lentement et soigneusement cinq coups avec le pilon A lâche sur la glace. Évitez de créer des bulles en utilisant les mouvements de torsion du pilon lorsque vous le tirez de haut en bas. Assurez-vous que le pilon se déplace soigneusement du haut vers le bas de l’homogénéisateur à chaque coup.
- Par la suite, effectuez 10 à 15 coups lents avec le pilon B serré sur la glace. Évitez de créer des bulles.
REMARQUE: Il est recommandé d’inspecter visuellement les noyaux au microscope après 10 coups avec le pilon B pour déterminer si d’autres coups sont nécessaires. Pour ce faire, utilisez une coloration au bleu de trypan (rapport 1:1 du trypan à l’échantillon) et un hémocytomètre manuel (p. ex., mélanger 10 μL de bleu de trypan avec 10 μL de suspension de noyaux, et utiliser 10 μL du mélange pour l’examen au microscope; Figure 2C). - Filtrer l’homogénat à travers une crépine cellulaire de 50 μm tout en le transférant dans un tube prérefroidi de 1,5 mL. Utiliser plus d’un filtre et/ou tube pour les homogénats contenant une grande quantité d’amas de tissu conjonctif.
- Rincer l’homogénéisateur et les filtres utilisés avec 0,5 à 1 mL supplémentaires de HB pour bien recueillir tout l’homogénat tissulaire. Passez à la centrifugation du gradient de densité.
- Couper un morceau de tissu de 20 à 30 mg (ou 5 mm3) avec un scalpel prérefroidi à l’intérieur de la boîte de Petri sur de la glace sèche. Ensuite, transférer immédiatement la boîte de Petri sur de la glace humide (4 °C). Ajouter 1 mL de HB et, à l’aide du scalpel froid, hacher le tissu autant que possible pour lui permettre d’être facilement aspiré avec un embout à orifice large de 1 mL.
- Centrifugation à gradient de densité
- Centrifuger l’homogénat filtré dans une centrifugeuse prérefroidie à angle fixe à 1 000 × g pendant 8 min à 4 °C.
- Pendant que l’échantillon tourne, préparer un tube de 1,5 mL contenant 250 μL de dilution d’iodixanol à 50 % et un tube de 2 mL contenant 500 μL de dilution d’iodixanol à 29 %. Gardez les deux tubes sur la glace.
- Après centrifugation, aspirer le surnageant sans déranger la pastille à l’aide d’une pompe à vide.
REMARQUE: L’utilisation d’un pipetage manuel compromettra la qualité de la suspension finale des noyaux. - Ajouter 250 μL de HB à la pastille à l’aide de l’embout de la pipette à orifice large de 1 mL et remettre en suspension très lentement.
- Transvaser 250 μL de la suspension de noyaux dans un tube prérefroidi de 1,5 mL contenant 250 μL de dilution d’iodixanol à 50 % et mélanger doucement mais soigneusement pour obtenir une suspension d’iodixanol/noyaux à 25 %.
- Transvaser 500 μL de la suspension d’iodixanol/noyaux à 25 % dans un tube prérefroidi de 2 mL contenant 500 μL de dilution d’iodixanol à 29 %.
REMARQUE : Les 500 μL de la suspension d’iodixanol/noyaux à 25 % doivent être déposés doucement sur les 500 μL de solution d’iodixanol à 29 % de sorte que les mélanges d’iodixanol à 25 %/29 % présentent une séparation de phase claire. Utilisez le côté de la paroi du tube avec l’embout de la pipette positionné à un angle de 45° pour créer cette interface de dégradé. Dès lors, le tube doit être manipulé avec douceur afin de ne pas perturber cette pente. - Centrifuger le tube dans une centrifugeuse à godet pivotant prérefroidie à 12 500 g pendant 20 minutes avec le frein réglé sur OFF.
- Juste avant la fin de l’étape de centrifugation, ajouter les inhibiteurs de l’ARNis au tampon NSB lorsque vous passez aux pipelines scRNA-seq (voir Tableau 1G).
- Après centrifugation, aspirer le surnageant sans déranger la pastille à l’aide d’une pompe à vide.
REMARQUE: L’utilisation d’un pipetage manuel compromettra la qualité de la suspension finale des noyaux. - À l’aide de l’embout de la pipette à orifice large de 1 mL, remettre doucement la pastille en suspension dans 100 à 300 μL de NSB et transférer la suspension de noyaux dans un tube propre et prérefroidi de 1,5 mL.
- Compter les noyaux à l’aide d’une solution de bleu de trypan (rapport 1:1 du trypan à l’échantillon) et d’un hémocytomètre manuel (p. ex., mélanger 10 μL de bleu de trypan avec 10 μL de suspension de noyaux, et utiliser 10 μL du mélange pour le comptage; Figure 2D).
- Utilisez immédiatement la suspension de noyaux obtenus pour le test génomique mononoyau.
REMARQUE : La suspension de noyaux dans NSB peut être conservée au réfrigérateur à 4 °C jusqu’à 1 semaine pour une analyse plus approfondie par cytométrie en flux et/ou imagerie en flux, mais pas pour le snRNA-seq ou le snATAC-seq.
3. Tri des noyaux pour le profilage de la ploïdie des hépatocytes ou des approches de séquençage bien fondées
- Pour le tri cellulaire basé sur la cytométrie en flux, filtrer la suspension de noyaux à travers un filtre de 50 μm dans un tube FACS prérefroidi de 5 mL.
- Utilisez un trieur de cytométrie en flux muni d’une buse de 100 μm. Chargez le tube FACS sur le trieur et prévisualisez l’échantillon.
- Configurez la stratégie de contrôle pour le tri des noyaux, en commençant par une porte de dispersion en traçant la zone de diffusion vers l’avant par rapport à la zone de dispersion latérale (FSC-A/SSC-A), suivie de Hoechst-Height versus Hoechst-Area (porte des noyaux), puis de Hoechst-Width par rapport à Hoechst-Area (porte singlets). Visualisez le profil de ploïdie des noyaux sur l’histogramme de la zone de Hoechst.
REMARQUE: Pour une meilleure résolution de crête, visualisez le canal Hoechst (450/50) sur l’échelle linéaire (Figure 3A). - Pour le tri en plaques de 96/384 puits, régler le délai de gouttelettes et optimiser l’alignement de la plaque à l’aide de la méthode colorimétrique avec substrat benzidine-peroxydase de raifort (TMB-HRP), comme décrit précédemment6.
- Réglez le refroidissement de l’échantillon et le porte-plaque pour qu’ils refroidissent à 4 °C, avec la rotation de l’échantillon activée à 300 tr/min.
- Trier les noyaux individuels à une concentration d’échantillon de ~1 x 105 noyaux/mL et avec un débit de 200-500 événements/s.
4. Inspection visuelle et quantification des paramètres nucléaires par cytométrie d’imagerie (facultatif)
- Charger un tube de 0,5 mL contenant 50 μL de noyaux en suspension à une concentration de 2 × 107 noyaux/mL.
- Mettre en place une porte tous événements basée sur le rapport d’aspect par rapport au canal de fluorescence de Hoechst pour visualiser le profil de ploïdie des noyaux (Figure 3B).
- Acquérir l’échantillon à un grossissement de 40x en utilisant le fond clair (BF) et le canal de fluorescence de Hoechst.
- Inspectez et quantifiez les noyaux 2n et 4n à l’aide de la mesure du fond clair et de l’intensité de fluorescence de Hoechst à l’aide d’un logiciel de cytométrie d’imagerie (Figure 3C).
5. Construction et séquençage de bibliothèques d’ARN mononoyau, d’ATAC ou multiome
- Pour les approches de séquençage d’ARNn à base de gouttelettes, chargez la suspension de noyaux purifiés directement dans le dispositif microfluidique pour un partitionnement parallèle automatisé et un codage à barres moléculaire17.
- Une fois l’exécution microfluidique terminée dans le dispositif de séparation unicellulaire, prélever les noyaux encapsulés dans des billes de gel, incuber et nettoyer comme décrit précédemment dans les directives du fabricant17.
- Effectuer 11 cycles de réaction en chaîne par polymérase (PCR) pour la préamplification de l’ADNc à l’aide du programme suivant : 3 min à 98 °C, (15 s à 98 °C, 20 s à 63 °C et 1 min à 72 °C) x 11, 1 min à 72 °C, et maintenir à 4 °C. Poursuivre avec une réparation finale et une étape de queue A et ligature de l’adaptateur, comme indiqué par le fabricant17. Pour la construction ultérieure de la bibliothèque finale d’expression génique, effectuer 10 cycles de PCR à l’aide du programme suivant : 45 s à 98 °C, (20 s à 98 °C, 30 s à 54 °C, 20 s à 72 °C) x 10, 1 min à 72 °C, et maintenir à 4 °C.
- Séquencez les bibliothèques obtenues à une profondeur de lecture de ~20 000-50 000 lectures moyennes par noyau.
- Pour le séquençage multiomique articulaire (ARN + ATAC) basé sur les gouttelettes, incuber les noyaux du foie dans un tampon de lyse pendant 5 min, puis les marquer pendant 1 h, comme décrit précédemment18.
- Chargez les noyaux marqués directement dans un dispositif microfluidique pour un partitionnement parallèle automatisé et un codage à barres moléculaire.
- Une fois l’exécution microfluidique terminée, prélever les noyaux encapsulés dans des billes de gel, incuber et nettoyer comme décrit par le fabricant18.
- Effectuer six cycles de PCR pour l’étape de préamplification de l’ADNc à l’aide du programme suivant : 5 min à 72 °C, 3 min à 98 °C, (20 s à 98 °C, 30 s à 63 °C, 1 min à 72 °C) x 6, 1 min à 72 °C, et maintenir à 4 °C.
- Prélever 35 μL de l’échantillon préamplifié et effectuer une amplification de l’ADNc comme suit : 3 min à 98 °C, (15 s à 98 °C, 20 s à 63 °C, 1 min à 72 °C) x 6, 1 min à 72 °C, et maintenir à 4 °C. Poursuivre la réparation finale et l’étape de dérive en A et la ligature de l’adaptateur, comme indiqué par le fabricant18 . Pour la PCR d’indexation finale de l’échantillon suivante, effectuer 15 cycles de PCR comme suit: 45 s à 98 °C, (20 s à 98 °C, 30 s à 54 °C, 20 s à 72 °C) x 15, 1 min à 72 °C, et maintenir à 4 °C.
- Pour la construction de bibliothèques ATAC, utiliser 40 μL et amplifier pendant six cycles de PCR pour l’indexation des échantillons à l’aide du programme suivant : 45 s à 98 °C, (20 s à 98 °C, 30 s à 67 °C, 20 s à 72 °C) x 6, 1 min à 72 °C, et maintenir à 4 °C.
- Séquencer les bibliothèques d’expression génique multiome obtenues à une profondeur de lecture minimale de 20 000 paires de lecture par noyau et les bibliothèques ATAC multiomes à une profondeur de lecture minimale de 25 000 paires de lecture par cellule, comme recommandé par le fabricant18.
Résultats
Ce flux de travail pour l’isolement mononucléaire à partir d’échantillons de foie congelés est adapté à la multiomique mononucléique et repose sur trois étapes principales, qui peuvent être résumées comme suit : i) prélèvement d’échantillons pour l’analyse parallèle de l’hétérogénéité cellulaire et de l’architecture tissulaire, ii) suspension mononucléocompacte et iii) multiomique mononoyau (Figure 1 ). Les foies extraits sont disséqués à partir de souris euthanasiées et coupés en morceaux pour une inspection histologique pour l’incorporation de paraffine, la cryosection, ou les deux. Les autres morceaux coupés sont immédiatement surgelés dans de l’azote liquide pour l’isolement mononucléaire en aval pour les analyses multiomiques. Ce système de collecte de tissus permet à l’utilisateur de valider davantage les données omiques mononucléiques sur des coupes de tissus du même individu, complétant ainsi l’ensemble de données par une transcriptomique spatiale ou par des analyses immunohistochimiques, si nécessaire.
L’examen microscopique des noyaux extraits de foies congelés avec la méthode décrite ici montre que l’étape de centrifugation du gradient de densité facilite grandement l’élimination des débris cellulaires et tissulaires indésirables (Figure 2C,D). De plus, cette méthodologie préserve tous les niveaux de ploïdie, qui peuvent être validés et quantifiés par des analyses cytométriques (Figure 3).
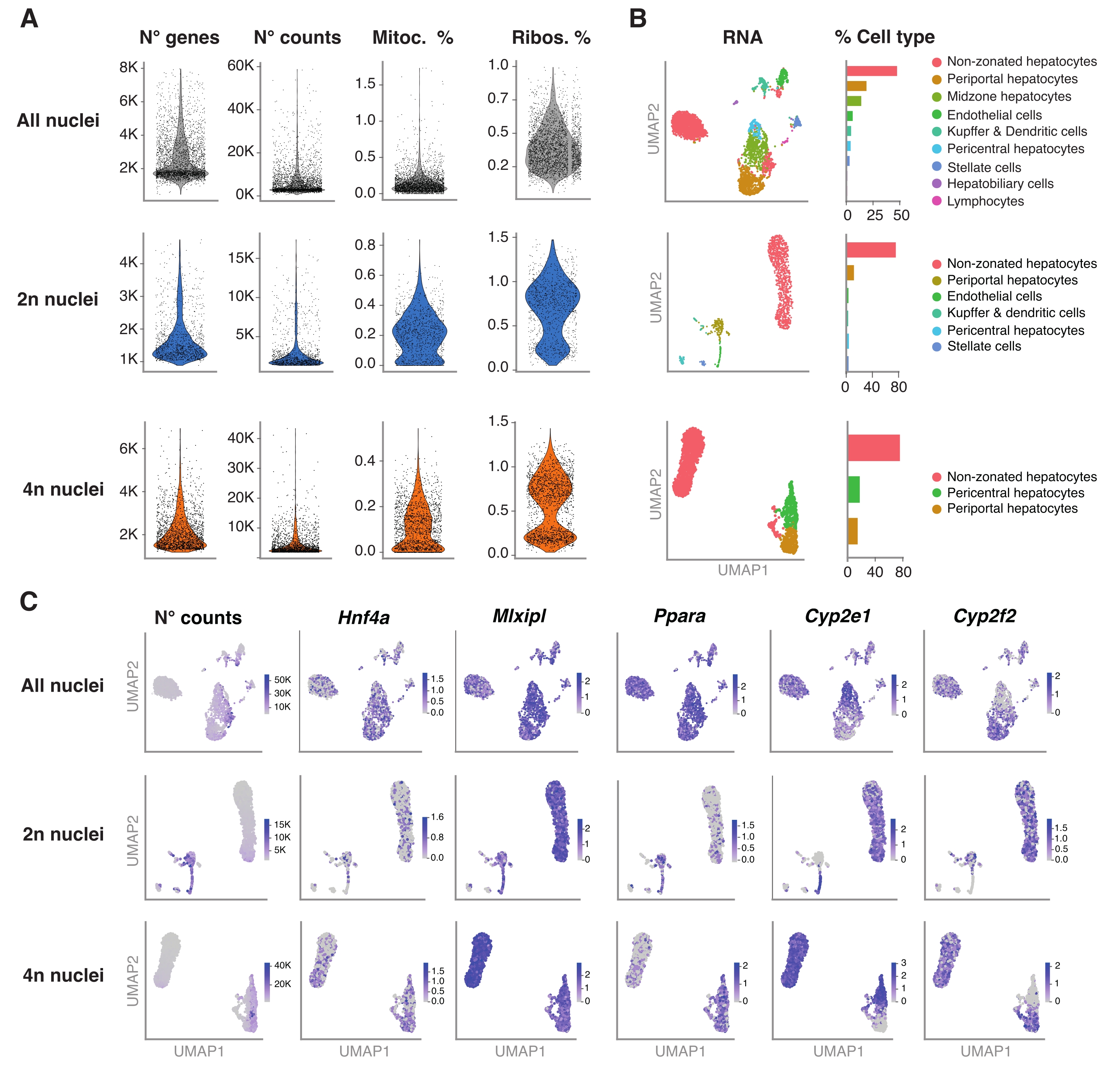
Pour valider davantage les performances de ce protocole, nous avons effectué un snRNA-seq à base de gouttelettes sur des noyaux non triés et triés par FACS et analysé les données suivant le pipeline Seurat. En bref, les noyaux uniques extraits ont été préparés pour le snRNA-seq à base de gouttelettes, comme décrit précédemment19. Pour le snRNA-seq des noyaux 2n et 4n ou des niveaux supérieurs de ploïdie, 11 cycles ont été utilisés pour l’étape d’amplification de l’ADNc et 10 cycles pour la construction finale de la bibliothèque d’expression génique. Les bibliothèques résultantes ont été séquencées à une profondeur de lecture de ~25 000-39 000 lectures moyennes par noyau. Les lectures de noyaux uniques obtenues ont été mappées au génome de souris GRCm39/mm39. Lors de l’exécution du pipeline de prétraitement, la commande - -include-intron a été ajoutée pour déterminer l’inclusion et la quantification de l’ARN messager non épissé (ARNm) présent dans les noyaux. L’algorithme EmptyDrops incorporé dans l’aligneur a filtré et supprimé les gouttelettes vides.
Le package R Seurat (version 4.1.1) a été utilisé pour calculer les mesures de contrôle de la qualité (CQ) à l’aide de la matrice de comptage de l’identifiant moléculaire unique (UMI) telle que sortie par le pipeline d’analyse snRNA-seq. Les comptes avec moins de 100 caractéristiques (gènes) et moins de 10 cellules ont été retirés. Les noyaux ont été filtrés selon des seuils de CQ identifiés : nombre minimum de gènes = 200 et nombre maximal de gènes = 8 000, fraction mitochondriale <1 % et fraction ribosomique <2 %. Les 3 000 principaux gènes hautement variables (HVG) ont été utilisés pour l’analyse en composantes principales (ACP), telle que mise en œuvre dans Seurat. Le clustering basé sur des graphiques a été effectué après la saisie des 15 principales dimensions du PC résultant de l’analyse PCA. Pour regrouper les cellules, nous avons appliqué la technique d’optimisation de la modularité (algorithme de Louvain) avec un paramètre de résolution défini sur 0,5. Pour visualiser et explorer cet ensemble de données, nous avons effectué une réduction dimensionnelle non linéaire, à savoir UMAP (Uniform Manifold Approximation and Projection). L’identité de chaque grappe a été attribuée en fonction de la connaissance préalable des gènes marqueurs 6,20,21.
La figure 4A montre des mesures de haute qualité à partir des données obtenues à l’aide de cette méthode d’extraction des noyaux. UMAP représente le nombre de numérations dans tous les noyaux et les principaux types de cellules qui ne peuvent être identifiés avec certitude qu’avec le transcriptome nucléaire et un séquençage relativement peu profond (~25 000-40 000 lectures moyennes par cellule) (Figure 4B). Cette approche permet d’étudier les facteurs de transcription spécifiques au foie tels que Hnf4a, Mlxipl et Ppara, ainsi que les gènes cibles en aval impliqués dans le métabolisme des xénobiotiques (c.-à-d. Cyp2e1 et Cyp2f2) (Figure 4C). Il convient de noter que les noyaux extraits ont conservé des informations critiques sur la zonation du foie, comme le montre le modèle complémentaire de gènes caractéristiques tels que le Cyp2e1 péricentral et le Cyp2f2 périportail (Figure 4C).
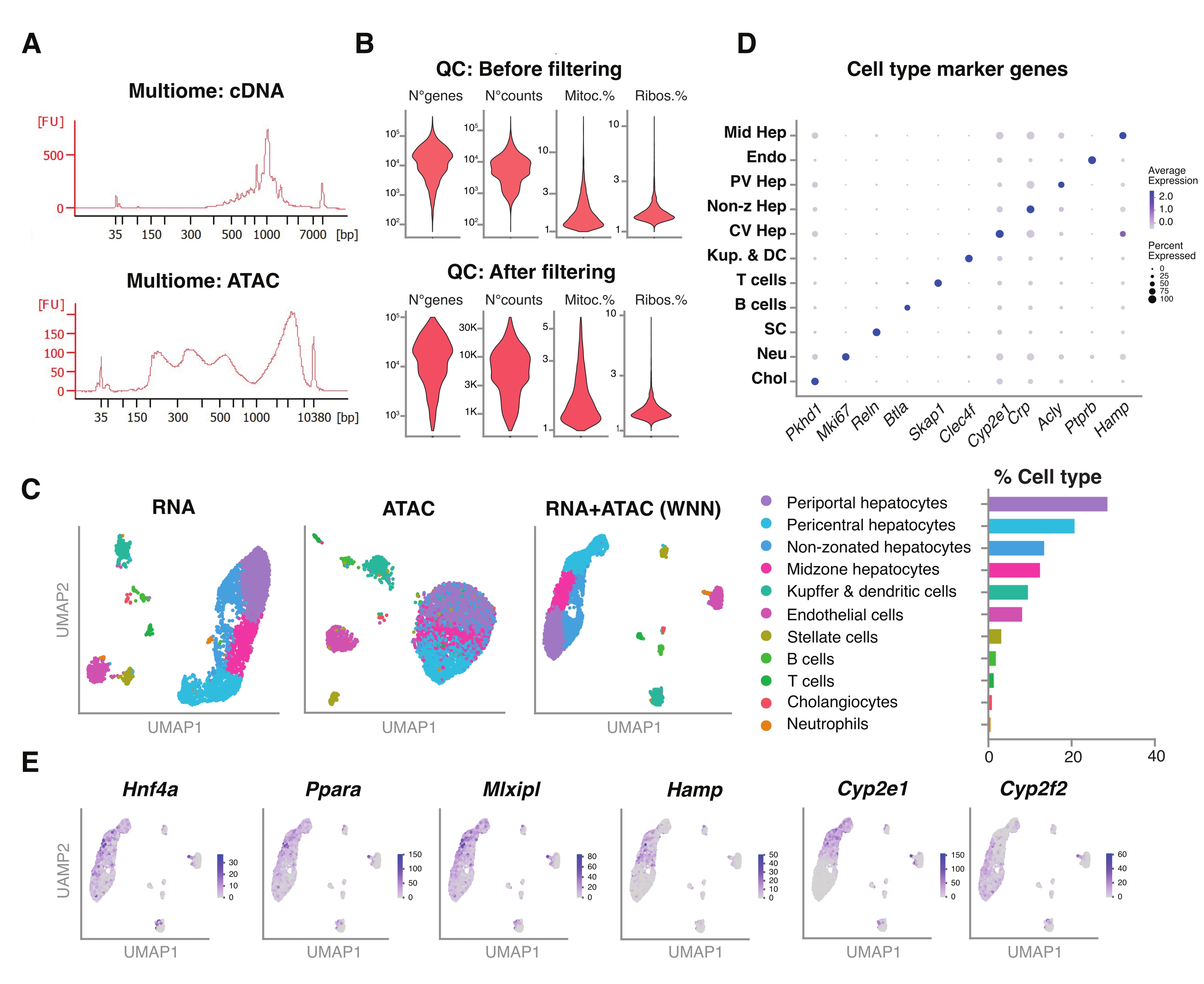
Nous avons en outre évalué la compatibilité des noyaux non triés extraits à l’aide de cette méthode avec des tests multiomiques plus récents pour le profilage simultané du paysage épigénomique (ATAC) et de l’expression génique (ARN) dans les mêmes noyaux uniques18. Nous avons optimisé le temps d’incubation de la lyse des noyaux à 5 minutes avant la transposition. Les bibliothèques de séquençage ont été construites en exécutant six cycles de PCR pour la préamplification de l’ADNc. À partir de l’échantillon préamplifié, 35 μL ont été prélevés et amplifiés par 15 cycles de PCR pour l’indexation de l’échantillon afin de construire la bibliothèque d’expression génique. Un électrophérogramme représentatif de la trace d’ADNc est illustré à la figure 5A (en haut). Pour la construction des bibliothèques ATAC, 40 μL d’échantillon préamplifié ont été utilisés et exécutés pendant six cycles de PCR supplémentaires pour l’indexation des échantillons, avec l’électrophérogramme représentatif de la trace d’ADN illustré à la figure 5A (en bas). La bibliothèque d’expression génique (ARN) résultante a été séquencée à une profondeur de 44 600 lectures par noyau et la bibliothèque ATAC à une profondeur de 43 500 lectures par noyau (Figure 5B).
À l’instar du protocole de séquençage à base de gouttelettes à modalité unique décrit ci-dessus, la cartographie des lectures, l’alignement, l’élimination des gouttes vides et le comptage des fragments ont été effectués conformément aux directives standard, comme décrit précédemment, en utilisant le génome de référence GRCm39/mm3918. À l’aide des packages Seurat et Signac22, nous avons effectué une analyse du « plus proche voisin pondéré » (WNN) pour de multiples mesures des deux modalités (ARN + ATAC) (Figure 5C), ce qui nous a permis d’identifier et d’annoter les types de cellules hépatiques majeures et mineures sans biais importants dus à la taille des cellules ou à la fragilité nucléaire (Figure 5D). Le pipeline, tel que publié par le laboratoire Satija22,23, comprend des étapes standard de contrôle qualité - prétraitement et réduction dimensionnelle - effectuées sur les deux tests indépendamment. Pour avoir une bonne représentation de la combinaison pondérée des modalités RNA-seq et ATAC-seq, le graphique WNN a été tracé et utilisé pour la visualisation, le regroupement et l’annotation UMAP basés sur les gènes marqueurs précédemment identifiés 6,20,21. À l’instar des tests utilisant une seule modalité, nous avons également détecté des régulateurs transcriptionnels en amont (Hnf4a, Ppara, Mlxipl) et des gènes caractéristiques de la zonation hépatique (Hamp, Cyp2e1 et Cyp2f2) (Figure 5E).

Figure 1 : Vue d’ensemble expérimentale, flux de travail et applications génomiques unicellulaires. (A) Représentation illustrative de l’échantillonnage tissulaire pour l’histologie (à gauche, trois sections sont sélectionnées pour l’enrobage de la paraffine et/ou la cryosection), collecte de tissus surgelés pour la génomique unicellulaire (au milieu) et analyses immunohistochimiques et immunofluorescence représentatives (à droite); barre d’échelle = 100 μm. (B) Étapes critiques pour des suspensions mononucléaires de haute qualité. (C) Les suspensions de noyaux peuvent être chargées sur une puce de chrome 10x ou utilisées pour le tri FACS et les approches basées sur des plaques. Abréviations : H & E = hématoxyline et éosine; DAPI = 4',6-diamidino-2-phénylindole; FACS = tri cellulaire activé par fluorescence. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2 : Dissection hépatique et homogénéisation tissulaire en verre de boudeur. (A) Foie murin représentatif d’une souris C57BL6/J âgée de 3 mois (à gauche); La section du foie utilisée pour les isolements mononucléaux avant (milieu) et après hachage du tissu avec le scalpel (à droite). Barres d’échelle = 1 cm. (B) Images illustratives de 2 mL d’homogénéisation du verre drôle avant les coups avec le pilon « lâche » A (à gauche) et après le pilon « serré » B (à droite). Surveillance de l’homogénéisation tissulaire à l’aide d’un hémocytomètre (C) avant centrifugation par gradient et (D) après centrifugation par gradient. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 3 : Tri cellulaire activé par fluorescence et cytométrie en flux d’imagerie pour la caractérisation des noyaux à haut débit. (A) Stratégie de contrôle pour le tri FACS mononoyau dans une plaque pour l’interrogation de différents niveaux de ploïdie. Barrière de diffusion basée sur la zone de diffusion vers l’avant par rapport à la zone de diffusion latérale définie pour exclure les débris; porte des noyaux basée sur la hauteur de Hoechst par rapport à la zone de Hoechst incorporant plusieurs populations de noyaux; porte singulière basée sur l’ensemble Hoechst-Width versus Hoechst-A pour la discrimination doublet; l’histogramme Hoechst-A permet de visualiser le profil de ploïdie des noyaux. (B) Quantification par cytométrie d’imagerie représentative de tous les événements (à gauche) et uniques (à droite), montrant les noyaux diploïdes et tétraploïdes. (C) Images en fond clair et en Hoechst des noyaux 2n et 4n et leur quantification par cytométrie d’imagerie. Abréviations : FACS = tri cellulaire activé par fluorescence; FSC-A = zone de diffusion vers l’avant; SSC-A = zone de diffusion latérale; Hoechst-H = Hoechst-Height; Hoechst-A = Hoechst-Area; Hoechst-W = Hoechst-Width. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 4 : Caractérisation profonde de la ploïdie des hépatocytes avec séquençage de l’ARNn. (A) Tracés de violon montrant le nombre de gènes, le nombre de gènes, le pourcentage de gènes mitochondriaux et le pourcentage de gènes ribosomales détectés. (B) UMAP démontrant les types de cellules détectés à l’aide de snRNA-seq (à gauche), avec la quantification exprimée en pourcentage de noyaux (à droite). (C) UMAP illustre le nombre de numérations et indique l’expression de gènes spécifiques aux hépatocytes. Tous les noyaux (rangée du haut), 2n noyaux (rangée du milieu) et 4n noyaux (rangée du bas). Abréviations : snRNA-seq = RNA-seq mononucléaire; UMAP = Approximation et projection de variétés uniformes; Mitoc. = gènes mitochondriaux; Ribos. = gènes ribosomiques. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 5 : Contrôle de la qualité et analyse de la multiomique (ARN-seq conjoint et ATAC-seq) à partir de jeunes foies congelés et archivés. (A) Traces électrophorétiques automatiques représentatives obtenues après le pipeline multiomique montrant le produit de poids moléculaire après synthèse d’ADNc et ATAC. (B) Tracé de violon montrant le nombre de comptes pour ATAC-seq, RNA-seq, le pourcentage de gènes mitochondriaux et le pourcentage de gènes ribosomales avant et après filtrage. (C) UMAP montre l’expression génique de RNA-seq (gauche), ATAC-seq (milieu) et modalités articulaires-RNA-seq et ATAC-seq (droite). (D) Différents types de cellules sont annotés de différentes couleurs, avec l’expression de gènes spécifiques aux hépatocytes indiqués. (E) Diagramme des caractéristiques montrant l’expression spécifique au groupe des gènes indiqués dans les types de cellules indiqués. Abréviations : ATAC-seq = dosage de la chromatine accessible par la transposase avec séquençage à haut débit; Hep moyenne = hépatocytes de la zone moyenne; Endo = cellules endothéliales; PV Hep = hépatocytes périportals; Hep non-z = hépatocytes non zonés; CV Hep = hépatocytes péricentraux; Kup & DC = Kupffer & cellules dendritiques; SC = cellules étoilées, Neu = neutrophiles; Chol = cholangiocytes; Mitoc. = gènes mitochondriaux; Ribos. = gènes ribosomiques. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
| Réactif | Stock | 10 mL | 15 mL | 50 mL | ||
| (A ) Milieu iodixanol (IDM) | ||||||
| 250 mM de saccharose | 1M | 12,5 mL | ||||
| 150 mM KCl | 2M | 3,75 mL | ||||
| 30 mM MgCl2 | 1M | 1,5 mL | ||||
| 60 mM tampon Tris pH 8,0 | 1M | 3 mL | ||||
| Eau sans RNase ultrapure | 29,25 mL | |||||
| b) 50 % de MNI | ||||||
| Iodixanol | 60% | 12,5 mL | ||||
| IDM | 2,5 mL | |||||
| (C) 29 % IDM | ||||||
| Iodixanol | 60% | 7,25 mL | ||||
| IDM | 7,75 mL | |||||
| d ) Milieu d’isolement des noyaux-1 (NIM-1) | ||||||
| 250 mM de saccharose | 1M | 12,5 mL | ||||
| 25 mM KCl | 2M | 0,625 mL | ||||
| 5 mM MgCl2 | 1M | 0,25 mL | ||||
| 10 mM tampon Tris pH 8,0 | 1M | 0,5 mL | ||||
| Eau ultrapure sans ARNASE | 36,125 mL | |||||
| e ) Milieu d’isolement des noyaux-2 (NIM-2) | ||||||
| Tampon NIM-1 | 9,99 mL | |||||
| Dithiothréitol (DTT) | 1 mM | 0,01 mL | ||||
| Comprimé inhibiteur de protéase (sans EDTA) | 1 | 1 comprimé | ||||
| f ) Tampon d’homogénéisation (HB) | ||||||
| Tampon NIM-2 | 9,697 mL | |||||
| Inhibiteur de la RNase recombinante | 40 U/μL | 0,1 mL | ||||
| Inhibiteur de l’ARNase à base de protéines (SUPERase•IN) | 20 U/μL | 0,1 mL | ||||
| 0,1 % Triton-X | 10% | 0,1 mL | ||||
| 3 μg/mL Hoechst 33342 | 10 mg/mL | 0,003 mL | ||||
| g ) Tampon de stockage des noyaux (NSB) | ||||||
| 166,5 mM de saccharose | 1M | 1,665 mL | ||||
| 5 mM MgCl2 | 1M | 0,05 mL | ||||
| 10 mM Tris pH 8,0 | 1M | 0,1 mL | ||||
| Inhibiteur de la RNase recombinante | 40 U/μL | 0,1 mL | ||||
| Inhibiteur de la RNase à base de protéines (SUPERase•IN) * | 20 U/μL | 0,1 mL | ||||
| Eau sans RNase ultrapure | 8,085 mL | |||||
| * Facultatif (pour le tri FACS uniquement) | ||||||
Tableau 1 : Recettes de solution. A) Préparation du milieu iodixanol (IDM); B) dilution à 50 % de la solution d’iodixanol; (C) dilution à 29 % de la solution d’iodixanol. D) Préparation du milieu d’isolement des noyaux-1 (NIM-1). E) Préparation du milieu d’isolement des noyaux-2 (NIM-2). F) Préparation du tampon d’homogénéisation (HB). G) Préparation du tampon de stockage des noyaux (NSB).
Discussion
La dissection de la composition cellulaire du foie par séquençage de l’ARN unicellulaire ou mononucléaire permet de mieux comprendre le développement et la progression de la maladie hépatique 3,4,5,24. L’isolement unicellulaire du foie prend beaucoup de temps et nécessite des protocoles qui impliquent une dissociation mécanique ou enzymatique sévère25,26,27. Il est largement admis que chaque tissu nécessite une évaluation systématique pour déterminer le protocole optimal de dissociation tissulaire, ainsi qu’une méthode de stockage appropriée pour capturer les types de cellules fragiles ou les noyaux28. Selon la disponibilité du tissu, la maladie d’intérêt, le stade de développement ou l’organisme modèle, la préparation d’une suspension mononucléaire pour le traitement en aval pourrait être une méthodologie plus appropriée que l’utilisation de suspensions unicellulaires. Il est important de noter que dans le foie, scRNA-seq et snRNA-seq ont montré une forte corrélation entre l’ARNm nucléaire et cytoplasmique, suggérant que les deux approches présentent des informations complémentaires 2,3,4,6,29.
Cet article fournit une isolation mononucléaire normalisée, robuste et reproductible à partir d’échantillons de foie congelés et archivés de souris et d’autres espèces, y compris les humains et les macaques. Cette méthode peut être utilisée pour les souris de type sauvage nourries avec du chow et un régime riche en graisses (HFD) et pour des modèles murins de fibrose hépatique utilisant à la fois des approches génomiques mononucléiques à base de plaques de puits et de gouttelettes6. Cette méthode repose sur le protocole décrit à l’origine pour le tissu cérébral par Krishnaswami et al.30 avec des modifications supplémentaires adaptées au foie surgelé. Une homogénéisation optimale libère la plupart des noyaux du tissu sans affecter négativement l’intégrité de la membrane nucléaire. Cependant, un dédoublement excessif peut endommager les noyaux fragiles et diminuer leur qualité globale. Les foies jeunes et / ou gras ne nécessitent généralement que 5 coups avec le pilon A et 10 coups avec le pilon B, tandis que les foies âgés et / ou fibrotiques peuvent nécessiter 15 coups avec le pilon B mais pas plus. Il n’est donc pas recommandé d’effectuer plus de coups au-delà des nombres indiqués ici. Un dépassement pourrait affecter négativement la qualité de la suspension monopolaire et augmenter la quantité d’ARN ambiant. Par la suite, cela pourrait entraîner la nécessité d’effectuer des étapes de filtrage informatique supplémentaires lors des analyses de données en aval.
Le protocole présenté ici est polyvalent et peut être ajusté à différentes affections hépatiques chez les souris jeunes (3 mois) et âgées (24 mois). Étant donné que nous avons constaté qu’une plus grande section du foie est nécessaire pour les tissus anciens, HFD et fibrotiques, la taille du tissu disponible pour le traitement peut constituer une limitation pour certains utilisateurs ayant de plus petites quantités de matériel biologique de départ. Cependant, la purification par gradient est fortement recommandée pour le traitement immédiat des échantillons avec des tests génomiques basés sur des gouttelettes. Si les noyaux doivent être triés FACS en plaques à 96/384 puits pour des essais bien basés, la purification du gradient peut être omise. Nous encourageons les utilisateurs à effectuer la purification par gradient s’il y a suffisamment d’échantillons de tissus pour obtenir la concentration recommandée de noyaux pour le tri FACS (c.-à-d. ~1 × 105 noyaux/mL).
Le foie est caractérisé par la nature polyploïde des hépatocytes9, mais le rôle de la ploïdie hépatocytes dans la physiologie normale et la maladie n’est pas encore clair. Il existe de plus en plus de preuves indiquant que la ploïdie fournit une variabilité génomique31, et il est bien connu que la ploïdie augmente avec l’âge32,33. Cependant, l’enrichissement des hépatocytes tétraploïdes mononucléés est également cliniquement associé à un mauvais pronostic dans le carcinome hépatocellulaire humain (CHC)34. De même, les changements dans les taux de ploïdie des hépatocytes sont liés à des maladies hépatiques chroniques liées au vieillissement telles que la stéatose hépatique non alcoolique (NAFLD)35,36,37. La ploïdie est la condition de posséder plus de deux copies du génome, qui peut être explorée en colorant le contenu du génome avec un colorant à ADN tel que Hoechst38. Le colorant Hoechst, qui est ajouté au HB avant l’extraction, marque tous les noyaux pendant le protocole d’isolement. Cela permet de distinguer les noyaux diploïdes et polyploïdes en fonction de leur teneur en ADN lorsqu’ils sont excités par un laser UV (350 nm) ou violet (450 nm) sur un instrument de cytométrie en flux. Grâce à la stratégie de contrôle présentée, les niveaux 2n, 4n, 8n et plus élevés de ploïdie hépatocytes peuvent être étudiés dans des foies congelés archivés afin de mieux comprendre le rôle de l’hétérogénéité cellulaire dans la fonction tissulaire1 (Figure 3A). De plus, la morphologie nucléaire, y compris la taille et le volume, peut être quantifiée à l’aide de la cytométrie en flux d’imagerie pour corréler les changements dans la taille du noyau avec les changements dans le nombre total de comptes ou le nombre de gènes en fonction du niveau de ploïdie (Figure 3B,C).
La mesure omique multimodale offre la possibilité d’étudier simultanément plusieurs couches d’organisation génomique. L’approche multiomique conjointe ARN + ATAC permet d’étudier les régulateurs en amont et les gènes métaboliques en aval, fournissant une approche globale pour étudier les réseaux transcriptionnels et l’architecture de la chromatine associée à la fonction hépatique à la résolution unicellulaire. En outre, avec les progrès des méthodes de calcul qui peuvent tenir compte de la rareté des données et de la réduction des coûts de séquençage, la multiomique unicellulaire est pionnière dans l’évaluation de plusieurs modalités à partir de la même cellule. Ce protocole d’isolement mononucléique est compatible avec l’évaluation individuelle et conjointe des ensembles de données d’expression et de chromatine. Nous avons utilisé des pipelines standard établis par Stuart et al.23 (package Signac) pour illustrer la qualité des données, tandis que plusieurs méthodes de calcul disponibles et alternatives peuvent être facilement adoptées pour les analyses en aval23,39,40,41.
Dans l’ensemble, la multiomique mononucléaire permet d’étudier les tissus hépatiques de souris FF, de primates humains et non humains bio-archivés en utilisant une très petite quantité d’échantillons de départ en mettant en œuvre le protocole d’extraction de noyau présenté ici. Cet outil inestimable permettra aux biologistes du foie d’interroger à la fois l’expression des gènes et l’accessibilité de la chromatine dans le contexte de diverses pathologies hépatiques. De plus, divers niveaux de ploïdie hépatocytes et l’ajustement de l’expression génique qui en résulte en fonction de leur emplacement dans le lobule hépatique pourraient révéler leur rôle dans les pathologies hépatiques. Par conséquent, nous prévoyons que l’étude de l’hétérogénéité cellulaire offrira de nouvelles opportunités pour le développement de la médecine de précision et des interventions ciblées contre des maladies telles que le CHC et la NAFLD.
Déclarations de divulgation
Les auteurs déclarent n’avoir aucun conflit d’intérêts.
Remerciements
Cette recherche a été soutenue par le Helmholtz Pioneer Campus (M.S., K.Y., C.P.M.-J.) et l’Institute of Computational Biology (C. T.-L.). Cette recherche a également été soutenue par AMED sous le numéro de subvention JP20jm0610035 (C.P.M.-J.). Nous remercions le Core Genomics de HMGU (I. de la Rosa) et le soutien de Bioinformatics (T. Walzthoeni), en particulier Xavier Pastor pour la formation et l’orientation. Nous remercions A. Feuchtinger, U. Buchholz, J. Bushe et tous les autres membres du personnel de l’installation centrale de pathologie et d’analyse tissulaire du HMGU pour leur soutien technique et scientifique, ainsi que J. Zorn, R. Erdelen, D. Würzinger, membres du personnel d’E-Streifen, ainsi que l’installation principale des Services aux animaux de laboratoire pour leur soutien scientifique continu et leurs discussions. Nous sommes reconnaissants envers l’analyse cellulaire de l’installation centrale de TranslaTUM (R. Mishra) et de Luminex, une société DiaSorin (P. Rein). Nous remercions le Dr I Deligiannis pour son soutien technique. Le Dr M. Hartman, le Dr A. Schröder et Mme A. Barden (Helmholtz Pioneer Campus) ont joué un rôle fondamental dans leur soutien juridique, managérial et administratif.
matériels
| Name | Company | Catalog Number | Comments |
| 10% Tween 20 - 5 mL | Bio-Rad | 1662404 | |
| 2100 Bioanalyzer Instrument | Agilent | G2939BA | |
| Adhesive PCR film | Thermo Fisher Scientific | AB0558 | |
| Bioanalyzer High Sensitivity DNA Analysis | Agilent | 5067-4626 | |
| C1000 Touch Thermal Cycler | Bio-Rad | 1851196 | |
| Cell Sorter | For fluoresence-activated cell sorting (FACS); e.g. BD FACSAria Cell Sorter. | ||
| Centrifuge 5430R | Eppendorf | 5428000619 | Use chilled at 4 °C |
| Chromium Controller & Next GEM Accessory Kit | 10X Genomics | 1000204 | |
| Chromium Next GEM Chip G Single Cell Kit | 10X Genomics | 1000127 | |
| Chromium Next GEM Chip J Single cell kit, 16 reactions | 10X Genomics | 1000230 | |
| Chromium Next GEM Single Cell 3' Kit v3.1, 4 reactions | 10X Genomics | 1000269 | |
| Chromium Next GEM Single Cell Multiome ATAC + Gene Expression Reagent Bundle, 4 reactions | 10X Genomics | 1000285 | |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Sigma Aldrich | 11873580001 | |
| Dithiothreitol (DTT) 1 M solution | Sigma Aldrich | 646563 | Make 1 mM stock solution and use at 1 µM final concentration. |
| DNA AWAY Surface Decontaminant | Thermo Fisher Scientific | 10223471 | Wipe surfaces and pipettes before start of experiment |
| DNA LoBind Tubes, 1.5 mL | Thermo Fisher Scientific | 16628742 | |
| DNA LoBind Tubes, 2.0 mL | Thermo Fisher Scientific | 16638742 | |
| Elution Buffer (EB) - 250 mL | Qiagen | 19086 | |
| Eppendorf ThermoMixer C | Thermo Fisher Scientific | 13527550 | |
| Eppendorf ThermoMixer C Accessory: Smartblock | Thermo Fisher Scientific | 13518470 | |
| Ethanol, Absolute (200 Proof), Molecular Biology Grade - 100 mL | Thermo Fisher Scientific | 10517694 | |
| Filters 50 µm, sterile | SYSMEX PARTEC - CELLTRICS | 04-004-2327 | Adjust filter diameter according with tissue and nuclei size |
| Glycerin (Glycerol), 50% (v/v) - 1 L | Ricca Chemical Company | 3290-32 | |
| Hard-Shell, 384-Well PCR Plates, thin wall, skirted, clear/white | Bio-Rad | HSP3905 | |
| Herenz Heinz ABS Forceps | Thermo Fisher Scientific | 1131884 | |
| Hoechst 33342, Trihydrochloride, Trihydrate, 10 mg/mL Solution in Water | Invitrogen | H3570 | Light-sensitive |
| Imaging Flow Cytometer | For imaging flow cytometry analysis, e.g. Luminex Amnis ImageStream | ||
| Invitrogen TE Buffer - 100 mL | Thermo Fisher Scientific | 11568846 | |
| KCl (2 M), RNase-free, 100 mL | Thermo Fisher Scientific | AM9640G | |
| MgCl2 (1 M), 100 mL | Thermo Fisher Scientific | AM9530G | |
| MicroAmp 8-Cap Strip, clear-300 strips | Thermo Fisher Scientific | 10209104 | |
| MicroAmp 8-Tube Strip, 0.2 mL-125 strips | Thermo Fisher Scientific | 10733087 | |
| Mörser 2 mL DOUNCE | Wagner & Munz GmbH | 9651632 | RNase zap and rinse with MillQ before use |
| MyFuge 12 Mini MicroCentrifuge C1012 | Benchmark Scientific | C1012 | Or any other strip and tube mini centrifuge |
| Neubauer Hemocytometer | OMNILAB LABORZENTRUM | 5435293 | Visualize and count nuclei under microscope |
| Nuclease-Free Water (not DEPC-Treated) | Thermo Fisher Scientific | AM9937 | |
| OptiPrep Density Gradient Medium | Sigma Aldrich | D1556 | |
| Pipette tips RT LTS 1000 µL, Wide-O | Mettler Toledo | 30389218 | |
| Pistill "A" 2 mL | Wagner & Munz GmbH | 9651621 | RNase zap and rinse with MillQ before use |
| Pistill "B" 2 mL | Wagner & Munz GmbH | 9651627 | RNase zap and rinse with MillQ before use |
| Polypropylene 15 mL Centrifuge Tube | Thermo Fisher Scientific | 10579691 | |
| Polystyrene Petri dish, 60 mm x 15 mm | Thermo Fisher Scientific | 10634141 | |
| Polystyrene Round-Bottom 5 mL FACS Tubes | Thermo Fisher Scientific | 10100151 | |
| Protector RNase inhibitor - 2,000 U | Sigma Aldrich | 3335399001 | Keep in -20 °C until use |
| Protein-based RNase Inhibitor SUPERase•In (20 U/μL) | Thermo Fisher Scientific | AM2696 | Keep in -20 °C until use |
| Recombinant RNase Inhibitor | Clontech Takara | 2313B | Keep in -20 °C until use |
| RNAse free microfuge tubes - 0.5 mL | Thermo Fisher Scientific | AM12450 | |
| RNaseZap RNase Decontamination Solution | Thermo Fisher Scientific | AM9780 | Wipe surfaces and pipettes before start of experiment |
| SPRIselect - 60 mL | Beckman Coulter | B23318 | Aliquot and store in 4 °C |
| Sucrose, 500 g | Sigma Aldrich | S0389-500G | Make a 1 M stock solution |
| Swann-Morton Sterile Disposable Stainless Steel Scalpels | Thermo Fisher Scientific | 11798343 | |
| Tris-HCI (1M), pH 8.0 | Invitrogen | 15568025 | |
| Triton X-100, 98%, 100 mL | Thermo Fisher Scientific | 10671652 | Make 10% stock solution. Keep at 4 °C and protect from light. |
| Trypan Blue Solution, 0.4% | Thermo Fisher Scientific | 11538886 | |
| Vortex- Mixer | VWR | 444-1372 | Or any other type of vortex |
Références
- Kamies, R., Martinez-Jimenez, C. P. Advances of single-cell genomics and epigenomics in human disease: where are we now. Mamm Genome. 31 (5-6), 170-180 (2020).
- Ramachandran, P., Matchett, K. P., Dobie, R., Wilson-Kanamori, J. R., Henderson, N. C. Single-cell technologies in hepatology: New insights into liver biology and disease pathogenesis. Nature Reviews Gastroenterology & Hepatology. 17 (8), 457-472 (2020).
- Ramachandran, P., et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. 575 (7783), 512-518 (2019).
- Andrews, T. S., et al. Single-cell, single-nucleus, and spatial RNA sequencing of the human liver identifies cholangiocyte and mesenchymal heterogeneity. Hepatology Communications. 6 (4), 821-840 (2022).
- Xiong, X., et al. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Molecular Cell. 75 (3), 644-660 (2019).
- Richter, M. L., et al. Single-nucleus RNA-seq2 reveals functional crosstalk between liver zonation and ploidy. Nature Communications. 12 (1), 4264 (2021).
- Matsumoto, T., Wakefield, L., Tarlow, B. D., Grompe, M. In vivo lineage tracing of polyploid hepatocytes reveals extensive proliferation during liver regeneration. Cell Stem Cell. 26 (1), 34-47 (2020).
- Chen, F., et al. Broad distribution of hepatocyte proliferation in liver homeostasis and regeneration. Cell Stem Cell. 26 (1), 27-33 (2020).
- Donne, R., Saroul-Ainama, M., Cordier, P., Celton-Morizur, S., Desdouets, C. Polyploidy in liver development, homeostasis and disease. Nature Reviews. Gastroenterology & Hepatology. 17 (7), 391-405 (2020).
- Lengefeld, J., et al. Cell size is a determinant of stem cell potential during aging. Science Advances. 7 (46), 0271 (2021).
- Lanz, M. C., et al. Increasing cell size remodels the proteome and promotes senescence. Mol Cell. 82 (17), 3255-3269 (2022).
- Kim, J. Y., et al. PIDDosome-SCAP crosstalk controls high-fructose-diet-dependent transition from simple steatosis to steatohepatitis. Cell Metabolism. 34 (10), 1548-1560 (2022).
- Padovan-Merhar, O., et al. Single mammalian cells compensate for differences in cellular volume and DNA copy number through independent global transcriptional mechanisms. Molecular Cell. 58 (2), 339-352 (2015).
- Miettinen, T. P., et al. Identification of transcriptional and metabolic programs related to mammalian cell size. Current Biology. 24 (6), 598-608 (2014).
- Vargas-Garcia, C. A., Ghusinga, K. R., Singh, A. Cell size control and gene expression homeostasis in single-cells. Current Opinion in Systems Biology. 8, 109-116 (2018).
- Knoblaugh, S. E., Randolph-Habecker, J. . Necropsy and histology. In Comparative Anatomy and Histology: A Mouse, Rat, and Human Atlas (Second Edition). , (2018).
- Chromium Next GEM Single Cell 3 Reagent Kits v3.1 User Guide. Document number CG000204 (Rev D). 10x Genomics Available from: https://www.10xgenomics.com/support/single-cell-gene-expression/documentation/steps/library-prep/chromium-single-cell-3-reagent-kits-user-guide-v-3-1-chemistry (2019)
- Chromium Next GEM Single Cell Multiome ATAC + Gene Expression User Guide. Document number CG000338 (Rev D). 10x Genomics Available from: https://www.10xgenomics.com/support/single-cell-multiome-atac-plus-gene-expression/documentation/steps/library-prep/chromium-next-gem-single-cell-multiome-atac-plus-gene-expression-reagent-kits-user-guide (2021)
- MacParland, S. A., et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nature Communications. 9 (1), 4383 (2018).
- Martinez-Jimenez, C. P., Kyrmizi, I., Cardot, P., Gonzalez, F. J., Talianidis, I. Hepatocyte nuclear factor 4alpha coordinates a transcription factor network regulating hepatic fatty acid metabolism. Molecular and Cell Biology. 30 (3), 565-577 (2010).
- Schmidt, D., et al. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science. 328 (5981), 1036-1040 (2010).
- Hao, Y., et al. Integrated analysis of multimodal single-cell data. Cell. 184 (13), 3573-3587 (2021).
- Stuart, T., Srivastava, A., Madad, S., Lareau, C. A., Satija, R. Single-cell chromatin state analysis with Signac. Nature Methods. 18 (11), 1333-1341 (2021).
- Sampaziotis, F., et al. Cholangiocyte organoids can repair bile ducts after transplantation in the human liver. Science. 371 (6531), 839-846 (2021).
- Gomez-Lechon, M. J., Donato, M. T., Castell, J. V., Jover, R. Human hepatocytes as a tool for studying toxicity and drug metabolism. Current Drug Metabolism. 4 (4), 292-312 (2003).
- Gomez-Lechon, M. J., Donato, M. T., Castell, J. V., Jover, R. Human hepatocytes in primary culture: The choice to investigate drug metabolism in man. Current Drug Metabolism. 5 (5), 443-462 (2004).
- vanden Brink, S. C., et al. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nature Methods. 14 (10), 935-936 (2017).
- Denisenko, E., et al. Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biology. 21 (1), 130 (2020).
- Nault, R., Fader, K. A., Bhattacharya, S., Zacharewski, T. R. Single-nuclei RNA sequencing assessment of the hepatic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Cellular and Molecular Gastroenterology and Hepatology. 11 (1), 147-159 (2021).
- Krishnaswami, S. R., et al. Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons. Nature Protocols. 11 (3), 499-524 (2016).
- Duncan, A. W., et al. Aneuploidy as a mechanism for stress-induced liver adaptation. Journal of Clinical Investigation. 122 (9), 3307-3315 (2012).
- Kreutz, C., et al. Hepatocyte ploidy is a diversity factor for liver homeostasis. Frontiers in Physiology. 8, 862 (2017).
- Hunt, N. J., Kang, S. W. S., Lockwood, G. P., Le Couteur, D. G., Cogger, V. C. Hallmarks of aging in the liver. Computational and Structural Biotechnology Journal. 17, 1151-1161 (2019).
- Bou-Nader, M., et al. Polyploidy spectrum: a new marker in HCC classification. Gut. 69 (2), 355-364 (2019).
- Gentric, G., et al. Oxidative stress promotes pathologic polyploidization in nonalcoholic fatty liver disease. Journal of Clinical Investigation. 125 (3), 981-992 (2015).
- Schwartz-Arad, D., Zajicek, G., Bartfeld, E. The streaming liver IV: DNA content of the hepatocyte increases with its age. Liver. 9 (2), 93-99 (1989).
- Kudryavtsev, B. N., Kudryavtseva, M. V., Sakuta, G. A., Stein, G. I. Human hepatocyte polyploidization kinetics in the course of life cycle. Virchows Archiv. B, Cell Pathology Including Molecular Pathology. 64 (6), 387-393 (1993).
- Duncan, A. W., et al. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature. 467 (7316), 707-710 (2010).
- Granja, J. M., et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nature Genetics. 53 (3), 403-411 (2021).
- Bredikhin, D., Kats, I., Stegle, O. MUON: Multimodal omics analysis framework. Genome Biology. 23 (1), 42 (2022).
- Velten, B., et al. Identifying temporal and spatial patterns of variation from multimodal data using MEFISTO. Nature Methods. 19 (2), 179-186 (2022).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.