
Method Article
Einfache und reproduzierbare Low-Density-Primärkultur mit gefrorenem Bestand embryonaler Hippocampus-Neuronen
In diesem Artikel
Zusammenfassung
Ein gebrauchsfertiger gefrorener Vorrat an Neuronen ist ein leistungsfähiges Werkzeug zur Bewertung synaptischer Funktionen. Hier stellen wir eine einfache Primärkultur mit geringer Dichte aus gefrorenen Beständen unter Verwendung einer 96-Well-Platte vor.
Zusammenfassung
Neuronale Kultur ist ein wertvolles System zur Bewertung synaptischer Funktionen und Medikamentenscreenings. Insbesondere eine Kultur mit geringer Dichte von primären Hippocampus-Neuronen ermöglicht die Untersuchung einzelner Neuronen oder subzellulärer Komponenten. Wir haben die Lokalisierung subzellulärer Proteine innerhalb eines Neurons durch Immunzytochemie, neuronale Polarität, synaptische Morphologie und ihre Entwicklungsänderung unter Verwendung einer primären Hippocampuskultur mit niedriger Dichte gezeigt. In jüngster Zeit sind gebrauchsfertige gefrorene Bestände von Neuronen kommerziell verfügbar geworden. Diese gefrorenen Neuronenbestände verkürzen den Zeitaufwand für die Vorbereitung von Tierversuchen und tragen auch dazu bei, die Anzahl der verwendeten Tiere zu reduzieren. Hier stellen wir eine reproduzierbare Primärkulturmethode mit niedriger Dichte unter Verwendung einer 96-Well-Platte vor. Wir verwendeten einen kommerziell erhältlichen gefrorenen Vorrat an Neuronen aus dem embryonalen Hippocampus der Ratte. Die Neuronen können langfristig ohne Medienveränderungen stabil kultiviert werden, indem das Wachstum von Gliazellen zu bestimmten Zeitpunkten reduziert wird. Dieser Hochdurchsatz-Assay mit Low-Density-Kultur ermöglicht reproduzierbare bildgebende Auswertungen der synaptischen Plastizität.
Einleitung
Die Entwicklung eines experimentellen In-vitro-Systems, das synaptische Funktionen beurteilen kann, die an Lernen und Gedächtnis beteiligt sind, ist wichtig. Neuronale Kultur ist ein wertvolles System zur Bewertung synaptischer Funktionen in vitro. Die neuronale Kulturtechnik wurde erstmals in den 1980er Jahren eingesetzt, und in den 1990er Jahren wurde eine Low-Density-Kultur von primären Hippocampus-Neuronen entwickelt 1,2,3 für die Untersuchung einzelner Neuronen in Bezug auf subzelluläre Lokalisation von Proteinkomponenten, Proteintransport, neuronale Polarität, Wirbelsäulenmorphologie, Synapsenentwicklung und Plastizität 4,5,6,7,8 . Bei dieser Technik sind jedoch viele Schritte erforderlich: Paarung der Tiere, Sezieren von Embryonen, Vorbereiten von Kulturgefäßen und Kultivieren von Zellen für 3 Wochen mit Medienwechsel einmal pro Woche. Darüber hinaus erfordert es fortgeschrittene Techniken3.
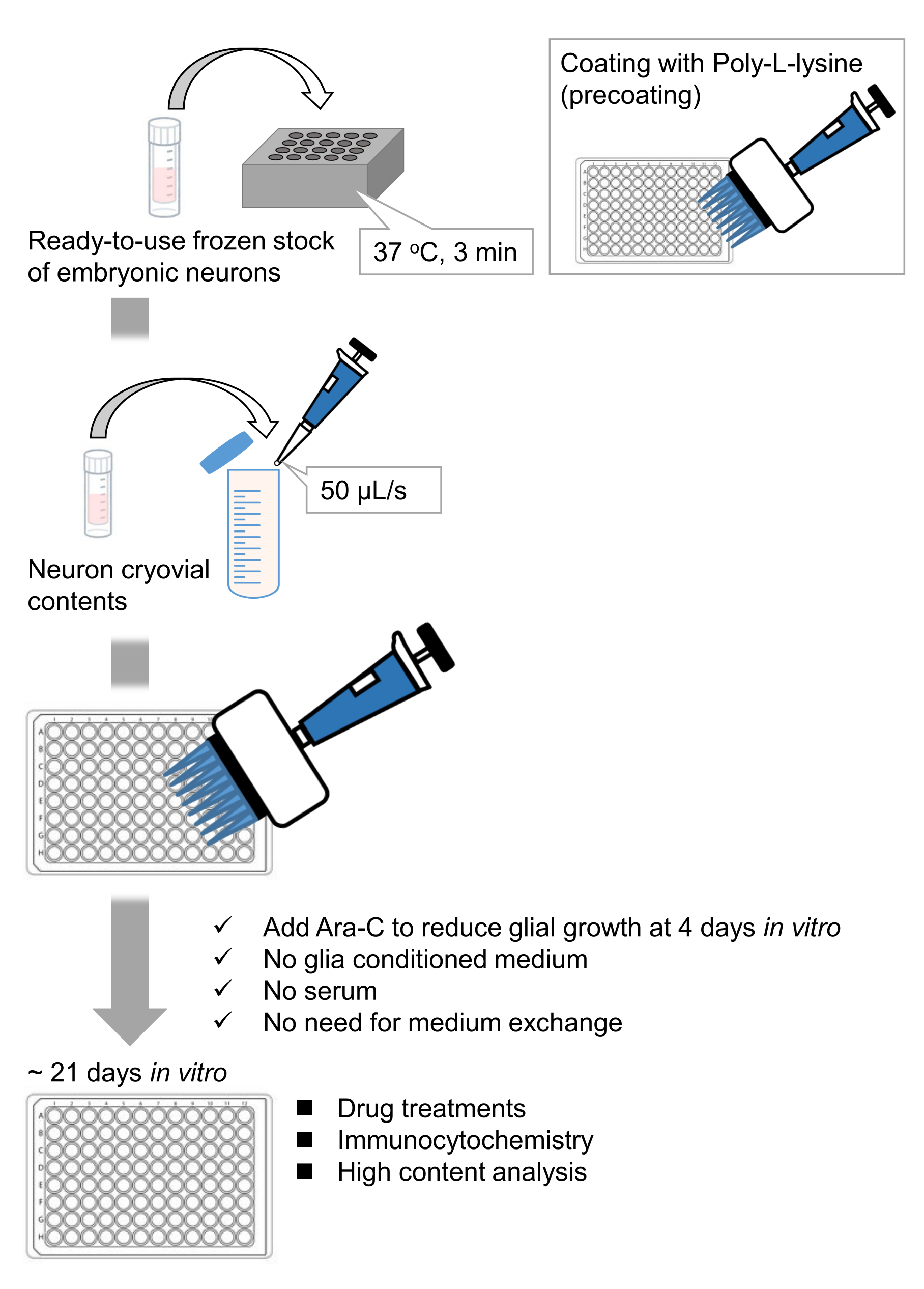
Wir haben gefrorene Bestände von dissoziierten Hippocampus-Neuronen aus Rattenembryonen 9,10 entwickelt. Die gefrorenen Neuronenvorräte sind gebrauchsfertig, und es sind keine fortgeschrittenen Techniken erforderlich, um die Zellen zu kultivieren11,12. Mit anderen Worten, die Kultivierung der Neuronen aus gefrorenen Beständen hängt nicht von der Technik eines Experimentators ab. Es macht Tierversuche überflüssig (z. B. Genehmigung von Tierversuchen, Vermittlung von zeitlich begrenzten trächtigen Tieren und Sezieren von Rattenembryonen), wodurch die Anzahl der verwendeten Tiere reduziert wird. In jüngster Zeit sind hochwertige, gebrauchsfertige gefrorene Bestände von Neuronen kommerziell verfügbar geworden. Hier verwendeten wir handelsübliche Tiefkühlbestände aus dem embryonalen Tag (E) 18 Rattenhippocampus13,14,15. Die Kultivierung der Neuronen aus einem gefrorenen Stamm erfordert keine glia-konditionierten Medien oder Co-Kultur mit Gliazellen. Gewöhnliche Primärkulturmedien ohne zusätzliches Serum können zur Kultivierung der Zellen verwendet werden; So können wir reproduzierbare Daten gewinnen. Darüber hinaus ist für 3 Wochen nach der Zellaussaat kein Medienaustausch erforderlich, da das Wachstum von Gliazellen reduziert ist (Abbildung 1).
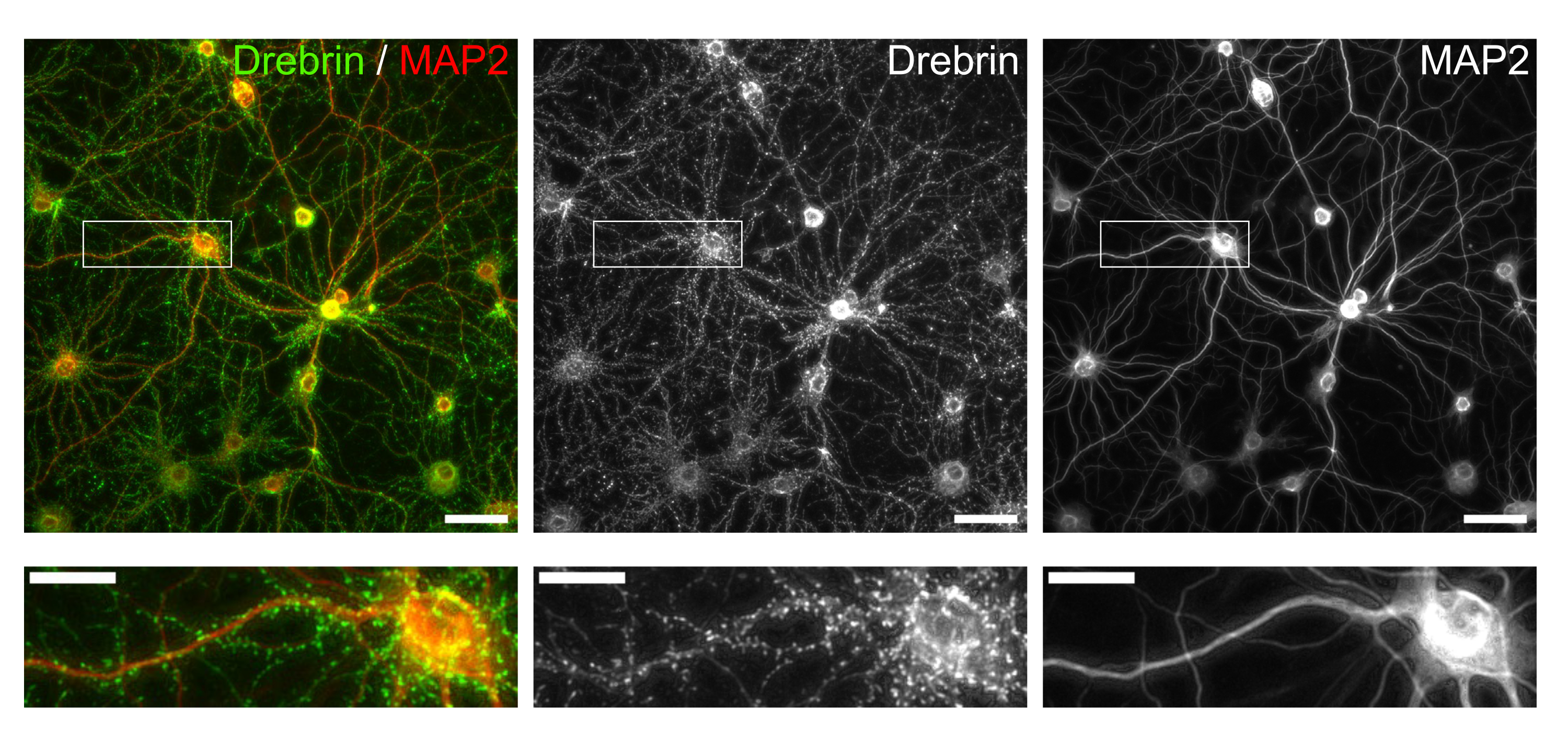
Dendritische Dornen sind das postsynaptische Kompartiment der meisten erregenden Synapsen. Sie enthalten Rezeptorproteine, postsynaptische Gerüstproteine und Aktin-Zytoskelettproteine. Wir konzentrierten uns auf ein Aktin-bindendes ProteinDrebrin 5,6,7,16,17,18. Drebrin reichert sich am Wirbelsäulenkopf in reifen Neuronenan 19, und wir berichteten über Drebrin als Marker für den synaptischen Zustand 15,17,20,21,22,23. Durch die Durchführung einer High-Content-Analyse mit Drebrin als Auslese haben wir kürzlich über die hemmenden Wirkungen von Phencyclidin-Analoga auf N-Methyl-D-Asparaginsäure-Glutamatrezeptoren (NMDARs)10 und die NMDAR-abhängigen Wirkungen von natürlichen Verbindungen und rohen Arzneimitteln auf synaptische Zustände berichtet15.
Hier beschreiben wir, wie gefrorene Bestände von Neuronen mit geringer Dichte kultiviert werden können. Darüber hinaus zeigen wir eine Drebrin-Imaging-basierte Bewertung des synaptischen Zustands unter Verwendung von 96-Well-Platten.
Protokoll
1. Plattenbeschichtung
- Eine 96-Well-Mikrotiterplatte wird mit Poly-L-Lysin (1 mg/ml, verdünnt in 0,1 M Boratpuffer [pH: 8,5]; 100 μL/Well) beschichtet und über Nacht bei 37 °C inkubiert.
HINWEIS: Beschichten Sie nur die Brunnen, die verwendet werden müssen. In den Experimenten, die hier durchgeführt werden, werden die mittleren 60 Vertiefungen verwendet. Der Boratpuffer wird durch Mischen von 50 mM Borsäure und 12 mM Borat in sterilisiertem Wasser hergestellt. - Waschen Sie die Platte zweimal mit sterilisiertem Wasser (250 μL/Well).
- Waschen Sie die Platte einmal mit frischem Nährmedium ohne Zusätze (250 μL/Well).
- Trocknen Sie die Platte auf einer sauberen Bank für 20 Minuten.
- Wickeln Sie die Platte mit Alufolie ein und halten Sie sie bis zur Verwendung bei 4 °C (gültig für 1 Monat).
2. Zell-Seeding
- Geben Sie 50 μL/Well des Kulturmediums auf die beschichtete Platte und bewahren Sie sie in einem 37 °C, 5% CO2 -Inkubator für 30 min bis 1 h auf. Füllen Sie die peripheren Vertiefungen mit sterilisiertem Wasser (200 μL/Well).
HINWEIS: Das Nährmedium wird durch Zugabe von 50x B-27, 400x Glutamax und 100 U/ml Penicillin/Streptomycin zum neurobasalen Medium hergestellt (Details finden Sie in der Materialtabelle ). - Entfernen Sie das Neuronen-Kryovial aus dem Flüssigstickstofftank. Bei den hier verwendeten Neuronen handelte es sich um DMSO-kryokonservierte Neuronen11.
- Tauchen Sie das Cryovial für bis zu 3 min in einen 37 °C heißen Heizblock und tauen Sie den Inhalt teilweise auf. Erwärmen Sie das Kryovial nicht zu lange. Den Inhalt in ein 50-ml-Röhrchen überführen, sobald es aufgetaut ist.
- Übertragen Sie den kryovialen Inhalt der Neuronen langsam auf ein steriles 50-ml-Röhrchen tropfenweise (50 μl/s) mit einer 1-ml-Pipette mit breitporiger Spitze.
- Spülen Sie das leere Kryovial mit 1 ml des Nährmediums (Raumtemperatur; RT). Dieses 1 mL des Kulturmediums wird tropfenweise (50 μL/s) aus dem kryovialen Röhrchen in das 50-ml-Röhrchen mit der Zellsuspension übertragen.
- Geben Sie 9 ml des Kulturmediums (RT) tropfenweise (0,5 ml/s) in das 50-ml-Röhrchen und füllen Sie das Volumen auf 11 ml auf. Wiederholen Sie das Pipettieren nicht, sondern mischen Sie die Zellsuspension langsam.
- Zählen Sie die Zellzahl (verwenden Sie einen Zellzähler oder ein Hämozytometer).
- Die gesamte Zellsuspension wird in ein Reservoir überführt und mit einer Mehrkanalpipette mit breitporigen Spitzen (1,0 x 104 Zellen/Well) auf die 96-Well-Platte abgegeben. Um die Verdunstung des Nährmediums zu reduzieren, füllen Sie die peripheren Vertiefungen mit sterilisiertem Wasser (Schritt 2.1).
HINWEIS: Diese Studie bestätigt, dass die Verdunstung des Kulturmediums für eine 3-wöchige Kultur ohne Mediumsaustausch gering ist. Die Reduktionsrate des Mediums beträgt 3,6% (n = 120 Wells). Somit wird die Osmolalitätsänderung während der 3-wöchigen Inkubationszeit nicht drastisch sein. - Inkubieren Sie die Neuronen für 1-2 h in einem 37 °C, 5% CO2 -Inkubator.
- Ersetzen Sie das Nährmedium durch 100 μL vorgewärmtes Nährmedium (37 °C) pro Vertiefung und geben Sie es zurück in einen 37 °C, 5% CO2 -Inkubator (während der Kultur ist kein Mediumwechsel erforderlich).
3. Ara-C-Behandlung
- Nach 4 Tagen in vitro (DIV) wird Cytosin β-D-Arabinofuranosid (Ara-C) zu einer Endkonzentration von 0,2 μM pro Vertiefung hinzugefügt, um das Wachstum von Gliazellen zu reduzieren.
4. Medikamentöse Behandlungen
- Behandeln Sie die Zellen nach 21 Tagen in vitro mit den interessierenden Medikamenten.
- Halten Sie die Temperatur der Platte während der medikamentösen Behandlung bei 37 ° C.
- Für eine Positivkontrolle werden die Zellen vor der Fixierung 10 min lang mit 100 μM Glutamat (pro Vertiefung für die Endkonzentration) behandelt.
5. Fixierung
- Für die Fixierung werden 4% Paraformaldehyd in 0,1 M Phosphatpuffer (100 μL/Well) verwendet.
- Nach ~20 min Fixierung werden die Vertiefungen mit phosphatgepufferter Kochsalzlösung (PBS; 250 μL/Well) 2x für jeweils 5 min gewaschen.
6. Immunzytochemie
- Waschen Sie die Zellen mit PBS (250 μL/Well) 1x für 5 min.
- Permeabilisieren Sie die Zellen mit 0,1% Triton X-100 (100 μL/Well) in PBS für 5 min.
- Waschen Sie die Zellen mit PBS (250 μL/Well) 3x für jeweils 5 Minuten.
- Zur Blockierung verwenden Sie 3% Rinderserumalbumin in PBS (PBSA; 100 μL/Well) für 1 h bei RT.
- Inkubieren Sie die Zellen mit Anti-Drebrin (1:1) und Anti-Microtubuli Associated Protein 2 (MAP2) (1:000) Antikörpern (60 μL/Well) bei 4 °C über Nacht.
- Waschen Sie die Zellen mit PBS (250 μL/Well) 4x für jeweils 5 min.
- Inkubieren Sie die Zellen mit geeigneten sekundären Antikörpern und 4′,6-Diamidino-2-phenylindol, Dihydrochlorid (DAPI; 1:1000) in PBSA (60 μL/Well) für 2 h bei RT.
- Waschen Sie die Zellen mit PBS (250 μL/Well) 4x für jeweils 5 min.
- Lagern Sie die Zellen in PBS, das 0,1% Natriumazid (150 μL/Well) enthält.
7. Bildaufnahme und -analyse
- Um die Bilder zu erfassen, verwenden Sie ein geeignetes Mikroskop.
- Um die Zellkörper von Neuronen zu identifizieren, verwenden Sie sowohl MAP2-positive als auch DAPI-positive Regionen.
- Um die Dendriten von Neuronen zu identifizieren, verwenden Sie MAP2-positive Signale ohne Zellkörper.
- Um Drebrin-Cluster zu identifizieren, verwenden Sie Drebrin-positive Signale entlang MAP2-positiver Dendriten.
Ergebnisse
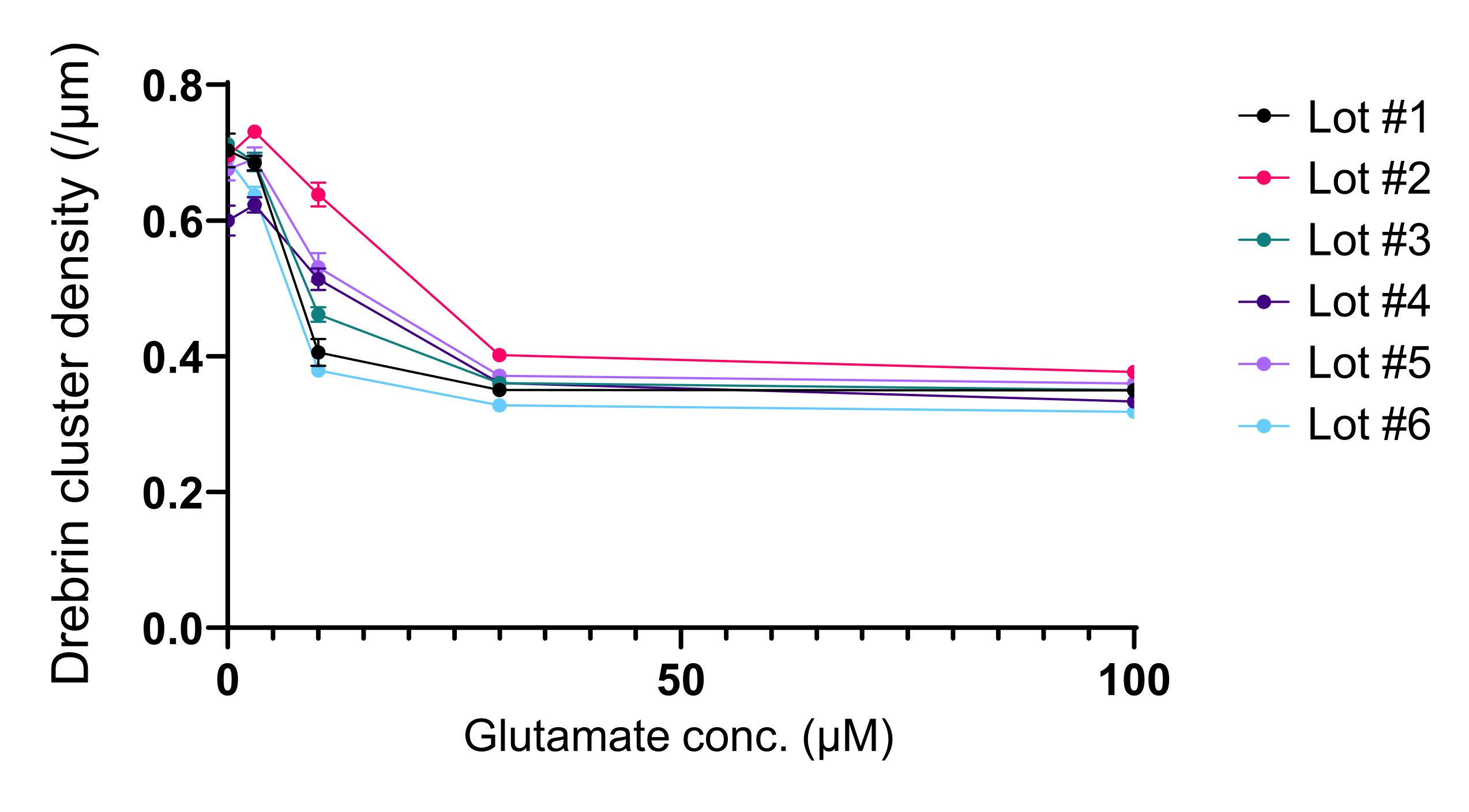
Nach dem Protokoll wurden die Neuronen 21 Tage lang in einer 96-Well-Platte kultiviert und dann mit Glutamat behandelt (Abbildung 1). Die Neuronen entwickelten sich 3 Wochen lang normal ohne Austausch des Kulturmediums (Abbildung 2). Wir behandelten die Zellen mit mehreren Konzentrationen von Glutamat (1 μM, 3 μM, 10 μM, 30 μM und 100 μM verdünnt in sterilisiertem Wasser) für 10 min und fixierten sie. Die Immunzytochemie wurde durchgeführt und Fluoreszenzbilder von Drebrin und MAP2 wurden mit einem automatisierten Fluoreszenzmikroskop mit einer sCMOS-Kamera aufgenommen. Wie in Abbildung 3 gezeigt, sind Drebrin-positive dendritische Stacheln entlang von MAP2-positiven Dendriten deutlich zu beobachten. Es hat sich gezeigt, dass die Glutamat-Stimulation einen Ca2+-Einstrom durch NMDAR hervorruft, der einen Drebrin Exodus aus dendritischen Stacheln verursacht, was zu einer Verringerung der Drebrin Cluster Dichten 5,17 führt. Dementsprechend beobachteten wir die dosisabhängige Reduktion der Drebrin-Cluster-Dichten gegen die Glutamat-Stimulation10 (Abbildung 4). Wie in Abbildung 5 gezeigt, ist diese Methode sehr gut reproduzierbar, wenn Drebrin als Marker für synaptische Zustände verwendet wird.

Abbildung 1: Schema der Methode. Die Neuronen wurden 21 Tage lang in einer 96-Well-Platte kultiviert und dann mit Glutamat behandelt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 2: Hellfeldaufnahmen von kultivierten Neuronen mit einer 96-Well-Platte. Phasenkontrastbilder wurden von jedem Entwicklungsstadium (DIV 1, 7, 14, 21) mit einem konfokalen quantitativen Bildzytometer erhalten. Maßstabsbalken: 50 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 3: Repräsentative Bilder von immungefärbten kultivierten Neuronen. (links) Fusionierte Fluoreszenzaufnahmen von Drebrin (grün) und MAP2 (rot). Jedes Fluoreszenzbild von Drebrin und MAP2 zeigte sich in der Mitte bzw. rechten Tafel. Weiße Rechtecke zeigen den Bereich unten vergrößert. Maßstabsbalken; Obere Platten: 50 μm, untere Platten: 20 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 4: Glutamat-abhängige Dosis-Wirkungs-Änderungen der normalisierten Drebrin-Cluster-Dichte . (A) Repräsentative Fluoreszenzbilder, immungefärbt mit Drebrin (grün) und MAP2 (rot) aus der Vertiefung, die mit 0 μM, 10 μM und 100 μM Glutamat (von links nach rechts) behandelt wird. Maßstabsbalken: 50 μm. (B) Die Dichte des Drebrin-Clusters wurde durch den Mittelwert der Kontrolle (0 μM) normalisiert. 0 μM, N = 58 Vertiefungen; 1 μM, N = 46; 3 μM, N = 54; 10 μM, N = 45; 30 μM, N = 54; 100 μM, N = 55, aus 13 Experimenten mit verschiedenen Chargen. ** P < 0,01 gegenüber der Kontrolle (0 μM) durch Dunnetts Mehrfachvergleichstest nach ANOVA. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 5: Glutamat-abhängige Dosis-Wirkungs-Änderungen der Drebrin-Cluster-Dichte. Die Rohdaten aus sechs Experimenten mit unterschiedlichen Chargen. N = 4 Vertiefungen für jede Konzentration (0 μM, 3 μM, 10 μM, 30 μM und 100 μM). Die Werte werden als Mittelwert ± SEM ausgedrückt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

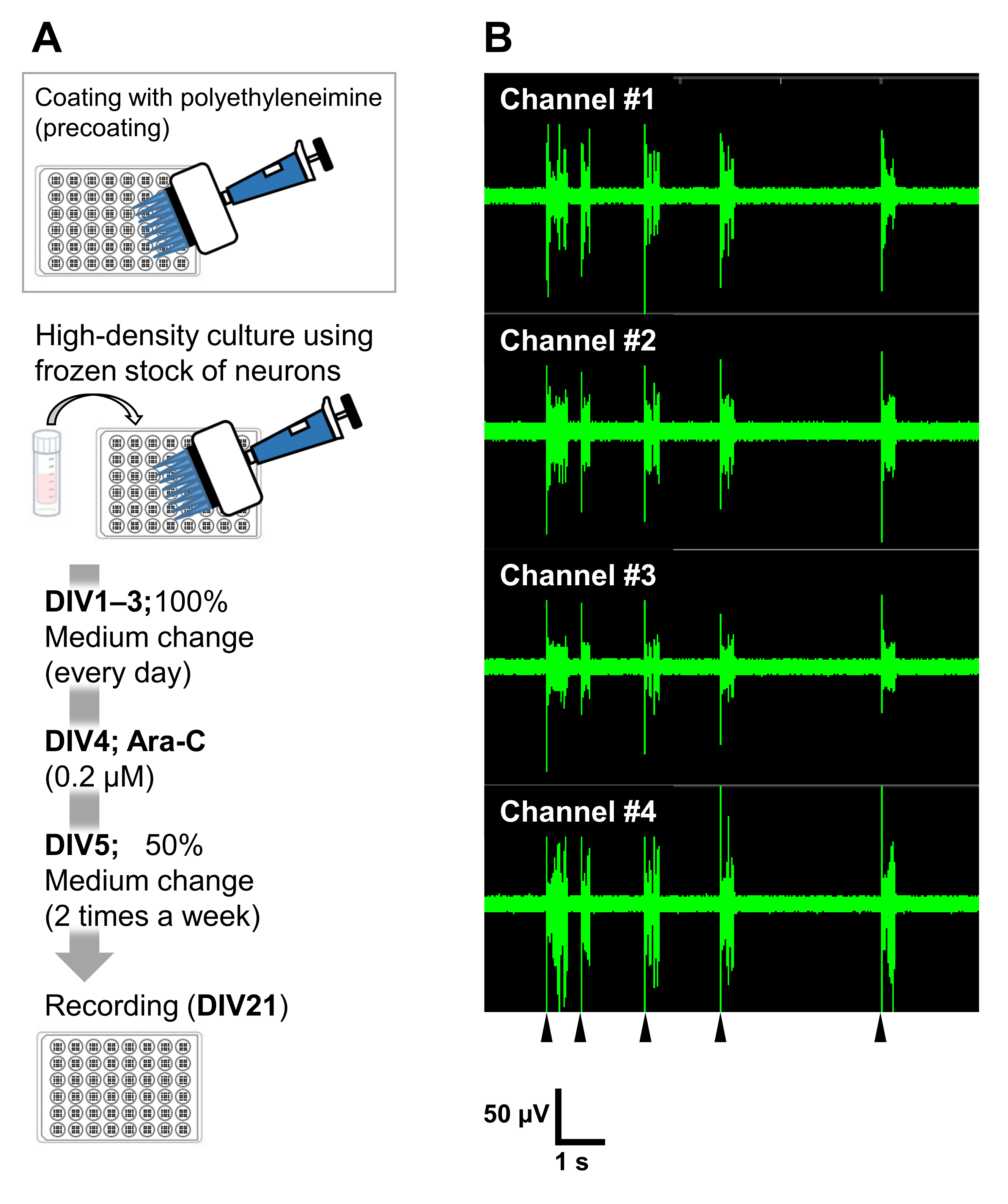
Abbildung 6: Anwendung der gefrorenen Neuronenbestände auf elektrophysiologische Experimente. (A) Ein Protokoll für elektrophysiologische Experimente unter Verwendung von Mikroelektroden-Array-Platten (MEA). Beschichtung: Einen Tag vor der Beschichtung der Zellen wurde jede 48-Well-MEA-Platte mit einer Polyethylenimin-Lösung (PEI: 0,1%) vorbeschichtet und für 1 h bei 37 °C inkubiert. Die MEA-Platte wurde dann 3x mit sterilisiertem Wasser gewaschen und für 1 h getrocknet. Dann wurde die MEA-Platte über Nacht bei 4 °C gehalten. Hochdichtekultur: 50.000 Zellen/Well der Neuronen wurden auf 48-Well-MEA-Platten plattiert. Der Zellimpfschritt wurde wie in Abschnitt 2 des oben beschriebenen Protokolls beschrieben durchgeführt. Laminin (20 μg/ml) zugesetztes Kulturmedium (2 v/v% B-27, 2,5 mM Glutamax und 100 μg/ml Penicillin/Streptomycin zum neurobasalen Medium) wurde verwendet, um die Neuronen zu beschichten. Danach wurden die Neuronen bei 37 °C kultiviert, 5%CO2 im Kulturmedium. Das Medium wurde vollständig auf DIV 1 mit dem Nährmedium bis DIV 3 ausgetauscht. Ara-C wurde an DIV 4 (final 0,2 μM) zugegeben. Ab DIV 5 und 2 mal pro Woche wurden 50% der Medien mit dem Kulturmedium gewechselt. Die Aktivität der Neuronen auf jeder Vertiefung der MEA-Platte wurde mit einem MEA-System aufgezeichnet. (B) Die spontane neuronale Aktivität wurde bei 37 °C unter einer 5%igen CO2 -Atmosphäre unter Verwendung eines MEA-Systems mit einer Abtastrate von 12,5 kHz / Kanal bei DIV 21 erfasst. Es werden Aufnahmen von 4 Kanälen aus den 16 Kanälen innerhalb eines Brunnens gezeigt. Für alle Aufnahmen wurde ein Butterworth-Bandpassfilter (200-3.000 Hz) verwendet. Pfeilspitzen zeigen den Zeitpunkt des synchronisierten Burst-Schusses an. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Diskussion
Ein kritischer Schritt bei dieser Methode ist das Auftauen der Zellsuspension. Der Transfer der Zellsuspension, bevor es zu warm wird, ist sehr wichtig. Um den schnellen Wechsel der Osmolalität zu vermeiden, sollte die Zellsuspension jedoch nicht auf einmal auf ein großes Volumen des Mediums übertragen werden. Die tropfenweise Zugabe des Nährmediums ist ebenfalls entscheidend, um plötzliche Änderungen des osmotischen Drucks zu vermeiden.
Die Neuronen können in anderen Kulturgefäßen kultiviert werden: 24-Well-Platten, 8-Well-Kammern, 60-mm-Schalen oder MED-Sonden. In diesen Fällen müssen jedoch die Endkonzentration und der Zeitpunkt der Zugabe von Ara-C angepasst werden. Darüber hinaus muss die Dichte der Neuronen in verschiedenen Arten von Experimenten optimiert werden. Zum Beispiel ist für elektrophysiologische Experimente eine Kultur mit hoher Dichte erforderlich, und in diesem Fall ist der Medienaustausch zweimal pro Woche erforderlich (Abbildung 6). Daher erfordert eine Kultur mit geringer Dichte weniger Schritte als eine Kultur mit hoher Dichte.
Neuronale Kulturen mit geringer Dichte erfordern oft fortschrittliche Techniken. Die Verwendung von gebrauchsfertiger Tiefkühlware löst dieses Problem jedoch. Die beschriebene Methode hängt nicht von den Fähigkeiten eines Experimentators ab. Die Qualität der Tiefkühlware ist stabil und kann stabil kultiviert werden, solange sie in flüssigem Stickstoff gelagert werden und Temperaturschwankungen für bis zu 4 Jahre vermieden werden.
Die Kultivierung der Zellen über 3 Wochen ohne Mediumsaustausch wirft die Frage auf, ob es zu signifikanten Osmolalitätsänderungen oder Nährmedienverdunstung kommt. Wir haben jedoch bestätigt, dass die Verdunstung von Nährmedien gering ist (Reduktionsrate von 3,6%). Die Lokalisation der synaptischen Proteine und die Morphologie der Neuronen erscheinen nach 3 Wochen normal. Daher verursacht eine 3-wöchige Kultur ohne Medienaustausch keine großen Osmolalitätsänderungen, die sich auf die Bedingungen der kultivierten Neuronen auswirken. Die Aufbewahrung der Platte in einem Inkubator nach der Ara-C-Behandlung ist ebenfalls ein wichtiger Punkt, der die Verdunstung minimiert.
Es gibt keine Einschränkung in Bezug auf die Verwendung der gefrorenen Stammneuronen. Es gibt jedoch einige Einschränkungen der Low-Density-Kulturmethode. Wir bestätigten, dass die Low-Density-Kultur für die morphologische Beobachtung von Neuronen, die Bewertung der synaptischen Funktion und die GFP-Transfektion eingesetzt werden kann. Wir haben jedoch die Bildgebung von lebenden Zellen nicht untersucht. Darüber hinaus ist, wie oben erwähnt, eine Kultur mit hoher Dichte erforderlich, um die Elektrophysiologie durchzuführen.
Die Synapsenreifung dauert in der Regel 3 Wochen7, und wir können bis zum Ende nicht bestätigen, dass die kultivierten Neuronen richtige Synapsen haben. Wenn die Synapsenreifung nach 3 Wochen nicht gut ist, müssten wir wieder kultivieren. Indem wir die Qualität der Neuronen kennen, bevor wir mit den Experimenten beginnen, können wir diese 3 Wochen sparen. Um Experimente effizient durchführen zu können, ist es daher am besten, die Qualität der Neuronen im Voraus zu überprüfen. Gefrorene Bestände ermöglichen es, die Qualität der Neuronen im Vorfeld zu überprüfen. Jede Charge von Tiefkühlbeständen wird aus einem Wurf Ratten erzeugt, und wir können einen der Bestände aus jeder Charge für eine Qualitätskontrolle verwenden. Drebrin ist ein guter Marker für die Qualitätskontrolle der Neuronen. Wie beschrieben, reichert sich Drebrin im Wirbelsäulenkopf in reifen Neuronen an und reagiert auf synaptische Stimulation. So können wir die Qualität der Neuronen in Tiefkühlbeständen überprüfen, indem wir Drebrin als Marker verwenden.
Diese Methode kann angewendet werden, um die Wirkung von Medikamenten auf den synaptischen Zustand zu bewerten. Der Drebrin-Exodus aus dendritischen Dornen tritt in den Anfangsstadien der synaptischen Plastizitätauf 22. Daher zeigt der Nachweis einer Drebrin-Cluster-Reduktion, die durch eine medikamentöse Behandlung hervorgerufen wird, dass das Medikament die Synapse stimuliert und synaptische Plastizität verursacht. Um festzustellen, ob die Reduktion NMDAR-abhängig ist, ist ein Experiment mit 2-Amino-5-Phosphonovaleriansäure (APV, ein NMDAR-Antagonist) nützlich. Mit Drebrin als Marker wird sogar eine NMDAR-Abhängigkeit eindeutig festgestellt10,15. Die beschriebene Methode ist nützlich bei Arzneimittel-Screenings, sicherheitspharmakologischen Studien und der Bewertung der synaptischen Funktion.
Offenlegungen
Tomoaki Shirao ist CEO von AlzMed, Inc. Die Studie wurde von AlzMed, Inc. finanziert (500.000 JPY an NK für das Projekt mit dem Titel "Hochdurchsatzanalyse der synaptischen Funktion").
Danksagungen
Wir danken Kazumi Kamiyama und Manami Kawada für die Unterstützung bei den Experimenten. Diese Arbeit wurde von JSPS KAKENHI (Fördernummer 19K08010 an N.K.) und Japan Agency for Medical Research and Development (AMED) (Fördernummer JP19bk0104077 und JP22bm0804024 an T.S.) unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| 96 well plate | Zeon Corporation | Gifted | |
| 96 well plate | greiner | 655986 | |
| Anti-drebrin antibody (M2F6) | MBL | D029-3 | Mouse monoclonal (dilution 1:1) |
| Anti-MAP2 antibody | Millipore | AB5622 | Rabbit (dilution 1:1000) |
| Anti-mouse Alexa Fluor 568 | Thermo Fisher Scientific | A11031 | Dilution 1: 500 |
| Anti-rabbit Alexa Fluor | Thermo Fisher Scientific | A11008 | Dilution 1: 500 |
| B-27 | Gibco | 17504-044 | 2 v/v% for MEA plates; 50x for normal plates |
| Borax | Sigma | B-9876 | Final concentration 12 mM |
| Boric acid | WAKO | 021-02195 | Final concentration 50 mM |
| Bovine serum albumin | Millipore | 12659-100G | Final concentration: 3% in PBS |
| Confocal quantitative image cytometer CellVoyager CQ1 | YOKOGAWA | Phase contrast images | |
| Cytosine β-D-arabino-furanoside (Ara-C) | Sigma | C-6645 | Diluted in dH2O (final concentration: 0.2 µM) |
| DAPI | FUJIFILM | 340-07971 | Dilution 1:1000 |
| GlutaMAX | Gibco | 35050-061 | 2.5 mM for MEA plates; 400x for normal plates |
| In Cell Analyzer 2200 | Cytiva | Fluorescence images | |
| Laminin | Sigma | 114956-81-9 | Final concentration: 20 µg/mL |
| Maestro | Axion Biosystems | MEA recordings | |
| MEA plate | Axion Biosystems | M768-tMEA-48W | |
| Neurobasal | Gibco | 21103-049 | |
| Paraformaldehyde | nacalai tesque | 26126-25 | Final concentration: 4% in PBS |
| Penicillin/Streptomycin | Gibco | 15140-122 | 100 U/mL for normal plates |
| Penicillin/Streptomycin | nacalai tesque | 26253-84 | 100 µg/mL for MEA plates |
| polyethyleimine | Sigma | 9002-98-6 | Final concentration: 0.1% |
| Poly-L-lysine | Sigma | P2636 | Diluted in the borate buffer (final concentration: 1 mg/mL) |
| SKY Neuron | AlzMed , Inc. | ARH001 | 1.0 x 106 cells/tube |
| Sodium azide | FUJIFILM | 195-11092 | 0.1% |
| SodiumL(+)-Glutamate monohydrate | WAKO | 194-02032 | Diluted in dH2O (final concentrations: 1 µM, 3 µM, 10 µM, 30 µM, 100 µM) |
Referenzen
- Banker, G. A., Cowan, W. M. Rat hippocampal neurons in dispersed cell culture. Brain Research. 126 (3), 397-342 (1977).
- Kaech, S., Banker, G. Culturing hippocampal neurons. Nature Protocols. 1 (5), 2406-2415 (2006).
- Roppongi, R. T., Champagne-Jorgensen, K. P., Siddiqui, T. J. Low-density primary hippocampal neuron culture. Journal of Visualized Experiments. (122), e55000(2017).
- Mizui, T., et al. Drebrin E is involved in the regulation of axonal growth through actin-myosin interactions. Journal of Neurochemistry. 109 (2), 611-622 (2009).
- Mizui, T., et al. Myosin II ATPase activity mediates the long-term potentiation-induced exodus of stable F-actin bound by drebrin A from dendritic spines. PLoS One. 9 (1), 85367(2014).
- Takahashi, H., Mizui, T., Shirao, T. Down-regulation of drebrin A expression suppresses synaptic targeting of NMDA receptors in developing hippocampal neurons. Journal of Neurochemistry. 97, 110-115 (2006).
- Takahashi, H., et al. Drebrin-dependent actin clustering in dendritic filopodia governs synaptic targeting of postsynaptic density-95 and dendritic spine morphogenesis. The Journal of Neuroscience. 23 (16), 6586-6595 (2003).
- Yamazaki, H., Sasagawa, Y., Yamamoto, H., Bito, H., Shirao, T. CaMKIIbeta is localized in dendritic spines as both drebrin-dependent and drebrin-independent pools. Journal of Neurochemistry. 146 (2), 145-159 (2018).
- Hanamura, K., et al. High-content imaging analysis for detecting the loss of drebrin clusters along dendrites in cultured hippocampal neurons. Journal of Pharmacological and Toxicological Methods. 99, 106607(2019).
- Mitsuoka, T., et al. Assessment of NMDA receptor inhibition of phencyclidine analogues using a high-throughput drebrin immunocytochemical assay. Journal of Pharmacological and Toxicological Methods. 99, 106583(2019).
- Ishizuka, Y., Bramham, C. R. A simple DMSO-based method for cryopreservation of primary hippocampal and cortical neurons. Journal of Neuroscience Methods. 333, 108578(2020).
- Pischedda, F., et al. Cryopreservation of primary mouse neurons: The benefit of neurostore cryoprotective medium. Frontiers in Cellular Neuroscience. 12, 81(2018).
- Kobayashi, Y., et al. Impairment of ciliary dynamics in an APP knock-in mouse model of Alzheimer's disease. Biochemical and Biophysical Research Communications. 610, 85-91 (2022).
- Kobayashi, Y., et al. Properties of primary cilia in melanin-concentrating hormone receptor 1-bearing hippocampal neurons in vivo and in vitro. Neurochemistry International. 142, 104902(2021).
- Koganezawa, N., et al. NMDA receptor-dependent and -independent effects of natural compounds and crude drugs on synaptic states as revealed by drebrin imaging analysis. The European Journal of Neuroscience. 53 (11), 3548-3560 (2021).
- Mizui, T., Takahashi, H., Sekino, Y., Shirao, T. Overexpression of drebrin A in immature neurons induces the accumulation of F-actin and PSD-95 into dendritic filopodia, and the formation of large abnormal protrusions. Molecular and Cellular Neurosciences. 30 (1), 149-157 (2005).
- Sekino, Y., et al. Activation of N-methyl-D-aspartate receptor induces a shift of drebrin distribution: disappearance from dendritic spines and appearance in dendritic shafts. Molecular and Cellular Neurosciences. 31 (3), 493-504 (2006).
- Takahashi, H., Yamazaki, H., Hanamura, K., Sekino, Y., Shirao, T. Activity of the AMPA receptor regulates drebrin stabilization in dendritic spine morphogenesis. Journal of Cell Science. 122, 1211-1219 (2009).
- Aoki, C., et al. Drebrin A is a postsynaptic protein that localizes in vivo to the submembranous surface of dendritic sites forming excitatory synapses. The Journal of Comparative Neurology. 483 (4), 383-402 (2005).
- Koganezawa, N., Hanamura, K., Sekino, Y., Shirao, T. The role of drebrin in dendritic spines. Molecular and Cellular Neurosciences. 84, 85-92 (2017).
- Shirao, T., et al. The role of drebrin in neurons. Journal of Neurochemistry. 141 (6), 819-834 (2017).
- Sekino, Y., Koganezawa, N., Mizui, T., Shirao, T. Role of drebrin in synaptic plasticity. Advances in Experimental Medicine and Biology. 1006, 183-201 (2017).
- Togo, K., et al. Postsynaptic structure formation of human iPS cell-derived neurons takes longer than presynaptic formation during neural differentiation in vitro. Molecular Brain. 14 (1), 149(2021).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten