
Method Article
Easy and Reproducible Low-Density Primary Culture using Frozen Stock of Embryonic Hippocampal Neurons
In This Article
Summary
A ready-to-use frozen stock of neurons is a powerful tool for evaluating synaptic functions. Here, we introduce an easy low-density primary culture from frozen stock using a 96-well plate.
Abstract
Neuronal culture is a valuable system for evaluating synaptic functions and drug screenings. In particular, a low-density culture of primary hippocampal neurons allows the study of individual neurons or subcellular components. We have shown subcellular protein localization within a neuron by immunocytochemistry, neuronal polarity, synaptic morphology, and its developmental change using a low-density primary hippocampal culture. Recently, ready-to-use frozen stocks of neurons have become commercially available. These frozen stocks of neurons reduce the time needed to prepare animal experiments and also contribute to the reduction of the number of animals used. Here, we introduce a reproducible low-density primary culture method using a 96-well plate. We used a commercially available frozen stock of neurons from the rat embryonic hippocampus. The neurons can be stably cultured long-term without media changes by reducing the growth of glial cells at particular timepoints. This high-throughput assay using low-density culture allows reproducible imaging-based evaluations of synaptic plasticity.
Introduction
The development of an in vitro experimental system that can assess synaptic functions involved in learning and memory is important. Neuronal culture is a valuable system for evaluating synaptic functions in vitro. The neuronal culture technique was first used in the 1980s, and in the 1990s, low-density culture of primary hippocampal neurons was developed1,2,3 for the study of individual neurons in terms of subcellular localization of protein components, protein trafficking, neuronal polarity, spine morphology, synapse development, and plasticity4,5,6,7,8. However, there are many steps involved in this technique: mating animals, dissecting embryos, preparing culture vessels, and culturing cells for 3 weeks with media changes once a week. In addition, it requires advance techniques3.
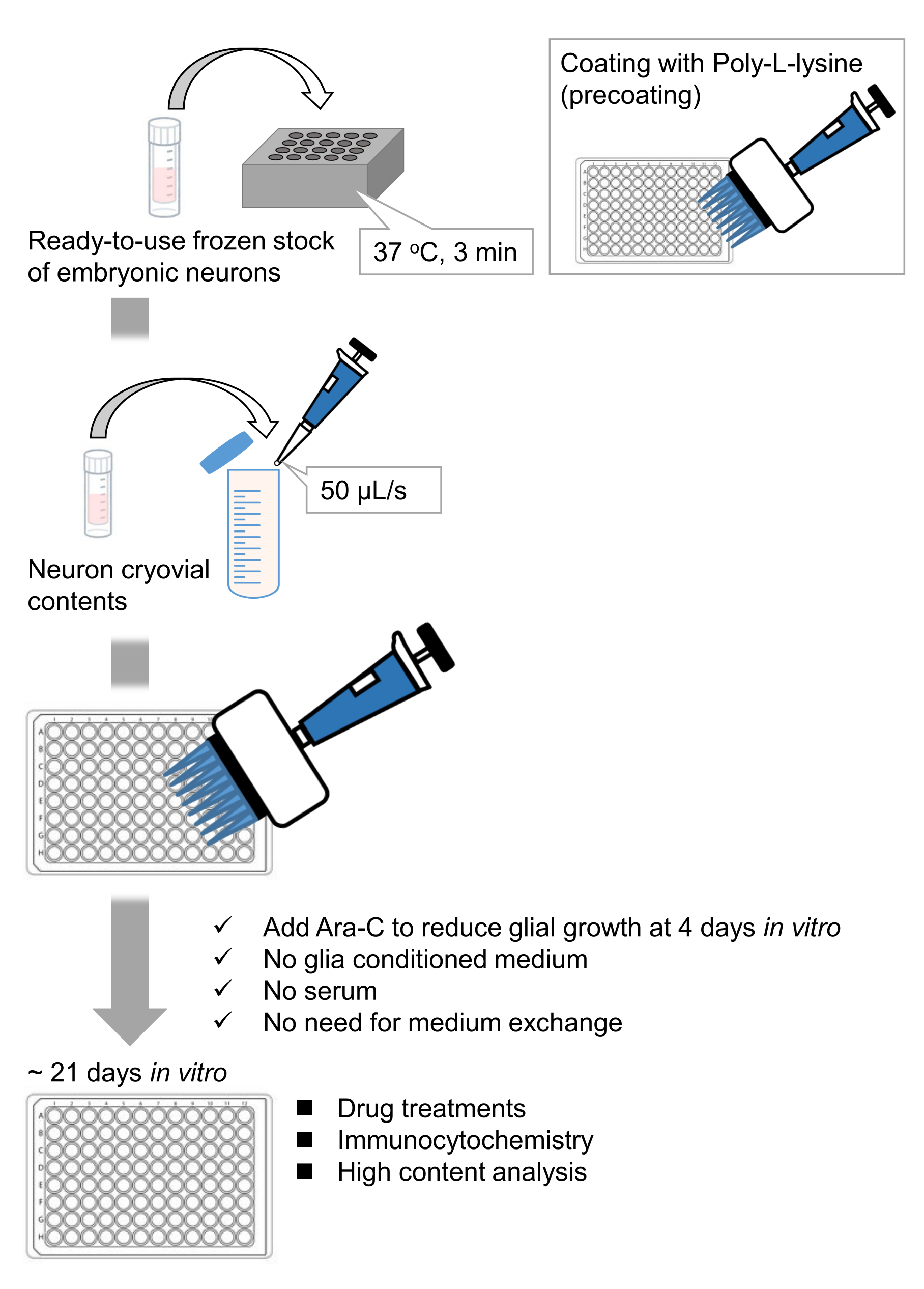
We have developed frozen stocks of dissociated hippocampal neurons from rat embryos9,10. The frozen stocks of neurons are ready-to-use, and no advance techniques are required for culturing the cells11,12. In other words, culturing the neurons from frozen stocks does not depend on the technique of an experimenter. It eliminates the need for animal experiments (e.g., permission for animal experiments, arranging timed pregnant animals, and dissecting rat embryos), thereby reducing the number of animals used. Recently, high-quality, ready-to-use frozen stocks of neurons have become commercially available. Here, we used commercially available frozen stocks from the embryonic day (E) 18 rat hippocampus13,14,15. Culturing the neurons from a frozen stock does not require glia-conditioned media or co-culture with glial cells. Ordinary primary culture media without additional serum can be used to culture the cells; hence we can acquire reproducible data. Furthermore, there is no need for media exchange for 3 weeks after the cell seeding as the growth of glial cells is reduced (Figure 1).
Dendritic spines are the postsynaptic compartment of most excitatory synapses. They contain receptor proteins, postsynaptic scaffold proteins, and actin cytoskeletal proteins. We focused on an actin-binding protein drebrin5,6,7,16,17,18. Drebrin accumulates at the spine head in mature neurons19, and we reported drebrin as a marker for the synaptic state15,17,20,21,22,23. By conducting a high-content analysis using drebrin as readout, we have recently reported the inhibitory effects of phencyclidine analogs on N-methyl-D-aspartic acid-type glutamate receptors (NMDARs)10 and the NMDAR-dependent effects of natural compounds and crude drugs on synaptic states15.
Here, we detail how to culture frozen stocks of neurons at low density. In addition, we show a drebrin imaging-based evaluation of the synaptic state using 96-well plates.
Protocol
1. Plate coating
- Coat a 96-well microplate with poly-L-lysine (1 mg/mL, diluted in 0.1 M borate buffer [pH: 8.5]; 100 µL/well) and incubate overnight at 37 °C.
NOTE: Only coat the wells that need to be used. In the experiments performed here, the middle 60 wells are used. The borate buffer is prepared by mixing 50 mM boric acid and 12 mM borate in sterilized water. - Wash the plate twice with sterilized water (250 µL/well).
- Wash the plate once with fresh culture medium without supplements (250 µL/well).
- Dry the plate on a clean bench for 20 min.
- Wrap the plate with aluminum foil and keep it at 4 °C until use (valid for 1 month).
2. Cell seeding
- Add 50 µL/well of the culture medium to the coated plate and keep it in a 37 °C, 5% CO2 incubator for 30 min to 1 h. Fill the peripheral wells with sterilized water (200 µL/well).
NOTE: The culture medium is prepared by adding 50x B-27, 400x Glutamax, and 100 U/mL of penicillin/streptomycin to the neurobasal medium (refer to the Table of Materials for details). - Remove the neuron cryovial from the liquid nitrogen tank. The neurons used here were DMSO-cryopreserved neurons11.
- Immerse the cryovial in a 37 °C heat block for up to 3 min and partially thaw the contents. Do not warm up the cryovial for too long. Transfer the contents to a 50 mL tube as soon as it is thawed.
- Slowly transfer the neuron cryovial contents to a sterile 50 mL tube drop-wise (50 µL/s) using a 1 mL pipette with a wide-pore tip.
- Rinse the empty cryovial with 1 mL of the culture medium (room temperature; RT). Transfer this 1 mL of the culture medium from the cryovial drop-wise (50 µL/s) to the 50 mL tube containing the cell suspension.
- Add 9 mL of the culture medium (RT) to the 50 mL tube drop-wise (0.5 mL/s) and make up the volume to 11 mL. Do not repeat pipetting, but mix the cell suspension slowly.
- Count the cell number (use a cell counter or a hemocytometer).
- Transfer all the cell suspension to a reservoir and dispense the cell suspension to the 96-well plate using a multichannel pipette with wide-pore tips (1.0 x 104 cells/well). To reduce the culture medium evaporation, fill the peripheral wells with sterilized water (step 2.1).
NOTE: This study confirms that the culture medium evaporation is small for a 3-week culture without medium exchange. The reduction rate of the medium is 3.6% (n = 120 wells). Thus, the osmolality change will not be drastic during the 3-week incubation period. - Incubate the neurons for 1-2 h in a 37 °C, 5% CO2 incubator.
- Replace the culture medium with 100 µL of preheated culture medium (37 °C) per well and put it back in a 37 °C, 5% CO2 incubator (no medium change is required during culture).
3. Ara-C treatment
- At 4 days in vitro (DIV), add cytosine β-D-arabino-furanoside (Ara-C) to a final concentration of 0.2 µM per well to reduce the growth of glial cells.
4. Drug treatments
- At 21 days in vitro, treat the cells with the drugs of interest.
- Keep the temperature of the plate at 37 °C during drug treatments.
- For a positive control, treat the cells with 100 µM glutamate (per well for final concentration) for 10 min before fixation.
5. Fixation
- For the fixation, use 4% paraformaldehyde in 0.1 M phosphate buffer (100 µL/well).
- After ~20 min of fixation, wash the wells with phosphate-buffered saline (PBS; 250 µL/well) 2x for 5 min each.
6. Immunocytochemistry
- Wash the cells with PBS (250 µL/well) 1x for 5 min.
- Permeabilize the cells with 0.1% Triton X-100 (100 µL/well) in PBS for 5 min.
- Wash the cells with PBS (250 µL/well) 3x for 5 min each.
- For blocking, use 3% bovine serum albumin in PBS (PBSA; 100 µL/well) for 1 h at RT.
- Incubate the cells with anti-drebrin (1:1) and anti-Microtubule Associated Protein 2 (MAP2) (1:000) antibodies (60 µL/well) at 4 °C overnight.
- Wash the cells with PBS (250 µL/well) 4x for 5 min each.
- Incubate the cells with appropriate secondary antibodies and 4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI; 1:1000) in PBSA (60 µL/well) for 2 h at RT.
- Wash the cells with PBS (250 µL/well) 4x for 5 min each.
- Store the cells in PBS containing 0.1% sodium azide (150 µL/well).
7. Image acquisition and analysis
- To acquire the images, use an appropriate microscope.
- To identify the cell bodies of neurons, use both MAP2-positive and DAPI-positive regions.
- To identify the dendrites of neurons, use MAP2-positive signals without cell bodies.
- To identify drebrin clusters, use drebrin-positive signals along MAP2 positive dendrites.
Results
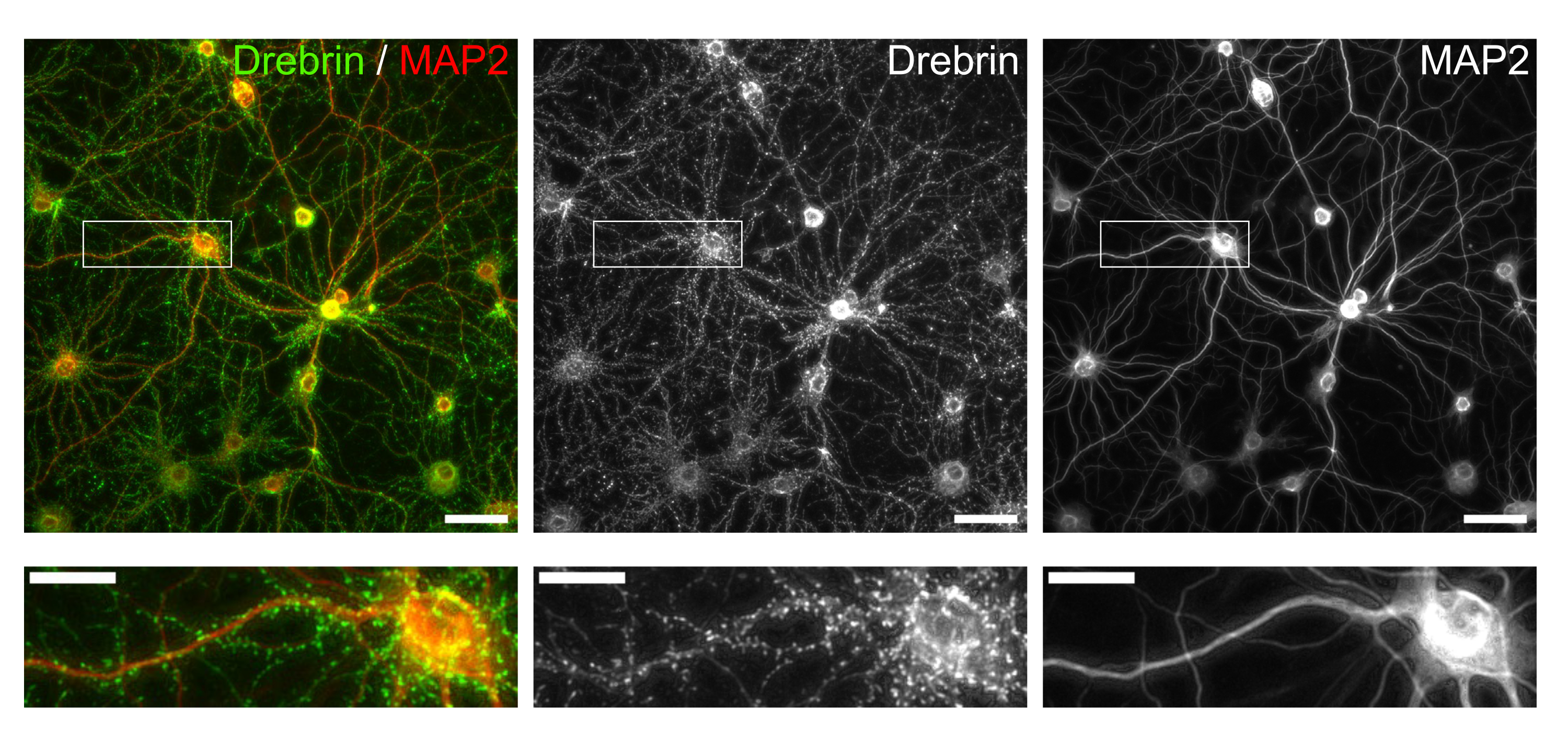
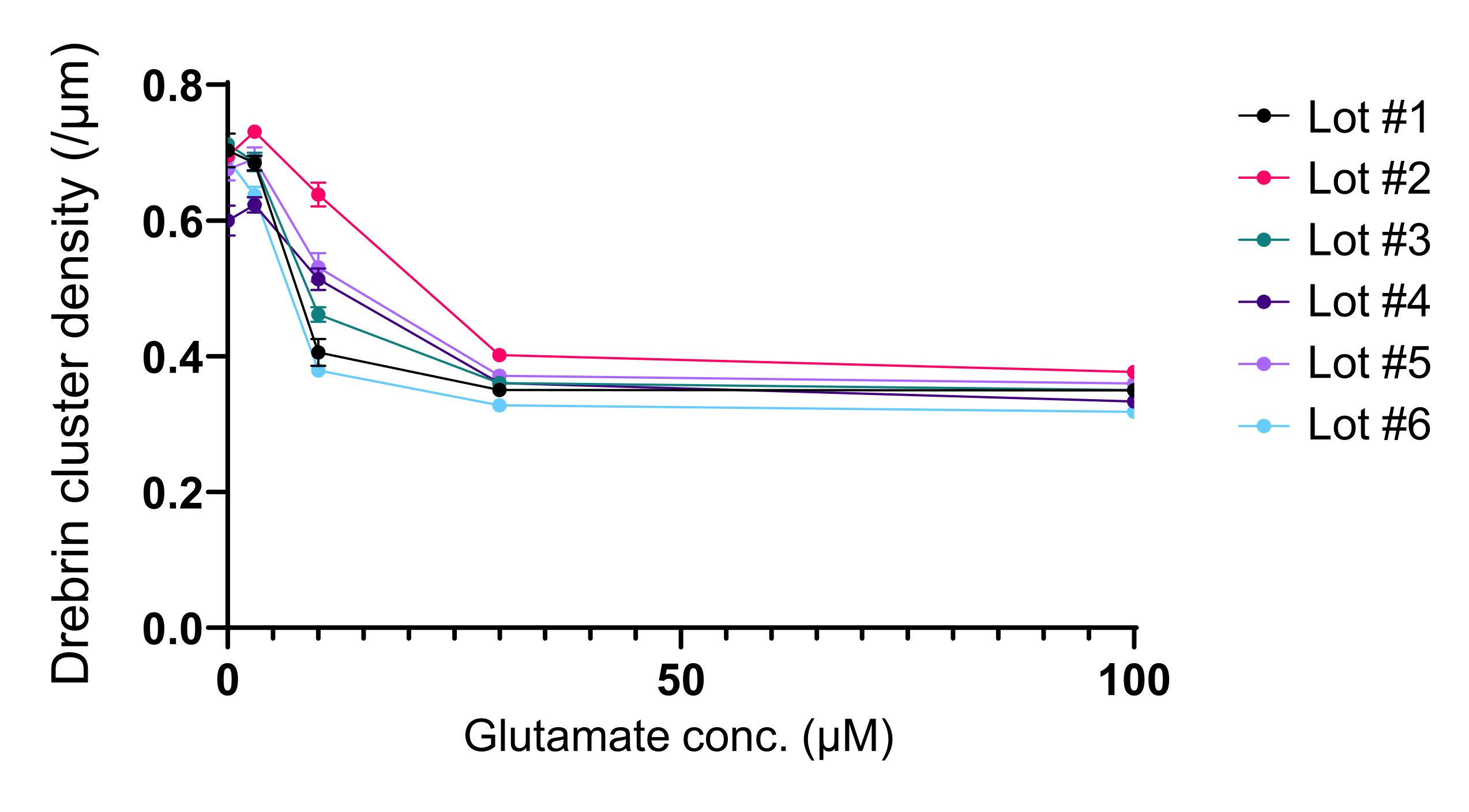
Following the protocol, the neurons were cultured in a 96-well plate for 21 days, and then treated with glutamate (Figure 1). The neurons developed normally without an exchange of the culture medium for 3 weeks (Figure 2). We treated the cells with several concentrations of glutamate (1 µM, 3 µM, 10 µM, 30 µM, and 100 µM diluted in sterilized water) for 10 min and fixed them. Immunocytochemistry was performed, and fluorescence images of drebrin and MAP2 were acquired using an automated fluorescence microscope with an sCMOS camera. As shown in Figure 3, drebrin-positive dendritic spines are clearly observed along MAP2-positive dendrites. It has been shown that glutamate stimulation elicits Ca2+ influx through NMDAR, which causes drebrin exodus from dendritic spines resulting in a reduction of drebrin cluster densities5,17. Accordingly, we observed the dose-dependent reduction of drebrin cluster densities against glutamate stimulation10 (Figure 4). As shown in Figure 5, this method is highly reproducible if drebrin is used as a marker for synaptic states.

Figure 1: Scheme of the method. The neurons were cultured in a 96-well plate for 21 days, and then treated with glutamate. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Bright-field images of cultured neurons using a 96-well plate. Phase contrast images were obtained from each developmental stage (DIV 1, 7, 14, 21) using a confocal quantitative image cytometer. Scale bar: 50 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Representative images of immunostained cultured neurons. (Left) Merged fluorescence images of drebrin (green) and MAP2 (red). Each fluorescence image of drebrin and MAP2 showed in the middle and right panels, respectively. White rectangles show the area magnified below. Scale bars; upper panels: 50 µm, lower panels: 20 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Glutamate-dependent dose-response changes in normalized drebrin cluster density. (A) Representative fluorescence images immunostained using drebrin (green) and MAP2 (red) from the well that is treated with 0 µM, 10 µM, and 100 µM glutamate (from left to right). Scale bar: 50 µm. (B) Drebrin cluster density was normalized by the average of control (0 µM). 0 µM, N = 58 wells; 1 µM, N = 46; 3 µM, N = 54; 10 µM, N = 45; 30 µM, N = 54; 100 µM, N = 55, from 13 experiments using different lots. ** P < 0.01 versus control (0 µM) by Dunnett's multiple comparisons test following ANOVA. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Glutamate-dependent dose-response changes in drebrin cluster density. The raw data from six experiments using different lots. N = 4 wells for each concentration (0 µM, 3 µM, 10 µM, 30 µM, and 100 µM). Values are expressed as mean ± SEM. Please click here to view a larger version of this figure.
{kind=link}

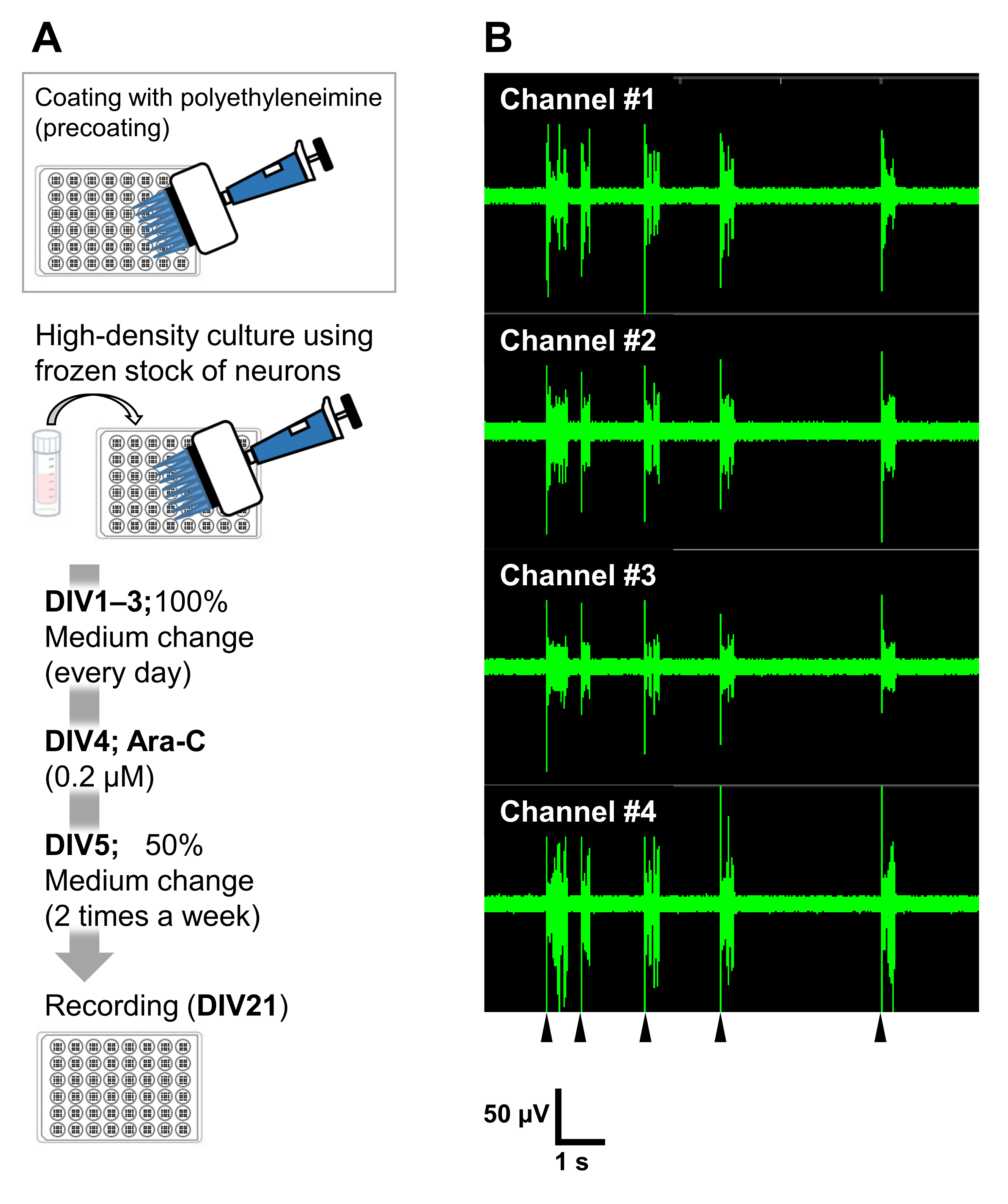
Figure 6: Application of the frozen stocks of neurons to electrophysiological experiments. (A) A protocol for electrophysiological experiments using microelectrode array (MEA) plates. Coating: One day before plating the cells, each 48-well MEA plate was pre-coated with a polyethyleneimine (PEI: 0.1%) solution and incubated for 1 h at 37 °C. The MEA plate was then washed 3x with sterilized water and dried for 1 h. Then, the MEA plate was kept at 4 °C overnight. High-density culture: 50,000 cells/well of the neurons were plated onto 48-well MEA plates. The cell seeding step was performed as described in section 2 of the above-described protocol. Laminin (20 µg/mL) added culture medium (add 2 v/v% B-27, 2.5 mM Glutamax, and 100 µg/mL of penicillin/streptomycin to the neurobasal medium) was used to plate the neurons. Thereafter, the neurons were cultured at 37 °C, 5% CO2 in the culture medium. The media was fully exchanged on DIV 1 with the culture medium up to DIV 3. Ara-C was added at DIV 4 (final 0.2 µM). From DIV 5 onward and 2 times a week, 50% of the media was changed with the culture medium. The activity of the neurons on each well of the MEA plate was recorded with an MEA system. (B) Spontaneous neuronal activity was acquired at 37 °C under a 5% CO2 atmosphere using an MEA system at a sampling rate of 12.5 kHz/channel at DIV 21. Recordings from 4 channels out of the 16 channels within a well are shown. For all the recordings, a Butterworth band-pass filter (200-3,000 Hz) was applied. Arrowheads show the timing of synchronized burst firing. Please click here to view a larger version of this figure.
{kind=link}
Discussion
A critical step in this method is to thaw the cell suspension. Transfer of the cell suspension before it becomes too warm is very important. To avoid the rapid change of osmolality however, do not transfer the cell suspension to a large volume of the medium at once. Drop-wise addition of the culture medium is also crucial to avoid sudden changes in osmotic pressure.
The neurons can be cultured in other culture vessels: 24-well plates, 8-well chambers, 60 mm dishes, or MED probes. In those cases, however, the final concentration and the timing of the addition of Ara-C need to be adjusted. In addition, the density of neurons needs to be optimized in different types of experiments. For example, high-density culture is required for electrophysiological experiments, and in that case, the media exchange is needed twice a week (Figure 6). Thus, a low-density culture requires fewer steps than a high-density culture.
Low-density neuronal culture often requires advance techniques; however, using ready-to-use frozen stock solves this problem. The described method does not depend on the skill of an experimenter. The quality of frozen stock is stable and can be stably cultured as long as they are stored in liquid nitrogen and avoid temperature changes for up to 4 years.
Culturing the cells for 3 weeks without medium exchange raises the question on whether there are significant osmolality changes or culture media evaporation. However, we have confirmed that the culture media evaporation is small (reduction rate of 3.6%). The localization of synaptic proteins and the morphology of the neurons appear normal after 3 weeks. Therefore, 3-week culture without media exchange does not cause large osmolality changes affecting the conditions of the cultured neurons. Keeping the plate in an incubator after Ara-C treatment is also an important point that minimizes evaporation.
There is no limitation regarding the use of the frozen stock neurons. However, there are some limitations of the low-density culture method. We confirmed that the low-density culture could be applied for morphological observation of neurons, evaluation of synaptic function, and GFP transfection. However, we have not examined live cell imaging. In addition, as mentioned above, high-density culture is required to perform electrophysiology.
Synapse maturation typically takes 3 weeks7, and we cannot confirm that the cultured neurons have proper synapses until the end. If the synapse maturation is not good after 3 weeks, we would have to culture again. By knowing the quality of the neurons before starting the experiments, we can save these 3 weeks. Therefore, to perform experiments efficiently, it is best to check the quality of the neurons in advance. Frozen stocks make it possible to check the quality of the neurons beforehand. Each batch of frozen stocks is generated from one litter of rats, and we can use one of the stocks from each batch for a quality check. Drebrin is a good marker for the quality check of the neurons. As described, drebrin accumulates in the spine head in mature neurons, and it reacts to synaptic stimulation. Thus, we can check the quality of the neurons in frozen stocks by using drebrin as a marker.
This method can be applied to evaluate the effect of drugs on the synaptic state. The drebrin exodus from dendritic spines occurs during the initial stages of synaptic plasticity22. Therefore, the detection of drebrin cluster reduction elicited by drug treatment shows that the drug stimulates the synapse and causes synaptic plasticity. Furthermore, to identify whether the reduction is NMDAR dependent, an experiment using 2-amino-5-phosphonovaleric acid (APV, an NMDAR antagonist) is useful. Using drebrin as a marker, even an NMDAR-dependency is clearly determined10,15. The method described is useful in drug screenings, safety pharmacological studies, and evaluation of synaptic function.
Disclosures
Tomoaki Shirao is the CEO of AlzMed, Inc. The study was funded by AlzMed, Inc. (500,000 JPY to NK for the project entitled "High-throughput analysis of synaptic function").
Acknowledgements
We thank Kazumi Kamiyama and Manami Kawada for assistance with experiments. This work was supported by JSPS KAKENHI (Grant Number 19K08010 to N.K.) and Japan Agency for Medical Research and Development (AMED) (Grant Number JP19bk0104077 and JP22bm0804024 to T.S.).
Materials
| Name | Company | Catalog Number | Comments |
| 96 well plate | Zeon Corporation | Gifted | |
| 96 well plate | greiner | 655986 | |
| Anti-drebrin antibody (M2F6) | MBL | D029-3 | Mouse monoclonal (dilution 1:1) |
| Anti-MAP2 antibody | Millipore | AB5622 | Rabbit (dilution 1:1000) |
| Anti-mouse Alexa Fluor 568 | Thermo Fisher Scientific | A11031 | Dilution 1: 500 |
| Anti-rabbit Alexa Fluor | Thermo Fisher Scientific | A11008 | Dilution 1: 500 |
| B-27 | Gibco | 17504-044 | 2 v/v% for MEA plates; 50x for normal plates |
| Borax | Sigma | B-9876 | Final concentration 12 mM |
| Boric acid | WAKO | 021-02195 | Final concentration 50 mM |
| Bovine serum albumin | Millipore | 12659-100G | Final concentration: 3% in PBS |
| Confocal quantitative image cytometer CellVoyager CQ1 | YOKOGAWA | Phase contrast images | |
| Cytosine β-D-arabino-furanoside (Ara-C) | Sigma | C-6645 | Diluted in dH2O (final concentration: 0.2 µM) |
| DAPI | FUJIFILM | 340-07971 | Dilution 1:1000 |
| GlutaMAX | Gibco | 35050-061 | 2.5 mM for MEA plates; 400x for normal plates |
| In Cell Analyzer 2200 | Cytiva | Fluorescence images | |
| Laminin | Sigma | 114956-81-9 | Final concentration: 20 µg/mL |
| Maestro | Axion Biosystems | MEA recordings | |
| MEA plate | Axion Biosystems | M768-tMEA-48W | |
| Neurobasal | Gibco | 21103-049 | |
| Paraformaldehyde | nacalai tesque | 26126-25 | Final concentration: 4% in PBS |
| Penicillin/Streptomycin | Gibco | 15140-122 | 100 U/mL for normal plates |
| Penicillin/Streptomycin | nacalai tesque | 26253-84 | 100 µg/mL for MEA plates |
| polyethyleimine | Sigma | 9002-98-6 | Final concentration: 0.1% |
| Poly-L-lysine | Sigma | P2636 | Diluted in the borate buffer (final concentration: 1 mg/mL) |
| SKY Neuron | AlzMed , Inc. | ARH001 | 1.0 x 106 cells/tube |
| Sodium azide | FUJIFILM | 195-11092 | 0.1% |
| SodiumL(+)-Glutamate monohydrate | WAKO | 194-02032 | Diluted in dH2O (final concentrations: 1 µM, 3 µM, 10 µM, 30 µM, 100 µM) |
References
- Banker, G. A., Cowan, W. M. Rat hippocampal neurons in dispersed cell culture. Brain Research. 126 (3), 397-342 (1977).
- Kaech, S., Banker, G. Culturing hippocampal neurons. Nature Protocols. 1 (5), 2406-2415 (2006).
- Roppongi, R. T., Champagne-Jorgensen, K. P., Siddiqui, T. J. Low-density primary hippocampal neuron culture. Journal of Visualized Experiments. (122), e55000 (2017).
- Mizui, T., et al. Drebrin E is involved in the regulation of axonal growth through actin-myosin interactions. Journal of Neurochemistry. 109 (2), 611-622 (2009).
- Mizui, T., et al. Myosin II ATPase activity mediates the long-term potentiation-induced exodus of stable F-actin bound by drebrin A from dendritic spines. PLoS One. 9 (1), 85367 (2014).
- Takahashi, H., Mizui, T., Shirao, T. Down-regulation of drebrin A expression suppresses synaptic targeting of NMDA receptors in developing hippocampal neurons. Journal of Neurochemistry. 97, 110-115 (2006).
- Takahashi, H., et al. Drebrin-dependent actin clustering in dendritic filopodia governs synaptic targeting of postsynaptic density-95 and dendritic spine morphogenesis. The Journal of Neuroscience. 23 (16), 6586-6595 (2003).
- Yamazaki, H., Sasagawa, Y., Yamamoto, H., Bito, H., Shirao, T. CaMKIIbeta is localized in dendritic spines as both drebrin-dependent and drebrin-independent pools. Journal of Neurochemistry. 146 (2), 145-159 (2018).
- Hanamura, K., et al. High-content imaging analysis for detecting the loss of drebrin clusters along dendrites in cultured hippocampal neurons. Journal of Pharmacological and Toxicological Methods. 99, 106607 (2019).
- Mitsuoka, T., et al. Assessment of NMDA receptor inhibition of phencyclidine analogues using a high-throughput drebrin immunocytochemical assay. Journal of Pharmacological and Toxicological Methods. 99, 106583 (2019).
- Ishizuka, Y., Bramham, C. R. A simple DMSO-based method for cryopreservation of primary hippocampal and cortical neurons. Journal of Neuroscience Methods. 333, 108578 (2020).
- Pischedda, F., et al. Cryopreservation of primary mouse neurons: The benefit of neurostore cryoprotective medium. Frontiers in Cellular Neuroscience. 12, 81 (2018).
- Kobayashi, Y., et al. Impairment of ciliary dynamics in an APP knock-in mouse model of Alzheimer's disease. Biochemical and Biophysical Research Communications. 610, 85-91 (2022).
- Kobayashi, Y., et al. Properties of primary cilia in melanin-concentrating hormone receptor 1-bearing hippocampal neurons in vivo and in vitro. Neurochemistry International. 142, 104902 (2021).
- Koganezawa, N., et al. NMDA receptor-dependent and -independent effects of natural compounds and crude drugs on synaptic states as revealed by drebrin imaging analysis. The European Journal of Neuroscience. 53 (11), 3548-3560 (2021).
- Mizui, T., Takahashi, H., Sekino, Y., Shirao, T. Overexpression of drebrin A in immature neurons induces the accumulation of F-actin and PSD-95 into dendritic filopodia, and the formation of large abnormal protrusions. Molecular and Cellular Neurosciences. 30 (1), 149-157 (2005).
- Sekino, Y., et al. Activation of N-methyl-D-aspartate receptor induces a shift of drebrin distribution: disappearance from dendritic spines and appearance in dendritic shafts. Molecular and Cellular Neurosciences. 31 (3), 493-504 (2006).
- Takahashi, H., Yamazaki, H., Hanamura, K., Sekino, Y., Shirao, T. Activity of the AMPA receptor regulates drebrin stabilization in dendritic spine morphogenesis. Journal of Cell Science. 122, 1211-1219 (2009).
- Aoki, C., et al. Drebrin A is a postsynaptic protein that localizes in vivo to the submembranous surface of dendritic sites forming excitatory synapses. The Journal of Comparative Neurology. 483 (4), 383-402 (2005).
- Koganezawa, N., Hanamura, K., Sekino, Y., Shirao, T. The role of drebrin in dendritic spines. Molecular and Cellular Neurosciences. 84, 85-92 (2017).
- Shirao, T., et al. The role of drebrin in neurons. Journal of Neurochemistry. 141 (6), 819-834 (2017).
- Sekino, Y., Koganezawa, N., Mizui, T., Shirao, T. Role of drebrin in synaptic plasticity. Advances in Experimental Medicine and Biology. 1006, 183-201 (2017).
- Togo, K., et al. Postsynaptic structure formation of human iPS cell-derived neurons takes longer than presynaptic formation during neural differentiation in vitro. Molecular Brain. 14 (1), 149 (2021).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved