
Method Article
Culture primaire de faible densité facile et reproductible utilisant un stock congelé de neurones embryonnaires de l’hippocampe
Dans cet article
Résumé
Un stock congelé de neurones prêt à l’emploi est un outil puissant pour évaluer les fonctions synaptiques. Ici, nous introduisons une culture primaire facile à faible densité à partir de stock congelé à l’aide d’une plaque de 96 puits.
Résumé
La culture neuronale est un système précieux pour évaluer les fonctions synaptiques et les dépistages de médicaments. En particulier, une culture de faible densité de neurones primaires de l’hippocampe permet l’étude de neurones individuels ou de composants subcellulaires. Nous avons montré la localisation des protéines subcellulaires dans un neurone par immunocytochimie, polarité neuronale, morphologie synaptique et son changement de développement en utilisant une culture hippocampique primaire de faible densité. Récemment, des stocks congelés de neurones prêts à l’emploi sont devenus disponibles dans le commerce. Ces stocks congelés de neurones réduisent le temps nécessaire à la préparation des expérimentations animales et contribuent également à la réduction du nombre d’animaux utilisés. Ici, nous introduisons une méthode de culture primaire reproductible à faible densité utilisant une plaque de 96 puits. Nous avons utilisé un stock congelé de neurones disponibles dans le commerce provenant de l’hippocampe embryonnaire du rat. Les neurones peuvent être cultivés de manière stable à long terme sans changements de milieu en réduisant la croissance des cellules gliales à des moments particuliers. Ce test à haut débit utilisant une culture de faible densité permet des évaluations reproductibles de la plasticité synaptique basées sur l’imagerie.
Introduction
Le développement d’un système expérimental in vitro capable d’évaluer les fonctions synaptiques impliquées dans l’apprentissage et la mémoire est important. La culture neuronale est un système précieux pour évaluer les fonctions synaptiques in vitro. La technique de culture neuronale a été utilisée pour la première fois dans les années 1980 et, dans les années 1990, une culture à faible densité de neurones primaires de l’hippocampe a été développée 1,2,3 pour l’étude des neurones individuels en termes de localisation subcellulaire des composants protéiques, de trafic de protéines, de polarité neuronale, de morphologie de la colonne vertébrale, de développement des synapses et de plasticité 4,5,6,7,8 . Cependant, cette technique comporte de nombreuses étapes: accouplement des animaux, dissection des embryons, préparation des vaisseaux de culture et culture de cellules pendant 3 semaines avec des changements de milieux une fois par semaine. De plus, il nécessite des techniques avancées3.
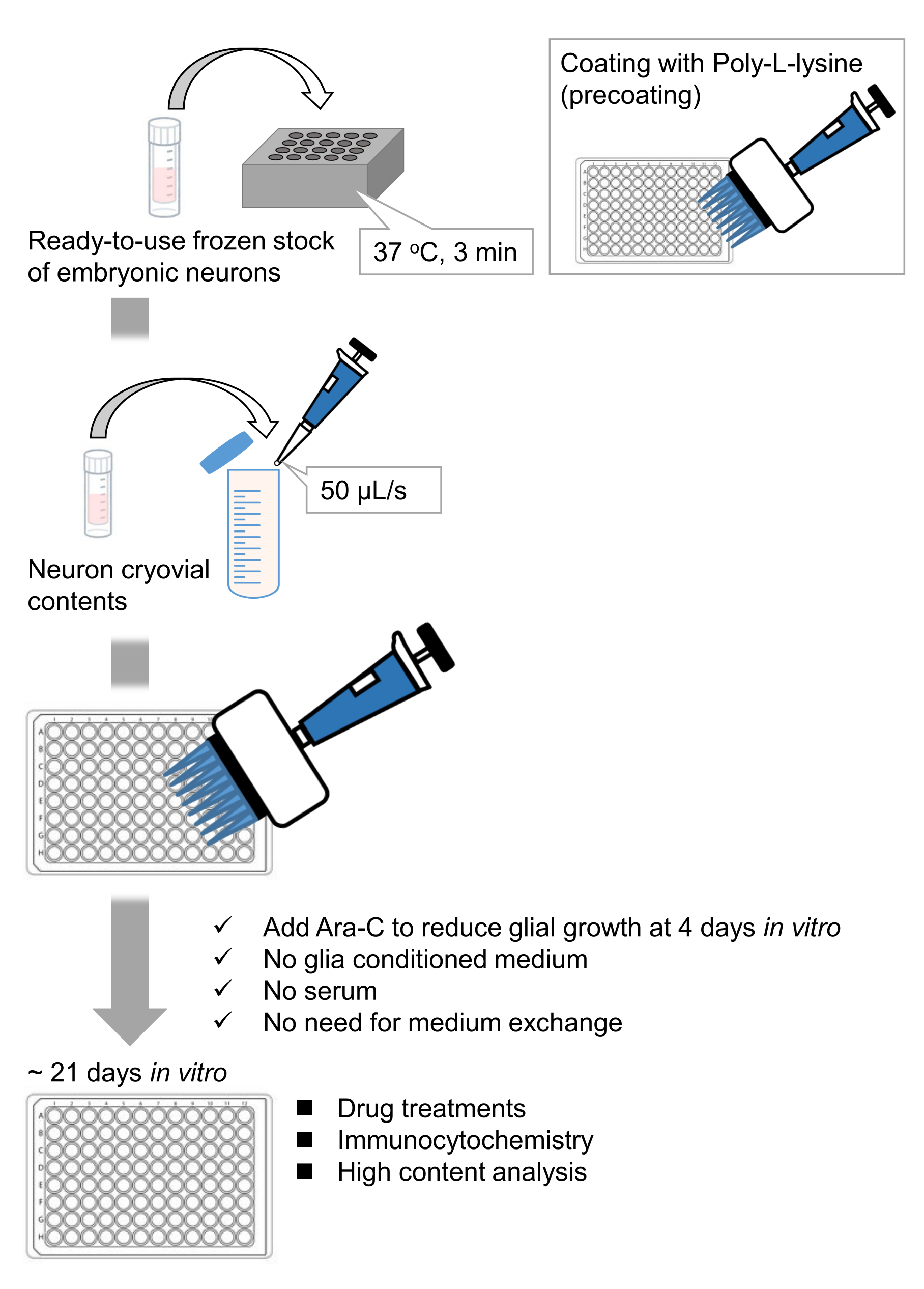
Nous avons développé des stocks congelés de neurones hippocampiques dissociés à partir d’embryons de rats 9,10. Les stocks congelés de neurones sont prêts à l’emploi et aucune technique avancée n’est nécessaire pour la culture des cellules11,12. En d’autres termes, la culture des neurones à partir de stocks congelés ne dépend pas de la technique d’un expérimentateur. Il élimine le besoin d’expériences sur les animaux (p. ex., autorisation d’expérimentation animale, organisation d’animaux gravides chronométrées et dissection d’embryons de rats), réduisant ainsi le nombre d’animaux utilisés. Récemment, des stocks congelés de neurones de haute qualité et prêts à l’emploi sont devenus disponibles dans le commerce. Ici, nous avons utilisé des stocks congelés disponibles dans le commerce de l’hippocampe de rat embryonnaire jour (E)18 13,14,15. La culture des neurones à partir d’un stock congelé ne nécessite pas de milieux conditionnés par la glie ou de co-culture avec des cellules gliales. Des milieux de culture primaires ordinaires sans sérum supplémentaire peuvent être utilisés pour cultiver les cellules; Nous pouvons donc acquérir des données reproductibles. De plus, il n’y a pas besoin d’échange de milieux pendant 3 semaines après l’ensemencement cellulaire car la croissance des cellules gliales est réduite (Figure 1).
Les épines dendritiques sont le compartiment postsynaptique de la plupart des synapses excitatrices. Ils contiennent des protéines réceptrices, des protéines d’échafaudage postsynaptiques et des protéines cytosquelettiques d’actine. Nous nous sommes concentrés sur une protéine liant l’actine drebrine 5,6,7,16,17,18. La drébrine s’accumule à la tête de la colonne vertébrale dans les neurones matures19, et nous avons rapporté la drébrine comme marqueur de l’état synaptique 15,17,20,21,22,23. En effectuant une analyse à haute teneur en utilisant la drebrine comme lecture, nous avons récemment rapporté les effets inhibiteurs des analogues de la phencyclidine sur les récepteurs du glutamate de type acide N-méthyl-D-aspartique (NMDAR)10 et les effets dépendants NMDAR des composés naturels et des médicaments bruts sur les états synaptiques15.
Ici, nous détaillons comment cultiver des stocks congelés de neurones à faible densité. De plus, nous montrons une évaluation de l’état synaptique basée sur l’imagerie de la drebrine à l’aide de plaques à 96 puits.
Protocole
1. Revêtement de plaque
- Enduire une microplaque de 96 puits de poly-L-lysine (1 mg/mL, diluée dans un tampon de borate de 0,1 M [pH : 8,5]; 100 μL/puits) et incuber pendant une nuit à 37 °C.
REMARQUE : Ne recouvrez que les puits qui doivent être utilisés. Dans les expériences effectuées ici, les 60 puits du milieu sont utilisés. Le tampon de borate est préparé en mélangeant 50 mM d’acide borique et 12 mM de borate dans de l’eau stérilisée. - Lavez l’assiette deux fois avec de l’eau stérilisée (250 μL/puits).
- Laver l’assiette une fois avec un milieu de culture frais sans suppléments (250 μL/puits).
- Sécher l’assiette sur un banc propre pendant 20 min.
- Envelopper la plaque de papier aluminium et la maintenir à 4 °C jusqu’à utilisation (valable 1 mois).
2. Ensemencement cellulaire
- Ajouter 50 μL/puits du milieu de culture à la plaque revêtue et la conserver dans un incubateur à 37 °C, 5 % de CO2 pendant 30 min à 1 h. Remplir les puits périphériques avec de l’eau stérilisée (200 μL/puits).
REMARQUE : Le milieu de culture est préparé en ajoutant 50x B-27, 400x Glutamax et 100 U/mL de pénicilline/streptomycine au milieu neurobasal (voir le tableau des matériaux pour plus de détails). - Retirez le neurone cryovial du réservoir d’azote liquide. Les neurones utilisés ici étaient des neurones cryoconservés par le DMSO11.
- Immerger le cryovial dans un bloc thermique à 37 °C jusqu’à 3 min et décongeler partiellement le contenu. Ne réchauffez pas le cryovial trop longtemps. Transférer le contenu dans un tube de 50 mL dès qu’il est décongelé.
- Transférer lentement le contenu cryovial des neurones dans un tube stérile de 50 mL goutte à goutte (50 μL/s) à l’aide d’une pipette de 1 mL munie d’une pointe à pores larges.
- Rincer le cryovial vide avec 1 mL du milieu de culture (température ambiante; RT). Transférer ce 1 mL du milieu de culture du cryovial goutte à goutte (50 μL/s) au tube de 50 mL contenant la suspension cellulaire.
- Ajouter 9 mL du milieu de culture (RT) dans le tube de 50 mL goutte à goutte (0,5 mL/s) et porter le volume à 11 mL. Ne répétez pas le pipetage, mais mélangez lentement la suspension cellulaire.
- Comptez le nombre de cellules (utilisez un compteur de cellules ou un hémocytomètre).
- Transférer toute la suspension cellulaire dans un réservoir et distribuer la suspension de cellule sur la plaque de 96 puits à l’aide d’une pipette multicanal à embouts à pores larges (1,0 x 104 cellules/puits). Pour réduire l’évaporation du milieu de culture, remplir les puits périphériques avec de l’eau stérilisée (étape 2.1).
NOTE: Cette étude confirme que l’évaporation du milieu de culture est faible pour une culture de 3 semaines sans échange de milieu. Le taux de réduction du milieu est de 3,6 % (n = 120 puits). Ainsi, le changement d’osmolalité ne sera pas drastique pendant la période d’incubation de 3 semaines. - Incuber les neurones pendant 1-2 h dans un incubateur à 37 °C, 5% CO2 .
- Remplacer le milieu de culture par 100 μL de milieu de culture préchauffé (37 °C) par puits et le remettre dans un incubateur à 37 °C, 5 % de CO2 (aucun changement de milieu n’est requis pendant la culture).
3. Traitement Ara-C
- À 4 jours in vitro (DIV), ajouter de la cytosine β-D-arabino-furanoside (Ara-C) à une concentration finale de 0,2 μM par puits pour réduire la croissance des cellules gliales.
4. Traitements médicamenteux
- À 21 jours in vitro, traiter les cellules avec les médicaments d’intérêt.
- Maintenez la température de la plaque à 37 °C pendant les traitements médicamenteux.
- Pour un contrôle positif, traiter les cellules avec 100 μM de glutamate (par puits pour la concentration finale) pendant 10 minutes avant la fixation.
5. Fixation
- Pour la fixation, utiliser du paraformaldéhyde à 4 % dans un tampon phosphate de 0,1 M (100 μL/puits).
- Après ~20 min de fixation, laver les puits avec une solution saline tamponnée au phosphate (PBS; 250 μL/puits) 2x pendant 5 min chacun.
6. Immunocytochimie
- Laver les cellules avec du PBS (250 μL/puits) 1x pendant 5 min.
- Perméabiliser les cellules avec 0,1% de Triton X-100 (100 μL/puits) dans du PBS pendant 5 min.
- Laver les cellules avec du PBS (250 μL/puits) 3x pendant 5 min chacune.
- Pour le blocage, utiliser de l’albumine sérique bovine à 3 % dans du PBS (PBSA; 100 μL/puits) pendant 1 h à TA.
- Incuber les cellules avec des anticorps anti-drébrine (1:1) et anti-Microtubule Associated Protein 2 (MAP2) (1:000) (60 μL/puits) à 4 °C pendant la nuit.
- Lavez les cellules avec du PBS (250 μL/puits) 4x pendant 5 min chacune.
- Incuber les cellules avec les anticorps secondaires appropriés et le 4′,6-diamidino-2-phénylindole, dichlorhydrate (DAPI; 1:1000) dans du PBSA (60 μL/puits) pendant 2 h à TA.
- Lavez les cellules avec du PBS (250 μL/puits) 4x pendant 5 min chacune.
- Entreposer les cellules dans du PBS contenant 0,1 % d’azoture de sodium (150 μL/puits).
7. Acquisition et analyse d’images
- Pour acquérir les images, utilisez un microscope approprié.
- Pour identifier les corps cellulaires des neurones, utilisez les régions MAP2-positives et DAPI-positives.
- Pour identifier les dendrites des neurones, utilisez des signaux MAP2-positifs sans corps cellulaire.
- Pour identifier les amas de drebrines, utilisez des signaux positifs à la drébrine le long des dendrites positives MAP2.
Résultats
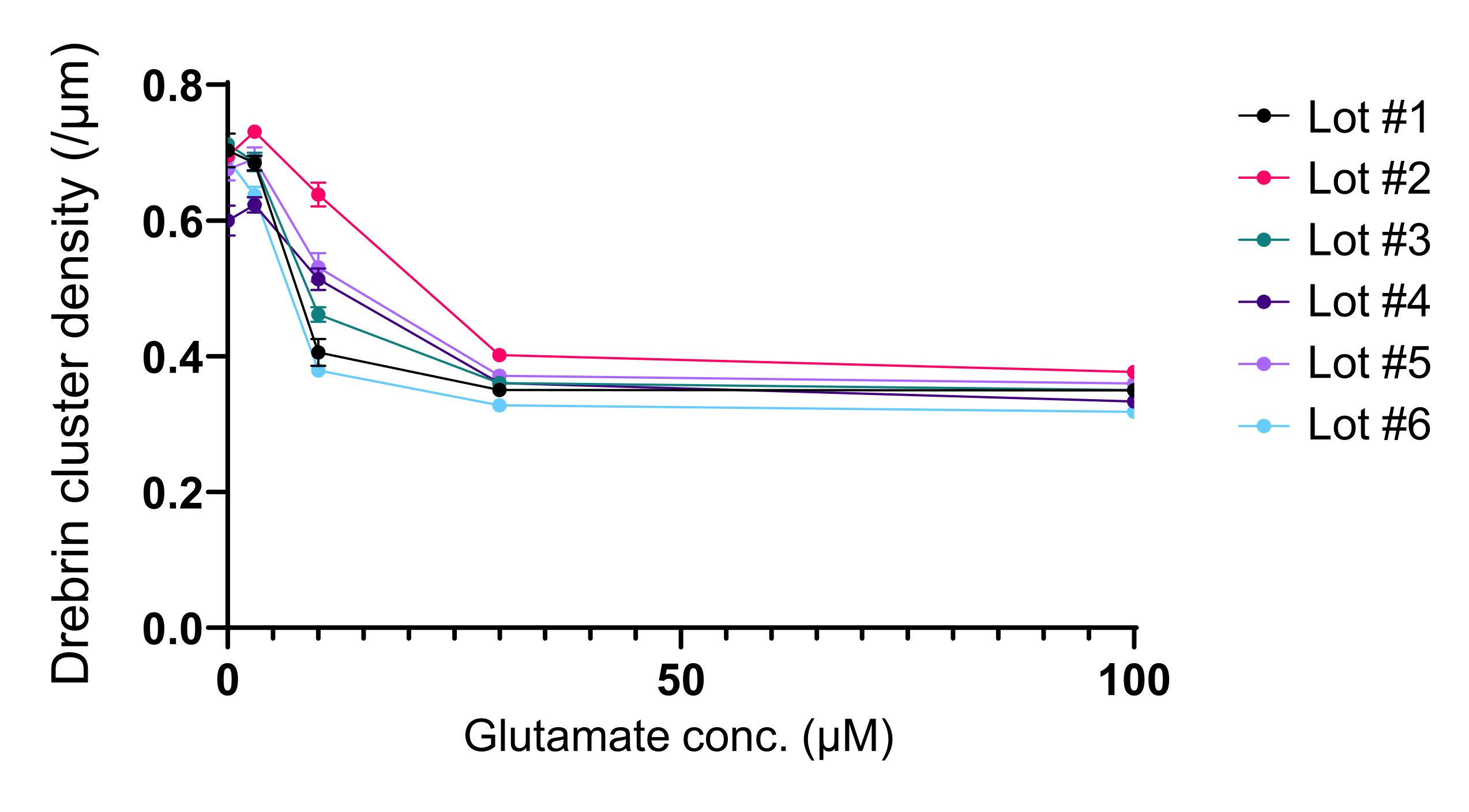
Conformément au protocole, les neurones ont été cultivés dans une plaque de 96 puits pendant 21 jours, puis traités au glutamate (Figure 1). Les neurones se sont développés normalement sans échange du milieu de culture pendant 3 semaines (Figure 2). Nous avons traité les cellules avec plusieurs concentrations de glutamate (1 μM, 3 μM, 10 μM, 30 μM et 100 μM diluées dans de l’eau stérilisée) pendant 10 min et les avons fixées. L’immunocytochimie a été réalisée et des images de fluorescence de la drebrine et de MAP2 ont été acquises à l’aide d’un microscope à fluorescence automatisé avec une caméra sCMOS. Comme le montre la figure 3, des épines dendritiques drebrines-positives sont clairement observées le long des dendrites MAP2-positives. Il a été démontré que la stimulation du glutamate provoque un afflux de Ca2+ à travers NMDAR, ce qui provoque l’exode de la drebrine des épines dendritiques entraînant une réduction des densités des amas de drebrine 5,17. En conséquence, nous avons observé la réduction dose-dépendante des densités des grappes de drebrine contre la stimulation du glutamate10 (Figure 4). Comme le montre la figure 5, cette méthode est hautement reproductible si la drébrine est utilisée comme marqueur des états synaptiques.

Figure 1 : Schéma de la méthode. Les neurones ont été cultivés dans une plaque de 96 puits pendant 21 jours, puis traités avec du glutamate. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2 : Images en fond clair de neurones en culture à l’aide d’une plaque à 96 puits. Des images de contraste de phase ont été obtenues à partir de chaque stade de développement (DIV 1, 7, 14, 21) à l’aide d’un cytomètre d’image confocale quantitative. Barre d’échelle: 50 μm. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

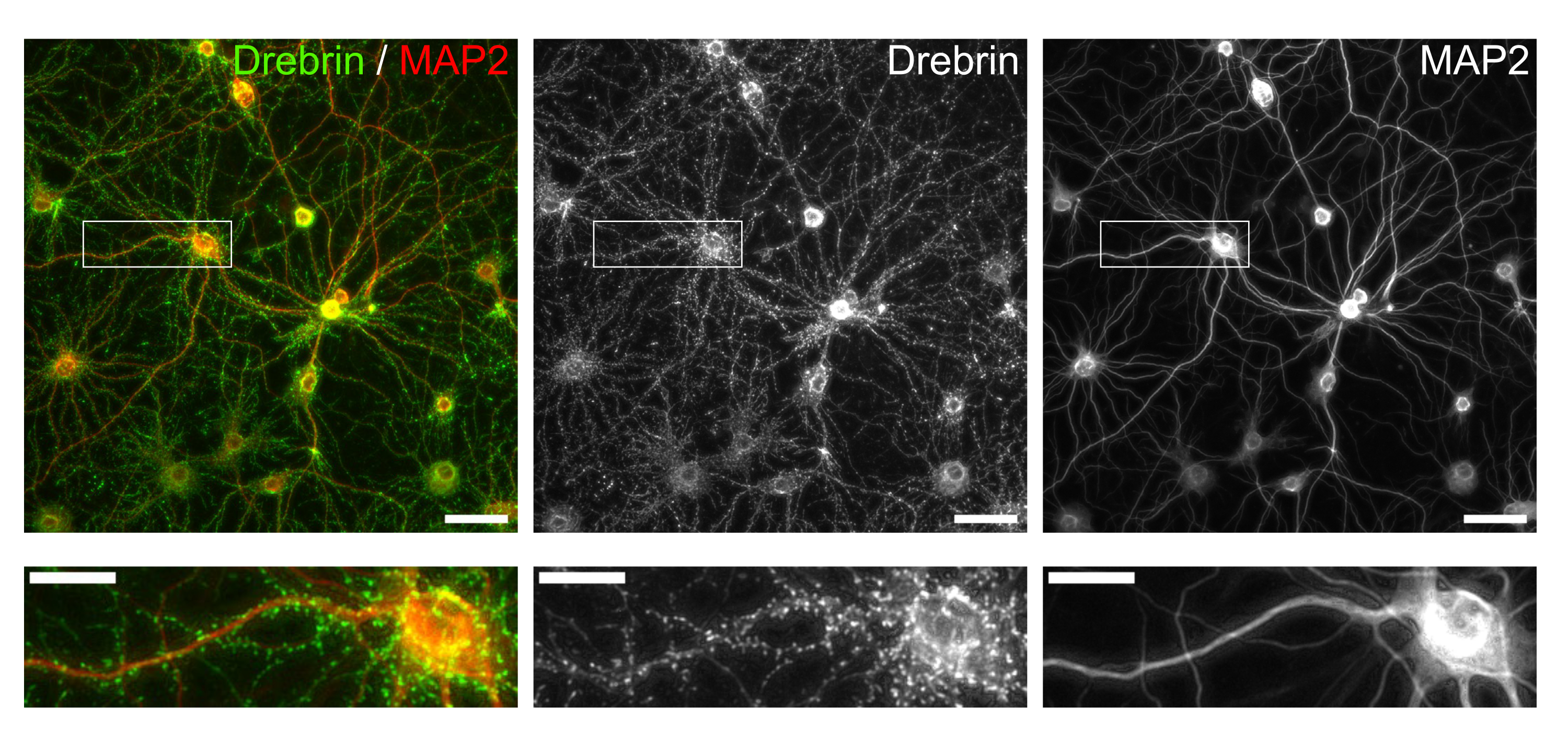
Figure 3 : Images représentatives de neurones en culture immunocolorés. (Gauche) Images de fluorescence fusionnées de la drebrine (vert) et de MAP2 (rouge). Chaque image de fluorescence de la drebrine et de MAP2 montrée dans les panneaux du milieu et de droite, respectivement. Des rectangles blancs montrent la zone agrandie ci-dessous. Barres d’échelle; panneaux supérieurs: 50 μm, panneaux inférieurs: 20 μm. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 4 : Changements dose-réponse dépendants du glutamate dans la densité normalisée des grappes de drebrine. (A) Images de fluorescence représentatives immunocolorées à l’aide de drebrine (vert) et de MAP2 (rouge) provenant du puits traité avec 0 μM, 10 μM et 100 μM de glutamate (de gauche à droite). Barre d’échelle : 50 μm. (B) La densité des grappes de drébrine a été normalisée par la moyenne du témoin (0 μM). 0 μM, N = 58 puits; 1 μM, N = 46; 3 μM, N = 54; 10 μM, N = 45; 30 μM, N = 54; 100 μM, N = 55, à partir de 13 expériences utilisant différents lots. ** P < 0,01 par rapport au contrôle (0 μM) par le test de comparaisons multiples de Dunnett après ANOVA. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 5 : Changements dose-réponse dépendants du glutamate dans la densité des grappes de drebrin. Les données brutes de six expériences utilisant différents lots. N = 4 puits pour chaque concentration (0 μM, 3 μM, 10 μM, 30 μM et 100 μM). Les valeurs sont exprimées sous forme de moyenne ± SEM. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

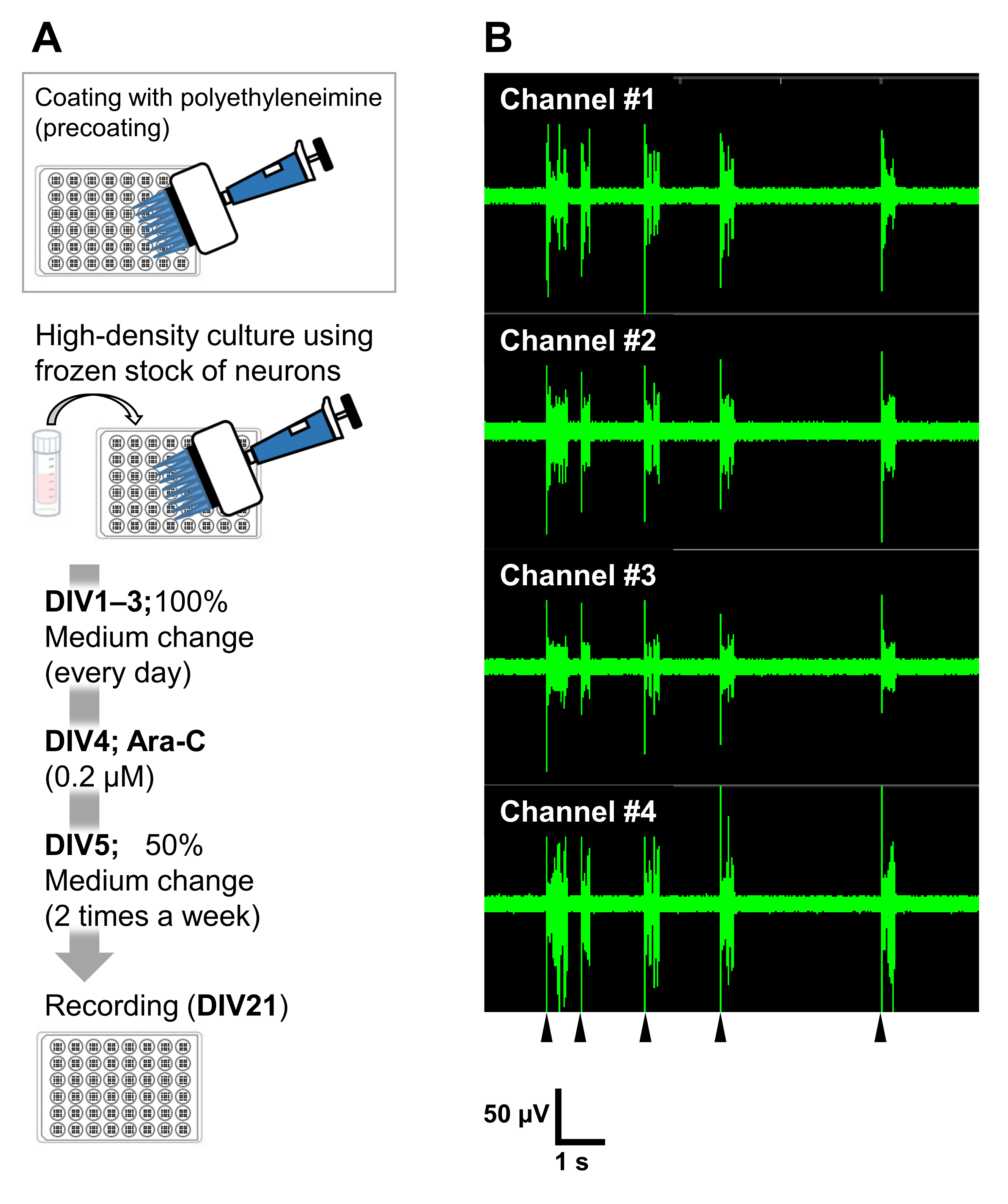
Figure 6 : Application des stocks congelés de neurones à des expériences électrophysiologiques. (A) Un protocole pour les expériences électrophysiologiques utilisant des plaques de microélectrodes (MEA). Revêtement : Un jour avant le placage des cellules, chaque plaque MEA de 48 puits a été pré-enduite d’une solution de polyéthylèneimine (PEI : 0,1%) et incubée pendant 1 h à 37 °C. La plaque MEA a ensuite été lavée 3x à l’eau stérilisée et séchée pendant 1 h. Ensuite, la plaque MEA a été maintenue à 4 °C pendant la nuit. Culture à haute densité : 50 000 cellules/puits de neurones ont été plaquées sur des plaques MEA à 48 puits. L’étape d’ensemencement cellulaire a été effectuée comme décrit à la section 2 du protocole décrit ci-dessus. La laminine (20 μg/mL) ajoutée au milieu de culture (ajouter 2 v/v% de B-27, 2,5 mM de glutamax et 100 μg/mL de pénicilline/streptomycine au milieu neurobasal) a été utilisée pour plaquer les neurones. Par la suite, les neurones ont été cultivés à 37 °C, 5% de CO2 dans le milieu de culture. Les médias ont été entièrement échangés sur DIV 1 avec le milieu de culture jusqu’à DIV 3. Ara-C a été ajouté à DIV 4 (0,2 μM final). À partir de DIV 5 et 2 fois par semaine, 50% des médias ont été changés avec le support culturel. L’activité des neurones sur chaque puits de la plaque MEA a été enregistrée avec un système MEA. (B) L’activité neuronale spontanée a été acquise à 37 °C sous atmosphère de 5 % de CO2 à l’aide d’un système MEA à une fréquence d’échantillonnage de 12,5 kHz/canal à DIV 21. Les enregistrements de 4 canaux sur les 16 canaux d’un puits sont affichés. Pour tous les enregistrements, un filtre passe-bande Butterworth (200-3 000 Hz) a été appliqué. Les pointes de flèches montrent le moment du tir synchronisé en rafale. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Discussion
Une étape critique de cette méthode consiste à décongeler la suspension cellulaire. Le transfert de la suspension cellulaire avant qu’elle ne devienne trop chaude est très important. Pour éviter le changement rapide de l’osmolalité cependant, ne transférez pas la suspension cellulaire sur un grand volume du milieu à la fois. L’ajout goutte à goutte du milieu de culture est également crucial pour éviter les changements brusques de la pression osmotique.
Les neurones peuvent être cultivés dans d’autres vaisseaux de culture : plaques à 24 puits, chambres à 8 puits, antennes de 60 mm ou sondes MED. Dans ces cas, cependant, la concentration finale et le moment de l’ajout d’Ara-C doivent être ajustés. En outre, la densité des neurones doit être optimisée dans différents types d’expériences. Par exemple, une culture à haute densité est nécessaire pour les expériences électrophysiologiques et, dans ce cas, l’échange de milieux est nécessaire deux fois par semaine (Figure 6). Ainsi, une culture de faible densité nécessite moins d’étapes qu’une culture à forte densité.
La culture neuronale de faible densité nécessite souvent des techniques avancées; Cependant, l’utilisation de produits congelés prêts à l’emploi résout ce problème. La méthode décrite ne dépend pas de l’habileté d’un expérimentateur. La qualité des animaux congelés est stable et peut être cultivée de manière stable tant qu’ils sont stockés dans de l’azote liquide et évitent les changements de température jusqu’à 4 ans.
La culture des cellules pendant 3 semaines sans échange de milieu soulève la question de savoir s’il y a des changements importants d’osmolalité ou une évaporation des milieux de culture. Cependant, nous avons confirmé que l’évaporation des milieux de culture est faible (taux de réduction de 3,6%). La localisation des protéines synaptiques et la morphologie des neurones semblent normales après 3 semaines. Par conséquent, une culture de 3 semaines sans échange de milieux ne provoque pas de grands changements d’osmolalité affectant les conditions des neurones cultivés. Garder la plaque dans un incubateur après le traitement Ara-C est également un point important qui minimise l’évaporation.
Il n’y a pas de limitation concernant l’utilisation des neurones de stock congelés. Cependant, la méthode de culture à faible densité présente certaines limites. Nous avons confirmé que la culture de faible densité pouvait être appliquée à l’observation morphologique des neurones, à l’évaluation de la fonction synaptique et à la transfection de la GFP. Cependant, nous n’avons pas examiné l’imagerie de cellules vivantes. En outre, comme mentionné ci-dessus, une culture à haute densité est nécessaire pour effectuer l’électrophysiologie.
La maturation des synapses prend généralement 3 semaines7, et nous ne pouvons pas confirmer que les neurones cultivés ont des synapses appropriées jusqu’à la fin. Si la maturation des synapses n’est pas bonne après 3 semaines, nous devrions refaire la culture. En connaissant la qualité des neurones avant de commencer les expériences, nous pouvons sauver ces 3 semaines. Par conséquent, pour effectuer des expériences efficacement, il est préférable de vérifier la qualité des neurones à l’avance. Les stocks congelés permettent de vérifier au préalable la qualité des neurones. Chaque lot de stocks congelés est généré à partir d’une portée de rats, et nous pouvons utiliser l’un des stocks de chaque lot pour un contrôle de qualité. La drébrine est un bon marqueur pour le contrôle de la qualité des neurones. Comme décrit, la drebrine s’accumule dans la tête de la colonne vertébrale dans les neurones matures et réagit à la stimulation synaptique. Ainsi, nous pouvons vérifier la qualité des neurones dans les stocks congelés en utilisant la drebrine comme marqueur.
Cette méthode peut être appliquée pour évaluer l’effet des médicaments sur l’état synaptique. L’exode de la drebrine des épines dendritiques se produit au cours des premiers stades de la plasticité synaptique22. Par conséquent, la détection de la réduction des grappes de drebrine provoquée par le traitement médicamenteux montre que le médicament stimule la synapse et provoque la plasticité synaptique. De plus, pour déterminer si la réduction est dépendante du NMDAR, une expérience utilisant l’acide 2-amino-5-phosphonovalérique (APV, un antagoniste NMDAR) est utile. En utilisant la drebrine comme marqueur, même une dépendance NMDAR est clairement déterminée10,15. La méthode décrite est utile dans les dépistages de médicaments, les études pharmacologiques d’innocuité et l’évaluation de la fonction synaptique.
Déclarations de divulgation
Tomoaki Shirao est le PDG d’AlzMed, Inc. L’étude a été financée par AlzMed, Inc. (500 000 JPY à NK pour le projet intitulé « High-throughput analysis of synaptic function »).
Remerciements
Nous remercions Kazumi Kamiyama et Manami Kawada pour leur aide dans les expériences. Ce travail a été soutenu par JSPS KAKENHI (numéro de subvention 19K08010 à N.K.) et l’Agence japonaise pour la recherche et le développement médicaux (AMED) (numéro de subvention JP19bk0104077 et JP22bm0804024 à T.S.).
matériels
| Name | Company | Catalog Number | Comments |
| 96 well plate | Zeon Corporation | Gifted | |
| 96 well plate | greiner | 655986 | |
| Anti-drebrin antibody (M2F6) | MBL | D029-3 | Mouse monoclonal (dilution 1:1) |
| Anti-MAP2 antibody | Millipore | AB5622 | Rabbit (dilution 1:1000) |
| Anti-mouse Alexa Fluor 568 | Thermo Fisher Scientific | A11031 | Dilution 1: 500 |
| Anti-rabbit Alexa Fluor | Thermo Fisher Scientific | A11008 | Dilution 1: 500 |
| B-27 | Gibco | 17504-044 | 2 v/v% for MEA plates; 50x for normal plates |
| Borax | Sigma | B-9876 | Final concentration 12 mM |
| Boric acid | WAKO | 021-02195 | Final concentration 50 mM |
| Bovine serum albumin | Millipore | 12659-100G | Final concentration: 3% in PBS |
| Confocal quantitative image cytometer CellVoyager CQ1 | YOKOGAWA | Phase contrast images | |
| Cytosine β-D-arabino-furanoside (Ara-C) | Sigma | C-6645 | Diluted in dH2O (final concentration: 0.2 µM) |
| DAPI | FUJIFILM | 340-07971 | Dilution 1:1000 |
| GlutaMAX | Gibco | 35050-061 | 2.5 mM for MEA plates; 400x for normal plates |
| In Cell Analyzer 2200 | Cytiva | Fluorescence images | |
| Laminin | Sigma | 114956-81-9 | Final concentration: 20 µg/mL |
| Maestro | Axion Biosystems | MEA recordings | |
| MEA plate | Axion Biosystems | M768-tMEA-48W | |
| Neurobasal | Gibco | 21103-049 | |
| Paraformaldehyde | nacalai tesque | 26126-25 | Final concentration: 4% in PBS |
| Penicillin/Streptomycin | Gibco | 15140-122 | 100 U/mL for normal plates |
| Penicillin/Streptomycin | nacalai tesque | 26253-84 | 100 µg/mL for MEA plates |
| polyethyleimine | Sigma | 9002-98-6 | Final concentration: 0.1% |
| Poly-L-lysine | Sigma | P2636 | Diluted in the borate buffer (final concentration: 1 mg/mL) |
| SKY Neuron | AlzMed , Inc. | ARH001 | 1.0 x 106 cells/tube |
| Sodium azide | FUJIFILM | 195-11092 | 0.1% |
| SodiumL(+)-Glutamate monohydrate | WAKO | 194-02032 | Diluted in dH2O (final concentrations: 1 µM, 3 µM, 10 µM, 30 µM, 100 µM) |
Références
- Banker, G. A., Cowan, W. M. Rat hippocampal neurons in dispersed cell culture. Brain Research. 126 (3), 397-342 (1977).
- Kaech, S., Banker, G. Culturing hippocampal neurons. Nature Protocols. 1 (5), 2406-2415 (2006).
- Roppongi, R. T., Champagne-Jorgensen, K. P., Siddiqui, T. J. Low-density primary hippocampal neuron culture. Journal of Visualized Experiments. (122), e55000 (2017).
- Mizui, T., et al. Drebrin E is involved in the regulation of axonal growth through actin-myosin interactions. Journal of Neurochemistry. 109 (2), 611-622 (2009).
- Mizui, T., et al. Myosin II ATPase activity mediates the long-term potentiation-induced exodus of stable F-actin bound by drebrin A from dendritic spines. PLoS One. 9 (1), 85367 (2014).
- Takahashi, H., Mizui, T., Shirao, T. Down-regulation of drebrin A expression suppresses synaptic targeting of NMDA receptors in developing hippocampal neurons. Journal of Neurochemistry. 97, 110-115 (2006).
- Takahashi, H., et al. Drebrin-dependent actin clustering in dendritic filopodia governs synaptic targeting of postsynaptic density-95 and dendritic spine morphogenesis. The Journal of Neuroscience. 23 (16), 6586-6595 (2003).
- Yamazaki, H., Sasagawa, Y., Yamamoto, H., Bito, H., Shirao, T. CaMKIIbeta is localized in dendritic spines as both drebrin-dependent and drebrin-independent pools. Journal of Neurochemistry. 146 (2), 145-159 (2018).
- Hanamura, K., et al. High-content imaging analysis for detecting the loss of drebrin clusters along dendrites in cultured hippocampal neurons. Journal of Pharmacological and Toxicological Methods. 99, 106607 (2019).
- Mitsuoka, T., et al. Assessment of NMDA receptor inhibition of phencyclidine analogues using a high-throughput drebrin immunocytochemical assay. Journal of Pharmacological and Toxicological Methods. 99, 106583 (2019).
- Ishizuka, Y., Bramham, C. R. A simple DMSO-based method for cryopreservation of primary hippocampal and cortical neurons. Journal of Neuroscience Methods. 333, 108578 (2020).
- Pischedda, F., et al. Cryopreservation of primary mouse neurons: The benefit of neurostore cryoprotective medium. Frontiers in Cellular Neuroscience. 12, 81 (2018).
- Kobayashi, Y., et al. Impairment of ciliary dynamics in an APP knock-in mouse model of Alzheimer's disease. Biochemical and Biophysical Research Communications. 610, 85-91 (2022).
- Kobayashi, Y., et al. Properties of primary cilia in melanin-concentrating hormone receptor 1-bearing hippocampal neurons in vivo and in vitro. Neurochemistry International. 142, 104902 (2021).
- Koganezawa, N., et al. NMDA receptor-dependent and -independent effects of natural compounds and crude drugs on synaptic states as revealed by drebrin imaging analysis. The European Journal of Neuroscience. 53 (11), 3548-3560 (2021).
- Mizui, T., Takahashi, H., Sekino, Y., Shirao, T. Overexpression of drebrin A in immature neurons induces the accumulation of F-actin and PSD-95 into dendritic filopodia, and the formation of large abnormal protrusions. Molecular and Cellular Neurosciences. 30 (1), 149-157 (2005).
- Sekino, Y., et al. Activation of N-methyl-D-aspartate receptor induces a shift of drebrin distribution: disappearance from dendritic spines and appearance in dendritic shafts. Molecular and Cellular Neurosciences. 31 (3), 493-504 (2006).
- Takahashi, H., Yamazaki, H., Hanamura, K., Sekino, Y., Shirao, T. Activity of the AMPA receptor regulates drebrin stabilization in dendritic spine morphogenesis. Journal of Cell Science. 122, 1211-1219 (2009).
- Aoki, C., et al. Drebrin A is a postsynaptic protein that localizes in vivo to the submembranous surface of dendritic sites forming excitatory synapses. The Journal of Comparative Neurology. 483 (4), 383-402 (2005).
- Koganezawa, N., Hanamura, K., Sekino, Y., Shirao, T. The role of drebrin in dendritic spines. Molecular and Cellular Neurosciences. 84, 85-92 (2017).
- Shirao, T., et al. The role of drebrin in neurons. Journal of Neurochemistry. 141 (6), 819-834 (2017).
- Sekino, Y., Koganezawa, N., Mizui, T., Shirao, T. Role of drebrin in synaptic plasticity. Advances in Experimental Medicine and Biology. 1006, 183-201 (2017).
- Togo, K., et al. Postsynaptic structure formation of human iPS cell-derived neurons takes longer than presynaptic formation during neural differentiation in vitro. Molecular Brain. 14 (1), 149 (2021).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.