
Method Article
Eine optimierte quantitative Pull-Down-Analyse von RNA-bindenden Proteinen unter Verwendung kurzer biotinylierter RNA
In diesem Artikel
Zusammenfassung
In dieser Arbeit stellen wir eine optimierte in vitro Methode vor, um Proteininteraktoren spezifischer RNA-Sequenzen aufzudecken, zu quantifizieren und zu validieren, wobei Gesamtproteinextrakt aus menschlichen Zellen, Streptavidin-Kügelchen, die mit biotinylierter RNA beschichtet sind, und Massenspektrometrie-Analysen verwendet werden.
Zusammenfassung
Protein-RNA-Interaktionen regulieren die Genexpression und zelluläre Funktionen auf transkriptioneller und posttranskriptioneller Ebene. Aus diesem Grund ist die Identifizierung der Bindungspartner einer RNA von großer Bedeutung, um die Mechanismen hinter vielen zellulären Prozessen aufzudecken. RNA-Moleküle können jedoch vorübergehend und dynamisch mit einigen RNA-bindenden Proteinen (RBPs) interagieren, insbesondere mit nicht-kanonischen Proteinen. Daher sind verbesserte Methoden zur Isolierung und Identifizierung solcher RBPs dringend erforderlich.
Um die Proteinpartner einer bekannten RNA-Sequenz effizient und quantitativ zu identifizieren, haben wir eine Methode entwickelt, die auf dem Pull-down und der Charakterisierung aller interagierenden Proteine, ausgehend von zellulärem Gesamtproteinextrakt, basiert. Wir optimierten den Protein-Pulldown mit biotinylierter RNA, die auf Streptavidin-beschichteten Beads vorgeladen war. Als Proof-of-Concept verwendeten wir eine kurze RNA-Sequenz, von der bekannt ist, dass sie das Neurodegeneration-assoziierte Protein TDP-43 bindet, und eine Negativkontrolle mit einer anderen Nukleotidzusammensetzung, aber gleicher Länge. Nachdem wir die Beads mit Hefe-tRNA blockiert hatten, luden wir die biotinylierten RNA-Sequenzen auf die Streptavidin-Beads und inkubierten sie mit dem Gesamtproteinextrakt aus HEK 293T-Zellen. Nach Inkubation und mehreren Waschschritten zur Entfernung unspezifischer Bindemittel eluierten wir die interagierenden Proteine mit einer Hochsalzlösung, die mit den am häufigsten verwendeten Proteinquantifizierungsreagenzien und mit der Probenvorbereitung für die Massenspektrometrie kompatibel ist. Wir quantifizierten die Anreicherung von TDP-43 im Pull-Down, der mit dem bekannten RNA-Binder durchgeführt wurde, im Vergleich zur Negativkontrolle mittels Massenspektrometrie. Wir verwendeten die gleiche Technik, um die selektiven Wechselwirkungen anderer Proteine zu verifizieren, von denen computergestützt vorhergesagt wurde, dass sie einzigartige Bindemittel unserer RNA von Interesse oder der Kontrolle sind. Schließlich validierten wir das Protokoll mittels Western Blot durch den Nachweis von TDP-43 mit einem geeigneten Antikörper.
Dieses Protokoll wird die Untersuchung der Proteinpartner einer RNA von Interesse unter nahezu physiologischen Bedingungen ermöglichen und dazu beitragen, einzigartige und unvorhergesehene Protein-RNA-Interaktionen aufzudecken.
Einleitung
RNA-bindende Proteine (RBPs) haben sich zu entscheidenden Akteuren bei der transkriptionellen und posttranskriptionellen Genregulation entwickelt, da sie an Prozessen wie dem Spleißen von mRNA, der zellulären Lokalisierung, Translation, Modifikation und dem Abbau von RNA beteiligt sind 1,2,3. Solche Wechselwirkungen zwischen den beiden Makromolekülen sind hochgradig koordiniert, präzise ausbalanciert und essentiell für die Bildung von Funktions- und Verarbeitungszentren. Variationen oder Dysregulationen innerhalb dieser Hubs haben das Potenzial, die fein regulierten Protein-RNA-Netzwerke zu stören und werden zunehmend mit einer Vielzahl menschlicher Krankheiten in Verbindung gebracht, darunter Krebs 4,5 und neurodegenerative Erkrankungen 6,7,8. Die Wechselwirkungen zwischen RNA-Molekülen und ihren Proteinbindungspartnern können entweder stabil und experimentell leicht zu validieren sein, oder hochdynamisch, transient und schwieriger zu charakterisieren sein.
In den letzten Jahren wurden intensive Anstrengungen unternommen, um diese Wechselwirkungen zu verstehen. Unter den etabliertesten Methoden sind Protein-Pull-Down-Assays (PDs) wahrscheinlich die am meisten geschätzten und am häufigsten verwendeten Ansätze, um die Hauptakteure zu entschlüsseln, aus denen Ribonukleoprotein-Komplexe (RNP) und andere Protein-RNA-Interaktionsnetzwerke bestehen 3,9,10. PDs umfassen ein breites Spektrum an informativen Techniken, wie z. B. die Immunpräzipitation entweder der RNA (RIP)11,12 oder des interessierenden Proteins (CLIP)13,14. Einige dieser RNA-PD-Protokolle verwenden eine bekannte RNA als Köder für Proteine15, am häufigsten unter Ausnutzung von Tags mit hoher Affinität wie Biotin. In diesem Fall können die Interaktionspartner einer biotinylierten RNA durch Verankerung der RNA auf Streptavidin-beschichteten Beads nachgewiesen werden, was eine effiziente Isolierung der RNPs ermöglicht. Die Haupteinschränkungen dieser Ansätze sind in der Regel das Design der biotinylierten Sonden und die Prüfung ihrer Fähigkeit, Zielproteine zu binden. Zu diesem Zweck könnte es sinnvoll sein, sich auf veröffentlichte CLIP-Daten des interessierenden Proteins zu stützen, sofern verfügbar, da sie mit hoher Präzision die kurzen RNA-Regionen zeigen, die den Peaks der Wechselwirkungen mit dem Zielprotein entsprechen13,16. Dieselben Regionen könnten für die Entwicklung von Sonden für Parkinson-Inson verwendet werden. Eine alternative Methode zur Entwicklung solcher RNA-Köder könnte die systematische Evolution von Liganden durch exponentielle Anreicherung (SELEX)17 sein, die das Design von Aptameren durch In-vitro-Selektion ermöglichen, ausgehend von einer umfassenden randomisierten Bibliothek und über eine Reihe von PCR-gesteuerten Optimierungszyklen. SELEX ist jedoch komplex und zeitaufwändig, und die Endergebnisse hängen stark von der ursprünglichen Bibliothek ab. Um den RNA-Köder auszuwählen, der in dem hier vorgestellten Protokoll verwendet werden soll, wurde ein weiterer Ansatz ausgenutzt, der darin besteht, einen RNA-Köder zu verwenden, der de novo mit Hilfe der Rechenleistung des Algorithmus catRAPID entwickelt wurde, der die bevorzugte Bindung eines gegebenen Proteins an bestimmte RNA-Sequenzen vorhersagt18,19,20.
Bei dem hier vorgestellten Protokoll handelt es sich um eine Version einer RNA-PD, die für die Eluierung spezifischer Proteinpartner unter nahezu physiologischen Bedingungen optimiert ist, ohne den Einsatz von Detergenzien, Denaturierungsmitteln oder hohen Temperaturen. Es beruht auf nano-superparamagnetischen Kügelchen, die kovalent mit hochreinem Streptavidin beschichtet sind, und der Verwendung einer spezifischen, in silico entwickelten biotinylierten RNA als Köder. Dieses Protokoll bietet eine schnelle und effiziente Methode, um die Bindungspartner von biotinylierten RNA-Molekülen unter nativen Bedingungen zu isolieren, und bietet das Potenzial für eine Vielzahl von nachgelagerten Anwendungen. Um dieses Protokoll zu testen, wurde eine 10-Nukleotid-Einzelstrang-RNA-Aptamer-Sequenz verwendet, die zuvor entwickelt wurde, um das Protein TAR DNA-binding protein 43 (TDP-43) mit hoher Affinität und Spezifität zu binden20. Ausgehend von HEK 293T-Zelllysaten wurden die Interaktoren des biotinylierten RNA-Aptamers mittels massenspektrometrischer Analyse an Proben identifiziert, die mit einem hypertonen Puffer vom RNA-Köder gelöst wurden. Diese Analyse bestätigte die erfolgreiche Identifizierung und Quantifizierung von TDP-43 als bevorzugtes Bindemittel.
Dieses Protokoll ermöglicht die erfolgreiche Identifizierung von Proteininteraktoren mit nur einem kurzen, in vitro synthetisierten RNA-Oligonukleotid. Darüber hinaus garantiert die Verwendung von in silico designten RNA-Aptameren als PD-Sonden21,22 Spezifität für die Targets bei deutlich reduzierten Kosten.
Protokoll
1. Allgemeine Methoden und Materialien
- Bereiten Sie das geeignete Medium für die gewählte Säugetierzellkultur vor und wärmen Sie es vor der Verwendung 20 min bei 37 °C vor.
- Bereiten Sie das benötigte Material im Voraus vor, wie in der Materialtabelle beschrieben. Autoklavieren Sie Glaswaren, Kunststoffwaren und Pufferbestände.
- Bereiten Sie die Puffer wie in Tabelle 1 beschrieben vor. Stellen Sie den pH-Wert der Stammlösungen mit konzentriertem HCl oder NaOH ein, bevor Sie die Komponenten auf ihr endgültiges Volumen verdünnen.
2. Präparation von Säugetierzelllinien
- Züchtung von HEK 293T-Zellen in Dulbeccos modifiziertem Eagle Medium (DMEM), ergänzt mit 10 % fötalem Kälberserum (FBS) und 100 μg/ml Penicillin/Streptomycin-Lösung. Inkubieren Sie sie bei 37 °C in einem befeuchteten Inkubator, der mit 5 % CO2 versorgt wird. Teilen Sie die Zellen routinemäßig auf.
- Spülen Sie die Zellen vor dem Abnehmen mit ausreichend phosphatgepufferter Kochsalzlösung (PBS) ab, um die Wachstumsoberfläche zu bedecken.
- Entfernen Sie das PBS und fügen Sie eine ultradünne Schicht Trypsin-EDTA-Lösung hinzu.
- Inkubieren Sie die Zellen bei 37 °C in einem befeuchteten Inkubator, der mit 5 % CO2 versehen ist, für 5 Minuten oder bis sich die Zellen gelöst haben (sie sollten unter mikroskopischer Beobachtung zerstreut aussehen).
- Verdünnen Sie die Trypsin-EDTA-Lösung um das Zehnfache, indem Sie vollständiges DMEM hinzufügen, um sie zu inaktivieren und die Zellen zu zählen.
- Platte 1,5 x 105 Zellen/ml in 6-Well-Platten unter Berücksichtigung von zwei Wells/zu testenden Bedingungen.
- Inkubieren Sie die Zellen bei 37 °C für 48 h in einem befeuchteten Inkubator mit 5% CO2.
HINWEIS: Überprüfen Sie die Anweisungen des Herstellers für die Art des Mediums und die für die Zelllinie geeignete Ergänzung. Auch die Menge und die Inkubationszeit von Trypsin-EDTA hängen von der Zelllinie ab. Einige Zelltypen wachsen schneller/langsamer als in diesem Protokoll beschrieben. Daher sollte die Aussaatkonzentration vorher geprüft werden.
3. Gesamte Proteinernte
- Entfernen Sie das Medium aus den Vertiefungen, in denen die Zellen wachsen.
- Waschen Sie jede der 6-Well-Platten mit 1 ml PBS.
- Verwerfen Sie das PBS.
- Bewegen Sie die Platten auf Eis und fahren Sie entweder mit Schritt 3.5 fort oder frieren Sie die trockenen Platten bei -80 °C ein, um die Lyse zu erleichtern.
- Geben Sie 200 μl Lysepuffer in jede Vertiefung.
- Verwenden Sie einen Zellschaber, um die Zellen zu lösen und zu brechen.
- Übertragen Sie den Zellextrakt aus zwei Vertiefungen in dasselbe 1,5-ml-Röhrchen.
- Legen Sie das Röhrchen mit dem Proteinextrakt für 30 Minuten auf Eis.
- Zentrifugieren Sie die Zelllysate bei 17.000 x g für 15 min bei 4 °C.
- Übertragen Sie jeden Überstand in ein vorgekühltes Röhrchen.
HINWEIS: Insgesamt werden 106-10 7 Zellen für jede PD-Bedingung empfohlen. Die Zelllyse und Proteinernte sollte mit eiskalten Puffern durchgeführt werden. Proteasehemmer sollten dem Lysepuffer zugesetzt werden, um einen Proteinabbau zu verhindern.
4. Bestimmung der Proteinkonzentration
- Bereiten Sie das Bradford-Reagenz nach Angaben des Herstellers vor, indem Sie es fünffach in dH2O verdünnen.
- Verteilen Sie 1 ml Reagenz in einer 1 cm großen Küvette, fügen Sie 1 μl der Probe hinzu und mischen Sie sie durch Inversion.
- Im Dunkeln bei Raumtemperatur 5-10 min inkubieren.
- Lesen Sie die Absorption bei 595 nm ab.
- Berechnen Sie das Volumen des Proteinextrakts, das 1,5 mg Proteinen entspricht, und bringen Sie alle Proben mit Lysepuffer auf ein Endvolumen von 600 μl.
- Bewahren Sie die Proben bis zur Verwendung auf Eis auf.
HINWEIS: Es kann jede andere Methode zur Bestimmung der Proteinkonzentration verwendet werden, wobei die Empfehlungen zur Pufferkompatibilität zu befolgen sind. In jedem Fall sollte der Lysepuffer als Rohling verwendet werden. Viele Reagenzien sind nicht mit Dithiothreitol (DTT) kompatibel. Es wird empfohlen, DTT oder andere Reduktionsmittel erst nach Proteinquantifizierung (DTT bis zu einer Endkonzentration von 1 mM) zuzugeben.
5. Perlenvorbereitung
- Mischen Sie die Perlen in ihrem Speicherpuffer, indem Sie das Röhrchen schnippen.
- Berechnen Sie 100 μl Aufschlämmungsmedium/Probe und legen Sie das Volumen in ein Magnetgestell.
- Perlenwäsche
- Entfernen Sie die Aufbewahrungslösung und waschen Sie die Kügelchen, indem Sie 1 ml Lysepuffer/Röhrchen hinzufügen und manuell invertieren.
- Entfernen Sie den Puffer mit dem Magnetgestell.
- Wiederholen Sie den Waschschritt.
- Fügen Sie ein Volumen des Lysepuffers hinzu, das dem ursprünglichen Volumen des Aufschlämmungsmediums entspricht, mischen Sie es durch Schnippen des Röhrchens und dosieren Sie das Medium gleichmäßig in so viele 1,5-ml-Röhrchen, wie Proben vorhanden sind.
- Perlen-Blockierung
- Entfernen Sie den Puffer mit dem Magnetgestell und fügen Sie 600 μl einer 0,25 mg/ml-Lösung von Hefe-tRNA hinzu, die im Lysepuffer hergestellt wurde.
- 1 h bei Raumtemperatur auf einem rotierenden Rad inkubieren.
- Entfernen Sie die tRNA-Lösung mit Hilfe des magnetischen Racks.
- Fügen Sie 600 μl Lysepuffer hinzu und waschen Sie sie durch manuelles Mischen.
- Wiederholen Sie den Waschschritt und entsorgen Sie den Puffer.
6. Beladung der Perlen
- Bereiten Sie 200 μg RNA-Oligonukleotid in 600 μl Lysepuffer für jedes Röhrchen vor, das das anfängliche 100-μl-Slurry-Medium enthält (jetzt blockierte Kügelchen).
- Das Oligo zu den Kügelchen geben und 1 h bei Raumtemperatur unter Rotation inkubieren.
- Entfernen Sie die Lösung, fügen Sie 600 μl Lysepuffer hinzu und waschen Sie die Kügelchen zweimal, indem Sie die Röhrchen 5 Minuten lang bei Raumtemperatur drehen.
- Verwerfen Sie den Puffer.
Anmerkungen: Wirbeln Sie die Perlen niemals, sondern schnippen Sie stattdessen. Begrenzen Sie die Anzahl der Pipettierschritte, sofern dies nicht erforderlich ist. Verwenden Sie nach Möglichkeit geschnittene 1-ml-Spitzen. Die Menge des Bead-Slurry-Mediums/der Probe hängt von der Bindungsfähigkeit der Beads und der Ausgangsmenge der Gesamtproteine ab. Wenn das RNA-Oligo eine signifikante Menge an Sekundärstruktur aufweist, empfehlen wir, es zuerst bei 80 °C für 10 min zu denaturieren und dann langsam bei Raumtemperatur abzukühlen oder es durch Inkubation bei 30 °C für 1 h neu zu falten. Es wird empfohlen, das RNA-Oligo nach der Beadung zu gewinnen und die verbleibende Konzentration zu bestimmen, um die für die Beladung erforderliche Menge zu optimieren und die Möglichkeit der Wiederverwendung der RNA zu bewerten.
7. Proteinbindung an Beads
Anmerkungen: Führen Sie von nun an, wenn möglich, die Schritte bei 4 °C durch.
- Nehmen Sie ein Volumen von 5 % aus der 600 μl Proteinlösung und bewahren Sie es als INPUT (IN) für die weitere Analyse auf (1,5 mg Proteine sind in 600 μl gelöst, 5 % entsprechen also 30 μl und 75 μg Proteinen).
- Die restliche Proteinmischung in jedes Röhrchen mit beladenen Kügelchen geben und über Nacht bei 4 °C langsam rotieren lassen.
8. Waschen von unspezifischen Bindemitteln
- Entfernen Sie die ungebundene Fraktion mit dem Magnetgestell. Speichern Sie 5 % des Volumens und kennzeichnen Sie es als FLOWTHROUGH (FT) (das ungebundene Volumen beträgt ca. 600 μL, also behalten Sie wieder 30 μL für die weitere Analyse).
- Geben Sie 1 ml Waschpuffer 1 zu den Kügelchen und lassen Sie ihn 5 Minuten lang bei 4 °C drehen.
- Verwerfen Sie den Puffer.
- Wiederholen Sie die Schritte 8.2 und 8.3.
- Geben Sie 1 ml Waschpuffer 2 zu den Kügelchen und lassen Sie ihn 5 Minuten lang bei 4 °C drehen.
- Verwerfen Sie den Überstand.
9. Elution spezifischer Bindemittel
- Geben Sie 100 μl Elutionspuffer 1 oder Elutionspuffer 2 zu den Kügelchen.
- Manuell durch Schnippen mischen und 5 Minuten bei Raumtemperatur inkubieren.
- Die Röhrchen in einen Thermomischer geben und 5 min bei 95 °C kräftig schütteln.
- Legen Sie das Röhrchen in das Magnetgestell und sammeln Sie die eluierte Fraktion in einem sauberen Röhrchen.
- Schleudern Sie die Kügelchen schnell mit einer Bankzentrifuge, um die Rückgewinnung des Eluats zu maximieren.
- Bewahren Sie 5 % des gesamten ELUATE (EL)-Volumens für weitere Analysen auf (Gesamtvolumen beträgt 100 μl, trennen Sie also 5 μl in ein anderes Röhrchen).
- Bei Bedarf kann die Proteinkonzentration wie in Abschnitt 4 bestimmt werden, indem Elutionspuffer als Leerzeichen verwendet wird.
HINWEIS: Es wird empfohlen, keine DTT in den Elutionspuffer zu geben, bis die Proteinkonzentration bestimmt wurde. Wenn keine Proteinquantifizierung erforderlich ist oder wenn reduktionsmittelkompatible Proteinquantifizierungskits verfügbar sind, kann dem Elutionspuffer von Anfang an 1 mM DTT zugesetzt werden. In diesem Protokoll wurden sowohl der Elutionspuffer 1 (mit 1 M NaCl) als auch der Elutionspuffer 2 (mit 2 M NaCl) getestet. Es wurde kein Unterschied in der Elutionseffizienz des Zielproteins mit erhöhter Ionenstärke beobachtet, es wird jedoch empfohlen, beide Bedingungen zu testen, bevor der am besten geeignete Puffer festgelegt wird. Wenn ein hoher Salzgehalt im Elutionspuffer eine Einschränkung für die weitere Analyse darstellt, ermöglicht die sehr geringe Menge an Detergenzien im Elutionspuffer einen Pufferaustausch. Alternativ kann das Eluat verdünnt werden, um die gewünschte Salzkonzentration zu erreichen.
10. Identifizierung von Proteinbindern mittels Massenspektrometrie
- Aceton-Fällung
- Konzentrieren Sie die eluierten Proteine, indem Sie sie vierfach in kaltem (-20 °C) Aceton verdünnen.
- Vortexieren und inkubieren Sie das Röhrchen bei -20 °C über Nacht.
- Bei 17.000 x g für 30 min bei 4 °C schleudern.
- Entfernen Sie vorsichtig den Überstand und lassen Sie das Aceton verdampfen, bis das Pellet vollständig getrocknet ist.
- Proteinverdauung in Lösung
- Lösen Sie das Proteinpellet durch Zugabe von 50 μL Denaturierungspuffer auf.
- DTT wird bis zu einer Endkonzentration von 5 mM zugegeben, so dass eine Proteinreduktion für 30 min bei 55 °C möglich ist.
- Kühlen Sie die Proben bei Raumtemperatur ab und fahren Sie mit der Proteinalkylierungsreaktion fort, indem Sie Iodacetamid (IAA) in einer Konzentration von 10 mM für 15 Minuten hinzufügen.
- Verdauen Sie die Proteine mit einem geeigneten Enzym (Trypsin, LysC) und inkubieren Sie die Proben über Nacht bei 37 °C.
- Stoppen Sie den Aufschluss durch Zugabe von 1 μl 10%iger Trifluoressigsäure (TFA).
- Reinigen und konzentrieren Sie die Peptide auf einer benutzerdefinierten Mikrosäule der umgekehrten Phase C18, wie zuvor beschrieben19.
- Elute Peptide aus der C18-Spitze mit Puffer B.
- Entfernen Sie die organische Komponente mit einer Vakuumzentrifuge und resuspendieren Sie die Peptide in 5 μl 0,1%iger Ameisensäure zur weiteren Analyse.
HINWEIS: Alternativ kann der Proteinaufschluss direkt nach dem Waschen unspezifischer Bindemittel "auf den Kügelchen" durchgeführt werden (Schritte 8.1-8.6), wodurch das Protokoll beschleunigt wird. Es ist jedoch ratsam, die Effizienz des Enzyms, das "auf Beads" immobilisierte Proteine arbeitet, im Vergleich zum Standard im Lösungsaufschluss zu testen, um eine optimale Abdeckung der experimentellen Proteinsequenz zu gewährleisten.
- Flüssigkeitschromatographie-Tandem-Massenspektrometrie (LC-MS/MS)
- Stecken Sie die analytische Säule (C18-stationäre Phase) ein und halten Sie sie während des Laufs bei 45 °C.
- Verbinden Sie die Säule mit dem Ausgang einer Sechs-Wege-Zellenradschleuse der LC-Pumpe über eine kapillare, fingerfeste Verschraubung (20 μm x 550 mm) in einer Einsäulenkonfiguration.
- Passen Sie die LC-Einstellungen wie folgt an:
- Laden Sie die Peptide unter kontrolliertem Druck (980 bar) in Puffer A.
- Wenden Sie einen Puffer-B-Gradienten von 5 % bis 20 % bei 300 ml/min über 59 Minuten an, gefolgt von einem Puffer-B-Gradienten von 20 % bis 30 % über 15 Minuten und einem Puffer-B-Gradienten von 30 % bis 65 % über 5 Minuten.
- Fügen Sie einen Waschschritt hinzu, indem Sie die Konzentration von Puffer B über 5 Minuten auf 95 % erhöhen, plus einen isokratischen Schritt von 5 Minuten bei 95 % Puffer B.
- Betreiben Sie das Massenspektrometer im datenabhängigen Erfassungsmodus (DDA), um automatisch zwischen MS- und MSMS-Ereignissen zu wechseln.
- Definieren Sie eine Schleifenanzahl von 15 mit einem Zielwert für die automatische Verstärkungsregelung (AGC) von 3 x 106 bzw. 1 x 105 für die MS- bzw. MSMS-Ereignisse.
- Stellen Sie die maximal zulässige Ionenakkumulationszeit auf 20 ms für MS mit einer Auflösung von 60 K und 100 ms für MSMS mit einer Auflösung von 15 K ein.
- Führen Sie ein HCD-Fragmentierungsexperiment (High Collision Dissociation) mit einer normalisierten Kollisionsenergie von 28 % und einer dynamischen Ausschlusszeit von 20 s durch.
- Bearbeiten Sie die Quellparameter wie folgt:
Sprühspannung: 1,7 kV
Kapillarspannung: 275 °C
Weder Mantel noch Hilfsgas verwendet
HINWEIS: In diesem Protokoll wurde die massenspektrometrische (MS) Analyse der Ultrahochleistungsflüssigkeitschromatographie (UHPLC) speziell unter Verwendung eines LC-Einsäulenaufbaus durchgeführt, der mit einem hybriden Dreifach-Quadrupol-Orbitrap-Instrument gekoppelt ist (Materialtabelle). Andere LCMS-Systeme können verwendet werden, eine Anpassung der Parameter wird jedoch empfohlen.
- Datenanalyse
- Verwenden Sie die Schaltfläche Laden , um die Rohdateien zu importieren.
- Definieren Sie die Namen der Experimente, indem Sie auf die Schaltfläche Experiment festlegen klicken.
- Geben Sie den Abschnitt gruppenspezifische Parameter ein, um alle Parameter anzugeben, die sich auf die Identifizierung beziehen:
Enzym für die Verdauung: Trypsin/P
Fehlende Dekolletés: bis zu drei
Behobene Modifikation: Carbamidomethylierung
Variable Modifikation: N-Acetyl (Protein), Oxidation (M) - Laden Sie eine aktualisierte FASTA-Datei hoch, die in öffentlichen Datenbanken wie UniprotKB verfügbar ist.
- Geben Sie die richtigen Analyseregeln entsprechend der Quelle der ausgewählten Datenbank an.
- Definieren Sie einen FDR-Wert (Parentage False Discovery Rate) = 1 für Proteine und Peptide.
- Fügen Sie die Option "Beschriftungsfreie Quantifizierung" (LFQ) auf der Registerkarte "Beschriftungsfreie Quantifizierung" hinzu.
- Halten Sie die minimale Anzahl des LFQ-Verhältnisses bei zwei.
HINWEIS: Hier beschreiben wir die Datenanalyse mit MaxQuant24 und der Perseus-Software25 zur Durchführung der Proteinquantifizierung bzw. der anschließenden statistischen Analyse. Die Datenanalyse kann jedoch mit jeder anderen kommerziell erhältlichen oder kostenlosen Bioinformatik durchgeführt werden. FDR wird unter Verwendung eines auf Target-Decoy-Datenbanken basierenden Ansatzesgeschätzt 26. Peptid- und Protein-FDR gleich 0,01 bedeutet, dass die identifizierten Peptide und Proteine voraussichtlich 1 % falsch positive Ergebnisse enthalten.
- Statistische Analyse
- Laden Sie die Datei proteingroups.txt, um statistische Analysen auf Proteinebene durchzuführen.
- Definieren Sie die LFQ-Werte als Hauptspalten.
- Entfernen Sie "umgekehrt" und "Verunreinigungen", indem Sie Zeilen basierend auf der kategorialen Spalte filtern.
- Verwenden Sie die kategorialen Anmerkungszeilen, um die verschiedenen experimentellen Bedingungen zu gruppieren.
- Reduzieren Sie die Datenmatrix, indem Sie die Anzahl der gültigen Werte in jeder der zuvor definierten Gruppen auswählen.
- Wählen Sie den statistischen Test aus, der besser zu den Versuchsbedingungen passt (z. B. t-Test, ANOVA des Mehrfachstichprobentests).
- Bestimmen Sie einen Grenzwert für signifikante Hinweise mit einer FDR-basierten Berechnung. In der Regel werden sowohl 0,01 als auch 0,05 als Schwellenwerte für den angepassten p-Wert akzeptiert.
- Visualisieren Sie die Ergebnisse von Differentialanalysen auf der Grundlage von t-Test-Statistiken mithilfe einer Vulkandiagrammdarstellung.
- Exportieren Sie die endgültige Matrix in .txt Format, um die endgültige Ergebnistabelle weiter zu bearbeiten.
HINWEIS: Der Konfigurationsordner enthält eine FASTA-Datei mit Proteinen wie Keratinen, die in globalen Proteomik-Experimenten als häufige Kontaminanten gelten, die in der Ausgabetabelle mit einem + gekennzeichnet sind. In der vorliegenden Studie wird mit zwei Stichprobenbedingungen ein t-Test für die statistische Auswertung verwendet.
11. Validierung der Ergebnisse durch Western Blot
- Probenvorbereitung
- Fügen Sie das entsprechende Volumen von 4x Probenladepuffer zu jedem Aliquot von IN, FT und EL hinzu.
- Kochen Sie die Proben 5 Minuten lang bei 95 °C.
- Schnelles Schleudern, um verdampfte Probe von der Oberseite der Röhrchen zu gewinnen.
- SDS-PAGE und Geltransfer
- Laden Sie die Proben auf ein 4%-12% denaturierendes Polyacrylamid-Gel.
- Lassen Sie das Gel mit MES SDS-Laufpuffer 1,5 h bei 120 V laufen.
- Übertragen Sie das Gel auf eine Nitrozellulosemembran mit einer halbtrockenen Transferkassette und befolgen Sie dabei die Anweisungen des Herstellers. Wir empfehlen einen 10-minütigen Transfer bei 15 V.
- Immundetektion
- Blockieren Sie die Membran mit 10 % Kälberserumalbumin (BSA) für 1 h bei Raumtemperatur unter sanftem Rühren.
- Fügen Sie den primären Antikörper, der in 5 % BSA hergestellt wurde, gemäß den Anweisungen des Herstellers hinzu. Über Nacht bei 4 °C oder 1 h bei Raumtemperatur unter leichtem Rühren stehen lassen.
- Waschen Sie die Membran dreimal mit TBST, jedes Mal für 5 Minuten.
- Fügen Sie den in TBST hergestellten Sekundärantikörper für 1 h bei Raumtemperatur unter Rühren hinzu.
- Waschen Sie die Membran dreimal mit TBST, jedes Mal für 5 Minuten.
- Visualisieren Sie die Ergebnisse mit einem Blot-Imager.
HINWEIS: Für den Nachweis von TDP-43, einem bekannten Bindemittel unserer RNA, wurde ein rekombinanter monoklonaler Kaninchen-Antikörper verwendet, der über Nacht bei 4 °C in der Membran belassen wurde. Als Sekundärantikörper wurde die Anti-Kaninchen-IgG-Meerrettichperoxidase (HRP) verwendet, aber auch ein fluoreszierender Sekundärantikörper würde funktionieren. Um die Antikörper auf der Membran sichtbar zu machen, wurde die Membran 1 min lang mit dem Clarity Western ECL-Substrat inkubiert, bevor sie mit dem ChemiDoc-Bildgebungssystem abgebildet wurde.
Ergebnisse
Um die Gültigkeit des vorgeschlagenen Protokolls zu überprüfen, wurden die hier vorgestellten PD-Experimente mit einem biotinylierten RNA-Aptamer durchgeführt, das in silico entwickelt wurde, um spezifisch TDP-4320 zu binden. Diese RNA bindet ihr Proteintarget mit hoher Bindungsaffinität (Kd = 90 nM)20. Hier wird diese RNA der Sequenz 5'-CGGUGUUGCU-3' mit dem Namen "+RNA" bezeichnet. Als Negativkontrolle wurde die umgekehrte komplementäre Sequenz von +RNA, die hier als "-RNA" bezeichnet wird, verwendet. Seine Sequenz ist 5'-AGCAACACCG-3'. -RNA zeigt eine signifikant geringere Bindungsaffinität gegenüber TDP-43 (Kd = 1,5 μM)19. Für die Zwecke des hier beschriebenen Protokolls wurden diese RNA-Oligonukleotide an ein Biotin-Molekül konjugiert gekauft, um eine Bindung an die Streptavidin-Kügelchen zu ermöglichen. +RNA wurde mit einem Biotin-TEG am 3'-Ende gekauft, das einen 15-Atom-Triethylenglykol-Spacer zwischen dem Biotin und der Phosphatgruppe der Nukleinsäure enthält; -RNA hatte stattdessen ein Biotin an ihrem 5'-Ende, das über einen Amino-C6-Linker an die Nukleinsäure konjugiert war. Wenn das Design des RNA-Köders jedoch robust ist und keine strukturellen oder chemischen Interferenzen zwischen dem Linker und der RNA bestehen, könnten andere Positionen für die Biotin-Konjugation und andere Linkerlängen verwendet werden.
Die Kenntnis der Identität des Hauptproteins, das nach dem PD an die +RNA-Sonde gebunden war, ermöglichte die Validierung des Protokolls durch die Identifizierung von TDP-43 im Eluat, sowohl mittels Massenspektrometrie (MS) als auch mit Western Blot (WB) (Abbildung 1).
Die MS-Analyse wurde an vier PD-Replikaten durchgeführt, die entweder mit +RNA oder -RNA durchgeführt wurden (Abbildung 2). Die Identifizierung der Interaktome von +RNA und -RNA würde den Rahmen dieses Protokolls sprengen, jedoch werden einige Ergebnisse berichtet, die die Genauigkeit des Protokolls bestätigen. Bemerkenswert ist, dass die Darstellung der signifikant angereicherten Proteine in einem Vulkandiagramm zeigte, dass der Gesamtproteingehalt und die angereicherten Proteine, die aus +RNA eluiert wurden, signifikant höher waren als das, was aus -RNA gewonnen wurde (Abbildung 2). Das bedeutet, dass +RNA bei gleicher Länge und gleichem Strukturgehalt (linear) eine höhere Anzahl spezifischer Wechselwirkungen aufbauen kann, die bis zum Elutionsschritt mit hohem Salzgehalt erhalten bleiben. Es ist wahrscheinlich, dass -RNA stattdessen eine höhere Anzahl unspezifischer Kontakte aufbaut, die während der Waschschritte unterbrochen werden. Wie erwartet, konnte TDP-43 als einzigartiger Interaktor von +RNA20 identifiziert werden; Die durchschnittliche markierungsfreie Quantifizierung (LFQ) für die vier PD-Replikate, die mit +RNA durchgeführt wurden, beträgt 31,96 ± 0,56, während das Protein nicht unter den Interaktoren von -RNA identifiziert wird. Darüber hinaus wurde festgestellt, dass TDP-43 unter allen einzigartigen Interaktoren von +RNA das am häufigsten angereicherte Protein ist.
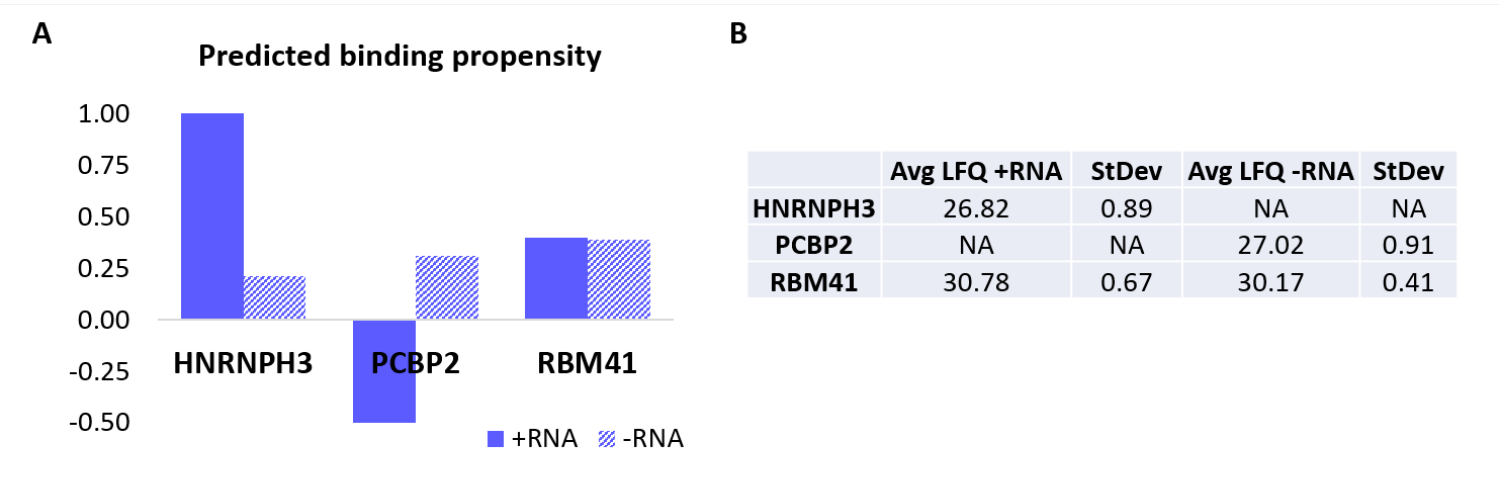
Um das Protokoll weiter zu validieren, wurde der hauseigene Algorithmus catRAPID 18,19 verwendet, um rechnerisch vorherzusagen, welche anderen Proteine spezifisch entweder +RNA oder -RNA binden würden. Insbesondere wurden die Interaktionswerte für +RNA und -RNA mit den Proteinen, aus denen sich das menschliche Proteom zusammensetzt, mit Hilfe der catRAPID-Funktion "Interaktionsneigung" berechnet, wie sie in unserer früheren Arbeit definiertwurde 27. Unter den Proteinen, die mit hoher Konfidenz bewertet wurden, wurde vorhergesagt, dass HNRNPH3 selektiv an +RNA bindet (+RNA-Interaktions-Score = 1,01; -RNA-Interaktions-Score = 0,21) und PCBP2 spezifisch mit -RNA interagiert (+RNA-Interaktions-Score = -0,5; -RNA-Interaktions-Score = 0,31) (Abbildung 3A). Darüber hinaus wurde vorhergesagt, dass das Protein RBM41 für beide RNA-Oligonukleotide promiskuitiv ist (+RNA-Interaktions-Score = 0,4; -RNA-Interaktions-Score = 0,39) (Abbildung 3A). Die MS-Analyse bestätigte tatsächlich das Vorhandensein von HNRNPH3 und PCBP2 in der PD von +RNA bzw. -RNA, während RBM41 mit beiden interagierte (Abbildung 3B).
WB wurde verwendet, um das Vorhandensein von TDP-43 zu detektieren, um die Ergebnisse weiter zu bestätigen und während der Protokolloptimierung (Abbildung 4). Bei dem hier beschriebenen Verfahren wurden verschiedene Proben in unterschiedlichen Stadien entnommen. Die Eingangsprobe (IN) bestand aus den gesamten Proteinen, die in Lysepuffer verdünnt wurden. Der Durchfluss (FT) wurde nach einer nächtlichen Inkubation der Gesamtproteine mit den Streptavidin-Kügelchen erhalten, die mit der biotinylierten RNA vorbeschichtet waren, was den Anteil der Proteine darstellt, die die RNA nicht binden. Schließlich enthielt das Eluat (EL) alle Proteine, die spezifisch die untersuchte RNA erkannten, da zwischen dem FT- und dem EL-Schritt drei Waschschritte mit 150 mM Salz und 0,1% Triton-X die schwächsten Wechselwirkungen entfernt haben sollten.
Für jedes Replikat wurde die gleiche Menge (5 % v/v) IN, FT und EL parallel auf einer SDS-PAGE durchgeführt und mit einem Anti-TDP-43-Antikörper gefärbt (Abbildung 4). Im Fall von +RNA wurde die Bande von TDP-43 in IN und EL beobachtet, was darauf hindeutet, dass das Protein, das von Anfang an im Gesamtproteinextrakt vorhanden ist, während der Waschschritte von +RNA zurückgehalten wird und erst am Ende mit einem hohen Salzpuffer eluiert wird. TDP-43 war auch in IN für -RNA vorhanden, jedoch ist die Bande, die dem Protein entspricht, auch in FT sichtbar, was darauf hindeutet, dass diese RNA nicht an TDP-43 bindet. Das Fehlen des TDP-43-Bandes in EL bestätigt dieses Ergebnis.
Bei der Optimierung des Protokolls wurde die Elution der spezifisch an die RNA-Sequenzen gebundenen Proteine sowohl mit einem Elutionspuffer mit 1 M NaCl (EB1) als auch mit einem Elutionspuffer mit 2 M NaCl (EB2) untersucht (Abbildung 4). Die mit beiden EB erhaltenen Eluate wurden auf einer SDS-PAGE verglichen und mit dem Anti-TDP-43-Antikörper blottiert. Die erhaltenen Bilder wurden dann mit BildJ28 analysiert, um einen Unterschied in der TDP-43-Menge zu quantifizieren, die mit den beiden Puffern eluiert wurde. Insgesamt wurde kein signifikanter Unterschied beobachtet, und wir kamen zu dem Schluss, dass innerhalb dieser Assays 1 M Salz ausreicht, um selbst die stärksten Protein-RNA-Interaktionen zu stören.
Insgesamt zeigen die hier berichteten Ergebnisse für MS und WBs, dass dieses Protokoll effizient ist, um die Proteininteraktoren einer bestimmten RNA auf eine bestimmte Weise zu erfassen, und dass es die Elution in Puffern ermöglicht, die mit der nachgelagerten Analyse kompatibel sind.

Abbildung 1: Skizze der experimentellen Pipeline, die im vorgeschlagenen Protokoll verwendet wird . (A) Das biotinylierte RNA-Oligonukleotid wird in Lysepuffer in der entsprechenden Konzentration hergestellt. (B) Magnetische Streptavidin-Kügelchen werden gewaschen, mit Hefe-tRNA blockiert und mit der biotinylierten RNA beladen. (C) Gesamtproteinextrakt aus kultivierten Säugetierzelllinien wird dem Beads-RNA-Gemisch zugesetzt. (D) Es werden mehrere Wäschen durchgeführt, um unspezifische Wechselwirkungen zu entfernen. (E) Die spezifischen Proteininteraktoren werden mit einer hypertonen Lösung von der RNA abgelöst. (F) Die Identität der Interaktoren wird durch Massenspektrometrie aufgedeckt, und spezifische Fälle werden durch Western Blot validiert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 2: Analytische Strategie zur markierungsfreien MS-basierten Proteinquantifizierung . (A) Eluierte Proteine werden über Nacht in kaltem Aceton ausgefällt. Die Proteine werden dann denaturiert und es wird ein Aufschluss in Lösung durchgeführt. Proteolytische Peptide werden konzentriert und entsalzt. (B) Peptide werden mittels LC-MS/MS mit einem "Shotgun-Ansatz" analysiert. (C) Die Rohdatenverarbeitung und -analyse erfolgt mit der Software MaxQuant bzw. Perseus. (D) Statistisch signifikant angereicherte Proteine werden in einem Vulkandiagramm dargestellt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 3: Korrelation zwischen prognostizierten Interaktionsneigungen und experimentell ermittelten Wechselwirkungen von +RNA und -RNA. (A) catRAPID-Interaktionswerte relativ zu HNRNPH3, PCBP2 und RBM41, was auf eine bevorzugte Bindung von HNRNPH3 für +RNA und von PCBP2 für -RNA hinweist, während RBM41 wahllos an beide RNA-Sequenzen bindet. (B) Markierungsfreie Quantifizierungsmittelwerte, die durch massenspektrometrische Analyse aus den mit +RNA und -RNA durchgeführten Pulldowns bestimmt wurden. Die Analyse bestätigt, dass HNRNPH3 ausschließlich +RNA, PCBP2 ausschließlich -RNA und RBM41 beide gleichermaßen bindet. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 4: Western-Blot-Validierung des Vorhandenseins/Fehlens von TDP-43 unter den Interaktoren ausgewählter RNA-Sequenzen. Die WB-Membran wurde mit Anti-TDP-43-Antikörpern behandelt. IN = Eingang; FT = Durchfluss; EL (EB1) = Elution mit Elutionspuffer 1; EL (EB2) = Elution mit Elutionspuffer 2; das Zeichen "+" kennzeichnet Proben, die aus dem mit +RNA durchgeführten Pulldown stammen; das Zeichen "-" kennzeichnet Proben, die aus dem mit -RNA durchgeführten Pull-Down stammen; Bahn 1 enthält eine Proteinleiter. TDP-43 ist durch einen Pfeil gekennzeichnet. Der WB deutet darauf hin, dass TDP-43 unter +RNA-Interaktoren, aber nicht unter -RNA-Interaktoren zu finden ist. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
| Name des Puffers | Zusammensetzung | |||||
| 10x Übertragungspuffer | 250 mM Tris, 1,92 M Glycin, 1% SDS, 20% Methanol. Vor Gebrauch 10-fach verdünnen | PD | ||||
| 20X MES SDS laufender Puffer | 1 M MES, 1 M Tris, 2 % SDS, 20 mM EDTA. Stellen Sie den pH-Wert auf 7,3 ein. Vor Gebrauch 20-fach verdünnen | |||||
| 4x Probenladepuffer | 0,25 M Trisbase, 0,28 M SDS, 40% Glycerin, 20% 2-Mercapto-Ethanol, 4 mg/ml Bromphenolblau | |||||
| Elutionspuffer 1 | 20 mM Phosphat pH 7,5, 1 M NaCl, 0,5 mM EDTA, 0,1 % Triton X-100, 1 mM DTT (nach Quantifizierung zuzugeben) | |||||
| Elutionspuffer 2 | 20 mM Phosphat pH 7,5, 2 M NaCl, 0,5 mM EDTA, 0,1 % Triton X-100, 1 mM DTT (nach Quantifizierung zuzugeben) | |||||
| Lyse-Puffer | 10 mM Tris-HCl pH 7,4, 150 mM NaCl, 0,5 mM EDTA, 0,1 % Triton X-100, 1 mM DTT und Proteaseinhibitoren | |||||
| Tris-gepufferte Kochsalzlösung mit Tween-20 | 1 M Tris-HCl pH 7,4, 3 M NaCl, 2,0 % Tween-20 | |||||
| Waschpuffer 1 | 10 mM Tris-HCl pH 7,4, 150 mM NaCl, 0,5 mM EDTA, 0,1 % Triton TM X-100, 1 mM DTT und Proteaseinhibitoren | |||||
| Waschpuffer 2 | 25 mM Hepes pH 8, 150 mM NaCl, 0,5 mM EDTA, 0,1 % Triton X-100, 1 mM DTT und Proteaseinhibitoren | |||||
| Puffer A | 0,1% Ameisensäure | FRAU | ||||
| Puffer B | 60% Acetonitril, 0,1% Ameisensäure | |||||
| Denaturierungspuffer | 8M Harnstoff, 50 mM Tris-HCl | |||||
Tabelle 1: PD- und MS-Puffer. Namen und Zusammensetzung der Puffer, die entweder für die Pull-Down-Experimente (PD) oder für die massenspektrometrische Analyse (MS) verwendet werden.
Diskussion
Diese Arbeit berichtet über die Optimierung eines PD-Protokolls, das mit biotinylierten RNA-Oligonukleotiden durchgeführt wurde, um deren Proteininteraktoren zu erfassen. Das hier beschriebene Protokoll ist einfach durchzuführen, benötigt wenig Material und liefert sehr zuverlässige Ergebnisse. Wichtig ist, dass die neuartigsten Aspekte dieses Protokolls in der Verwendung eines RNA-Köders bestehen, der vollständig in silico entwickelt wurde und spezifisch für das Proteinziel ist, und der Elution aller Proteine, die an den RNA-Köder gebunden sind, indem ihre Wechselwirkungen mit einer Lösung mit hohem Salzgehalt direkt unterbrochen werden, anstatt das Streptavidin mit Detergens und Hochtemperaturbehandlung vom Biotin zu dissoziieren.
Dieses Protokoll macht sich die Stärke der Bindung zwischen Biotin und Streptavidinzunutze 29,30. Entsprechend den gewählten Streptavidin-Kügelchen muss die Beladung der biotinylierten RNA getestet und quantifiziert werden, bevor fortgefahren wird. Außerdem könnte die dreidimensionale Faltung der RNA die Beladungseffizienz der Beads beeinflussen, da sie die Exposition des Biotins gegenüber dem Streptavidin begrenzen könnte. Das Blockieren der Beads mit nicht-biotinylierter tRNA verbessert die Reinheit der Ergebnisse, indem unspezifische Wechselwirkungen mit den Beads begrenzt werden. Der Beladungspuffer und der Elutionspuffer müssen in Abhängigkeit von den nachgeschalteten Anwendungen gewählt werden. Hier wurden sehr milde Bedingungen vorgeschlagen, die für die Mehrzahl der Anwendungen geeignet sind und entwickelt wurden, um potentielle Proteinkomplexe zu erhalten. Diese Methode ist jedoch sehr anpassungsfähig; Der Benutzer kann eine beliebige Zelllinie und eine beliebige RNA-Größe auswählen und sich entscheiden, das Protokoll nach dem Falten/Entfalten der RNA zu wiederholen, um den Einfluss der Struktur auf die Bindungseigenschaften zu bestimmen.
Ein weiterer origineller Aspekt dieses Protokolls ist die Verwendung von In-silico-Vorhersagewerkzeugen, um die Korrektheit der Ergebnisse sicherzustellen20. Im Voraus zu wissen, welche Proteine als Interaktoren der interessierenden RNA identifiziert werden sollten, bietet den beispiellosen Vorteil, die technischen Aspekte des Protokolls zu validieren. Mit einer einfachen WB-Analyse ist es beispielsweise möglich, das Vorhandensein eines bekannten Proteinziels in den Proben zu überprüfen, die aus den verschiedenen Schritten des Protokolls stammen, bevor mit der MS-Analyse fortgefahren wird, was spezielle Instrumente erfordert und teurer ist. Darüber hinaus wurde kürzlich über eine Methode berichtet, mit der cat RAPID20, ein interner Protein-RNA-Vorhersagealgorithmus, verwendet werden kann, um de novo RNA spezifisch für ein Zielprotein zu entwerfen. Bis vor kurzem war die einzige verfügbare Pipeline zur Entwicklung von DNA/RNA-Aptameren für ein Zielprotein der SELEX-Ansatz (systematische Evolution von Liganden durch exponentielle Anreicherung)31. Die In-silico-Methode ermöglicht ein wesentlich schnelleres und kostengünstigeres Design von RNA-Aptameren.
Die Haupteinschränkungen dieser Methode sind mit der Notwendigkeit verbunden, in nukleasefreien Puffern und Werkzeugen zu arbeiten. Wenn es als notwendig erachtet wird, die Bindung zwischen einer de novo-designten RNA und einem Zielprotein vor der Parkinson-Krankheit in vitro zu bestätigen, muss das Protein hergestellt und gereinigt und die Bindung mit biophysikalischen Ansätzen bestimmt werden. Dies ist eine Einschränkung, die mit der Produktion von monoklonalen Antikörpern geteilt wird.
Trotz dieser kleineren Probleme können zuverlässige Methoden zur Kartierung von RNA-Protein-Interaktionen, wie die hier vorgestellte, Wissenschaftler näher an die Enthüllung makromolekularer Netzwerke und komplexer Hauptakteure vieler physiologischer und pathologischer Mechanismen bringen, wie z. B. solche, die an Krebs, Kardiomyopathien, Diabetes, mikrobiellen Infektionen sowie genetischen und neurodegenerativen Erkrankungen beteiligt sind.
Offenlegungen
Die Autoren haben keine konkurrierenden finanziellen Interessen oder andere Interessenkonflikte.
Danksagungen
Die Autoren bedanken sich bei der Forschungsgruppe von Prof. Tartaglia und Dr. Cuomo für die angebotene Unterstützung. Das E.Z. erhielt eine Förderung aus dem MINDED-Stipendium des Forschungs- und Innovationsprogramms Horizon 2020 der Europäischen Union im Rahmen der Marie-Skłodowska-Curie-Fördervereinbarung Nr. 754490.
Materialien
| Name | Company | Catalog Number | Comments |
| 6-well tissue culture plates | VWR | 10861-554 | CELLS |
| Cell scrapers | BIOSIGMA | 10153 | CELLS |
| Dulbecco′s Modified Eagle′s Medium (DMEM) | Thermo Fisher Scientfic | 11995065 | CELLS |
| Fetal Bovine Serum, qualified, heat inactivated, Brazil | Thermo Fisher Scientfic | 10500064 | CELLS |
| Phosphate Buffer Saline (PBS, Waltham, MA) | Thermo Fisher Scientfic | 14190169 | CELLS |
| Trypsin (0.25%), phenol red | Thermo Fisher Scientfic | 15050065 | CELLS |
| Anti-rabbit IgG horseradish peroxidase (HRP) | Cellsignal | 7070 | PD |
| Biotinylated RNA | Eurofins | Custom RNA oligonucleotides | PD |
| Bovine serum albumin | Sigma-Aldrich | A9418 | PD |
| Clarity Western ECL Substrate, 500 ml | Biorad | 1705061 | PD |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Merck - Sigma Aldrich | 5056489001 | PD |
| NuPAGE 4 to 12%, Bis-Tris, 1.0 mm, Mini Protein Gel, 10-well | Invitrogen | NP0321BOX | PD |
| Recombinant anti-TDP43 antibody | Abcam | ab109535 | PD |
| Ribonucleic acid, transfer from baker's yeast (S. cerevisiae) | Merck - Sigma Aldrich | R5636-1ML | PD |
| Streptavidin Mag Sepharose | Merck - Sigma Aldrich | GE28-9857-99 | PD |
| Trans-Blot Turbo RTA Mini 0.2 µm PVDF Transfer Kit | Biorad | 1704272 | PD |
| Acetone | Thermo Fisher Scientfic | 022928.K2 | MS |
| C18 cartridge | Thermo Fisher Scientfic | 13-110-018 | MS |
| Dithiothreitol (DTT) | Thermo Fisher Scientfic | 20290 | MS |
| EASY-Spray HPLC Columns | Thermo Scientific | ES902 | MS |
| iodoacetamide (IAA) | Sigma Aldrich S.r.l. | I6125 | MS |
| Lys-C/Trypsin | Promega | V5073 | MS |
| Trifluoroacetic acid (TFA) | Thermo Fisher Scientfic | 28904 | MS |
| Urea | Thermo Fisher Scientfic | J75826.A7 | MS |
| Equipment | |||
| ChemiDoc imaging system | Bio-Rad | CELLS | |
| Dyna Mag -2 , Magnetic rack | Invitrogen | CELLS | |
| Forma Series 3 water jacketed C02 incubator | Thermo Scientific | PD | |
| PROTEAN II xi cell , power supply for PAGE applications | Bio-Rad | PD | |
| Rotating wheel, rotator SB3 | Stuart | PD | |
| Water bath set at 37 °C | VWR | PD | |
| XCell SureLock Mini-Cell electrophoresis system | ThermoFisher Scientific | MS | |
| Easy-nLC 1200 UHPLC | Thermo Scientific | MS | |
| Q exactive Mass Spectrometer | Thermo Scientific | MS | |
| Software | Version | ||
| MaxQuant | 2.0.3.0 | MS | |
| Perseus | 1.6.14.0 | MS |
Referenzen
- Gebauer, F., Schwarzl, T., Valcárcel, J., Hentze, M. W. RNA-binding proteins in human genetic disease. Nature Reviews Genetics. 22 (3), 185-198 (2021).
- Hentze, M. W., Castello, A., Schwarzl, T., Preiss, T. A brave new world of RNA-binding proteins. Nature Reviews Molecular Cell Biology. 19 (5), 327-341 (2018).
- Castello, A., et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 149 (6), 1393-1406 (2012).
- Cooper, T. A., Wan, L., Dreyfuss, G. RNA and disease. Cell. 136 (4), 777-793 (2009).
- Qin, H., et al. RNA-binding proteins in tumor progression. Journal of Hematology & Oncology. 13 (1), 90 (2020).
- Nussbacher, J. K., Tabet, R., Yeo, G. W., Lagier-Tourenne, C. Disruption of RNA metabolism in neurological diseases and emerging therapeutic interventions. Neuron. 102 (2), 294-320 (2019).
- Duan, R., Sharma, S., Xia, Q., Garber, K., Jin, P. Towards understanding RNA-mediated neurological disorders. Journal of Genetics and Genomics. 41 (9), 473-484 (2014).
- Maziuk, B., Ballance, H. I., Wolozin, B. Dysregulation of RNA binding protein aggregation in neurodegenerative disorders. Frontiers in Molecular Neuroscience. 10, 89 (2017).
- Zielinski, J., et al. In vivo identification of ribonucleoprotein-RNA interactions. Proceedings of the National Academy of Sciences. 103 (5), 1557-1562 (2006).
- Armaos, A., Zacco, E., Sanchez de Groot, N., Tartaglia, G. G. RNA-protein interactions: Central players in coordination of regulatory networks. BioEssays. 43 (2), 2000118 (2021).
- Weidmann, C. A., Mustoe, A. M., Jariwala, P. B., Calabrese, J. M., Weeks, K. M. Analysis of RNA-protein networks with RNP-MaP defines functional hubs on RNA. Nature Biotechnology. 39 (3), 347-356 (2021).
- Graindorge, A., et al. In-cell identification and measurement of RNA-protein interactions. Nature Communications. 10 (1), 5317 (2019).
- Ule, J., Hwang, H. W., Darnell, R. B. The future of cross-linking and immunoprecipitation (CLIP). Cold Spring Harbor Perspectives in Biology. 10 (8), 032243 (2018).
- Ascano, M., Hafner, M., Cekan, P., Gerstberger, S., Tuschl, T. Identification of RNA-protein interaction networks using PAR-CLIP. Wiley Interdisciplinary Reviews. RNA. 3 (2), 159-177 (2012).
- McHugh, C. A., Russell, P., Guttman, M. Methods for comprehensive experimental identification of RNA-protein interactions. Genome Biology. 15 (1), 203 (2014).
- Sugimoto, Y., et al. Analysis of CLIP and iCLIP methods for nucleotide-resolution studies of protein-RNA interactions. Genome Biology. 13 (8), (2012).
- Bayat, P., et al. SELEX methods on the road to protein targeting with nucleic acid aptamers. Biochimie. 154, 132-155 (2018).
- Armaos, A., Colantoni, A., Proietti, G., Rupert, J., Tartaglia, G. G. CatRAPID omics v2.0: Going deeper and wider in the prediction of protein-RNA interactions. Nucleic Acids Research. 49, 72-79 (2021).
- Agostini, F., et al. CatRAPID omics: A web server for large-scale prediction of protein-RNA interactions. Bioinformatics. 29 (22), 2928-2930 (2013).
- Zacco, E., et al. Probing TDP-43 condensation using an in silico designed aptamer. Nature Communications. 13 (1), 3306 (2022).
- Leppek, K., Stoecklin, G. An optimized streptavidin-binding RNA aptamer for purification of ribonucleoprotein complexes identifies novel ARE-binding proteins. Nucleic Acids Research. 42 (2), 13 (2014).
- Zhang, Y., Lai, B. S., Juhas, M. Recent advances in aptamer discovery and applications. Molecules. 24 (5), 941 (2019).
- UniProt Consortium. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Research. 51, 523-531 (2023).
- Tyanova, S., Temu, T., Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nature Protocols. 11 (12), 2301-2319 (2016).
- Tyanova, S., et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nature Methods. 13 (9), 731-740 (2016).
- Rappsilber, J., Mann, M., Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nature Protocols. 2 (8), 1896-1906 (2007).
- Bellucci, M., Agostini, F., Masin, M., Tartaglia, G. G. Predicting protein associations with long noncoding RNAs. Nature Methods. 8 (6), 444-445 (2011).
- Gallo-Oller, G., Ordoñez, R., Dotor, J. A new background subtraction method for Western blot densitometry band quantification through image analysis software. Journal of Immunological Methods. 457, 1-5 (2018).
- Weissinger, R., Heinold, L., Akram, S., Jansen, R. P., Hermesh, O. RNA proximity labeling: A new detection tool for RNA-protein interactions. Molecules. 26 (8), 2270 (2021).
- Hirsch, J. D., et al. Easily reversible desthiobiotin binding to streptavidin, avidin, and other biotin-binding proteins: Uses for protein labeling, detection, and isolation. Analytical Biochemistry. 308 (2), 343-357 (2002).
- Sefah, K., Shangguan, D., Xiong, X., O'Donoghue, M. B., Tan, W. Development of DNA aptamers using cell-SELEX. Nature Protocols. 5 (6), 1169-1185 (2010).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten