Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Aufbau von Stoffwechselnetzwerken außerhalb des Gleichgewichts in nano- und mikrometergroßen Vesikeln
In diesem Artikel
Zusammenfassung
Wir stellen ein Protokoll zur Rekonstitution von Membranproteinen und zur Verkapselung von Enzymen und anderen wasserlöslichen Komponenten in Lipidvesikeln von Submikrometer- und Mikrometergröße vor.
Zusammenfassung
Wir stellen eine Methode vor, um komplexe Proteinnetzwerke in Vesikel einzubauen, an denen integrale Membranproteine, Enzyme und fluoreszenzbasierte Sensoren unter Verwendung gereinigter Komponenten beteiligt sind. Diese Methode ist relevant für das Design und den Bau von Bioreaktoren und die Untersuchung komplexer metabolischer Reaktionsnetzwerke außerhalb des Gleichgewichts. Wir beginnen mit der Rekonstitution von (mehreren) Membranproteinen zu großen unilamellären Vesikeln (LUVs) nach einem zuvor entwickelten Protokoll. Anschließend verkapseln wir ein Gemisch aus gereinigten Enzymen, Metaboliten und fluoreszenzbasierten Sensoren (fluoreszierende Proteine oder Farbstoffe) mittels Gefrier-Auftau-Extrusion und entfernen nicht eingebaute Komponenten durch Zentrifugation und/oder Größenausschlusschromatographie. Die Leistung der Stoffwechselnetzwerke wird in Echtzeit gemessen, indem das ATP/ADP-Verhältnis, die Metabolitenkonzentration, der interne pH-Wert oder andere Parameter durch Fluoreszenzauslesung überwacht werden. Unsere Membranprotein-haltigen Vesikel mit einem Durchmesser von 100-400 nm können mit bestehenden, aber optimierten Verfahren in riesig-unilamelläre Vesikel (GUVs) umgewandelt werden. Der Ansatz ermöglicht den Einbau löslicher Komponenten (Enzyme, Metaboliten, Sensoren) in mikrometergroße Vesikel, wodurch das Volumen der Bioreaktoren um Größenordnungen vergrößert wird. Das metabolische Netzwerk, das GUVs enthält, wird in mikrofluidischen Geräten gefangen, um sie mit optischer Mikroskopie zu analysieren.
Einleitung
Der Bereich der synthetischen Biologie von unten konzentriert sich auf die Konstruktion von (minimalen) Zellen 1,2 und metabolischen Bioreaktoren für biotechnologische 3,4 oder biomedizinische Zwecke 5,6,7,8. Die Konstruktion synthetischer Zellen bietet eine einzigartige Plattform, die es Forschern ermöglicht, (Membran-)Proteine unter genau definierten Bedingungen zu untersuchen, die denen der natürlichen Umgebung nachahmen, und ermöglicht so die Entdeckung von emergenten Eigenschaften und verborgenen biochemischen Funktionen von Proteinen und Reaktionsnetzwerken9. Als Zwischenschritt hin zu einer autonom funktionierenden synthetischen Zelle werden Module entwickelt, die wesentliche Merkmale lebender Zellen wie metabolische Energieerhaltung, Protein- und Lipidsynthese sowie Homöostase erfassen. Solche Module erweitern nicht nur unser Verständnis des Lebens, sondern haben auch Anwendungsmöglichkeiten in den Bereichen Medizin8 und Biotechnologie10.
Transmembranproteine sind das Herzstück praktisch jedes Stoffwechselnetzwerks, da sie Moleküle in oder aus der Zelle transportieren, Signale senden und auf die Qualität der Umwelt reagieren und zahlreiche biosynthetische Rollen spielen. Daher erfordert das Engineering von Stoffwechselmodulen in synthetischen Zellen in den meisten Fällen die Rekonstitution von integralen und/oder peripheren Membranproteinen zu einer Membrandoppelschicht, die aus spezifischen Lipiden besteht und eine hohe Integrität (geringe Permeabilität) aufweist. Der Umgang mit diesen Membranproteinen ist anspruchsvoll und erfordert spezifisches Wissen und experimentelle Fähigkeiten.
Es wurden mehrere Methoden entwickelt, um Membranproteine in Phospholipid-Vesikeln zu rekonstituieren, meist mit dem Ziel, die Funktion11,12, die Regulation13, die kinetischen Eigenschaften14,15, die Lipidabhängigkeit15,16 und/oder die Stabilität17 eines spezifischen Proteins zu untersuchen. Diese Verfahren umfassen die rasche Verdünnung von durch Detergenzien gelöstem Protein in wässrigen Medien in Gegenwart von Lipiden18, die Entfernung von Detergenzien durch Inkubation von durch Detergenzien gelöstes Protein mit detergenzient destabilisierten Lipidvesikeln und die Absorption des Detergens bzw. der Detergenzien auf Polystyrolkügelchen19 oder das Entfernen von Detergenzien durch Dialyse oder Größenausschlusschromatographie20. Organische Lösungsmittel wurden verwendet, um Lipidvesikel zu bilden, z. B. durch die Bildung von Öl-Wasser-Interphasen21, aber die Mehrzahl der integralen Membranproteine wird inaktiviert, wenn sie solchen Lösungsmitteln ausgesetzt werden.
In unserem Labor rekonstituieren wir hauptsächlich Membranproteine mit der Detergenz-Absorptionsmethode zu großen unilamellären Vesikeln (LUVs)19. Dieses Verfahren ermöglicht die Co-Rekonstitution mehrerer Membranproteine und die Verkapselung von Enzymen, Metaboliten und Sonden im Vesikellumen22,23. Die Membranprotein-haltigen LUVs können in riesen-unilamelläre Vesikel (GUVs) mit/ohne Verkapselung wasserlöslicher Komponenten umgewandelt werden, wobei entweder die Elektroformation24 oder die gelgestützte Quellung25 und spezifische Bedingungen zur Erhaltung der Integrität der Membranproteine26 verwendet werden.
In dieser Arbeit wird ein Protokoll für die Rekonstitution eines außerhalb des Gleichgewichts befindlichen metabolischen Netzwerks in LUVs vorgestellt, das ATP durch den Abbau von L-Arginin in L-Ornithin regeneriert27. Die Bildung von ATP ist an die Produktion von Glycerin-3-phosphat (G3P) gekoppelt, einem wichtigen Baustein für die Phospholipidsynthese22,28. Der Stoffwechselweg besteht aus zwei integralen Membranproteinen, einem Arginin/Ornithin (ArcD) und einem G3P/Pi-Antiporter (GlpT). Darüber hinaus werden drei lösliche Enzyme (ArcA, ArcB, ArcC) für das Recycling von ATP benötigt, und GlpK wird verwendet, um Glycerin in Glycerin-3-phosphat umzuwandeln, wobei das ATP aus dem Abbau von L-Arginin verwendet wird, siehe Abbildung 1 für einen schematischen Überblick über den Weg. Dieses Protokoll stellt einen guten Ausgangspunkt für den zukünftigen Aufbau noch komplexerer Reaktionsnetzwerke dar - für die Synthese von Lipiden oder Proteinen oder die Teilung von Zellen. Die Lipidzusammensetzung der Vesikel unterstützt die Aktivität einer Vielzahl von integralen Membranproteinen und wurde für den Transport verschiedener Moleküle in oder aus den Vesikeln optimiert 27,29,30.

Abbildung 1: Überblick über den Weg der ATP-Produktion und der Glycerin-3-phosphat-Synthese und -Ausscheidung. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Kurz gesagt, gereinigte Membranproteine (solubilisiert in Dodecyl-β-D-Maltosid, DDM) werden zu vorgeformten Lipidvesikeln hinzugefügt, die mit Triton X-100 destabilisiert wurden, was die Insertion der Proteine in die Membran ermöglicht. Die Detergensmoleküle werden anschließend (langsam) durch die Zugabe von aktivierten Polystyrolkügelchen entfernt, was zur Bildung von gut verschlossenen Proteoliposomen führt. Lösliche Bestandteile können dann zu den Vesikeln hinzugefügt und über Gefrier-Tau-Zyklen verkapselt werden, wodurch die Moleküle im Prozess der Membranfusion eingefangen werden. Die gewonnenen Vesikel sind sehr heterogen und viele sind multilamell. Sie werden dann durch einen Polycarbonatfilter mit einer Porengröße von 400, 200 oder 100 nm extrudiert, was zu gleichmäßiger großen Vesikeln führt; Je kleiner die Porengröße, desto homogener und unilamellär sind die Vesikel, jedoch zum Preis eines kleineren inneren Volumens. Nicht eingebaute Proteine und kleine Moleküle werden durch Größenausschlusschromatographie aus der externen Lösung entfernt. Die proteoLUVs können durch gelgestütztes Aufquellen in mikrometergroße Vesikel umgewandelt werden, und diese proteoGUVs werden dann gesammelt und in einem mikrofluidischen Chip zur mikroskopischen Charakterisierung und Manipulation gefangen. Abbildung 2 zeigt einen schematischen Überblick über das gesamte Protokoll.

Abbildung 2: Überblick über das Protokoll zur Rekonstitution von Membranproteinen und zur Verkapselung von Enzymen und wasserlöslichen Komponenten in Lipidvesikeln mit Submikrometer- (LUVs) und Mikrometergröße (GUVs). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Die Rekonstitutions- und Verkapselungsprotokolle funktionieren gut und die Funktionalität der Proteine bleibt erhalten, aber die proteoLUVs und proteoGUVs sind heterogen groß. Mikrofluidische Ansätze 31,32 ermöglichen die Bildung von mikrometergroßen Vesikel, die in ihrer Größe homogener sind, aber eine funktionelle Rekonstitution von Membranproteinen ist im Allgemeinen nicht möglich, da das restliche Lösungsmittel in der Doppelschicht die Proteine inaktiviert. Die ProteoLUVs haben eine Größe von 100 bis 400 nm, und bei niedrigen Enzymkonzentrationen kann die Verkapselung zu Vesikeln mit unvollständigen Stoffwechselwegen führen (stochastische Effekte; siehe Abbildung 3). LUVs sind ideal für den Aufbau spezifischer Stoffwechselmodule, wie hier gezeigt, für die Produktion von ATP und Bausteinen wie G3P. Solche proteoLUVs können möglicherweise in GUVs eingekapselt werden und als organellenähnliche Kompartimente für die Wirtsvesikel dienen.

Abbildung 3: Anzahl der Moleküle pro Vesikel mit einem Durchmesser von 100, 200 oder 400 nm. (A) Wenn die verkapselten Proteine (Enzyme, Sonden) im Bereich von 1-10 μM liegen. (B) Die Rekonstitution erfolgt bei 1 bis 1.000, 1 bis 10.000 und 1 bis 100.000 Membranproteinen pro Lipid (mol/mol). Wir gehen davon aus, dass Moleküle in den angegebenen Konzentrationen verkapselt und bei diesen Protein-Lipid-Verhältnissen in die Membran eingebaut werden. Bei einigen Enzymen haben wir gesehen, dass sie an Membranen binden, was ihre scheinbare Konzentration in den Vesikeln erhöhen kann. Abkürzung: LPR = Lipid-Protein-Ratio Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
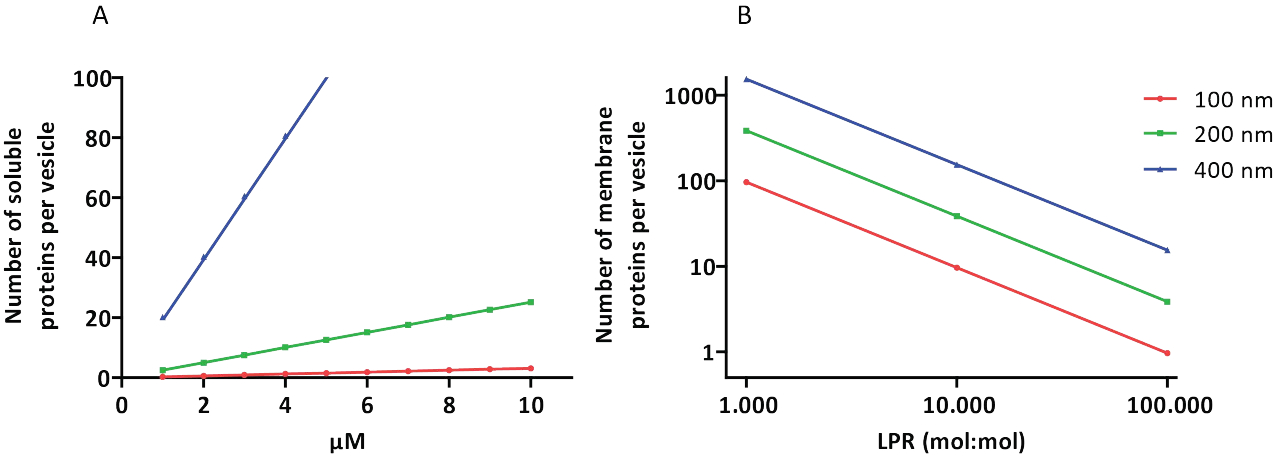{kind=link}
Protokoll
1. Allgemeine Vorbereitung
- Chemikalien
- Lösen Sie Lipide (in Pulverform) auf 25 mg/ml in CHCl3 auf, um vorgeformte Liposomen herzustellen.
HINWEIS: Es ist vorzuziehen, frische Lipide herzustellen, aber die Stammlösungen können auch einige Wochen bei -20 °C gelagert werden. Die Arbeit mit Lipiden in Pulverform ist genauer als die Verwendung von Lipiden, die bereits in CHCl3 solubilisiert sind. CHCl3 sollte mit Glaspipetten und/oder Spritzen gehandhabt und in Glasbehältern aufbewahrt werden, da CHCl3 Kunststoffe auflöst. - Kleine Moleküle (Nukleotide, Aminosäuren, Fluoreszenzsonden) für das Verkapselungsverfahren in 50 mM KPi (Puffer A, siehe Tabelle 1) auflösen und den pH-Wert auf 7,00 ± 0,01 einstellen. MgCl2 in deionisiertem Wasser auflösen, um die Bildung von Magnesiumphosphatausfällungen zu vermeiden.
HINWEIS: Stammlösungen können einige Wochen lang bei -20 °C gelagert werden, mit Ausnahme von DVB-T, das am Tag des Versuchs frisch zubereitet wird. - Ionophore (z. B. Valinomycin, Nigericin) in DMSO oder EtOH in einer Stammkonzentration von 100-500 μM auflösen. Einige Wochen bei -20 °C lagern; Verdunstung vermeiden.
HINWEIS: DMSO ist nicht flüchtig und daher gegenüber EtOH bevorzugt. Verwenden Sie Glas- anstelle von Plastikfläschchen, um ein Anhaften der Ionophore an der Oberfläche der Fläschchen zu vermeiden.
- Lösen Sie Lipide (in Pulverform) auf 25 mg/ml in CHCl3 auf, um vorgeformte Liposomen herzustellen.
- Puffer
- Bereiten Sie am Tag des Versuchs frische Puffer vor (Tabelle 1). Nicht länger als 24 h lagern.
- Reinigung löslicher Proteine
- Express ArcA, ArcB, ArcC (wir verwenden eine spezielle Variante namens ArcC1), PercevalHR und GlpK, wie zuvor beschrieben 27,28,33. Stellen Sie sicher, dass Sie dem Zelllysepuffer 10 % v/v Glycerin hinzufügen, was die Stabilität der Proteine erhöht. Reinigen Sie die löslichen Proteine, wiebeschrieben 27,28,33 und kurz nachstehend berichtet.
- Tauen Sie 10 mL Zelllysat (~5 g Nassgewicht) in einem Eiswasserbad auf. Tragen Sie in der Zwischenzeit 2 mL (1 CV) Ni2+-Sepharoseharz auf eine Schwerkraftflusssäule (20 mL Kapazität) mit deionisiertem Wasser (12 CVs) und Puffer B (4 CVs) zum Waschen auf. Übertragen Sie das aufgetaute Lysat auf Eis; Arbeiten Sie auf Eis, sofern nicht anders angegeben.
- Geben Sie Imidazol bis zu einer Endkonzentration von 10 mM in das aufgetaute Lysat und gießen Sie dann die Lösung auf die Schwerkraftsäule. 1 Stunde lang bei 4 °C unter sanfter Nutation inkubieren.
- Nach 1 h verwerfen Sie den Durchfluss und waschen Sie das Harz mit Puffer C (20 CVs).
- Eluieren Sie das Protein mit Puffer D. Verwenden Sie 60 % CV für den ersten Elutionsschritt, gefolgt von 4-6 Schritten mit 40 % CVs.
- Bestimmen Sie die Proteinkonzentration und fügen Sie Na-EDTA bis zu einer Endkonzentration von 5 mM hinzu.
- Schleudern Sie das gereinigte Protein in einer gekühlten Tischzentrifuge herunter (max. Drehzahl, 10 Minuten, 4 °C). Aufreinigung durch Größenausschlusschromatographie mit Puffer E. Die Elutionsfraktionen werden zusammengefasst und mit einem Konzentrationsfilter mit einem Cutoff-Wert von 30 kDa auf ~10 mg/ml konzentriert. Aliquots geeigneter Größe (~ 20 μl) vorbereiten, mit flüssigem Stickstoff schockfrosten und für die spätere Verwendung bei -80 °C lagern.
HINWEIS: Es ist wichtig, die Enzyme auf 50-100 μM zu konzentrieren, um das für die Verkapselung benötigte Volumen zu minimieren.
- Aufreinigung von Membranproteinen
- Überexpress ArcD und GlpT wie zuvor beschrieben 22,27,33. Stellen Sie sicher, dass Sie dem Zelllysepuffer 10 % v/v Glycerin hinzufügen. Für die Aufreinigung von ArcD sind 2 mM Reduktionsmittel (z. B. DTT) in den Puffer zu geben. Aufreinigung der affinitätsmarkierten Proteine durch Ni2+-Sepharosechromatographie.
- Ein Aliquot von rohen Membranvesikeln (10-20 mg Gesamtmembranprotein) wird in einem Eiswasserbad aufgetaut.
HINWEIS: Nach dem Auftauen immer auf Eis arbeiten, sofern nicht anders angegeben. In Tabelle 1 finden Sie die in diesem Abschnitt verwendeten Puffer. - Geben Sie die Membranvesikel in Puffer F (ArcD) oder Puffer G (GlpT) bis zu einem Endvolumen von 6 mL. Die Probe wird 1 h lang bei 4 °C unter sanfter Nutation inkubiert.
- Trennen Sie die löslichen Membranproteine durch Ultrazentrifugation (337.000 × g, 30 min, 4 °C) von den Membranresten. In der Zwischenzeit tragen Sie 0,25 mL (1 CV) Ni2+-Sepharoseharz auf eine Schwerkraftflusssäule (10 mL Kapazität) mit deionisiertem Wasser (40 CVs) und 20 CVs Puffer H (ArcD) oder Puffer I (GlpT) auf.
- Gießen Sie das lösliche Protein auf die Schwerkraftflusssäule und fügen Sie Imidazol bis zu einer Endkonzentration von 10 mM hinzu. 1 h bei 4 °C unter sanfter Nutation inkubieren.
- Nach 1 h verwerfen Sie den Durchfluss und waschen Sie das Harz mit 20 CVs Buffer J (ArcD) oder Buffer K (GlpT).
- Eluieren Sie das Membranprotein in Schritten von 60 % CV (1st) und 40 % CV (2nd-6 th) mit Buffer L (ArcD) oder Buffer M (GlpT).
- Die Proteinkonzentration ist zu bestimmen und mit Abschnitt 2.2 fortzufahren. für die Rekonstitution der Membran.
HINWEIS: Die Größenausschlussreinigung wird nicht unbedingt für Membranproteine durchgeführt, da die Membranrekonstitution eine ähnliche Reinigung ergibt. Die Schritte 1.4 und 2.2 können an 1 Arbeitstag durchgeführt werden. Beginnen Sie mit der Proteinreinigung (Schritt 1.4) am Morgen und fahren Sie mit der Rekonstitution (Schritt 2.2) am Nachmittag fort. Die Rekonstitution endet am folgenden Tag (siehe Abschnitt 2.2 für Details). HALTEPUNKT: Aufgereinigtes und DDM-solublisiertes ArcD und GlpT können für die spätere Verwendung bei -80 °C gelagert werden, dies gilt jedoch nicht für alle Membranproteine. Aliquots geeigneter Größe (50-200 μl) vorbereiten, mit flüssigem Stickstoff schockfrosten und für die spätere Verwendung bei -80 °C lagern. Diese Proteine sind mehrere Monate lang aktiv, wenn sie bei -80 °C in Gegenwart von 10 % v/v Glycerin gelagert werden.
- Ein Aliquot von rohen Membranvesikeln (10-20 mg Gesamtmembranprotein) wird in einem Eiswasserbad aufgetaut.
- Überexpress ArcD und GlpT wie zuvor beschrieben 22,27,33. Stellen Sie sicher, dass Sie dem Zelllysepuffer 10 % v/v Glycerin hinzufügen. Für die Aufreinigung von ArcD sind 2 mM Reduktionsmittel (z. B. DTT) in den Puffer zu geben. Aufreinigung der affinitätsmarkierten Proteine durch Ni2+-Sepharosechromatographie.
- Vorbereitung von β-Kasein für Passivierungszwecke
- 100 mg β-Kasein in 20 ml deionisiertem Wasser resuspendieren und mit 1 M NaOH titrieren, bis das β-Kasein vollständig aufgelöst ist. Fügen Sie dann 1 M Essigsäure hinzu, um den pH-Wert auf 7,0 einzustellen, und füllen Sie das Volumen auf 50 mL mit deionisiertem Wasser. Die Lösung wird durch einen 0,2 μm Spritzenvorsatzfilter filtriert und Aliquote von 500 μl hergestellt.
HINWEIS: Das β-Kasein kann 6 Monate bei -20 °C gelagert werden. Es wird empfohlen, das β-Kasein vor der Verwendung erneut zu filtrieren, um zu verhindern, dass β-Kasein-Aggregate den Mikrofluidik-Chip verstopfen.
- 100 mg β-Kasein in 20 ml deionisiertem Wasser resuspendieren und mit 1 M NaOH titrieren, bis das β-Kasein vollständig aufgelöst ist. Fügen Sie dann 1 M Essigsäure hinzu, um den pH-Wert auf 7,0 einzustellen, und füllen Sie das Volumen auf 50 mL mit deionisiertem Wasser. Die Lösung wird durch einen 0,2 μm Spritzenvorsatzfilter filtriert und Aliquote von 500 μl hergestellt.
| Puffer | Zusammensetzung | ||
| Puffer A | 50 mM KPi pH 7,0 | ||
| Puffer B | 50 mM KPi, 100 mM KCl, 10 % v/v Glycerin, 10 mM Imidazol, pH 7,5 | ||
| Puffer C | 50 mM KPi, 100 mM KCl, 10 % v/v Glycerin, 50 mM Imidazol, pH 7,5 | ||
| Puffer D | 50 mM KPi, 100 mM KCl, 10 % v/v Glycerin, 500 mM Imidazol, pH 7,5 | ||
| Puffer E | 50 mM KPi, 100 mM KCl, 10 % v/v Glycerin, pH 7,0 | ||
| Puffer F | 50 mM KPi, 100 mM KCl, 0,5 % w/v DDM, 10 % v/v Glycerin, 2 mM β-Mercaptoethanol, pH 7,5 | ||
| Puffer G | 50 mM Tris-HCl, 0,5 % w/v DDM, 20 % v/v Glycerin, pH 8 | ||
| Puffer H | 50 mM KPi, 100 mM KCl, 0,02 % w/v DDM, 10 % v/v Glycerin, 2 mM β-Mercaptoethanol, 10 mM Imidazol, pH 7,5 | ||
| Puffer I | 50 mM Tris-HCl, 0,04 % w/v DDM, 20 % v/v Glycerin, 10 mM Imidazol, pH 8,0 | ||
| Puffer J | 50 mM KPi, 200 mM KCl, 0,02 % w/v DDM, 10 % v/v Glycerin, 2 mM β-Mercaptoethanol, 50 mM Imidazol, pH 7,5 | ||
| Puffer K | 50 mM Tris-HCl, 0,04 % w/v DDM, 20 % v/v Glycerin, 50 mM Imidazol, pH 8 | ||
| Puffer L | 50 mM KPi, 200 mM KCl, 0,02 % w/v DDM, 10 % v/v Glycerin, 2 mM β-Mercaptoethanol, 500 mM Imidazol, pH 7,5 | ||
| Puffer M | 50 mM Tris-HCl, 0,04 % w/v DDM, 20 % v/v Glycerin, 500 mM Imidazol, pH 8 | ||
| Puffer N | 50 mM KPi, 58 mM NaCl, 2 mM DTT, pH 7,0 | ||
| Puffer O | 50 mM KPi, 0,5 mM L-Ornithin, 10 mM Na-ADP, 10 mM MgCl2, 2 mM DTT, pH 7,0 | ||
| Puffer P | 50 mM KPi pH 7,0, 2 mM DTT, x mM Glukose (x wird variiert, um der Osmolarität des externen und internen Mediums zu entsprechen) | ||
| Puffer Q | 50 mM KPi pH 7,0, 0,5 mM Saccharose, 2 mM DTT | ||
| Puffer R | 50 mM KPi pH 7,0, 2 mM DTT, 10 mM L-Arginin, x mM Glukose | ||
Tabelle 1: In diesem Protokoll verwendete Puffer.
2. Proteoliposomen: Rekonstitution von gereinigten Membranproteinen in vorgeformte Lipidvesikel
- Tag 1
- Präparation von vorgeformten Lipidvesikeln
- Wählen Sie die Lipidzusammensetzung (z.B. synthetische Phospholipide, E. coli polare Lipide) auf Basis der Anforderungen der Membranproteine.
HINWEIS: Eine Mischung aus DOPE, DOPG und DOPC (25:25:50 mol%) ist ein guter Ausgangspunkt, aber für einige Proteine können Sterole oder Cardiolipin erforderlich sein; Für Hefe-Plasmamembranproteine schließen wir Palmitoyl-Oleoyl-Lipide anstelle von Dioleoyl30 ein. Ein DOPE-, DOGG- und DOPC-Gemisch (25:25:50 mol%) reicht für die Rekonstitution von ArcD und GlpT aus. - Mischen Sie die gewünschten Lipide (solubilisiert inCHCl 3) und verdampfen Sie das CHCl3 in einem Rotationsverdampfer, bis sich ein Lipidfilm bildet. Waschen Sie die Lipide, indem Sie Diethylether in gleichem Volumen wieCHCl 3 hinzufügen. Verdampfen Sie den Diethylether und erhalten Sie einen trockenen Lipidfilm.
- Resuspendieren Sie den Lipidfilm auf 20 mg/ml Gesamtlipide in wässrigen Medien (Puffer A). Beginnen Sie mit der Hälfte des Gesamtvolumens und schütteln Sie es leicht; Füllen Sie dann die Lipide vorsichtig in ein sauberes Röhrchen oder eine Flasche mit ausreichender Größe um. Frischer Puffer A in den Kolben geben und den Vorgang wiederholen, um die restlichen Lipide aufzulösen und in ein neues Gefäß zu überführen. Fügen Sie zusätzlichen Puffer A hinzu, um eine Endkonzentration von 20 mg/ml zu erreichen.
- Beschallung der resuspendierten Lipide mit einem Sonden-Ultraschallgerät. Für eine Ultraschalldüse mit einem Durchmesser von 6 mm sind folgende Parameter zu verwenden: 4 μm Intensität, 70 % Amplitude, 5 s an, 45 s aus, 16 Zyklen. Tauchen Sie die Lipide in ein Eiswasserbad, das EtOH enthält, um eine Überhitzung durch Ultraschall zu vermeiden.
- Die beschallte Probe (Volumen von 40 mL in einem 50 mL-Zentrifugenröhrchen) wird in flüssigem Stickstoff schockgefroren und die Probe in einem Wasserbad bei Raumtemperatur aufgetaut. Einmal wiederholen; dann aliquotieren Sie die Liposomen in den gewünschten Volumina (z. B. 1 ml oder 20 mg Gesamtlipide in einem 1,5 ml-Kunststoffröhrchen).
STOPPPUNKT: An dieser Stelle kann der Vorgang gestoppt werden. Jedes Aliquot noch einmal schockfrosten (dritter Zyklus) und bis zu einigen Monaten in flüssigem Stickstoff lagern. Achten Sie darauf, die Tubendeckel zweimal mit einer Nadel zu durchstechen, um eine Explosion der Tube beim schnellen Sieden des flüssigen Stickstoffs zu vermeiden.
- Wählen Sie die Lipidzusammensetzung (z.B. synthetische Phospholipide, E. coli polare Lipide) auf Basis der Anforderungen der Membranproteine.
- Präparation von vorgeformten Lipidvesikeln
- Tag 2
- Rekonstitution von gereinigten Membranproteinen in vorgeformte Liposomen
- Tauen Sie ein Aliquot von Liposomen (20 mg Gesamtlipide) in einem Wasserbad bei Raumtemperatur auf. Bereiten Sie in der Zwischenzeit einen Extruder vor, indem Sie einen Filter Ihrer Wahl auftragen (z. B. Polycarbonat, Porendurchmesser von 400 nm); den Extruder mit Puffer A voräquilibrieren; und laden Sie die aufgetauten Liposomen ("Milchlösung") in den Extruder und passieren Sie sie 13x durch den Filter. Sammeln Sie die extrudierten Liposomen (jetzt große unilamelläre Vesikel; "undurchsichtige Lösung") in einem Glas- oder Kunststoffgefäß ausreichender Größe (z. B. 15 ml). Verdünnen Sie die Liposomen auf 4 mg/ml mit Puffer A, der mit 2 mM DTT ergänzt wird.
- Übertragen Sie 1 ml 4 mg/ml-Liposomen in eine transparente 1-ml-Küvette. Messen Sie in einem Spektralphotometer die anfängliche optische Dichte bei 540 nm. Gießen Sie die gemessene Probe zurück und geben Sie 50 μl 10 % v/v Triton X-100 zu den Liposomen.
HINWEIS: Ein Titrationsvolumen von 50 μl 10 % Triton-X100 ist für 20 mg Lipide in einem Volumen von 5 ml geeignet; die Zugabe von Triton X-100 verdünnt die Lipide um ~5%. Passen Sie das Titrationsvolumen an, wenn Sie mit unterschiedlichen Mengen an Liposomen arbeiten. Für stabile optische Dichtesignale titrieren Sie die Liposomen mit Triton X-100 bei Raumtemperatur. - Wiederholen Sie Schritt 2.2.1.2 und notieren Sie, wann eine maximale optische Dichte (Rsat) erreicht ist. Fahren Sie mit der Titration fort, bis eine optische Dichte von etwa 60 % Rsat erreicht ist (Abbildung 4). Gießen Sie die durch das Reinigungsmittel destabilisierten Vesikel zurück in das Glas-/Kunststoffröhrchen (das endgültige Volumen beträgt jetzt ca. 5,2 ml), geben Sie die Probe auf Eis und lassen Sie sie abkühlen.
- Fügen Sie das/die gereinigte(n) Membranprotein(e) zu den destabilisierten Liposomen hinzu, um ein gewünschtes Lipid-Protein-Verhältnis (w/w) zu erreichen. Verwenden Sie ein Verhältnis von 400:1 bis 100:1 w/w; Da sowohl ArcD als auch GlpT ein Molekulargewicht von ~55 kDa haben, entspricht ein Lipid-Protein-Verhältnis von 400:1 w/w ~30.000 Lipiden pro Protein und jeweils ~ 50 Molekülen ArcD und GlpT pro Vesikel mit einem Durchmesser von 400 nm.
HINWEIS: Hier verwenden wir 400:1 w/w, was 50 μg jedes Proteins pro 20 mg Lipide entspricht. - Die Proben werden 15 Minuten lang bei 4 °C nutatiert, damit sich die Membranproteine in die destabilisierte liposomale Membran einfügen können.
- Um das Reinigungsmittel zu entfernen, fügen Sie 200 mg trockene Polystyrolkügelchen hinzu, die gemäß den Anweisungen des Herstellers zubereitet wurden. Bei 4 °C weitere 15 Min. erhitzen.
HINWEIS: Eine Menge von 200 mg Trockenkügelchen ist für 20 mg Lipide geeignet, sollte jedoch angepasst werden, wenn eine andere Probengröße verwendet wird. - Schritt 2.2.1.6 2x wiederholen, um insgesamt drei Zusätze von Polystyrolkügelchen zu erhalten. Dann über Nacht bei 4 °C unter sanfter Nutation inkubieren.
- Rekonstitution von gereinigten Membranproteinen in vorgeformte Liposomen
- Tag 3
- Schritt 2.2.1.6 wiederholen; Diesmal jedoch 1 h lang bei 4 °C nutatieren.
- Befestigen Sie eine leere Schwerkraft-Flow-Säule (10 mL Kapazität) über einem leeren 6,5 mL Ultrazentrifugationsröhrchen auf Eis. Gießen Sie die Probe in die Säule und sammeln Sie die Proteoliposomen im Ultrazentrifugationsröhrchen (die Kügelchen werden in der Säule zurückgehalten).
- Waschen Sie die Kügelchen mit 0,5 mL Puffer A, ergänzt mit 2 mM DTT, und sammeln Sie das Filtrat im Ultrazentrifugationsröhrchen.
- Konzentrieren Sie die Proteoliposomen durch Ultrazentrifugation (337.000 × g, 30 min, 4 °C). Resuspendieren Sie die Proteoliposomen in einem Gesamtvolumen von 200 μl (100 mg Lipid/ml) in Puffer A, ergänzt mit 2 mM DTT; Das Trockenvolumen der Vesikel nach der Zentrifugation beträgt ~40 bis 120 μL27. Aufgeteilt in Aliquots der gewünschten Größe (z. B. drei Aliquots mit 6,66 mg Gesamtlipiden).
HINWEIS: Die aliquote Größe ist willkürlich, wirkt sich aber in späteren Schritten auf die Pelletgröße aus (siehe Schritt 3.2.3.1). STOPPPUNKT: Hier kann der Vorgang gestoppt werden. Jedes Aliquot schockfrosten und bis zu einigen Wochen in flüssigem Stickstoff lagern. Achten Sie darauf, die Tubendeckel zweimal mit einer Nadel zu durchstechen, um eine Explosion der Tube beim schnellen Sieden des flüssigen Stickstoffs zu vermeiden.

Abbildung 4: Titration von vorgeformten Liposomen mit Triton X-100. Liposomen mit 5 mg Lipiden/ml werden durch einen Polycarbonatfilter (400 nm) in 50 mM KPi (pH 7,0) extrudiert und dann mit Triton X-100 titriert (Protokollschritt 2.2.1.2). Die Trübung der Vesikel wird bei A540 gemessen. Der Pfeil zeigt die Triton X-100-Konzentration an, bei der die Vesikel ausreichend destabilisiert sind, um eine spontane Insertion von Membranproteinen zu ermöglichen, wie in19 beschrieben. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
3. Verkapselung eines metabolischen Netzwerks für ATP-Recycling und Glycerin-3-P-Synthese in submikrongroßen Vesikeln
- Tag 1
- Mischen von Komponenten
- Für eine Standardverkapselung verwenden Sie 66,6 μl Proteoliposomen in einem Endvolumen von 200 μl (33,33 mg/ml Gesamtlipide). Berechnen Sie das Volumen jeder Komponente (Enzym, Cofaktor), das benötigt wird, um die gewünschte Konzentration zu erreichen, fügen Sie diese Komponenten zu den Proteoliposomen hinzu und stellen Sie das Volumen mit Puffer A auf 200 μl ein (Tabelle 2).
HINWEIS: Die Proteinkonzentration kann basierend auf dem Versuchsaufbau angepasst werden. Es sollte jedoch darauf geachtet werden, dass durchschnittlich mehrere Kopien (>10) jedes Enzyms pro Vesikel vorhanden sind, um unerwünschte stochastische Effekte zu vermeiden. Konzentrationen > 1 μM sind im Allgemeinen sicher; 1 μM in einem Vesikel mit einem Durchmesser von 400 nm entspricht etwa 20 Kopien (Abbildung 3). - Pipettieren Sie Puffer A in ein leeres 1,5-ml-Röhrchen und fügen Sie DTT, Na-ADP, MgCl2, L-Ornithin (und Pyranin, falls für interne pH-Messungen erforderlich) hinzu. Als nächstes die Enzyme hinzufügen und vorsichtig mischen. Geben Sie die Lösung auf die vorgeformten Proteoliposomen und wirbeln Sie sie kurz bei niedriger Geschwindigkeit durch.
HINWEIS: Es ist wichtig, Na-ADP vor MgCl2 hinzuzufügen, um die unerwünschte Bildung von Magnesiumphosphatausfällungen zu vermeiden. Für das richtige Mischen der viskosen Lösung ist ein Vortexen erforderlich. Minimieren Sie jedoch die Dauer und Geschwindigkeit, um mechanische Schäden an den Proteinen zu vermeiden. PercevalHR und Pyranin können nicht gemeinsam verkapselt werden, da sich ihre Spektren überlappen.
- Für eine Standardverkapselung verwenden Sie 66,6 μl Proteoliposomen in einem Endvolumen von 200 μl (33,33 mg/ml Gesamtlipide). Berechnen Sie das Volumen jeder Komponente (Enzym, Cofaktor), das benötigt wird, um die gewünschte Konzentration zu erreichen, fügen Sie diese Komponenten zu den Proteoliposomen hinzu und stellen Sie das Volumen mit Puffer A auf 200 μl ein (Tabelle 2).
- Bestimmung der internen Osmolalität
- Bereiten Sie eine 50-μl-Lösung wie in Schritt 3.1.1.1 beschrieben, jedoch ohne Proteoliposomen, vor und messen Sie die Osmolalität mit einem Gefrierpunktosmometer.
- Erstellen Sie eine Kalibrierkurve mit Puffer (50 mM KPi, pH 7,0) und unterschiedlichen Konzentrationen von Salz (z. B. NaCl oder NaCl) oder Zucker. Bestimmen Sie die Osmolytkonzentration, die der internen Osmolalität (Puffer N) entspricht.
HINWEIS: Membranpermeable Komponenten (z. B. Glycerin) können nicht verwendet werden, um die interne Osmolalität anzupassen. Für Proteine, die in glycerinhaltigem Puffer (z. B. Puffer E) solubilisiert sind, sollte derselbe Puffer ohne Glycerin verwendet werden. Der für die Herstellung eines isoosmotischen externen Puffers gewählte Osmolyt sollte membranundurchlässig sein und nicht in das metabolische Netzwerk eingreifen.
- Frost-Auftauen
- Die Proteoliposomen werden zusammen mit den löslichen Bestandteilen schockgefroren (in flüssigem Stickstoff) und in einem Wasser-Eis-Bad bei ca. 10 °C aufgetaut.
- Wiederholen Sie Schritt 3.1.3.1 für insgesamt 5x.
STOPPPUNKT: Hier kann der Vorgang gestoppt werden. Überspringen Sie den letzten Auftauschritt und lagern Sie die gefrorene Probe 1-3 Tage lang in flüssigem Stickstoff. Achten Sie darauf, die Tubendeckel 2x mit einer Nadel zu durchstechen, um eine Explosion der Tube beim schnellen Sieden des flüssigen Stickstoffs zu vermeiden. Die Lagerung in flüssigem Stickstoff wird über -80 °C bevorzugt, um die Lipidoxidation zu minimieren.
- Mischen von Komponenten
- Tag 2
- Extrusion
HINWEIS: Alle Extrusionsschritte werden bei Raumtemperatur durchgeführt, da sonst die gasdichten Spritzen undicht werden.- Bereiten Sie einen Extruder vor, indem Sie einen Filter Ihrer Wahl anwenden (z. B. Polycarbonat, Porendurchmesser von 400 nm). Waschen Sie den Extruder mit einer Lösung, die den gleichen Puffer und die gleichen Metaboliten enthält, die für die Verkapselung der Vesikel verwendet wurden (z. B. Puffer O plus 0,1 mM Pyranin).
HINWEIS: Verwenden Sie einen speziellen Extruder für die Beladung von Proteoliposomen mit Pyranin, da der Farbstoff an der Extruderoberfläche haftet und in späteren Proben zu einer Kontamination führen kann. - Laden Sie das Verkapselungsgemisch in den Extruder und leiten Sie es 13x durch den Filter. Sammeln Sie die extrudierte Lösung in einem 1,5-ml-Röhrchen.
- Bereiten Sie einen Extruder vor, indem Sie einen Filter Ihrer Wahl anwenden (z. B. Polycarbonat, Porendurchmesser von 400 nm). Waschen Sie den Extruder mit einer Lösung, die den gleichen Puffer und die gleichen Metaboliten enthält, die für die Verkapselung der Vesikel verwendet wurden (z. B. Puffer O plus 0,1 mM Pyranin).
- Größenausschlusschromatographie (optional)
HINWEIS: Dieser Schritt wird durchgeführt, um externe Moleküle wie Farbstoffe wie Pyranin durch Größenausschlusschromatographie zu entfernen. Wenn keine Farbstoffe im System vorhanden sind und andere Komponenten bei niedrigen Konzentrationen nicht stören (beachten Sie, dass die Vesikel anschließend auch durch Ultrazentrifugation gewaschen werden), dann Schritt 3.2.2. kann übersprungen werden.- Rehydrieren Sie das Harz Sephadex G-75 und gießen Sie es in eine Glassäule (22 cm lang, 1,5 cm breit). Äquilibrieren Sie das Harz mit einem Überschuss an externem Puffer (z. B. Puffer N).
- Die extrudierten Proteoliposomen aus Schritt 3.2.1.2 werden auf die präequilibrierte Größenausschlusschromatographiesäule geladen und eine Schwerkraftströmung aus externem Puffer angelegt. Entsorgen Sie das Hohlraumvolumen (ca. 7 ml); Sammeln Sie dann zehn 1-ml-Aliquots. Visualisieren Sie die Proteoliposomen-haltigen Aliquots durch kurze Einwirkung einer UV-Lampe. Bündeln Sie die Fraktionen, die die meisten Proteoliposomen (2-4 mL) enthalten.
- Waschen und Resuspension
- Waschen Sie die extrudierten Proteoliposomen durch Ultrazentrifugation. Füllen Sie ein 6,5-ml-Ultrazentrifugationsröhrchen mit 5,8 mL Puffer N und tragen Sie die extrudierte Probe darauf auf. Wenn eine Größenausschlusschromatographie durchgeführt wurde, füllen Sie das Röhrchen mit den gepoolten Elutionsproben (2-4 mL) und fügen Sie Puffer N zu einem Endvolumen von 6 mL hinzu.
- Zentrifugieren bei 337.000 × g, 30 min, 4 °C. Entsorgen Sie den Überstand und trocknen Sie das Ultrazentrifugationsröhrchen gründlich mit einem staubfreien Tuch ab, wobei Sie darauf achten, das Pellet nicht zu berühren. Resuspendieren Sie das Pellet in einem kleinen Volumen Puffer N (200 μl). Wenn das Pellet vollständig resuspendiert ist, füllen Sie das Röhrchen bis zu 6 mL mit Puffer N.
HINWEIS: Das Wiederaufhängen des Pellets nimmt einige Zeit in Anspruch und sollte sorgfältig durchgeführt werden. - Die Schritte 3.2.3.1 bis 3.2.3.2 2x für insgesamt drei Wäschen wiederholen, es sei denn, es wird eine Größenausschlusschromatographie durchgeführt (in diesem Fall genügt ein Zentrifugationsschritt). Letztendlich resuspendieren Sie das Pellet auf eine gewünschte Konzentration (z. B. 5,55 mg/ml Gesamtlipid), indem Sie ein angemessenes Volumen Puffer N hinzufügen.
STOPPPUNKT: Die Proteoliposomen können sofort verwendet oder mindestens 48 h bei 4 °C gelagert werden. Die Größe der Ultrazentrifugationsröhrchen sollte auf der Grundlage der Probengröße gewählt werden. Für ein Pellet mit 6,66 mg Gesamtlipiden ist ein 6,5-ml-Röhrchen geeignet. Das Waschen von 200 μl Proteoliposomen (insgesamt 33,33 mg Lipide/ml; Trockenpelletvolumen ~40 μl)28 für 3 x 6 mL Puffer verdünnt die externen Komponenten für jeden Waschschritt um den Faktor 100. Wenn Tuben unterschiedlicher Größe verwendet werden, ist es wünschenswert, die Anzahl der Wäschen entsprechend anzupassen.
- Extrusion
- Tag 3
- Nachweis der ATP-Synthese durch Fluoreszenz
- Mischen Sie die Reaktionskomponenten zu einem Endvolumen von 120 μl (Tabelle 3) in einer schwarzen Quarzküvette mit einem Fenster von 3 x 5 mm und einem minimalen Innenvolumen von 100 μl.
HINWEIS: Ionophore können hinzugefügt werden, um elektrochemische Ionengradienten zu dissipieren. Ein Gemisch aus Valinomycin und Nigericin (je 1 μM) löst effektiv alle Protonen- und Kaliumgradienten auf. - Die Probe wird in einem auf 30 °C eingestellten Fluorometer vorgewärmt und Anregungsspektren von PercevalHR aufgenommen (Anregung 400-520 nm, Bandbreite 5 nm; Emission 550 nm, Bandbreite 5 nm). Sobald das Sondensignal konstant ist, wird das metabolische Netzwerk durch Zugabe eines Überschusses an L-Arginin (5-10 mM) und Glycerin (400 μM) gestartet, wenn das Recycling von ATP an die Synthese von Glycerin-3-phosphat gekoppelt ist. Verfolgen Sie die Reaktion im Laufe der Zeit.
- Mischen Sie die Reaktionskomponenten zu einem Endvolumen von 120 μl (Tabelle 3) in einer schwarzen Quarzküvette mit einem Fenster von 3 x 5 mm und einem minimalen Innenvolumen von 100 μl.
- Datenanalyse
- Plotten Sie das Verhältnis F500/F430 als Funktion der Zeit, das ein qualitativer Indikator für das ATP/ADP-Verhältnis ist. Für eine quantitativere Beurteilung der ATP-Bildung kann eine Kalibrierungskurve in Proteoliposomen erstellt werden oder ein komplementärer Ansatz verwendet werden (z. B. ATP-Quantifizierung durch Chemilumineszenz28).
- Nachweis der ATP-Synthese durch Fluoreszenz
| Bestandteil | Endkonzentration |
| Puffer A | 50 mM KPi pH 7,0 |
| DVB-T | 2 mM |
| Na-ADP | ca. 10 mM |
| MgCl2 | ca. 10 mM |
| L-Ornithin | 0,5 Mio. m |
| Fluoreszierende Sonde (PercevalHR oder Pyranin) | 5,8 μM bzw. 0,1 mM |
| ArcA (Arginin Deiminase) | 1 μM |
| ArcB (Ornithin-Carbamoyltransferase) | 2 μM |
| ArcC1 (Carbamat-Kinase) | 5 μM |
| GlpK (Glycerinkinase) | 1,6 μM |
| Proteoliposomen | 33,33 mg/ml Gesamtlipide |
Tabelle 2: Verkapselungskomponenten. Die Komponenten werden in der Reihenfolge ihrer Addition aufgelistet. Lösliche Proteine befinden sich in Puffer E; alle anderen Komponenten (außer MgCl2 in deionisiertem Wasser) befinden sich in Puffer A.
| Bestandteil | Endkonzentration |
| Puffer K | 50 mM KPi pH 7,0, 58 mM NaCl, 2 mM DTT, pH 7,0 |
| Proteoliposomen (5,55 mg/ml Lipide) | 2,7 mg/ml Lipide |
| Ionophore (Valinomycin, Nigericin) | je 1 μM |
Tabelle 3: Experimentelle Bedingungen. Die Komponenten werden in der Reihenfolge ihrer Addition aufgelistet. Proteoliposomen befinden sich in Puffer N, Ionophore in DMSO oder EtOH.
4. Upscaling eines metabolischen Netzwerks für mikrometergroße Vesikel
- Tag 1
- Mikrofluidische Chipvorbereitung
HINWEIS: In diesem Experiment wird ein mikrofluidisches Gerät verwendet, das von Robinson et al.34 entwickelt wurde. Andere Designs von mikrofluidischen Chips sind verfügbar 35,36,37 und können in diesem Protokoll leicht implementiert werden.- Schneiden Sie eine 200-μl-Pipettenspitze ein Drittel von der Unterseite ab und führen Sie den unteren Teil der Spitze in eine vorgefertigte mikrofluidische Auffangvorrichtung ein.
- In das Einlassreservoir des Chips werden 400 μl β-Kaseinlösung (2 mg/ml; siehe Schritt 1.5) gegeben, wobei darauf zu achten ist, dass bei diesem Schritt keine Luft eingebracht wird. Stellen Sie einen 96-Well-Platteneimer auf und geben Sie ein Laborgewebe in eine konische Tischröhrchenzentrifuge. Legen Sie den Chip auf das Gewebe und zentrifugieren Sie ihn 6 Minuten lang bei 900 × g , um die Passivierung des mikrofluidischen Chips zu ermöglichen. Nach dem Zentrifugationsschritt sollte der Flüssigkeitsstand am Einlassbehälter und an der Auslassspitze des mikrofluidischen Chips gleich sein. Überprüfen Sie, ob der Chip undicht ist. Inkubieren Sie die β-Kasein-Lösung mindestens 30 Minuten lang im Mikrofluidik-Chip.
- Entfernen Sie den größten Teil der Passivierungslösung, ohne Luft hinzuzufügen, und fügen Sie 400 μl Waschpuffer (Puffer P) hinzu. Legen Sie den Chip auf den 96-Well-Platteneimer und zentrifugieren Sie ihn 6 Minuten lang bei 900 × g . Lassen Sie den Chip bis zum Gebrauch in der Waschlösung (maximal 4 Stunden lang).
- Mikrofluidische Chipvorbereitung
- Gelpräparation für die Herstellung von Proteo-GUVs
HINWEIS: Die folgende Beschreibung wurde von25,38 übernommen und angepasst.- 0,5 % (w/w) einer Agarose mit niedriger Geliertemperatur (LGT) werden in deionisiertem Wasser gelöst, indem die Lösung in der Mikrowelle erhitzt wird; Stellen Sie sicher, dass sich die Agarose vollständig aufgelöst hat und vermeiden Sie es, die Lösung zu kochen. Bewahren Sie die Agarose bis zur weiteren Verwendung bei 50 °C auf.
HINWEIS: Gelöste Agarose kann mehrere Wochen bei Raumtemperatur gelagert werden. Um die Agarose wiederzuverwenden, schmelzen Sie das Gel einfach in der Mikrowelle. - Nehmen Sie zwei Objektivfolien und zeichnen Sie den Umriss des Abstandshalters auf die Folien. Machen Sie die Objektträger hydrophil durch Plasmareinigung mit Plasma mit hohem Sauerstoffgehalt für 1 min.
- Geben Sie LGT Agarose auf den Objektträger, bis er vollständig mit Agarose (~500 μL) bedeckt ist; Kippen Sie dann den Objektträger in einem Winkel von 90° und lassen Sie überschüssige Agarose auf ein Taschentuch abtropfen. Lassen Sie die Objektträger 30 min bei 50 °C ruhen.
- In flüssigem Stickstoff gelagerte Proteoliposomen (Abschnitt 3) entnehmen und auf Eis auftauen. Verdünnen Sie die Vesikel mit Puffer Q auf 5 mg/ml Lipide.
HINWEIS: Die in Abschnitt 3 hergestellten Proteoliposomen enthalten keine löslichen Proteine und können weit im Voraus hergestellt werden, wenn sie in flüssigem Stickstoff gelagert werden. Achten Sie bei der Arbeit mit proteoGUVs darauf, die Arbeitslösungen immer zu filtern, um ein Verstopfen des mikrofluidischen Geräts zu vermeiden. - Beschallung der Proteo-Liposomen mit einem tragbaren Sonden-Ultraschallgerät mit einer 1-mm-Sonde. Beschallung für 10 Zyklen von 0,5 s an und 0,5 s aus bei 70 % Amplitude. Halten Sie die Vesikel, im Folgenden als ProteoSUVs bezeichnet, 30 s lang auf Eis und wiederholen Sie den Ultraschallprozess 5x.
- Füllen Sie eine 100-μl-Spritze (in einem tragbaren LCP-Dispenser) mit der proteo-SUV-Suspension. 0,5 μl Tröpfchen von Proteo-SUVs auf das zuvor vorbereitete Agarosegel ablegen; Achten Sie darauf, die Agaroseschicht nicht zu stören und genügend Abstand zu halten, um ein Verschmelzen von Tröpfchen zu verhindern.
HINWEIS Die Verwendung eines tragbaren LCP-Dispensers ermöglicht eine reproduzierbare Tröpfchenabscheidung, aber es können auch alternative Pipettiersysteme verwendet werden. Die Schmierblutung mit einer Glaskapillare ist weniger geeignet, da sie das getrocknete Agarosegel stört. - Trocknen Sie die SUV-Tröpfchen in ~10 Minuten mit einem Stickstoffstrom anstelle von Druckluft, um die Möglichkeit einer Oxidation der Lipide zu verringern.
- 1 ml eines 1,25x konzentrierten Puffers O (Tabelle 4) in Puffer A vorbereiten und durch einen 0,2 μm Celluloseacetatfilter passieren. Nehmen Sie 800 μl der 1,25-fach konzentrierten Lösung und fügen Sie die löslichen Enzyme und Sonden bis zu einer in Tabelle 4 angegebenen Konzentration hinzu. Verwenden Sie gefiltertes deionisiertes Wasser, um das Endvolumen auf 1 ml zu erhöhen.
- Bereiten Sie 100 μl Quelllösung vor, die alle Bestandteile der Tabelle 4 mit Ausnahme von Proteinen und Glycerin enthält. Messen Sie die Osmolalität der Quelllösung mit einem kalibrierten 3-Punkt-Gefrierpunktosmometer.
HINWEIS: Bereiten Sie 100 μl Quelllösung ohne Protein und Glycerin vor, um die Osmolalität dieser Lösung genau zu bestimmen. Glycerin, das in der Proteinlösung vorhanden ist, beeinflusst die Osmolalität, aber in GUVs diffundiert Glycerin schnell durch die Membran und führt nur zu vorübergehenden osmotischen Unterschieden. - Bauen Sie die GUV-Quellkammer zusammen, indem Sie ein Sandwich aus zwei Objektivgläsern mit dem Gel und getrockneten SUVs mit einem 1,5 oder 3,0 mm dicken Teflon-Abstandshalter dazwischen herstellen. Geben Sie dann die Quelllösung mit einer Spritze und einer Nadel durch das kleine Loch in der Seite in die Kammer.
HINWEIS: Das Volumen der Quelllösung kann durch Variieren der Abstandshalter von 1,5 bis 3,0 mm eingestellt werden.
- 0,5 % (w/w) einer Agarose mit niedriger Geliertemperatur (LGT) werden in deionisiertem Wasser gelöst, indem die Lösung in der Mikrowelle erhitzt wird; Stellen Sie sicher, dass sich die Agarose vollständig aufgelöst hat und vermeiden Sie es, die Lösung zu kochen. Bewahren Sie die Agarose bis zur weiteren Verwendung bei 50 °C auf.
- Aufblähung von Vesikeln und Entnahme von GUVs
- Lassen Sie das Aufquellen der Vesikel zu, indem Sie die Kammer mindestens 30 Minuten lang auf 22 °C stellen.
HINWEIS: Die Schwellung der Vesikel kann durch Lichtmikroskopie (z. B. ein Tisch-Phasenkontrastmikroskop oder ein Weitfeld-Fluoreszenzmikroskop) verfolgt werden, wenn die Proteine oder Lipide fluoreszenzmarkiert sind. - Ernten Sie die GUVs aus dem Gel, indem Sie die Kammer sanft physikalisch bewegen, indem Sie die Kammer auf eine feste Oberfläche (z. B. Labortisch) klopfen; Nehmen Sie ein Drittel des Volumens heraus und verwenden Sie die resultierende Luftblase, um die verbleibende Flüssigkeit sanft in Bewegung zu bringen, wodurch sich die GUVs vom Gel lösen.
- Während die GUVs quellen, bereiten Sie den Puffer P vor und passen Sie die Osmolalität an die Quelllösung an, indem Sie die Glukosekonzentration anpassen. Den Puffer durch einen 0,2 μm Spritzenvorsatzfilter filtrieren.
HINWEIS: Im Allgemeinen sollte die Wasch- und Substratlösung innerhalb von ± 5 mosmol/kg gehalten werden. Jede Lösung, die durch den Chip fließt, sollte sehr sauber sein, da Verunreinigungen die Kanäle verstopfen können. Stellen Sie jedes Mal frische Puffer her oder lagern Sie die vorbereiteten Puffer bei -20 °C und filtern Sie sie vor der Verwendung.
- Lassen Sie das Aufquellen der Vesikel zu, indem Sie die Kammer mindestens 30 Minuten lang auf 22 °C stellen.
- Einfangen der GUVs für Mikroskopie-Experimente
- Montieren Sie den passivierten Mikrofluidik-Chip auf dem Probentisch des Mikroskops. Auf mögliche Defekte prüfen (z. B. Undichtigkeiten, Lufteingeschlossenheit, verstopfte Kanäle).
HINWEIS: Die Überprüfung des Chips kann lange vor der Verwendung erfolgen, damit bei Bedarf ein neuer Chip vorbereitet werden kann. - Verbinden Sie den Schlauch mit einer gebogenen Nadel mit dem Auslass des Reservoirs und verbinden Sie das andere Ende mit einer 1-ml-Spritze. Montieren Sie die Spritze an einer Pumpe und stellen Sie die Durchflussrate auf maximal 10 μl/min ein.
- Den Waschpuffer aus dem Behälter nehmen (Schritt 4.1.1.3) und durch frisches Medium (Puffer P) ersetzen. Starten Sie den Pufferfluss durch den Chip, indem Sie über die Spritze bei 1-10 μl/min infundieren. Mit mindestens 80 μl osmotisch ausgeglichenem Waschpuffer (Puffer P) waschen.
- Entfernen Sie überschüssigen Waschpuffer aus dem Reservoir und geben Sie die (Proteo)GUVs in das Reservoir. Stellen Sie die Durchflussrate auf 0,1 bis 1 μl/min ein, damit die GUVs durch den Chip fließen können. Überwachen Sie den Chip im Laufe der Zeit, bis genügend GUVs im Chip eingeschlossen sind.
HINWEIS: Vesikel mit relativ großen Mengen (>20 mol%) an geladenen Lipiden wie Phosphatidylglycerin (PG), Phosphatidylserin (PS) oder Phosphatidsäure (PA) oder nicht doppelschichtbildenden Lipiden wie Phosphatidylethanolamin (PE) werden mit einer geringeren Flussrate durch den Chip eingeführt, um ein Platzen zu verhindern. Vesikel, die aus reinem Phosphatidylcholin (PC) bestehen, sind tendenziell stabiler. - Entfernen Sie überschüssige GUV-Lösung und fügen Sie den Waschpuffer P mit einem konstanten Fluss von 0,1-1 μl/min hinzu, um das externe Medium auszutauschen, und waschen Sie die eingeschlossenen GUVs mindestens 1 h lang, um nicht verkapselte Verbindungen zu entfernen und die Hintergrundfluoreszenz zu verringern, wenn ein Fluorophor verkapselt ist. Überwachen Sie die Hintergrundfluoreszenz im Laufe der Zeit; Wenn die Hintergrundfluoreszenz nicht abnimmt, kann der Chip blockiert sein.
- Lokalisieren Sie Fallen mit ausreichenden Mengen an Vesikeln und speichern Sie deren Positionen. Wenden Sie die Einstellungen am Mikroskop an (z. B. Laserintensität, Verstärkung, Wellenlänge) und starten Sie ein Zeitreihenexperiment.
- Geben Sie osmotisch ausgeglichene Substratlösung (Puffer R) in das Reservoir und starten Sie eine Durchflussrate von 0,5 μL/min.
- Montieren Sie den passivierten Mikrofluidik-Chip auf dem Probentisch des Mikroskops. Auf mögliche Defekte prüfen (z. B. Undichtigkeiten, Lufteingeschlossenheit, verstopfte Kanäle).
| Komponenten von Buffer L | ||
| Bestandteil | 1,25 x Konzentration | Arbeitskonzentration |
| Puffer A | 62,5 mM KPi pH 7,0 | 50 mM KPi pH 7,0 |
| Saccharose | 125 mM | ca. 100 mM |
| DVB-T | 2,5 Mio. M. | 2 mM |
| Na-ADP | 12,5 Mio. M. | ca. 10 mM |
| MgCl2 | 12,5 Mio. M. | ca. 10 mM |
| L-Ornithin | 0,625 mM | 0,5 Mio. m |
| Komponenten für die Verkapselung | ||
| Pyranin oder PercevalHR | 1 mM oder 20 μM | |
| ArcA (Arginin Deiminase) | 1 μM | |
| ArcB (Ornithin-Carbamoyltransferase) | 2 μM | |
| ArcC1 (Carbamat-Kinase) | 5 μM | |
Tabelle 4: Puffer O und Verkapselungskomponenten. Die Komponenten werden in der Reihenfolge ihrer Addition aufgelistet. Alle Komponenten (mit Ausnahme von MgCl2 in deionisiertem Wasser) befinden sich in Puffer A. Die Verkapselungskomponenten sind in der Reihenfolge ihrer Zugabe aufgeführt. Alle wasserlöslichen Proteine befinden sich in Puffer E.
Ergebnisse
Die Rekonstitution von solubilisierten Membranproteinen in Liposomen erfordert die Destabilisierung von vorgeformten Vesikel. Die Zugabe geringer Mengen an Triton X-100 führt zunächst zu einer Erhöhung der Absorption bei 540 nm (A540) aufgrund einer Zunahme der Lichtstreuung durch die Schwellung der Vesikel (Abbildung 4). Der maximaleA-540-Wert ist der Punkt, an dem die Liposomen mit Detergens (Rsat) gesättigt sind, wonach jede weitere Zugabe von...
Diskussion
Wir stellen ein Protokoll für die Synthese von (Membran-)Proteinen vor, die submikrometergroße Lipidvesikel (proteoLUVs) enthalten, und die Umwandlung von proteoLUVs in riesen-unilamelläre Vesikel (proteoGUVs). Das Protokoll sollte anwendbar sein für die Rekonstitution anderer Membranproteine13, 19, 30, 40 und die Verkapselung anderer Stoffwechselnetzwerke als der hier vorgestellten L-Argin...
Offenlegungen
Die Autoren erklären, dass keine konkurrierenden finanziellen Interessen bestehen.
Danksagungen
Die Autoren danken Aditya Iyer für die Klonierung des pBAD-PercevalHR-Gens und Gea Schuurman-Wolters für die Unterstützung bei der Proteinproduktion und -reinigung. Die Forschung wurde durch das NWO-Gravitationsprogramm "Building a Synthetic Cell" (BaSyC) gefördert.
Materialien
| Name | Company | Catalog Number | Comments |
| Agarose | Sigma Aldrich | A9414-25g | |
| Amicon cut-off filter | Sigma Aldrich | Milipore centrifugal filter units Amicon Ultra | |
| BioBeads | BioRad | 152-3920 | |
| CHCl3 | Macron Fine Chemicals | MFCD00000826 | |
| D(+)-Glucose | Formedium | - | |
| D(+)-Sucrose | Formedium | - | |
| DDM | Glycon | D97002 -C | |
| Diethyl Ether | Biosolve | 52805 | |
| DMSO | Sigma-Aldrich | 276855-100ml | |
| DOPC | Avanti | 850375P-1g | |
| DOPE | Avanti | 850725P-1g | |
| DOPG | Avanti | 840475P-1g | |
| DTT | Formedium | DTT005 | |
| EtOH | J.T.Baker Avantor | MFCD00003568 | |
| Extruder | Avestin Inc | LF-1 | |
| Fluorimeter | Jasco | Spectrofluorometer FP-8300 | |
| Glycerol | BOOM | 51171608 | |
| Gravity flow column | Bio-Rad | 732-1010 | |
| Hamilton syringe 100 µL | Hamilton | 7656-01 | |
| Hamilton syringe 1000 µL | Hamilton | 81320 | |
| Handheld LCP dispenser | Art Robbins Instruments | 620-411-00 | |
| Handheld Sonicator | Hielscher Ultrasound Technology | UP50H | |
| HCl | BOOM | x76021889.1000 | |
| Imidazole | Roth | X998.4-250g | |
| K2HPO4 | Supelco | 1.05099.1000 | |
| KCl | BOOM | 76028270.1 | |
| KH2PO4 | Supelco | 1.04873.1000 | |
| Kimwipe | Kimtech Science | 7552 | |
| Large Falcon tube centrifuge | Eppendorf | Centrifuge 5810 R | |
| L-Arginine | Sigma-Aldrich | A5006-100G | |
| Light microscope | Leica | DM LS2 | |
| L-Ornithine | Roth | T204.1 | |
| LSM Laser Scanning Confocal Microscope | Zeiss | LSM 710 ConfoCor 3 | |
| MgCl2 | Sigma-Aldrich | M2670-1KG | |
| Microfluidic chip | Homemade | PDMS based | DOI: https://doi.org/10.1039/C8LC01275J |
| Na-ADP | Sigma-Aldrich | A2754-1G | |
| NaCl | Supelco | 1.06404.1000 | |
| Nanodrop Spectrometer | Isogen Life Science | ND-1000 spectrophotometer NanoDrop | |
| NaOH | Supelco | 1.06498.1000 | |
| Needles for GUVs | Henke-Ject | 14-14575 | 27 G x 3/4'' 0.4 x 20 mm |
| Needles for microfluidics | Henke-Ject | 14-15538 | 18 G x 1 1/2'' 1.2 x 40 mm |
| Ni2+ Sepharose | Cytiva | 17526802 | |
| Nigericin | Sigma-Aldrich | N7143-5MG | |
| Nutator | VWR | 83007-210 | |
| Osmolality meter | Gonotec Salmenkipp | Osmomat 3000 basic freezing point osmometer | |
| Plasmacleaner | Plasma Etch | PE-Avenger | |
| Polycarbonate filter | Cytiva Whatman | Nuclepor Track-Etch Membrane Product: 10417104 | 0.4 µm |
| Polycarbonate ultracentrifuge tube | Beckman Coulter | 355647 | |
| Pyranine | Acros Organics | H1529-1G | |
| Quartz cuvette (black) | Hellma Analytics | 108B-10-40 | |
| Sephadex G-75 resin | GE Healthcare | 17-0050-01 | |
| Sonicator | Sonics Sonics & Materials INC | Sonics vibra cell | |
| Syringe filter | Sarstedt | Filtropur S plus 0.2 | 0.2 µm |
| Syringe pump | Harvard Apparatus | A-42467 | |
| Tabletop centrifuge | Eppendorf | centrifuge 5418 | |
| Teflon spacer | Homemade | Teflon based | 45 x 26 x 1.5 or 45 x 26 x 3 or 20 x 20 x 3 mm |
| Tris | PanReac AppliChem | A1086.1000 | |
| Triton X-100 | Sigma Aldrich | T8787-100 ml | |
| Ultracentrifuge | Beckman Coulter | Optima Max-E | |
| UV lamp | Spectroline | ENB-280C/FE | |
| UV/VIS Spectrometer | Jasco | V730 spectrophotometer | |
| Valinomycin | Sigma-Aldrich | V0627-10MG | |
| Widefield fluorescence microscope | Zeiss | AxioObserver | |
| β-Casein | Sigma Aldrich | C5890-500g |
Referenzen
- Hirschi, S., Ward, T. R., Meier, W. P., Müller, D. J., Fotiadis, D. Synthetic biology: bottom-up assembly of molecular systems. Chem Rev. 122 (21), 16294-16328 (2022).
- Ivanov, I., et al. Bottom-up synthesis of artificial cells: recent highlights and future challenges. Annu Rev Chem Biomol. Eng. 12 (1), 287-308 (2021).
- Clomburg, J. M., Crumbley, A. M., Gonzalez, R. Industrial biomanufacturing: The future of chemical production. Science. 355 (6320), (2017).
- Shi, T., Han, P., You, C., Zhang, Y. -. H. P. J. An in vitro synthetic biology platform for emerging industrial biomanufacturing: Bottom-up pathway design. Synth Syst Biotechnol. 3 (3), 186-195 (2018).
- Wang, A., et al. Liver-target and glucose-responsive polymersomes toward mimicking endogenous insulin secretion with improved hepatic glucose utilization. Adv Funct Mater. 30 (13), 1910168 (2020).
- Kanter, G., et al. Cell-free production of scFv fusion proteins: an efficient approach for personalized lymphoma vaccines. Blood. 109 (8), 3393-3399 (2007).
- Zeltins, A. Construction and characterization of virus-like particles: a review. Mol Biotechnol. 53 (1), 92-107 (2013).
- Jain, K. K. Synthetic biology and personalized medicine. Med Princ Pract. 22 (3), 209-219 (2013).
- Schwille, P., Frohn, B. P. Hidden protein functions and what they may teach us. Trends Cell Biol. 32 (2), 102-109 (2022).
- Sachsenmeier, P. Industry 5.0-The relevance and implications of bionics and synthetic biology. Engineering. 2 (2), 225-229 (2016).
- Schmidt, D., Jiang, Q. -. X., MacKinnon, R. Phospholipids and the origin of cationic gating charges in voltage sensors. Nature. 444 (7120), 775-779 (2006).
- Godoy-Hernandez, A., et al. Rapid and highly stable membrane reconstitution by LAiR enables the study of physiological integral membrane protein functions. ACS Cent Sci. 9 (3), 494-507 (2023).
- Sikkema, H. R., et al. Gating by ionic strength and safety check by cyclic-di-AMP in the ABC transporter OpuA. Sci Adv. 6 (47), 7697 (2020).
- Foucaud, C., Poolman, B. Lactose transport system of Streptococcus thermophilus. Functional reconstitution of the protein and characterization of the kinetic mechanism of transport. J Biol Chem. 267 (31), 22087-22094 (1992).
- Yoneda, J. S., Sebinelli, H. G., Itri, R., Ciancaglini, P. Overview on solubilization and lipid reconstitution of Na,K-ATPase: enzyme kinetic and biophysical characterization. Biophys Rev. 12 (1), 49-64 (2020).
- Simidjiev, I., et al. Self-assembly of large, ordered lamellae from non-bilayer lipids and integral membrane proteins in vitro. Proc Natl Acad Sci. 97 (4), 1473-1476 (2000).
- Harris, N. J., Booth, P. J. Folding and stability of membrane transport proteins in vitro. Biochim Biophys Acta BBA - Biomembr. 1818 (4), 1055-1066 (2012).
- Jackson, M. L., Litman, B. J. Rhodopsin-egg phosphatidylcholine reconstitution by an octyl glucoside dilution procedure. Biochim Biophys Acta BBA - Biomembr. 812 (2), 369-376 (1985).
- Geertsma, E. R., Nik Mahmood, N. A. B., Schuurman-Wolters, G. K., Poolman, B. Membrane reconstitution of ABC transporters and assays of translocator function. Nat Protoc. 3 (2), 256-266 (2008).
- Rigaud, J. -. L., Pitard, B., Levy, D. Reconstitution of membrane proteins into liposomes: application to energy-transducing membrane proteins. Biochim Biophys Acta BBA - Bioenerg. 1231 (3), 223-246 (1995).
- Szoka, F., Papahadjopoulos, D. Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation. Proc Natl Acad Sci. 75 (9), 4194-4198 (1978).
- . Synthetic Organelles for Energy Conservation and Delivery of Building Blocks for Lipid Biosynthesis Available from: https://www.researchsquare.com/article/rs-3385355/v1 (2023)
- Lee, K. Y., et al. Photosynthetic artificial organelles sustain and control ATP-dependent reactions in a protocellular system. Nat Biotechnol. 36 (6), 530-535 (2018).
- Méléard, P., Bagatolli, L. A., Pott, T. Giant unilamellar vesicle electroformation. Methods in Enzymology. , 161-176 (2009).
- Garten, M., Aimon, S., Bassereau, P., Toombes, G. E. S. Reconstitution of a transmembrane protein, the voltage-gated ion channel, KvAP, into giant unilamellar vesicles for microscopy and patch clamp studies. J. Vis. Exp. (95), e52281 (2015).
- Doeven, M. K., et al. lateral mobility and function of membrane proteins incorporated into giant unilamellar vesicles. Biophys J. 88 (2), 1134-1142 (2005).
- Pols, T., et al. A synthetic metabolic network for physicochemical homeostasis. Nat Commun. 10 (1), 4239 (2019).
- Bailoni, E., Poolman, B. ATP recycling fuels sustainable glycerol 3-phosphate formation in synthetic cells fed by dynamic dialysis. ACS Synth Biol. 11 (7), 2348-2360 (2022).
- Van Der Heide, T. On the osmotic signal and osmosensing mechanism of an ABC transport system for glycine betaine. EMBO J. 20 (24), 7022-7032 (2001).
- Van'T Klooster, J. S., et al. Membrane lipid requirements of the lysine transporter Lyp1 from Saccharomyces cerevisiae. J Mol Biol. 432 (14), 4023-4031 (2020).
- Lou, G., Anderluzzi, G., Woods, S., Roberts, C. W., Perrie, Y. A novel microfluidic-based approach to formulate size-tuneable large unilamellar cationic liposomes: Formulation, cellular uptake and biodistribution investigations. Eur J Pharm Biopharm. 143, 51-60 (2019).
- Weiss, M., et al. Sequential bottom-up assembly of mechanically stabilized synthetic cells by microfluidics. Nat Mater. 17 (1), 89-96 (2018).
- Pols, T., Singh, S., Deelman-Driessen, C., Gaastra, B. F., Poolman, B. Enzymology of the pathway for ATP production by arginine breakdown. FEBS J. 288 (1), 293-309 (2021).
- Yandrapalli, N., Robinson, T. Ultra-high capacity microfluidic trapping of giant vesicles for high-throughput membrane studies. Lab Chip. 19 (4), 626-633 (2019).
- Elias, M., et al. Microfluidic characterization of biomimetic membrane mechanics with an on-chip micropipette. Micro Nano Eng. 8, 100064 (2020).
- Robinson, T., Kuhn, P., Eyer, K., Dittrich, P. S. Microfluidic trapping of giant unilamellar vesicles to study transport through a membrane pore. Biomicrofluidics. 7 (4), 044105 (2013).
- Cooper, A., Girish, V., Subramaniam, A. B. Osmotic Pressure Enables High-Yield Assembly of Giant Vesicles in Solutions of Physiological Ionic Strengths. Langmuir. 39 (15), 5579-5590 (2023).
- Tantama, M., Martínez-François, J. R., Mongeon, R., Yellen, G. Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nat Commun. 4 (1), 2550 (2013).
- Setyawati, I., et al. In vitro reconstitution of dynamically interacting integral membrane subunits of energy-coupling factor transporters. eLife. 9, e64389 (2020).
- Oropeza-Guzman, E., Ríos-Ramírez, M., Ruiz-Suárez, J. C. Leveraging the coffee ring effect for a defect-free electroformation of giant unilamellar vesicles. Langmuir. 35 (50), 16528-16535 (2019).
- Estes, D. J., Mayer, M. Electroformation of giant liposomes from spin-coated films of lipids. Colloids Surf B Biointerfaces. 42 (2), 115-123 (2005).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten