Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Construction de réseaux métaboliques hors équilibre dans des vésicules de taille nanométrique et micrométrique
Dans cet article
Résumé
Nous présentons un protocole pour reconstituer les protéines membranaires et encapsuler des enzymes et d’autres composants solubles dans l’eau dans des vésicules lipidiques de taille submicrométrique et micrométrique.
Résumé
Nous présentons une méthode pour incorporer dans les vésicules des réseaux de protéines complexes, impliquant des protéines membranaires intégrales, des enzymes et des capteurs basés sur la fluorescence, en utilisant des composants purifiés. Cette méthode est pertinente pour la conception et la construction de bioréacteurs et l’étude de réseaux de réactions métaboliques complexes hors équilibre. Nous commençons par reconstituer des protéines membranaires (multiples) en grandes vésicules unilamellaires (LUV) selon un protocole préalablement développé. Nous encapsulons ensuite un mélange d’enzymes purifiées, de métabolites et de capteurs basés sur la fluorescence (protéines fluorescentes ou colorants) par congélation-décongélation-extrusion et éliminons les composants non incorporés par centrifugation et/ou chromatographie d’exclusion stérique. La performance des réseaux métaboliques est mesurée en temps réel en surveillant le rapport ATP/ADP, la concentration en métabolites, le pH interne ou d’autres paramètres par lecture de fluorescence. Nos vésicules contenant des protéines membranaires de 100 à 400 nm de diamètre peuvent être converties en vésicules unilamellaires géantes (GUV), en utilisant des procédures existantes mais optimisées. L’approche permet d’inclure des composants solubles (enzymes, métabolites, capteurs) dans des vésicules de taille micrométrique, augmentant ainsi le volume des bioréacteurs de plusieurs ordres de grandeur. Le réseau métabolique contenant les GUVs est piégé dans des dispositifs microfluidiques pour être analysé par microscopie optique.
Introduction
Le domaine de la biologie synthétique ascendante se concentre sur la construction de cellules (minimales) 1,2 et de bioréacteurs métaboliques à des fins biotechnologiques 3,4 ou biomédicales 5,6,7,8. La construction de cellules synthétiques fournit une plate-forme unique qui permet aux chercheurs d’étudier les protéines (membranaires) dans des conditions bien définies imitant celles des environnements natifs, permettant la découverte de propriétés émergentes et de fonctions biochimiques cachées des protéines et des réseaux de réactions9. En tant qu’étape intermédiaire vers une cellule synthétique fonctionnant de manière autonome, des modules sont développés qui capturent les caractéristiques essentielles des cellules vivantes telles que la conservation de l’énergie métabolique, la synthèse des protéines et des lipides et l’homéostasie. De tels modules améliorent non seulement notre compréhension de la vie, mais ont également des applications potentielles dans les domaines de la médecine8 et de la biotechnologie10.
Les protéines transmembranaires sont au cœur de pratiquement tous les réseaux métaboliques, car elles transportent des molécules à l’intérieur ou à l’extérieur de la cellule, signalent et répondent à la qualité de l’environnement et jouent de nombreux rôles biosynthétiques. Ainsi, l’ingénierie de modules métaboliques dans des cellules synthétiques nécessite dans la plupart des cas la reconstitution de protéines membranaires intégrales et/ou périphériques en une bicouche membranaire composée de lipides spécifiques et d’une grande intégrité (faible perméabilité). La manipulation de ces protéines membranaires est un défi et nécessite des connaissances spécifiques et des compétences expérimentales.
Plusieurs méthodes ont été développées pour reconstituer des protéines membranaires au sein des vésicules phospholipidiques, le plus souvent dans le but d’étudier la fonction11,12, la régulation13, les propriétés cinétiques14,15, la dépendance lipidique15,16 et/ou la stabilité17 d’une protéine spécifique. Ces méthodes impliquent la dilution rapide des protéines solubilisées au détergent dans un milieu aqueux en présence de lipides18, l’élimination des détergents par incubation de protéines solubilisées au détergent avec des vésicules lipidiques déstabilisées par le détergent et l’absorption du ou des détergents sur des billes de polystyrène19, ou l’élimination des détergents par dialyse ou chromatographie d’exclusion stérique20. Des solvants organiques ont été utilisés pour former des vésicules lipidiques, par exemple, via la formation d’interphases huile-eau21, mais la majorité des protéines membranaires intégrales sont inactivées lorsqu’elles sont exposées à de tels solvants.
Dans notre laboratoire, nous reconstituons principalement les protéines membranaires par la méthode d’absorption détergente pour former de grandes vésicules unilamellaires (LUVs)19. Cette méthode permet la co-reconstitution de plusieurs protéines membranaires et l’encapsulation dans la lumière vésiculaire d’enzymes, de métabolites et de sondes22,23. Les LUV contenant des protéines membranaires peuvent être convertis en vésicules unilamellaires géantes (GUV) avec/sans encapsulation de composants solubles dans l’eau, en utilisant soit l’électroformation24, soit le gonflement assisté par gel25 et des conditions spécifiques pour préserver l’intégrité des protéines membranaires26.
Cet article présente un protocole pour la reconstitution dans les LUV d’un réseau métabolique hors équilibre qui régénère l’ATP par la décomposition de la L-arginine en L-ornithine27. La formation d’ATP est couplée à la production de glycérol-3-phosphate (G3P), un élément constitutif important pour la synthèse des phospholipides22,28. La voie métabolique se compose de deux protéines membranaires intégrales, une arginine/ornithine (ArcD) et un antiporteur G3P/Pi (GlpT). De plus, trois enzymes solubles (ArcA, ArcB, ArcC) sont nécessaires au recyclage de l’ATP, et GlpK est utilisé pour convertir le glycérol en glycérol 3-phosphate, en utilisant l’ATP issu de la dégradation de la L-arginine, voir la figure 1 pour une vue d’ensemble schématique de la voie. Ce protocole représente un bon point de départ pour la construction future de réseaux réactionnels encore plus complexes, pour la synthèse de lipides ou de protéines ou la division des cellules. La composition lipidique des vésicules soutient l’activité d’une grande variété de protéines membranaires intégrales et a été optimisée pour le transport de diverses molécules dans ou hors des vésicules 27,29,30.

Figure 1 : Vue d’ensemble de la voie de production de l’ATP et de la synthèse et de l’excrétion du glycérol 3-phosphate. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
En bref, des protéines membranaires purifiées (solubilisées dans du dodécyl-β-D-maltoside, DDM) sont ajoutées à des vésicules lipidiques préformées qui ont été déstabilisées avec Triton X-100, ce qui permet l’insertion des protéines dans la membrane. Les molécules de détergent sont ensuite (lentement) éliminées par l’ajout de billes de polystyrène activées, ce qui entraîne la formation de protéoliposomes bien scellés. Des composants solubles peuvent ensuite être ajoutés aux vésicules et encapsulés via des cycles de gel-dégel, ce qui piège les molécules dans le processus de fusion membranaire. Les vésicules obtenues sont très hétérogènes et beaucoup sont multilamellaires. Ils sont ensuite extrudés à travers un filtre en polycarbonate avec une taille de pores de 400, 200 ou 100 nm, ce qui donne des vésicules de taille plus uniforme ; Plus la taille des pores est petite, plus les vésicules sont homogènes et unilamellaires, mais au prix d’un volume interne plus petit. Les protéines non incorporées et les petites molécules sont éliminées de la solution externe par chromatographie d’exclusion stérique. Les proteoLUV peuvent être convertis en vésicules de taille micrométrique par gonflement assisté par gel, et ces proteoGUVs sont ensuite collectés et piégés dans une puce microfluidique pour la caractérisation et la manipulation microscopiques. La figure 2 présente une vue d’ensemble schématique de l’ensemble du protocole.

Figure 2 : Vue d’ensemble du protocole de reconstitution des protéines membranaires et d’encapsulation d’enzymes et de composants hydrosolubles dans des vésicules lipidiques submicrométriques (LUV) et micrométriques (GUV). Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
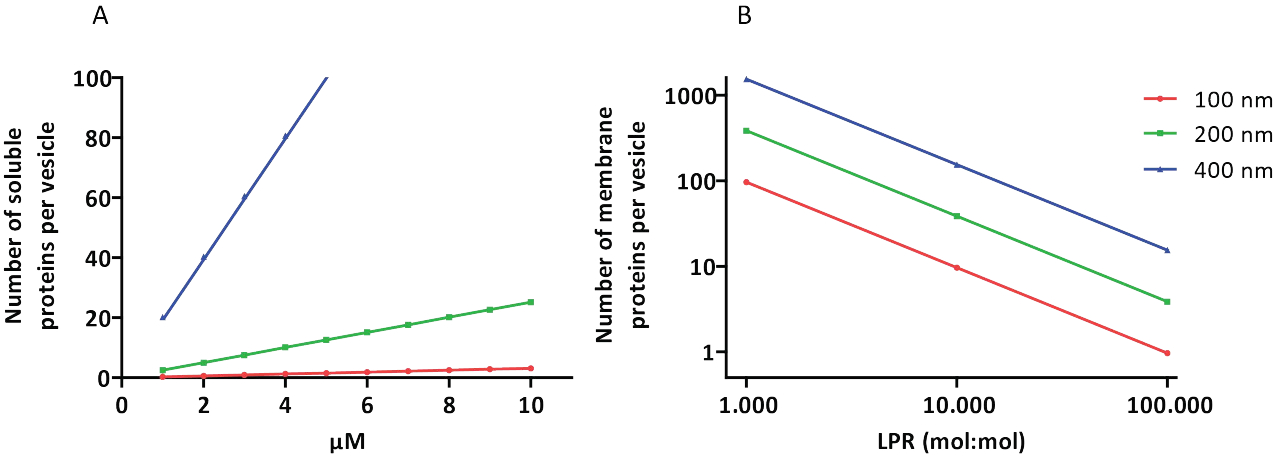
Les protocoles de reconstitution et d’encapsulation fonctionnent bien et la fonctionnalité des protéines est conservée, mais les proteoLUVs et les proteoGUVs sont de taille hétérogène. Les approches microfluidiques31,32 permettent la formation de vésicules de taille micrométrique de taille plus homogène, mais la reconstitution fonctionnelle des protéines membranaires n’est généralement pas possible car le solvant résiduel dans la bicouche inactive les protéines. La taille des protéoLUV varie de 100 à 400 nm, et à de faibles concentrations d’enzymes, l’encapsulation peut conduire à des vésicules avec des voies métaboliques incomplètes (effets stochastiques ; voir Figure 3). Les LUV sont idéaux pour construire des modules métaboliques spécifiques, comme le montre ici la production d’ATP et de blocs de construction comme le G3P. De tels protéoLUV peuvent potentiellement être encapsulés dans des GUV et servir de compartiments semblables à des organites pour les vésicules hôtes.

Figure 3 : Nombre de molécules par vésicule d’un diamètre de 100, 200 ou 400 nm. (A) Lorsque les protéines encapsulées (enzymes, sondes) sont dans la gamme de 1 à 10 μM. (B) La reconstitution se fait à 1 à 1 000, 1 à 10 000 et 1 à 100 000 protéines membranaires par lipide (mol/mol). Nous faisons l’hypothèse que les molécules sont encapsulées aux concentrations indiquées et incorporées dans la membrane à ces ratios protéines/lipides. Pour certaines enzymes, nous avons vu qu’elles se lient aux membranes, ce qui peut augmenter leur concentration apparente dans les vésicules. Abréviation : LPR = Lipid-Protein-Ratio Veuillez cliquer ici pour voir une version agrandie de ce chiffre.
{kind=link}
Protocole
1. Préparation générale
- Produits chimiques
- Dissoudre les lipides (sous forme de poudre) à 25 mg/mL dans CHCl3 pour la fabrication de liposomes préformés.
REMARQUE : Il est préférable de préparer des stocks de lipides frais, mais les solutions mères peuvent également être conservées à -20 °C pendant quelques semaines. Travailler avec des lipides sous forme de poudre est plus précis que d’utiliser des lipides déjà solubilisés dans CHCl3. Le CHCl3 doit être manipulé à l’aide de pipettes et/ou de seringues en verre et stocké dans des récipients en verre, car le CHCl3 dissout les plastiques. - Dissoudre les petites molécules (nucléotides, acides aminés, sondes de fluorescence) pour la procédure d’encapsulation dans 50 mM KPi (tampon A, voir tableau 1) et ajuster le pH à 7,00 ± 0,01. Dissoudre le MgCl2 dans de l’eau déminéralisée pour éviter la formation de précipités de phosphate de magnésium.
REMARQUE : Les solutions mères peuvent être conservées à -20 °C pendant quelques semaines, à l’exception du DTT, qui est fraîchement préparé le jour de l’expérience. - Dissoudre les ionophores (p. ex. valinomycine, nigéricine) dans le DMSO ou l’EtOH à une concentration de stock de 100 à 500 μM. Conserver à -20 °C pendant quelques semaines ; éviter l’évaporation.
REMARQUE : Le DMSO n’est pas volatil et est donc préféré à l’EtOH. Utilisez du verre plutôt que des flacons en plastique pour éviter l’adhérence des ionophores à la surface des flacons.
- Dissoudre les lipides (sous forme de poudre) à 25 mg/mL dans CHCl3 pour la fabrication de liposomes préformés.
- Tampons
- Préparez de nouveaux tampons le jour de l’expérience (tableau 1). Ne pas conserver plus de 24 h.
- Purification des protéines solubles
- Express ArcA, ArcB, ArcC (nous utilisons une variante spécifique nommée ArcC1), PercevalHR et GlpK comme décrit précédemment 27,28,33. Assurez-vous d’ajouter 10 % v/v de glycérol au tampon de lyse cellulaire, ce qui améliore la stabilité des protéines. Purifier les protéines solubles comme décrit27, 28, 33 et brièvement rapporté ci-après.
- Décongeler 10 mL de lysat cellulaire (~5 g de poids humide) dans un bain d’eau glacée. Entre-temps, appliquer 2 mL (1 CV) de résine Ni2+-Sepharose sur une colonne d’écoulement par gravité (capacité de 20 mL) avec de l’eau désionisée (12 CV) et un tampon B (4 CV) pour laver. Transférer le lysat décongelé dans de la glace ; travail sur glace, sauf indication contraire.
- Ajouter de l’imidazole à une concentration finale de 10 mM au lysat décongelé, puis verser la solution sur la colonne d’écoulement par gravité. Incuber pendant 1 heure à 4 °C avec une nutation douce.
- Après 1 h, jetez le flux et lavez la résine avec le tampon C (20 CV).
- Éluer la protéine avec le tampon D. Utilisez 60 % de CV pour la première étape d’élution, suivie de 4 à 6 étapes de 40 % de CV.
- Déterminer la concentration en protéines et ajouter du Na-EDTA à une concentration finale de 5 mM.
- Faites tourner la protéine purifiée dans une centrifugeuse de table réfrigérée (vitesse maximale, 10 minutes, 4 °C). Purifier par chromatographie d’exclusion stérique à l’aide du tampon E. Regrouper les fractions d’élution et les concentrer à ~10 mg/mL avec un filtre concentrateur avec une coupure de 30 kDa. Préparez des aliquotes de taille appropriée (~ 20 μL), congelez-les avec de l’azote liquide et conservez-les à -80 °C pour une utilisation ultérieure.
REMARQUE : Il est important de concentrer les enzymes à 50-100 μM pour minimiser le volume nécessaire à l’encapsulation.
- Purification des protéines membranaires
- Surexpression de l’ArcD et du GlpT comme décrit précédemment 22,27,33. Assurez-vous d’ajouter 10 % v/v de glycérol au tampon de lyse cellulaire ; pour la purification de l’ArcD, inclure 2 mM d’agent réducteur (p. ex., DTT) dans la mémoire tampon. Purifier les protéines marquées par affinité par chromatographie Ni2+-sépharose.
- Décongeler une aliquote de vésicules membranaires brutes (10 à 20 mg de protéines membranaires totales) dans un bain d’eau glacée.
REMARQUE : Une fois décongelé, travaillez toujours sur de la glace, sauf indication contraire. Voir le Tableau 1 pour connaître les tampons utilisés dans cette section. - Ajouter les vésicules membranaires dans le tampon F (ArcD) ou le tampon G (GlpT) jusqu’à un volume final de 6 ml. Incuber l’échantillon pendant 1 h à 4 °C avec une nutation douce.
- Séparer les protéines membranaires solubilisées des débris membranaires par ultracentrifugation (337 000 × g, 30 min, 4 °C). Entre-temps, appliquer 0,25 mL (1 CV) de résine Ni2+-sépharose sur une colonne d’écoulement par gravité (capacité de 10 mL) avec de l’eau désionisée (40 CV) et 20 CV de tampon H (ArcD) ou de tampon I (GlpT).
- Verser la protéine solubilisée sur la colonne d’écoulement par gravité et ajouter de l’imidazole à une concentration finale de 10 mM. Incuber pendant 1 h à 4 °C avec une nutation douce.
- Après 1 h, jetez le flux continu et lavez la résine avec 20 CV de tampon J (ArcD) ou de tampon K (GlpT).
- Éluer la protéine membranaire par paliers de 60 % de CV (1ère) et de 40 % de CV (2e-6 e) avec le tampon L (ArcD) ou le tampon M (GlpT).
- Déterminer la concentration en protéines et passer à la section 2.2. pour la reconstitution membranaire.
REMARQUE : La purification par exclusion stérique n’est pas nécessairement effectuée pour les protéines membranaires car la reconstitution membranaire produit une purification similaire. Les étapes 1.4 et 2.2 peuvent être effectuées en 1 jour de travail. Commencez par la purification des protéines (étape 1.4) le matin et poursuivez avec la reconstitution (étape 2.2) l’après-midi. La reconstitution se termine le lendemain (voir section 2.2 pour plus de détails). POINT D’ARRÊT : Les ArcD et GlpT purifiés et solublés au DDM peuvent être stockés à -80 °C pour une utilisation ultérieure, mais ce n’est pas le cas pour toutes les protéines membranaires. Préparez des aliquotes de taille appropriée (50-200 μL), congelez-les avec de l’azote liquide et conservez-les à -80 °C pour une utilisation ultérieure. Ces protéines sont actives pendant plusieurs mois lorsqu’elles sont stockées à -80 °C en présence de glycérol à 10% v/v.
- Décongeler une aliquote de vésicules membranaires brutes (10 à 20 mg de protéines membranaires totales) dans un bain d’eau glacée.
- Surexpression de l’ArcD et du GlpT comme décrit précédemment 22,27,33. Assurez-vous d’ajouter 10 % v/v de glycérol au tampon de lyse cellulaire ; pour la purification de l’ArcD, inclure 2 mM d’agent réducteur (p. ex., DTT) dans la mémoire tampon. Purifier les protéines marquées par affinité par chromatographie Ni2+-sépharose.
- Préparation de β-caséine à des fins de passivation
- Mettre de nouveau en suspension 100 mg de β-caséine dans 20 mL d’eau désionisée et titrer avec 1 M de NaOH jusqu’à ce que la β-caséine soit complètement dissoute. Ensuite, ajoutez 1 M d’acide acétique pour ajuster le pH à 7,0 et remplissez le volume à 50 ml avec de l’eau désionisée. Filtrer la solution à l’aide d’un filtre à seringue de 0,2 μm et faire des aliquotes de 500 μL.
REMARQUE : La β-caséine peut être conservée à -20 °C pendant 6 mois. Il est recommandé de filtrer à nouveau la β-caséine avant utilisation pour éviter que les agrégats de β-caséine n’obstruent la puce microfluidique.
- Mettre de nouveau en suspension 100 mg de β-caséine dans 20 mL d’eau désionisée et titrer avec 1 M de NaOH jusqu’à ce que la β-caséine soit complètement dissoute. Ensuite, ajoutez 1 M d’acide acétique pour ajuster le pH à 7,0 et remplissez le volume à 50 ml avec de l’eau désionisée. Filtrer la solution à l’aide d’un filtre à seringue de 0,2 μm et faire des aliquotes de 500 μL.
| Tampon | Composition | ||
| Tampon A | 50 mM KPi pH 7,0 | ||
| Tampon B | 50 mM KPi, 100 mM KCl, 10 % v/v de glycérol, 10 mM d’imidazole, pH 7,5 | ||
| Tampon C | 50 mM KPi, 100 mM KCl, 10 % v/v de glycérol, 50 mM d’imidazole, pH 7,5 | ||
| Tampon D | 50 mM KPi, 100 mM KCl, 10 % v/v de glycérol, 500 mM d’imidazole, pH 7,5 | ||
| Tampon E | 50 mM KPi, 100 mM KCl, 10 % v/v de glycérol, pH 7,0 | ||
| Tampon F | 50 mM KPi, 100 mM KCl, 0,5 % p/v DDM, 10 % v/v glycérol, 2 mM β-mercaptoéthanol, pH 7,5 | ||
| Tampon G | 50 mM de Tris-HCl, 0,5 % p/v DDM, 20 % v/v de glycérol, pH 8 | ||
| Tampon H | 50 mM KPi, 100 mM KCl, 0,02 % p/v DDM, 10 % v/v glycérol, 2 mM β-mercaptoéthanol, 10 mM imidazole, pH 7,5 | ||
| Tampon I | 50 mM de Tris-HCl, 0,04 % p/v de DDM, 20 % v/v de glycérol, 10 mM d’imidazole, pH 8,0 | ||
| Tampon J | 50 mM KPi, 200 mM KCl, 0,02 % p/v DDM, 10 % v/v glycérol, 2 mM β-mercaptoéthanol, 50 mM d’imidazole, pH 7,5 | ||
| Tampon K | 50 mM de Tris-HCl, 0,04 % p/v de DDM, 20 % v/v de glycérol, 50 mM d’imidazole, pH 8 | ||
| Tampon L | 50 mM KPi, 200 mM KCl, 0,02 % p/v DDM, 10 % v/v glycérol, 2 mM β-mercaptoéthanol, 500 mM imidazole, pH 7,5 | ||
| Tampon M | 50 mM de Tris-HCl, 0,04 % p/v de DDM, 20 % v/v de glycérol, 500 mM d’imidazole, pH 8 | ||
| Zone tampon N | 50 mM KPi, 58 mM NaCl, 2 mM DTT, pH 7,0 | ||
| Tampon O | 50 mM de KPi, 0,5 mM de L-ornithine, 10 mM de Na-ADP, 10 mM de MgCl2, 2 mM de DTT, pH 7,0 | ||
| Tampon P | 50 mM KPi pH 7,0, 2 mM DTT, x mM glucose (x est varié pour correspondre à l’osmolarité du milieu externe et interne) | ||
| Tampon Q | 50 mM KPi pH 7,0, 0,5 mM Saccharose, 2 mM DTT | ||
| Tampon R | 50 mM KPi pH 7,0, 2 mM DTT, 10 mM L-arginine, x mM glucose | ||
Tableau 1 : Tampons utilisés dans ce protocole.
2. Protéoliposomes : reconstitution de protéines membranaires purifiées en vésicules lipidiques préformées
- Jour 1
- Préparation de vésicules lipidiques préformées
- Choisissez la composition lipidique (par exemple, phospholipides synthétiques, lipides polaires d’E. coli ) en fonction des besoins des protéines membranaires.
REMARQUE : Un mélange de DOPE, DOPG et DOPC (25:25:50 % molaire) est un bon point de départ, mais des stérols ou de la cardiolipine peuvent être nécessaires pour certaines protéines ; Pour les protéines de membrane plasmique de levure, nous incluons des lipides de palmitoyl-oléoyle au lieu de dioleoyl30. Un mélange de DOPE, de DOPG et de DOPC (25:25:50 % molaire) suffit pour la reconstitution d’ArcD et de GlpT. - Mélangez les lipides souhaités (solubilisés dans le CHCl3) et évaporez le CHCl3 dans un évaporateur rotatif jusqu’à formation d’un film lipidique. Laver les lipides en ajoutant de l’éther diéthylique à volume égal à celui du CHCl3. Évaporer l’éther diéthylique et obtenir un film lipidique sec.
- Remettre en suspension le film lipidique à 20 mg/mL de lipides totaux en milieu aqueux (tampon A). Commencez avec la moitié du volume total et secouez doucement ; Ensuite, transférez soigneusement les lipides dans un tube ou une bouteille propre de taille adéquate. Ajoutez le tampon A frais dans le flacon et répétez la procédure pour dissoudre les lipides restants et les transférer dans un nouveau récipient. Ajouter du tampon A supplémentaire pour atteindre une concentration finale de 20 mg/mL.
- Sonicer les lipides remis en suspension à l’aide d’une sonde sonicatrice. Utilisez les paramètres suivants pour une pointe de sonicateur d’un diamètre de 6 mm : intensité de 4 μm, amplitude de 70 %, 5 s de marche, 45 s de désactivation, 16 cycles. Plongez les lipides dans un bain d’eau glacée contenant de l’EtOH pour éviter la surchauffe par sonication.
- Congelez l’échantillon soniqué (volume de 40 mL dans un tube à centrifuger de 50 mL) dans de l’azote liquide et décongelez l’échantillon dans un bain-marie à température ambiante. Répétez une fois ; ensuite, aliquote les liposomes dans les volumes souhaités (par exemple, 1 mL ou 20 mg de lipides totaux dans un tube en plastique de 1,5 mL).
POINT D’ARRÊT : La procédure peut être arrêtée à ce stade. Congelez à nouveau chaque aliquote (troisième cycle) et stockez-la dans de l’azote liquide jusqu’à quelques mois. Prenez soin de percer les couvercles du tube deux fois avec une aiguille pour éviter l’explosion du tube lors de l’ébullition rapide de l’azote liquide.
- Choisissez la composition lipidique (par exemple, phospholipides synthétiques, lipides polaires d’E. coli ) en fonction des besoins des protéines membranaires.
- Préparation de vésicules lipidiques préformées
- Jour 2
- Reconstitution de protéines membranaires purifiées en liposomes préformés
- Décongeler une aliquote de liposomes (20 mg de lipides totaux) dans un bain-marie à température ambiante. Pendant ce temps, préparez une extrudeuse en appliquant un filtre de votre choix (par exemple, polycarbonate, diamètre des pores de 400 nm) ; pré-équilibrer l’extrudeuse avec le tampon A ; et chargez les liposomes décongelés (« solution laiteuse ») dans l’extrudeuse et passez-les 13 fois à travers le filtre. Recueillir les liposomes extrudés (maintenant de grandes vésicules unilamellaires ; « solution opaque ») dans un récipient en verre ou en plastique de taille adéquate (p. ex., 15 ml). Diluer les liposomes à 4 mg/mL avec le tampon A complété par 2 mM de DTT.
- Transvaser 1 mL de liposomes à 4 mg/mL dans une cuvette transparente de 1 mL. Dans un spectrophotomètre, mesurez la densité optique initiale à 540 nm. Versez l’échantillon mesuré et ajoutez 50 μL de Triton X-100 à 10 % v/v dans les liposomes.
REMARQUE : Un volume de titrage de 50 μL de 10 % de Triton-X100 convient à 20 mg de lipides dans un volume de 5 mL ; l’ajout de Triton X-100 va diluer les lipides de ~5%. Ajustez le volume de titrage lorsque vous travaillez avec différentes quantités de liposomes. Pour des signaux de densité optique stables, titrez les liposomes avec Triton X-100 à température ambiante. - Répétez l’étape 2.2.1.2 et notez quand une densité optique maximale est atteinte (Rsat). Poursuivez le titrage jusqu’à ce qu’une densité optique d’environ 60 % de Rsat soit atteinte (Figure 4). Versez les vésicules déstabilisées par le détergent dans le tube en verre/plastique (le volume final est maintenant d’environ 5,2 ml), transférez l’échantillon dans de la glace et laissez-le refroidir.
- Ajoutez la ou les protéines membranaires purifiées aux liposomes déstabilisés pour atteindre un rapport lipide/protéine souhaité (p/p). Utilisez un rapport de 400:1 à 100:1 p/p ; puisque ArcD et GlpT ont tous deux des poids moléculaires de ~55 kDa, un rapport lipides/protéines de 400:1 p/p correspond à ~30 000 lipides par protéine, et ~ 50 molécules d’ArcD et de GlpT chacune par vésicule d’un diamètre de 400 nm.
REMARQUE : Ici, nous utilisons 400:1 p/p, ce qui correspond à 50 μg de chaque protéine pour 20 mg de lipides. - Nuter les échantillons à 4 °C pendant 15 min pour permettre aux protéines membranaires de s’insérer dans la membrane liposomale déstabilisée.
- Pour enlever le détergent, ajoutez 200 mg de billes de polystyrène sèches, préparées selon les instructions du fabricant. Nuter à 4 °C pendant 15 minutes supplémentaires.
REMARQUE : Une quantité de 200 mg de billes sèches convient pour 20 mg de lipides, mais doit être ajustée lorsqu’une taille d’échantillon différente est utilisée. - Répétez l’étape 2.2.1.6 2 fois, pour un total de trois ajouts de billes de polystyrène. Ensuite, incuber toute la nuit à 4 °C avec une légère nutation.
- Reconstitution de protéines membranaires purifiées en liposomes préformés
- Jour 3
- Répétez l’étape 2.2.1.6 ; cependant, cette fois-ci, nuter à 4 °C pendant 1 h.
- Fixez une colonne d’écoulement par gravité vide (capacité de 10 ml) au-dessus d’un tube d’ultracentrifugation vide de 6,5 ml sur de la glace. Versez l’échantillon dans la colonne et récupérez les protéoliposomes dans le tube d’ultracentrifugation (les billes sont retenues dans la colonne).
- Laver les billes avec 0,5 mL de tampon A complété par 2 mM de DTT et recueillir le filtrat dans le tube d’ultracentrifugation.
- Concentrer les protéoliposomes par ultracentrifugation (337 000 × g, 30 min, 4 °C). Remettre en suspension les protéoliposomes dans un volume total de 200 μL (100 mg de lipides/mL) dans le tampon A complété par 2 mM de DTT ; le volume sec des vésicules après centrifugation est de ~40 à 120 μL27. Diviser en aliquotes de la taille désirée (p. ex., trois aliquotes de 6,66 mg de lipides totaux).
REMARQUE : La taille de l’aliquote est arbitraire mais affectera la taille des granulés dans les étapes ultérieures (voir étape 3.2.3.1). POINT D’ARRÊT : La procédure peut être arrêtée ici. Congelez chaque aliquote et conservez-la dans de l’azote liquide jusqu’à quelques semaines. Prenez soin de percer les couvercles du tube deux fois avec une aiguille pour éviter l’explosion du tube lors de l’ébullition rapide de l’azote liquide.

Figure 4 : Titrage de liposomes préformés avec Triton X-100. Les liposomes à 5 mg de lipides/mL sont extrudés à travers un filtre en polycarbonate (400 nm) dans 50 mM KPi (pH 7,0), puis titrés avec Triton X-100 (étape du protocole 2.2.1.2). La turbidité des vésicules est mesurée à A540. La flèche indique la concentration de Triton X-100 à laquelle les vésicules sont suffisamment déstabilisées pour l’insertion spontanée de protéines membranaires comme décrit en19. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
3. Encapsulation d’un réseau métabolique pour le recyclage de l’ATP et la synthèse du glycérol 3-P dans des vésicules de taille submicronique
- Jour 1
- Mélange de composants
- Pour une encapsulation standard, utilisez 66,6 μL de protéoliposomes dans un volume final de 200 μL (33,33 mg/mL de lipides totaux). Calculez le volume de chaque composant (enzyme, cofacteur) nécessaire pour atteindre la concentration souhaitée, ajoutez ces composants aux protéoliposomes et ajustez le volume à 200 μL avec le tampon A (tableau 2).
REMARQUE : La concentration en protéines peut être ajustée en fonction de la configuration expérimentale. Cependant, il faut veiller à ce que plusieurs copies (>10) de chaque enzyme soient en moyenne présentes par vésicule, afin d’éviter des effets stochastiques indésirables. Les concentrations > 1 μM sont généralement sans danger ; 1 μM dans une vésicule d’un diamètre de 400 nm correspond à environ 20 copies (Figure 3). - Pipeter le tampon A dans un tube vide de 1,5 mL et ajouter le DTT, le Na-ADP, le MgCl2, la L-ornithine (et la pyranine, si nécessaire pour les mesures de pH internes). Ensuite, ajoutez les enzymes et mélangez doucement. Ajoutez la solution sur les protéoliposomes préformés et agitez brièvement à basse vitesse.
REMARQUE : Il est important d’ajouter du Na-ADP avant le MgCl2 pour éviter la formation indésirable de précipités de phosphate de magnésium. Le vortex est nécessaire pour un bon mélange de la solution visqueuse ; Cependant, minimisez la durée et la vitesse pour éviter d’endommager mécaniquement les protéines. PercevalHR et la pyranine ne peuvent pas être co-encapsulés car leurs spectres se chevauchent.
- Pour une encapsulation standard, utilisez 66,6 μL de protéoliposomes dans un volume final de 200 μL (33,33 mg/mL de lipides totaux). Calculez le volume de chaque composant (enzyme, cofacteur) nécessaire pour atteindre la concentration souhaitée, ajoutez ces composants aux protéoliposomes et ajustez le volume à 200 μL avec le tampon A (tableau 2).
- Détermination de l’osmolalité interne
- Préparez une solution de 50 μL comme décrit à l’étape 3.1.1.1, mais sans protéoliposomes, et mesurez l’osmolalité à l’aide d’un osmomètre à point de congélation.
- Préparez une courbe d’étalonnage à l’aide d’un tampon (50 mM KPi pH 7,0) et de différentes concentrations de sel (p. ex., NaCl ou NaCl) ou de sucre. Déterminez la concentration d’osmolyte qui correspond à l’osmolalité interne (tampon N).
REMARQUE : Les composants perméables à la membrane (par exemple, le glycérol) ne peuvent pas être utilisés pour correspondre à l’osmolalité interne. Pour les protéines solubilisées dans un tampon contenant du glycérol (par exemple, le tampon E), le même tampon doit être utilisé sans glycérol. L’osmolyte choisi pour la préparation d’un tampon externe iso-osmotique doit être imperméable à la membrane et ne pas interférer avec le réseau métabolique.
- Gel-dégel
- Congeler (dans de l’azote liquide) les protéoliposomes avec les composants solubles et les décongeler dans un bain d’eau glacée à environ 10 °C.
- Répétez l’étape 3.1.3.1 pour un total de 5 fois.
POINT D’ARRÊT : La procédure peut être arrêtée ici. Sautez la dernière étape de décongélation et conservez l’échantillon congelé dans de l’azote liquide pendant 1 à 3 jours. Prenez soin de percer les couvercles du tube 2x avec une aiguille pour éviter l’explosion du tube lors de l’ébullition rapide de l’azote liquide. Le stockage dans l’azote liquide est préférable à -80 °C pour minimiser l’oxydation des lipides.
- Mélange de composants
- Jour 2
- Extrusion
REMARQUE : Toutes les étapes d’extrusion sont effectuées à température ambiante, sinon les seringues étanches au gaz fuiraient.- Préparez une extrudeuse en appliquant le filtre de votre choix (par exemple, polycarbonate, diamètre des pores de 400 nm). Laver l’extrudeuse avec une solution contenant le même tampon et les mêmes métabolites que ceux utilisés pour l’encapsulation des vésicules (par exemple, le tampon O plus 0,1 mM de pyranine).
REMARQUE : Utilisez une extrudeuse dédiée pour le chargement des protéoliposomes avec de la pyranine car le colorant adhère à la surface de l’extrudeuse et peut provoquer une contamination dans les échantillons ultérieurs. - Chargez le mélange d’encapsulation dans l’extrudeuse et passez-le à travers le filtre 13x. Recueillir la solution extrudée dans un tube de 1,5 mL.
- Préparez une extrudeuse en appliquant le filtre de votre choix (par exemple, polycarbonate, diamètre des pores de 400 nm). Laver l’extrudeuse avec une solution contenant le même tampon et les mêmes métabolites que ceux utilisés pour l’encapsulation des vésicules (par exemple, le tampon O plus 0,1 mM de pyranine).
- Chromatographie d’exclusion stérique (en option)
REMARQUE : Cette étape est effectuée pour éliminer les molécules externes telles que les colorants comme la pyranine par chromatographie d’exclusion stérique. Si les colorants ne sont pas présents dans le système et que d’autres composants n’interfèrent pas à de faibles concentrations (notez que les vésicules sont ensuite également lavées par ultracentrifugation), alors étape 3.2.2. peut être ignoré.- Réhydratez la résine Sephadex G-75 et versez-la dans une colonne de verre (22 cm de long, 1,5 cm de large). Équilibrez la résine avec un excès de tampon externe (par exemple, tampon N).
- Chargez les protéoliposomes extrudés de l’étape 3.2.1.2 sur la colonne de chromatographie d’exclusion stérique prééquilibrée et appliquez un écoulement gravitaire de tampon externe. Jeter le volume du vide (environ 7 ml) ; puis, prélever dix aliquotes de 1 mL. Visualisez les aliquotes contenant des protéoliposomes par une courte exposition à une lampe UV. Mettez en commun les fractions contenant la plupart des protéoliposomes (2-4 mL).
- Lavage et remise en suspension
- Laver les protéoliposomes extrudés par ultracentrifugation. Remplissez un tube d’ultracentrifugation de 6,5 ml avec 5,8 ml de tampon N et appliquez l’échantillon extrudé sur le dessus. Si une chromatographie d’exclusion stérique a été effectuée, remplissez le tube avec les échantillons d’élution regroupés (2 à 4 ml) et ajoutez le tampon N à un volume final de 6 ml.
- Centrifugeuse à 337 000 × g, 30 min, 4 °C. Jetez le surnageant et séchez soigneusement le tube d’ultracentrifugation avec un chiffon sans poussière tout en faisant attention à ne pas toucher la pastille. Remettre la pastille en suspension dans un petit volume de tampon N (200 μL). Lorsque la pastille est complètement remise en suspension, remplissez le tube jusqu’à 6 ml avec le tampon N.
REMARQUE : la remise en suspension du granulé prendra du temps et doit être effectuée avec soin. - Répéter les étapes 3.2.3.1 à 3.2.3.2 2 fois, pour un total de trois lavages, à moins qu’une chromatographie d’exclusion stérique ne soit effectuée (auquel cas une seule étape de centrifugation suffit). En fin de compte, remettre la pastille en suspension à la concentration désirée (p. ex., 5,55 mg/mL de lipides totaux) en ajoutant un volume approprié de tampon N.
POINT D’ARRÊT : Les protéoliposomes peuvent être utilisés immédiatement ou stockés à 4 oC pendant au moins 48 h. La taille des tubes d’ultracentrifugation doit être choisie en fonction de la taille de l’échantillon. Pour une pastille de 6,66 mg de lipides totaux, un tube de 6,5 mL est approprié. Le lavage de 200 μL de protéoliposomes (33,33 mg de lipides/mL au total ; volume de granulés secs ~40 μL)28 pour 3 x 6 mL de tampon dilue les composants externes d’un facteur 100 pour chaque étape de lavage. Si des tubes de différentes tailles sont utilisés, il est souhaitable d’ajuster le nombre de lavages en conséquence.
- Extrusion
- Jour 3
- Détection de la synthèse d’ATP par fluorescence
- Mélanger les composants de la réaction jusqu’à un volume final de 120 μL (tableau 3) dans une cuvette en quartz noir avec une fenêtre de 3 x 5 mm et un volume interne minimal de 100 μL.
REMARQUE : Des ionophores peuvent être ajoutés pour dissiper les gradients d’ions électrochimiques. Un mélange de valinomycine et de nigéricine (1 μM chacun) dissipe efficacement tous les gradients de protons et de potassium. - Préchauffez l’échantillon dans un fluorimètre réglé à 30 °C et acquérez les spectres d’excitation de PercevalHR (excitation 400-520 nm, bande passante 5 nm ; émission 550 nm, bande passante 5 nm). Une fois que le signal de la sonde est constant, démarrer le réseau métabolique par l’ajout d’un excès de L-arginine (5-10 mM), et de glycérol (400 μM) lorsque le recyclage de l’ATP est couplé à la synthèse du glycérol 3-phosphate. Suivez la réaction au fil du temps.
- Mélanger les composants de la réaction jusqu’à un volume final de 120 μL (tableau 3) dans une cuvette en quartz noir avec une fenêtre de 3 x 5 mm et un volume interne minimal de 100 μL.
- Analyse des données
- Tracez le rapport F500/F430 en fonction du temps, qui est un indicateur qualitatif du rapport ATP/ADP. Pour une évaluation plus quantitative de la formation de l’ATP, construisez une courbe d’étalonnage dans les protéoliposomes ou utilisez une approche complémentaire (par exemple, la quantification de l’ATP par chimiluminescence28).
- Détection de la synthèse d’ATP par fluorescence
| Composant | Concentration finale |
| Tampon A | 50 mM KPi pH 7,0 |
| La TNT | 2 millions d’euros |
| Na-ADP | 10 millions de kilomètres |
| MgCl2 | 10 millions de kilomètres |
| L-ornithine | 0,5 million d’euros |
| Sonde fluorescente (PercevalHR ou pyranine) | 5,8 μM ou 0,1 mM, respectivement |
| ArcA (arginine déiminase) | 1 μM |
| ArcB (ornithine carbamoyltransférase) | 2 μM |
| ArcC1 (carbamamate kinase) | 5 μM |
| GlpK (glycérol kinase) | 1,6 μM |
| Protéoliposomes | 33,33 mg/mL de lipides totaux |
Tableau 2 : Composants d’encapsulation. Les composants sont répertoriés par ordre d’ajout. Les protéines solubles se trouvent dans le tampon E ; tous les autres composants (à l’exception du MgCl2, dans l’eau désionisée) se trouvent dans la zone tampon A.
| Composant | Concentration finale |
| Tampon K | 50 mM KPi pH 7,0, 58 mM NaCl, 2 mM DTT, pH 7,0 |
| Protéoliposomes (5,55 mg/mL de lipides) | 2,7 mg/mL de lipides |
| Ionophores (valinomycine, nigéricine) | 1 μM chacun |
Tableau 3 : Conditions expérimentales. Les composants sont répertoriés par ordre d’ajout. Les protéoliposomes sont dans le tampon N, les ionophores dans le DMSO ou l’EtOH.
4. Mise à l’échelle d’un réseau métabolique pour des vésicules de taille micrométrique
- Jour 1
- Préparation de la puce microfluidique
REMARQUE : Cette expérience utilise un dispositif microfluidique mis au point par Robinson et al.34. D’autres conceptions de puces microfluidiques sont disponibles 35,36,37 et peuvent facilement être mises en œuvre dans ce protocole.- Coupez une pointe de pipette de 200 μL à un tiers du bas et insérez la partie inférieure de la pointe dans un dispositif de piégeage microfluidique préfabriqué.
- Dans le réservoir d’entrée de la puce, ajoutez 400 μL de solution de β-caséine (2 mg/mL ; voir étape 1.5), en prenant soin d’éviter l’introduction d’air à cette étape. Placez un seau à plaques de 96 puits et ajoutez un mouchoir de laboratoire dans une centrifugeuse à tube conique de table. Placez la puce sur le tissu et centrifugez pendant 6 minutes à 900 × g pour permettre la passivation de la puce microfluidique. Après l’étape de centrifugation, le niveau de liquide doit être égal sur le réservoir d’entrée et l’extrémité de sortie de la puce microfluidique ; Vérifiez qu’il n’y a pas de fuite de la puce. Incuber la solution de β-caséine dans la puce microfluidique pendant au moins 30 min.
- Retirez la majeure partie de la solution de passivation sans introduire d’air et ajoutez 400 μl de tampon de lavage (tampon P). Placez la puce sur le godet à plaques à 96 puits et centrifugez à 900 × g pendant 6 min. Laissez la puce dans la solution de lavage jusqu’à l’utilisation (pendant 4h maximum).
- Préparation de la puce microfluidique
- Préparation de gel pour la fabrication de protéo-GUVs
NOTE : La description suivante est tirée et adaptée de 25,38.- Dissoudre 0,5 % (p/p) d’une agarose à basse température gélifiante (LGT) dans de l’eau désionisée en chauffant la solution au micro-ondes ; Assurez-vous que l’agarose est complètement dissoute et évitez de faire bouillir la solution. Maintenez l’agarose à 50 °C jusqu’à nouvel ordre.
REMARQUE : L’agarose dissoute peut être conservée à température ambiante pendant plusieurs semaines. Pour réutiliser l’agarose, il suffit de faire fondre le gel à l’aide d’un micro-ondes. - Prenez deux diapositives d’objectif et dessinez le contour de l’entretoise sur les diapositives. Rendez les lames hydrophiles par nettoyage au plasma, en utilisant du plasma à haute teneur en oxygène pendant 1 min.
- Ajouter de l’agarose LGT sur le dessus de la lame jusqu’à ce qu’elle soit entièrement recouverte d’agarose (~500 μL) ; Ensuite, inclinez la glissière à un angle de 90 ° et égouttez l’excès d’agarose sur un mouchoir. Laissez les lames pendant 30 min à 50 °C.
- Prendre les protéoliposomes stockés dans de l’azote liquide (section 3) et les décongeler sur de la glace. Diluer les vésicules à 5 mg/mL de lipides à l’aide du tampon Q.
REMARQUE : Les protéoliposomes préparés dans la section 3 ne contiennent pas de protéines solubles et peuvent être préparés bien à l’avance s’ils sont stockés dans de l’azote liquide. Lorsque vous travaillez avec des proteoGUVs, assurez-vous de toujours filtrer les solutions de travail pour éviter le colmatage du dispositif microfluidique. - Sonicer les protéo-liposomes à l’aide d’une sonde portative avec une sonde de 1 mm. Sonicate pendant 10 cycles de 0,5 s et 0,5 s de repos à 70 % d’amplitude. Maintenez les vésicules, ci-après appelées protéoSUVs, sur la glace pendant 30 s et répétez le processus de sonication 5 fois.
- Remplissez une seringue de 100 μL (dans un distributeur LCP portable) avec la suspension proteo-SUV. Déposer 0,5 μL de gouttelettes de protéo-SUV sur le gel d’agarose préalablement préparé ; Veillez à ne pas perturber la couche d’agarose et à garder une distance suffisante pour éviter que les gouttelettes ne fusionnent.
REMARQUE L’utilisation d’un distributeur LCP portatif permet de déposer des gouttelettes de manière reproductible, mais d’autres systèmes de pipetage peuvent également être utilisés. Le spotting avec un capillaire de verre est moins adapté car il perturbe le gel d’agarose séché. - Séchez les gouttelettes SUV en ~10 min en utilisant un flux d’azote au lieu d’air comprimé pour réduire la possibilité d’oxydation des lipides.
- Préparez 1 mL d’un tampon O concentré 1,25x (tableau 4) dans le tampon A et passez-le à travers un filtre en acétate de cellulose de 0,2 μm. Prélever 800 μL de la solution concentrée 1,25x et ajouter les enzymes solubles et les sondes à la concentration indiquée dans le tableau 4. Utilisez de l’eau désionisée filtrée pour obtenir un volume final de 1 ml.
- Préparez 100 μL de solution gonflante contenant tous les composants du tableau 4, à l’exception des protéines et du glycérol. Mesurez l’osmolalité de la solution gonflante à l’aide d’un osmomètre à point de congélation calibré en 3 points.
REMARQUE : Préparez 100 μL de solution gonflante sans protéines ni glycérol pour déterminer avec précision l’osmolalité de cette solution. Le glycérol, qui est présent dans la solution protéique, affecte l’osmolalité, mais, dans les GUV, le glycérol se diffuse rapidement à travers la membrane et n’entraîne que des différences osmotiques transitoires. - Assemblez la chambre de gonflage GUV en fabriquant un sandwich de deux verres d’objectif contenant le gel et des SUV séchés avec une entretoise en téflon de 1,5 ou 3,0 mm d’épaisseur entre les deux. Ensuite, ajoutez la solution de gonflement dans la chambre à travers le petit trou sur le côté à l’aide d’une seringue et d’une aiguille.
REMARQUE : Le volume de la solution de gonflement peut être ajusté en faisant varier les entretoises de 1,5 à 3,0 mm.
- Dissoudre 0,5 % (p/p) d’une agarose à basse température gélifiante (LGT) dans de l’eau désionisée en chauffant la solution au micro-ondes ; Assurez-vous que l’agarose est complètement dissoute et évitez de faire bouillir la solution. Maintenez l’agarose à 50 °C jusqu’à nouvel ordre.
- Gonflement des vésicules et prélèvement des GUV
- Permettre le gonflement des vésicules en mettant la chambre à 22 °C pendant au moins 30 min.
REMARQUE : Le gonflement des vésicules peut être suivi d’une microscopie optique (par exemple, un microscope à contraste de phase de table ou un microscope à fluorescence à grand champ) si les protéines ou les lipides sont marqués par fluorescence. - Récoltez les GUV du gel en appliquant une légère agitation physique en tapotant la chambre sur une surface solide (p. ex., une paillasse de laboratoire) ; retirez un tiers du volume et utilisez la bulle d’air résultante pour induire doucement un mouvement vers le liquide restant, détachant ainsi les GUV du gel.
- Pendant que les GUV gonflent, préparez le tampon P et faites correspondre l’osmolalité avec la solution gonflante en ajustant la concentration de glucose. Filtrez le tampon à l’aide d’un filtre à seringue de 0,2 μm.
REMARQUE : En général, la solution de lavage et de substrat doit être maintenue dans ± 5 mosmol/kg. Toute solution passant à travers la puce doit être très propre car les contaminants peuvent obstruer les canaux. Préparez de nouveaux tampons à chaque fois ou stockez les tampons préparés à -20 °C et filtrez avant utilisation.
- Permettre le gonflement des vésicules en mettant la chambre à 22 °C pendant au moins 30 min.
- Piégeage des GUV pour des expériences de microscopie
- Montez la puce microfluidique passivée sur la platine d’échantillonnage du microscope. Vérifiez s’il y a d’éventuels défauts (p. ex., fuites, air emprisonné, canaux obstrués).
REMARQUE : La vérification de la puce peut être effectuée bien avant de l’utiliser afin qu’une nouvelle puce puisse être préparée si nécessaire. - Raccordez le tube à l’aide d’une aiguille pliée à la sortie du réservoir et connectez l’autre extrémité à une seringue de 1 ml. Montez la seringue sur une pompe et réglez le débit à un maximum de 10 μL/min.
- Retirez le tampon de lavage du réservoir (étape 4.1.1.3) et remplacez-le par un fluide frais (tampon P). Démarrer l’écoulement du tampon à travers la puce en perfusant à l’aide de la seringue à 1-10 μL/min. Laver avec au moins 80 μL de tampon de lavage osmotiquement équilibré (tampon P).
- Retirez l’excès de tampon de lavage du réservoir et ajoutez les (proteo)GUVs dans le réservoir. Ajustez le débit à 0,1 à 1 μL/min pour permettre aux GUV de circuler à travers la puce. Surveillez la puce au fil du temps jusqu’à ce qu’un nombre suffisant de GUV aient été piégés dans la puce.
REMARQUE : Les vésicules contenant des quantités relativement importantes (>20 % molaire) de lipides chargés tels que le phosphatidylglycérol (PG), le phosphatidylsérine (PS) ou l’acide phosphatidique (PA) ou des lipides non bicouches comme la phosphatidyléthanolamine (PE) sont introduites à un débit plus faible à travers la puce pour éviter l’éclatement. Les vésicules composées de phosphatidylcholine pure (PC) ont tendance à être plus stables. - Éliminer l’excès de solution GUV et ajouter le tampon de lavage P, en utilisant un débit constant de 0,1 à 1 μL/min, pour remplacer le milieu externe et laver les GUV piégés pendant au moins 1 h pour éliminer les composés non encapsulés et réduire la fluorescence de fond lorsqu’un fluorophore est encapsulé. Surveiller la fluorescence de fond au fil du temps ; Si la fluorescence de fond ne diminue pas, la puce peut être bloquée.
- Localisez les pièges avec des quantités suffisantes de vésicules et enregistrez leurs positions. Appliquez les paramètres du microscope (par exemple, intensité laser, gain, longueur d’onde) et démarrez une expérience de série chronologique.
- Ajouter une solution de substrat osmotiquement équilibrée (tampon R) dans le réservoir et démarrer un débit de 0,5 μL/min.
- Montez la puce microfluidique passivée sur la platine d’échantillonnage du microscope. Vérifiez s’il y a d’éventuels défauts (p. ex., fuites, air emprisonné, canaux obstrués).
| Composants du tampon L | ||
| Composant | 1,25 fois la concentration | Concentration au travail |
| Tampon A | 62,5 mM KPi pH 7,0 | 50 mM KPi pH 7,0 |
| Saccharose | 125 millions d’euros | 100 millions de mètres |
| La TNT | 2,5 millions d’euros | 2 millions d’euros |
| Na-ADP | 12,5 millions d’euros | 10 millions de kilomètres |
| MgCl2 | 12,5 millions d’euros | 10 millions de kilomètres |
| L-ornithine | 0,625 million de kilomètres | 0,5 million d’euros |
| Composants d’encapsulation | ||
| Pyranine ou PercevalHR | 1 mM ou 20 μM | |
| ArcA (arginine déiminase) | 1 μM | |
| ArcB (ornithine carbamoyltransférase) | 2 μM | |
| ArcC1 (carbamamate kinase) | 5 μM | |
Tableau 4 : Composants de tampon O et d’encapsulation. Les composants sont répertoriés par ordre d’ajout. Tous les composants (à l’exception du MgCl2, dans l’eau désionisée) se trouvent dans le tampon A. Les composants d’encapsulation sont énumérés par ordre d’addition. Toutes les protéines hydrosolubles se trouvent dans le tampon E.
Résultats
La reconstitution des protéines membranaires solubilisées dans les liposomes nécessite la déstabilisation des vésicules préformées. L’ajout de faibles quantités de Triton X-100 entraîne initialement une augmentation de l’absorbance à 540 nm (A540) en raison d’une augmentation de la diffusion de la lumière par le gonflement des vésicules (Figure 4). La valeur maximale de A540 est le point où les liposomes sont saturés de détergent (Rsat
Discussion
Nous présentons un protocole pour la synthèse de protéines (membranaires) contenant des vésicules lipidiques de taille submicrométrique (proteoLUVs), et la conversion de proteoLUVs en vésicules géantes-unilamellaires (proteoGUVs). Le protocole devrait être applicable à la reconstitution d’autres protéines membranaires 13,19,30,40 et à l’encapsulation de réseaux métaboliques autres que les voies de dégradation de la L-arginine et de synthèse du glycérol 3-phosphate présentées ...
Déclarations de divulgation
Les auteurs ne déclarent aucun intérêt financier concurrent.
Remerciements
Les auteurs remercient Aditya Iyer pour le clonage du gène pBAD-PercevalHR et Gea Schuurman-Wolters pour son aide à la production et à la purification des protéines. La recherche a été financée par le programme de gravitation du NWO « Building a Synthetic Cell » (BaSyC).
matériels
| Name | Company | Catalog Number | Comments |
| Agarose | Sigma Aldrich | A9414-25g | |
| Amicon cut-off filter | Sigma Aldrich | Milipore centrifugal filter units Amicon Ultra | |
| BioBeads | BioRad | 152-3920 | |
| CHCl3 | Macron Fine Chemicals | MFCD00000826 | |
| D(+)-Glucose | Formedium | - | |
| D(+)-Sucrose | Formedium | - | |
| DDM | Glycon | D97002 -C | |
| Diethyl Ether | Biosolve | 52805 | |
| DMSO | Sigma-Aldrich | 276855-100ml | |
| DOPC | Avanti | 850375P-1g | |
| DOPE | Avanti | 850725P-1g | |
| DOPG | Avanti | 840475P-1g | |
| DTT | Formedium | DTT005 | |
| EtOH | J.T.Baker Avantor | MFCD00003568 | |
| Extruder | Avestin Inc | LF-1 | |
| Fluorimeter | Jasco | Spectrofluorometer FP-8300 | |
| Glycerol | BOOM | 51171608 | |
| Gravity flow column | Bio-Rad | 732-1010 | |
| Hamilton syringe 100 µL | Hamilton | 7656-01 | |
| Hamilton syringe 1000 µL | Hamilton | 81320 | |
| Handheld LCP dispenser | Art Robbins Instruments | 620-411-00 | |
| Handheld Sonicator | Hielscher Ultrasound Technology | UP50H | |
| HCl | BOOM | x76021889.1000 | |
| Imidazole | Roth | X998.4-250g | |
| K2HPO4 | Supelco | 1.05099.1000 | |
| KCl | BOOM | 76028270.1 | |
| KH2PO4 | Supelco | 1.04873.1000 | |
| Kimwipe | Kimtech Science | 7552 | |
| Large Falcon tube centrifuge | Eppendorf | Centrifuge 5810 R | |
| L-Arginine | Sigma-Aldrich | A5006-100G | |
| Light microscope | Leica | DM LS2 | |
| L-Ornithine | Roth | T204.1 | |
| LSM Laser Scanning Confocal Microscope | Zeiss | LSM 710 ConfoCor 3 | |
| MgCl2 | Sigma-Aldrich | M2670-1KG | |
| Microfluidic chip | Homemade | PDMS based | DOI: https://doi.org/10.1039/C8LC01275J |
| Na-ADP | Sigma-Aldrich | A2754-1G | |
| NaCl | Supelco | 1.06404.1000 | |
| Nanodrop Spectrometer | Isogen Life Science | ND-1000 spectrophotometer NanoDrop | |
| NaOH | Supelco | 1.06498.1000 | |
| Needles for GUVs | Henke-Ject | 14-14575 | 27 G x 3/4'' 0.4 x 20 mm |
| Needles for microfluidics | Henke-Ject | 14-15538 | 18 G x 1 1/2'' 1.2 x 40 mm |
| Ni2+ Sepharose | Cytiva | 17526802 | |
| Nigericin | Sigma-Aldrich | N7143-5MG | |
| Nutator | VWR | 83007-210 | |
| Osmolality meter | Gonotec Salmenkipp | Osmomat 3000 basic freezing point osmometer | |
| Plasmacleaner | Plasma Etch | PE-Avenger | |
| Polycarbonate filter | Cytiva Whatman | Nuclepor Track-Etch Membrane Product: 10417104 | 0.4 µm |
| Polycarbonate ultracentrifuge tube | Beckman Coulter | 355647 | |
| Pyranine | Acros Organics | H1529-1G | |
| Quartz cuvette (black) | Hellma Analytics | 108B-10-40 | |
| Sephadex G-75 resin | GE Healthcare | 17-0050-01 | |
| Sonicator | Sonics Sonics & Materials INC | Sonics vibra cell | |
| Syringe filter | Sarstedt | Filtropur S plus 0.2 | 0.2 µm |
| Syringe pump | Harvard Apparatus | A-42467 | |
| Tabletop centrifuge | Eppendorf | centrifuge 5418 | |
| Teflon spacer | Homemade | Teflon based | 45 x 26 x 1.5 or 45 x 26 x 3 or 20 x 20 x 3 mm |
| Tris | PanReac AppliChem | A1086.1000 | |
| Triton X-100 | Sigma Aldrich | T8787-100 ml | |
| Ultracentrifuge | Beckman Coulter | Optima Max-E | |
| UV lamp | Spectroline | ENB-280C/FE | |
| UV/VIS Spectrometer | Jasco | V730 spectrophotometer | |
| Valinomycin | Sigma-Aldrich | V0627-10MG | |
| Widefield fluorescence microscope | Zeiss | AxioObserver | |
| β-Casein | Sigma Aldrich | C5890-500g |
Références
- Hirschi, S., Ward, T. R., Meier, W. P., Müller, D. J., Fotiadis, D. Synthetic biology: bottom-up assembly of molecular systems. Chem Rev. 122 (21), 16294-16328 (2022).
- Ivanov, I., et al. Bottom-up synthesis of artificial cells: recent highlights and future challenges. Annu Rev Chem Biomol. Eng. 12 (1), 287-308 (2021).
- Clomburg, J. M., Crumbley, A. M., Gonzalez, R. Industrial biomanufacturing: The future of chemical production. Science. 355 (6320), (2017).
- Shi, T., Han, P., You, C., Zhang, Y. -. H. P. J. An in vitro synthetic biology platform for emerging industrial biomanufacturing: Bottom-up pathway design. Synth Syst Biotechnol. 3 (3), 186-195 (2018).
- Wang, A., et al. Liver-target and glucose-responsive polymersomes toward mimicking endogenous insulin secretion with improved hepatic glucose utilization. Adv Funct Mater. 30 (13), 1910168 (2020).
- Kanter, G., et al. Cell-free production of scFv fusion proteins: an efficient approach for personalized lymphoma vaccines. Blood. 109 (8), 3393-3399 (2007).
- Zeltins, A. Construction and characterization of virus-like particles: a review. Mol Biotechnol. 53 (1), 92-107 (2013).
- Jain, K. K. Synthetic biology and personalized medicine. Med Princ Pract. 22 (3), 209-219 (2013).
- Schwille, P., Frohn, B. P. Hidden protein functions and what they may teach us. Trends Cell Biol. 32 (2), 102-109 (2022).
- Sachsenmeier, P. Industry 5.0-The relevance and implications of bionics and synthetic biology. Engineering. 2 (2), 225-229 (2016).
- Schmidt, D., Jiang, Q. -. X., MacKinnon, R. Phospholipids and the origin of cationic gating charges in voltage sensors. Nature. 444 (7120), 775-779 (2006).
- Godoy-Hernandez, A., et al. Rapid and highly stable membrane reconstitution by LAiR enables the study of physiological integral membrane protein functions. ACS Cent Sci. 9 (3), 494-507 (2023).
- Sikkema, H. R., et al. Gating by ionic strength and safety check by cyclic-di-AMP in the ABC transporter OpuA. Sci Adv. 6 (47), 7697 (2020).
- Foucaud, C., Poolman, B. Lactose transport system of Streptococcus thermophilus. Functional reconstitution of the protein and characterization of the kinetic mechanism of transport. J Biol Chem. 267 (31), 22087-22094 (1992).
- Yoneda, J. S., Sebinelli, H. G., Itri, R., Ciancaglini, P. Overview on solubilization and lipid reconstitution of Na,K-ATPase: enzyme kinetic and biophysical characterization. Biophys Rev. 12 (1), 49-64 (2020).
- Simidjiev, I., et al. Self-assembly of large, ordered lamellae from non-bilayer lipids and integral membrane proteins in vitro. Proc Natl Acad Sci. 97 (4), 1473-1476 (2000).
- Harris, N. J., Booth, P. J. Folding and stability of membrane transport proteins in vitro. Biochim Biophys Acta BBA - Biomembr. 1818 (4), 1055-1066 (2012).
- Jackson, M. L., Litman, B. J. Rhodopsin-egg phosphatidylcholine reconstitution by an octyl glucoside dilution procedure. Biochim Biophys Acta BBA - Biomembr. 812 (2), 369-376 (1985).
- Geertsma, E. R., Nik Mahmood, N. A. B., Schuurman-Wolters, G. K., Poolman, B. Membrane reconstitution of ABC transporters and assays of translocator function. Nat Protoc. 3 (2), 256-266 (2008).
- Rigaud, J. -. L., Pitard, B., Levy, D. Reconstitution of membrane proteins into liposomes: application to energy-transducing membrane proteins. Biochim Biophys Acta BBA - Bioenerg. 1231 (3), 223-246 (1995).
- Szoka, F., Papahadjopoulos, D. Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation. Proc Natl Acad Sci. 75 (9), 4194-4198 (1978).
- . Synthetic Organelles for Energy Conservation and Delivery of Building Blocks for Lipid Biosynthesis Available from: https://www.researchsquare.com/article/rs-3385355/v1 (2023)
- Lee, K. Y., et al. Photosynthetic artificial organelles sustain and control ATP-dependent reactions in a protocellular system. Nat Biotechnol. 36 (6), 530-535 (2018).
- Méléard, P., Bagatolli, L. A., Pott, T. Giant unilamellar vesicle electroformation. Methods in Enzymology. , 161-176 (2009).
- Garten, M., Aimon, S., Bassereau, P., Toombes, G. E. S. Reconstitution of a transmembrane protein, the voltage-gated ion channel, KvAP, into giant unilamellar vesicles for microscopy and patch clamp studies. J. Vis. Exp. (95), e52281 (2015).
- Doeven, M. K., et al. lateral mobility and function of membrane proteins incorporated into giant unilamellar vesicles. Biophys J. 88 (2), 1134-1142 (2005).
- Pols, T., et al. A synthetic metabolic network for physicochemical homeostasis. Nat Commun. 10 (1), 4239 (2019).
- Bailoni, E., Poolman, B. ATP recycling fuels sustainable glycerol 3-phosphate formation in synthetic cells fed by dynamic dialysis. ACS Synth Biol. 11 (7), 2348-2360 (2022).
- Van Der Heide, T. On the osmotic signal and osmosensing mechanism of an ABC transport system for glycine betaine. EMBO J. 20 (24), 7022-7032 (2001).
- Van'T Klooster, J. S., et al. Membrane lipid requirements of the lysine transporter Lyp1 from Saccharomyces cerevisiae. J Mol Biol. 432 (14), 4023-4031 (2020).
- Lou, G., Anderluzzi, G., Woods, S., Roberts, C. W., Perrie, Y. A novel microfluidic-based approach to formulate size-tuneable large unilamellar cationic liposomes: Formulation, cellular uptake and biodistribution investigations. Eur J Pharm Biopharm. 143, 51-60 (2019).
- Weiss, M., et al. Sequential bottom-up assembly of mechanically stabilized synthetic cells by microfluidics. Nat Mater. 17 (1), 89-96 (2018).
- Pols, T., Singh, S., Deelman-Driessen, C., Gaastra, B. F., Poolman, B. Enzymology of the pathway for ATP production by arginine breakdown. FEBS J. 288 (1), 293-309 (2021).
- Yandrapalli, N., Robinson, T. Ultra-high capacity microfluidic trapping of giant vesicles for high-throughput membrane studies. Lab Chip. 19 (4), 626-633 (2019).
- Elias, M., et al. Microfluidic characterization of biomimetic membrane mechanics with an on-chip micropipette. Micro Nano Eng. 8, 100064 (2020).
- Robinson, T., Kuhn, P., Eyer, K., Dittrich, P. S. Microfluidic trapping of giant unilamellar vesicles to study transport through a membrane pore. Biomicrofluidics. 7 (4), 044105 (2013).
- Cooper, A., Girish, V., Subramaniam, A. B. Osmotic Pressure Enables High-Yield Assembly of Giant Vesicles in Solutions of Physiological Ionic Strengths. Langmuir. 39 (15), 5579-5590 (2023).
- Tantama, M., Martínez-François, J. R., Mongeon, R., Yellen, G. Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nat Commun. 4 (1), 2550 (2013).
- Setyawati, I., et al. In vitro reconstitution of dynamically interacting integral membrane subunits of energy-coupling factor transporters. eLife. 9, e64389 (2020).
- Oropeza-Guzman, E., Ríos-Ramírez, M., Ruiz-Suárez, J. C. Leveraging the coffee ring effect for a defect-free electroformation of giant unilamellar vesicles. Langmuir. 35 (50), 16528-16535 (2019).
- Estes, D. J., Mayer, M. Electroformation of giant liposomes from spin-coated films of lipids. Colloids Surf B Biointerfaces. 42 (2), 115-123 (2005).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.