
Method Article
Enhanced reducido Representación bisulfito secuenciación para la Evaluación de la metilación del ADN en la Resolución Base Par
* Estos autores han contribuido por igual
En este artículo
Resumen
Enhanced Reduced Representation Bisulfite Sequencing is a method for the preparation of sequencing libraries for DNA methylation analysis based on restriction enzyme digestion combined with cytosine bisulfite conversion. This protocol requires 50 ng of starting material and yields base pair resolution data at GC-rich genomic regions.
Resumen
ADN mapeo patrón de metilación se estudia en gran medida en los tejidos normales y enfermos. Una variedad de métodos se han establecido para interrogar a los patrones de metilación de citosina en las células. Representación reducida de toda la secuenciación del genoma de bisulfito fue desarrollado para detectar pares de bases patrones de metilación de citosina resolución cuantitativos en loci genómica GC-ricos. Esto se logra mediante la combinación del uso de una enzima de restricción seguido por la conversión de bisulfito. Enhanced representación reducida bisulfito de secuenciación (ERRBS) aumenta el loci genómico biológicamente relevante cubierto y se ha utilizado para perfil de metilación de citosina en el ADN de humano, ratón y otros organismos. ERRBS inicia con la restricción de la digestión enzimática de ADN para generar fragmentos de bajo peso molecular para uso en la preparación de la biblioteca. Estos fragmentos se someten a la construcción de bibliotecas estándar para la secuenciación de próxima generación. Bisulfito de conversión de citosinas no metiladas antes de la última amplificatide paso permite la resolución de base cuantitativa de los niveles de metilación de citosina en loci genómicos cubierto. El protocolo puede ser completada dentro de cuatro días. A pesar de baja complejidad en los primeros tres bases secuenciadas, bibliotecas ERRBS producen datos de alta calidad cuando se utiliza un carril de control de secuencia designada. Análisis de la cartografía y la bioinformática se realiza entonces y produce datos que se puede integrar fácilmente con una variedad de plataformas en todo el genoma. ERRBS puede utilizar pequeñas cantidades de material de entrada por lo que es factible para procesar las muestras clínicas humanas y aplicable en una amplia gama de aplicaciones de investigación. El video producido demuestra los pasos críticos del protocolo ERRBS.
Introducción
La metilación del ADN en citosina (5-metilcitosina) es una marca epigenética crítico en células de mamífero para una variedad de procesos biológicos, incluyendo, pero no limitado a la impresión, la inactivación del cromosoma X, el desarrollo, y la regulación de la expresión génica 1-8. El estudio de los patrones de metilación del ADN en los trastornos malignos y otros ha determinado los patrones específicos de la enfermedad y ha contribuido a la comprensión de la patogénesis de la enfermedad y los posibles descubrimientos de biomarcadores 9-17. Hay muchos protocolos que interrogan el epigenoma de estado de metilación del ADN. Estos se pueden dividir en basado afinidad, la enzima de restricción-basan, y ensayos basados conversión-bisulfito que utilizan microarrays o plataformas de secuenciación de aguas abajo. Además, hay algunos protocolos que unen estas categorías generales incluyendo, pero no limitado a, análisis de restricción combinado bisulfito de 18 y representación reducida bisulfito de secuenciación (RRB 19).
RRB fue descrito originalmente por Meissner et al. 19,20. El protocolo introdujo un paso para enriquecer regiones genómicas ricas en GC, seguido por secuenciación de bisulfito, lo que resultó en datos de resolución de pares de bases cuantitativa que es rentable 21,22. Las regiones ricas en GC son el blanco de la enzima de restricción MspI (C ^ CGG), y la metilación de citosina se resuelve mediante la conversión de bisulfito de citosinas (desaminación de citosinas no modificados a uracilo), seguido de la reacción en cadena de la polimerasa (PCR). RRB cubrieron la mayoría de los promotores de genes y las islas CpG en una fracción de la secuencia requerida para todo un genoma; sin embargo RRBs tenían cobertura limitada de costas CpG y otras regiones intergénicas de relevancia biológica. Varios grupos han publicado actualizado protocolos RRBs desde el informe original que mejorar en la metodología y cobertura resultante de estas regiones genómicas 23-25. Enhanced Representación reducido bisulfito Sequferencias (ERRBS) incluye modificaciones de preparación de biblioteca y una alineación de datos alternativa de aproximación 26 cuando se compara con los RRB. ERRBS resultó en un mayor número de CpG representadas en los datos generados y el aumento de la cobertura de todas las regiones genómicas interrogado 26. Este método ha sido utilizado para resolver los patrones de metilación del ADN en paciente humano y de otros especímenes de animales 26-30.
El protocolo descrito ERRBS ofrece detalles sobre todos los pasos necesarios para la terminación y los datos se generó utilizando ADN humano representante (muestras se obtuvieron a partir de informes anteriores, las muestras de pacientes de-identificado 31, y una muestra de médula ósea CD34 + a partir de un donante humano normal). El protocolo incluye un proceso de selección de tamaño automático, lo que reduce el tiempo de procesamiento por muestra y permite una mayor precisión en la selección de tamaño de la biblioteca. El protocolo combina una serie de técnicas de biología molecular establecidas. ADN de alto peso molecular se digiere wITH una enzima de restricción insensible a metilación (MspI), seguido de la reparación final, A-tizón, y ligación de adaptadores metilados. Selección del tamaño de los fragmentos ricos en GC es seguido por la conversión de bisulfito y la amplificación PCR antes de la secuenciación. Conversión de bisulfito se ha descrito previamente 32 y revisión detallada de análisis y aplicaciones de datos está más allá del alcance de este trabajo, sin embargo se incluyen recomendaciones y referencias para el uso de los lectores. El protocolo se puede realizar en cuatro días y es susceptible de pequeña entrada (50 ng o menos) las cantidades de material. El protocolo ya que los rendimientos descritos de datos con alta cobertura por sitio CpG suficiente no sólo para las determinaciones diferenciales sitio de metilación y la región, sino también para la detección de polimorfismo epigenética como se describe por Landan, et al. 33.
Protocolo
Todos los procedimientos ejecutados son aprobados por la Escuela de Medicina Institucional Cuidado de Animales y el empleo Comisión de la Universidad de Indiana y siguen Instituto Nacional de Salud directrices.
1. Técnica quirúrgica
- Mantenga una técnica aséptica durante este procedimiento mediante el uso de guantes estériles, instrumentos, y un campo quirúrgico estéril de acuerdo con las directrices del NIH 25. Esterilizar las herramientas antes de comenzar la cirugía mediante autoclave (véase la Tabla de específico Reactivos / Equipo para la lista completa). Utilice un esterilizador de perlas de vidrio para esterilizar las herramientas durante la operación.
2. Anestesia y Preparación
- Anestesiar al ratón en una caja de la anestesia con una mezcla de 0,9 L / min de oxígeno y 2,5% de isoflurano usando un sistema de vaporizador de isoflurano veterinaria. Asegúrese de que el ratón no responde a los cambios en la posición del cuerpo antes de sacarlo de la caja.
- Aplicar pomada oftálmica al mouLos ojos de sí para protegerlos de la desecación.
- Cambie el flujo de gas de la caja para el cono de la nariz. Coloque el ratón en ángulo recto en su lado izquierdo en una almohadilla de cubierta climatizada con una almohadilla quirúrgica y papel absorbente banco con su nariz y la boca en el interior del cono. Monitorear continuamente el ritmo respiratorio del ratón y la velocidad y ajustar los niveles de isoflurano según sea necesario (entre 2,5-3% isoflurano) para mantener un nivel adecuado de anestesia, y usar el pellizco reflejo dedo para confirmar la sedación total.
3. Abordaje quirúrgico
- Alinear y centrar el estereoscopio con el campo quirúrgico. Ajuste el cono de la nariz y la cinta hacia abajo para que se coloca a lo largo del borde del campo visual.
- Con el ratón acostado en su lado izquierdo, con cinta adhesiva el borde de la oreja derecha al cono de nariz, la exposición del área detrás de la oreja donde se realizará la incisión. Asegúrese de que la vena auricular posterior se desplaza horizontalmente a través de la oreja. Tenga en cuenta que la colocación correcta de tque los animales y la grabación de la oreja son cruciales para encontrar rápidamente el nervio facial.
- Humedezca la piel en y detrás de la oreja con etanol al 70% y afeitar el sitio quirúrgico utilizando una hoja de afeitar o bisturí. Pre-humedecer la piel hace que el afeitado más fácil en esta localización anatómica.
- Limpiar la piel con una solución de yodo, tales como lavado quirúrgico Betadine (7,5% de povidona-yodo), seguido por etanol al 70%. Repita esta limpieza dos veces más para desinfectar a fondo el área.
- Para determinar dónde hacer la incisión, traza la vena auricular posterior de la oreja caudalmente a la zona posterior a la protuberancia oído. Usando tijeras de primavera, hacer una incisión de 4 mm 2 - 3 mm posterior a la protuberancia.
- Diseccionar a través de la grasa subcutánea y la máscara con disección roma. Evite el corte directo con las tijeras porque los vasos sanguíneos o tejido muscular podrían dañarse fácilmente.
- Si se produce sangrado, aplique presión en el sitio de la cirugía con un hisopo de algodón estérildurante al menos 30 seg. Si se produce la pérdida de fluido significativa, inyectar el ratón por vía intraperitoneal con hasta 0,5 ml de solución salina al 0,9% estéril usando una aguja G 25 o 27.
- Utilice varios hitos clave, el nervio espinal accesorio, canal auditivo, y anterior del músculo digástrico (descrito más adelante), para localizar el nervio facial. Diseccionar alrededor de estos puntos de referencia hasta que se visualizan las ramas del nervio facial. El nervio aparecerá como una estructura de sólido blanco significativa cuando se revela y una capa de fascia se adhiere a las estructuras subyacentes.
- Encuentra el nervio espinal accesorio, que viaja de la porción caudal del cráneo para inervar el músculo trapecio, una vez que la grasa subcutánea y la fascia se han diseccionado. El nervio facial es profundo en el nervio espinal accesorio.
- Encuentra el conducto auditivo cartilaginoso que se ve de color blanco nacarado y se puede ver rostral al nervio facial.
- Encontrar el vientre muscular del músculo digástrico anterior que se encuentra en la parte superior de y caudal al nervio facial.
- Cuando se visualizan las principales ramas del nervio facial, rastrearlos dorsal para encontrar su origen en el agujero estilomastoideo. Utilizando pinzas de punta fina Dumont # 5/45 para mantener el lugar de la cirugía abierta, avanzar en las puntas de las tijeras de primavera siguientes trayectoria del nervio, a continuación, mueva las pinzas dorsal para mantener el área recién abierta avanzada.
- Visualice el tronco del nervio facial con el cigomático, bucal, y las ramas mandibulares marginales en este punto.
NOTA: La rama temporal se encontrará más cerca del agujero. Las ramas del nervio mandibular marginal en sus partes superior e inferior más cerca de la mandíbula, por lo tanto aquellas ramas nerviosas no serán visibles en este nivel.- Si se realiza un corte transversal del nervio, estabilizar el nervio suavemente con las pinzas de punta fina y cortar el nervio con las tijeras de primavera. Evite aplicar demasiada tracción en el nervio con las pinzas para evitar avulsing el nervio del tronco cerebral. Empujarlos tocones de distancia el uno del otro, o cortar y eliminar una porción del nervio distal para asegurar que no se produzca ninguna reconexión.
- Si la realización de una lesión por aplastamiento, utilice Dumont # 5/45 pinzas para comprimir el nervio durante 30 segundos usando una presión constante para cortar todos los axones, repita este flechazo en un segundo ángulo perpendicular al primer sitio de aplastamiento. Evite aplicar cantidades variables de presión durante la aglomeración 30 segundos, de lo contrario el daño será inconsistente entre los animales.
4. Cierre y Recuperación
- Vuelva a colocar la grasa y los músculos de las estructuras subyacentes.
- Aproximar los bordes de la incisión y cerrar la herida usando un clip herida 7,5 mm. Las suturas o pegamento también son aceptables para el cierre de la herida. Analgésicos posquirúrgicas se pueden proporcionar en este momento.
- Retire la cinta de la oreja del ratón. Apague el flujo isoflurano y permitir que el ratón para respirar oxígeno puro durante 30 segundos a 1 minuto. Place el ratón en una jaula vacía, sin ropa de cama para recuperarse de la anestesia.
- Cuando se recupera el ratón, examine su comportamiento en busca de signos de confirmación de la parálisis facial. Los bigotes se paralizaron y en ángulo hacia la mejilla, la nariz se produce una desviación, y el ojo no parpadeará en respuesta a un soplo de aire.
- Animales Casa en conjunto después de la cirugía si son de sexo femenino. Evite de ratones machos de forma conjunta, ya que son más agresivos y tienden a retirar por la fuerza clips de la herida de su cagemate, lo cual lleva a la infección. Proporcionar analgésicos posquirúrgicas en este momento, si es necesario.
- Monitorear los ratones una vez al día durante varios días después de la operación para asegurarse de que no hay infección u otra complicación se produce después de la operación. Retire los clips de la herida 7-10 días después de la cirugía si no han caído por su propia cuenta.
- Aplicar lubricante pomada ocular al ojo afectado diariamente para prevenir complicaciones de la córnea, o bien hasta que el reflejo del parpadeo del ojo es recubierta o hasta la eutanasia.
Resultados
La Figura 1 proporciona una visión general de ERRBS, destacando los pasos clave, que se explican a lo largo del protocolo descrito. Bibliotecas ERRBS se prepararon utilizando ADN de entrada 50 ng.
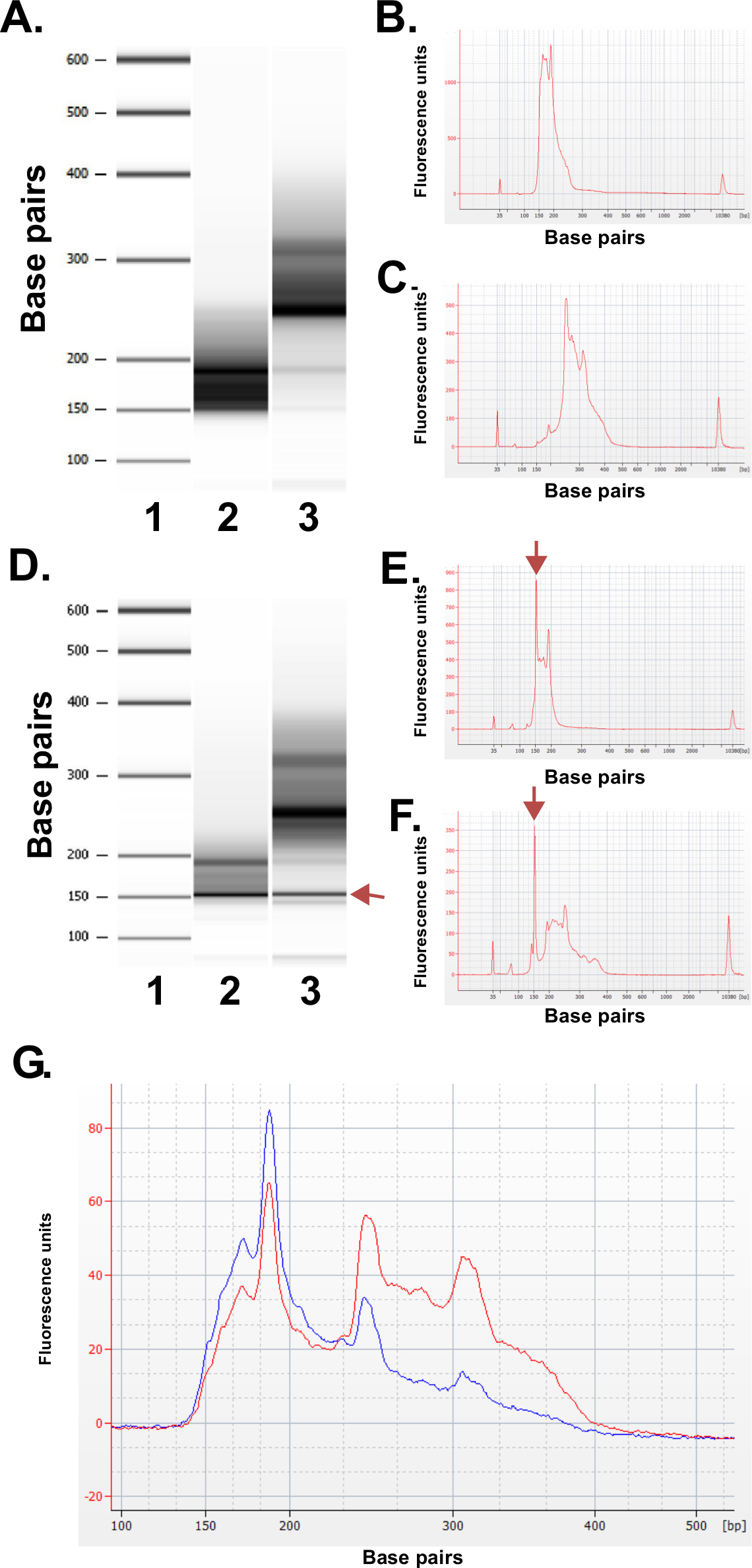
Evaluar la calidad de las bibliotecas preparadas. Producción Biblioteca produce rutinariamente tamaños de fracción de 150-250 pb y 250-400 pb (Figura 3A-C). Se espera que pequeñas diferencias en las distribuciones de tamaño de la biblioteca entre las muestras. Tenga en cuenta que en las dos fracciones de la biblioteca más bajos y más altos hay tamaños de ADN muy intensos, indicativos de enriquecimiento de una secuencia particular. MspI resultados digestión en el enriquecimiento de una familia de secuencias repetitivas de ADN presente en el genoma humano a 190 pb, 250 pb y 310 pb en las bibliotecas ERRBS. Estas tres repeticiones representan una firma característica de una biblioteca ERRBS 20 (véanse las Figuras 3A-C y 3G). Bibliotecas representativas fueron secuenciados en una secuencia de próxima generaciónr utilizando single-end lee. Al cargar a la concentración recomendada en una biblioteca Illumina HiSeq 2500 secuenciador, se espera que las densidades de racimo de 500.000-700.000 por mm2. A esta densidad de agrupamiento, 81.6% ± 3,14% (n = 81) de los grupos de filtro de paso (Figura 4A). Debido a la finalización de baja complejidad de los insertos de biblioteca (MspI sitio de reconocimiento: C ^ CGG), los valores de intensidad y niveles de calidad, grabadas durante la secuenciación son muy variables en los primeros tres bases (Figura 4B-C), sin embargo, si un carril de control independiente se incluye (véase el debate), el 85% de las bases tendrá puntuaciones de calidad de 30 o mayores (valores Q30; Figura 4D).
Alineación de datos y la determinación de metilación de citosina como se describe en los datos de protocolo de resolución de los rendimientos de pares de bases (Tabla 7). Para el genoma humano, un 51-ciclo secuenciación carrera sola lectura de una biblioteca ERRBS en un carril de un HiSeq 2500 en modo de alta salida normally genera 153194882 ± 12.918.302 Total de Lecturas que después de la filtración de la calidad y el adaptador de recorte rendimientos 152231183 ± 13189678 lee para la entrada en el análisis de tuberías. La eficiencia media de mapeo para una biblioteca ERRBS es típicamente 62,95% ± 5,92% con representación de 3.183.594 ± 713.547 CpG con una cobertura mínima por CpG de 10x y una cobertura promedio por CpG de 84,94 ± 16,29 (n = 100).
El protocolo ERRBS es susceptible de multiplexación (ver archivo Suplementaria 1: adaptación Protocolo para la secuenciación multiplexado). Los datos de secuenciación representante ejecuta se resume en la Figura 5 Los datos de carreras de secuenciación multiplexados (51-ciclo de ejecución de secuenciación de una sola lectura;. N = 128 para dos bibliotecas por carril; n = 11 para tres bibliotecas por carril; n = 11 para cuatro bibliotecas por carril) se compararon con un carril completo secuenciación de una biblioteca ERRBS (51 carreras de ciclo de una sola lectura de secuenciación; n = 100), así como la disminución de resolución un solo carril a Simulate 50%, 33% y 25% de lee por carril (2, 3, y 4 muestra la multiplexación por carril, respectivamente; n = 3). Como el número de lecturas por muestra disminuye con el factor de multiplexación, el número de CpG cubierto en una cobertura mínima de 10 veces y la cobertura por CpG también disminuye (Figura 5 y Tabla 8). Las tasas de conversión media de los sitios no CpG esperados son 99,85% ± 0,04% (n = 400). Las tasas de conversión más bajos del 99% pueden indicar menos de conversión de bisulfito óptima que puede resultar en altas tasas de los niveles de metilación falsas.
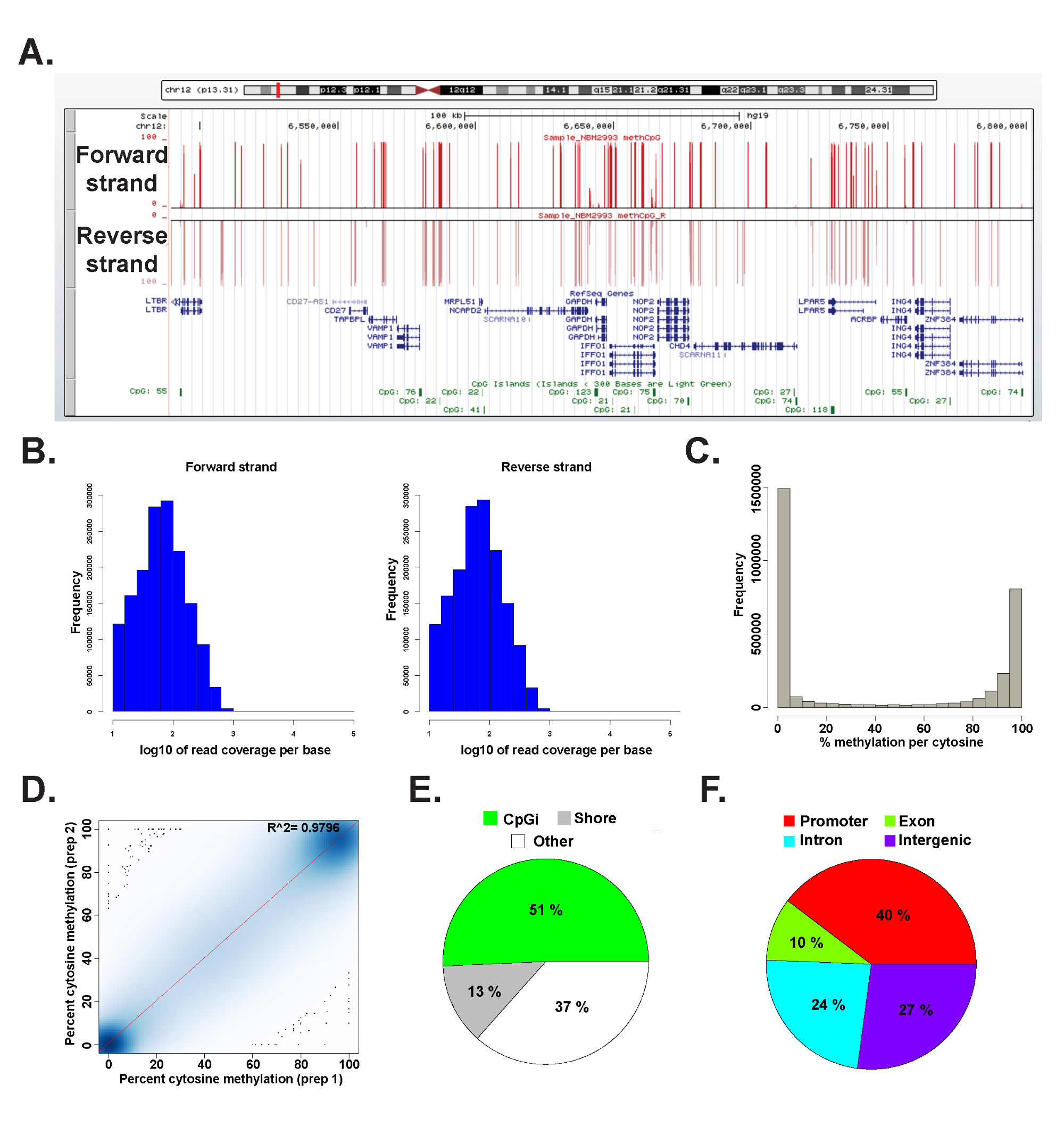
Los datos de una biblioteca ERRBS preparado a partir de un ADN genómico humano representante se analizó en I 2.15.2 45 utilizando el paquete methylKit 26 (ver archivo de código suplementario 1 para más detalles de mando). Los datos se pueden visualizar en el genoma navegadores de uso común (Figura 6A). Los datos de metilación de citosina es igualmente deriva de ambas cadenas (Figura 6B) y los rangos de todoespectro de los niveles de metilación de citosina potenciales (Figura 6c). Análisis de repeticiones técnica de un representante de los rendimientos de muestra de ADN humano de alta concordancia entre los resultados de los datos (Figura 6D) y cubre los CpG en un amplio espectro de loci del genoma (Figura 6E y F y como se ha descrito previamente 26). Mientras réplicas técnicas producirán alta R 2 valores (mayor que 97%), réplicas biológica producirá R 2 valores que van 0,92-,96 26, y que comparan diferentes tipos de células humanas producirá R 2 valores inferiores a 0,86 (datos no mostrados).

Figura 1: Diagrama de flujo de los pasos del protocolo ERRBS. Gráfico representa pasos, que se pueden completar en un día de trabajo tradicional. * Indica un punto de pausa potencial (siga inmediatamente ligadura ing limpieza y antes de la selección de tamaño, paso protocolo 5) en el que las muestras se puede congelar a -20 ° C antes de proceder con la duración de protocolo.

Figura 2: protocolo de selección Tamaño. (A) Captura de pantalla de la configuración utilizados en el protocolo ERRBS Pippin Prep (ver sección protocolo 5.1.2 - 5.1.6): (1) Seleccione el tipo de casete. (2) Seleccione el estándar que se utilizará. (3) Seleccione el modo de recogida para cada carril. (4) Introduzca los rangos colección pb. (5) Guarde el protocolo (B) Etapas de la extracción manual de gel utilizado en la sección 5.2 del protocolo:. (1) Las escaleras de gel visualizados. (2) tamaños para la selección por tamaño usando una cuchilla de afeitar Marcado. (3) Imagen de muestras extirpadas (fracción menor: 150-250 pb y una mayor fracción: 250-400 pb)."> Haga clic aquí para ver una versión más grande de esta figura.

Figura 3: Resultados de control de calidad para las bibliotecas ERRBS representativos preparados a partir de muestras de ADN humano utilizando una máquina bioanalizador. (A) Gel-como imagen que muestra una escalera estándar (1), fracción biblioteca inferior (135-240 pb fracción de Pippin Prep); 2) y la fracción de la biblioteca superior (240-410 pb fracción de Pippin Prep); . 3) (B) Bioanalyzer electroferograma de la fracción inferior biblioteca esperado (C) Bioanalyzer electroferograma de la mayor fracción de la biblioteca esperado D -.. F) Los datos representativos de una preparación de la biblioteca de mala calidad. -Gel como la imagen (D) de la escalera estándar (1), fracción de biblioteca inferior (2) y la fracción de la biblioteca superior (3). La banda a 150 pb marcado con un arrow indica cantidades excesivas de adaptador. Electroferograma de la inferior (E) y las fracciones de la biblioteca superiores (F) con los picos de adaptador en exceso de 150 pb (marcados con flechas). (G) Bioanalyzer electroferograma de una biblioteca ERRBS agrupado para la secuenciación. Rastro rojo representa una alta calidad biblioteca combinada con la igualdad de representación de las fracciones superiores e inferiores. Rastro azul representa una biblioteca agrupada no adecuado para la secuenciación debido a la falta de igualdad de representación de las fracciones superiores e inferiores. Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: La secuenciación de cartas para un representante ERRBS 51-ciclo de secuenciación de una sola lectura corrida en un secuenciador HiSeq 2500 en alto rendimientode modo. (A) la densidad de Racimo (K / mm 2 = 1.000 racimos por milímetro cuadrado; azul). Y densidades de racimo que pasan filtro (verde) en dos carriles con bibliotecas ERRBS (B) intensidades típicos observados en los primeros 30 ciclos en un carril con una biblioteca ERRBS. Nota la firma CGG de la digestión MspI en las intensidades de los tres primeros ciclos. (C) Porcentaje de bases con una puntuación de calidad de 30 o más (%> Q30) para cada ciclo en un carril ERRBS. (D) la distribución de puntuación de calidad todos los ciclos en un carril ERRBS. Azul = menos de Q30, Verde = mayor que o igual a Q30. En este carril, el 84,7% de las bases tenían puntuaciones de calidad de 30 o superior.

Figura 5: La secuenciación de los resultados de salida. Los diagramas de caja de los datos experimentales de la muestra multiplexados y solo por secuenciación carril ru ns (mostradas como cuadros de color verde) y de los datos derivados de la disminución de resolución simulada de carreras de secuenciación de tres ERRBS bibliotecas (aparece como cajas azules, muestra cinco veces en cada serie de secuenciación) de 51-ciclo de secuenciación de una sola lectura corre El factor de multiplexación corresponde. el número de bibliotecas ERRBS secuenciado por carril. 1 = conjunto de carril o 100% de lecturas y representa los datos de una sola biblioteca ERRBS por carril; 2 = 50% de carril y representa los datos de dos ERRBS bibliotecas por carril; 3 = 33% de un carril y representa los datos de tres ERRBS bibliotecas por carril; y, 4 = 25% de un carril y representa los datos de cuatro ERRBS bibliotecas por carril. (A) Los recuentos de lectura, o el número de secuencias analizadas, por factor de multiplexación. (B) El número de CpG de cubierta por los datos de secuenciación por multiplexación factor. (C) La cobertura media por CpG por factor de multiplexación._blank "> Haga clic aquí para ver una versión más grande de esta figura.

Figura 6: Los datos representativos de una biblioteca ERRBS preparada a partir de ADN genómico humano (A) de la Universidad de California, Santa Cruz (UCSC) genoma navegador 43 imagen de los datos representativos de un carril de secuenciación ERRBS.. La barra de escala eje y representa 0-100% de metilación de citosina en cada cubierta con un mínimo de 10 veces. La pista personalizada superior representa la cadena directa y la pista personalizada inferior representa la cadena inversa. Se muestra es chr12:.. 6,489,523-6,802,422 (hg19) incluyen todos los genes refseq e islas CpG en esta región genómica histogramas (B) Distribución de la cobertura a lo largo de CpG avance y retroceso hebras en un CD34 + humanas de médula ósea muestra representativa (C) histograma de distribuciónde los niveles de metilación CpG a lo largo de ambas cadenas en un CD34 + humanas de médula ósea muestra representativa. (D) Correlación parcela de los niveles de metilación CpG de una réplica técnico representante de una muestra de ADN humano. tabla (E) Pie ilustra las proporciones de las GPC cubiertos en ERRBS que anotadas, en las islas CpG (verde claro), CpG costas (gris) y otras regiones (blanco) en una muestra representativa preparada a partir de ADN genómico humano. tabla (F) Pie ilustra las proporciones de las GPC cubiertos en ERRBS que anotados a los promotores de genes (rojo ), los exones (verde), intrones (azules) y las regiones intergénicas (morado). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| Reactivo | Volumen | Comentario |
| 10x T4 ADN ligasa de tampón de reacción | 10 l | |
| Trifosfato Deoxynucleotide (dNTP) Solución Mix | 4 l | mezclar de 10 mM de cada nucleótido |
| T4 ADN polimerasa | 5 l | 3000 unidades / ml |
| ADN polimerasa I grande (Klenow) Fragmento | 1 l | 5.000 unidades / ml |
| T4 polinucleótido quinasa | 5 l | 10.000 unidades / ml |
| Agua libre de DNasa | 45 l |
Tabla 1: Fin. Reactivos de la reacción de reparación de nombres de reactivo y cantidades utilizadas en la reparación de reacción final (paso protocolo 2.1).
| Reactivo | Volumen | Comentario |
| Tampón de reacción 10x | 5 l | por ejemplo, NEBuffer 2 |
| 1 mM de 2'-desoxiadenosina 5'-trifosfato (dATP) | 10 l | |
| Fragmento Klenow (3 '→ 5' exo-) | 3 l | 5.000 unidades / ml |
Tabla 2:. A-tizón reactivos de reacción nombres de reactivo y cantidades utilizadas en la reacción A-tizón (paso protocolo 3.1).
| Reactivo | Volumen | Comentario |
| 15 mM adaptadores recocidos en agua libre de DNasa | 3 l | PE adaptador de 1,0 y PE adaptador de 2,0; véase la Tabla 4 para las secuencias y referencia |
| Tampón de reacción 10x T4 ADN ligasa | 581; l | |
| T4 ADN ligasa | 1 l | 2.000.000 unidades / ml |
| Agua libre de DNasa | 31 l |
Tabla 3: reactivos de ligación de adaptadores de reacción nombres de reactivo y cantidades utilizadas en la reacción de ligación del adaptador (paso protocolo 4.2)..

Tabla 4:. Oligos usados en el protocolo ERRBS Lista de oligos utilizados en todo el protocolo de ERRBS en la reacción de ligación (paso protocolo 4) y etapas de amplificación de PCR (paso 7) de protocolo.
| Reactivo | Volumen | Comentario |
| 10x FastStart Alta Fidelidad Reaction Buffer wiº mM de cloruro de magnesio 18 | 20 l | |
| 10 mM dNTP Mix Solución | 5 l | |
| 25 mM PCR PE imprimación 1.0 | 4 l | Ver Tabla 4 |
| 25 mM PCR PE imprimación 2.0 | 4 l | Ver Tabla 4 |
| Comienzo Acelerado High Fidelity Enzyme | 2 l | 5 unidades / l FastStart Taq ADN polimerasa |
| Agua libre de DNasa | 125 l |
Tabla 5: reactivos de la reacción de PCR nombres de reactivo y cantidades utilizadas en la reacción de amplificación PCR (protocolo de paso 7.1)..
| Paso Protocolo | Reactivo / detalle protocolo | Cantidad de ADN de entrada | ||
| 5-10 ng | 25 ng | 50 ng | ||
| 1 | Enzima MspI | 1 l | 2 l | 2 l |
| Volumen de reacción Mspl digest | 50 | 100 | 100 | |
| 4 | Adaptadores de reacción de ligación | 1 l | 2 l | 3 l |
| Volumen de reacción de ligación | 20 l | 25 l | 50 l | |
| 5 | Protocolo de selección Tamaño | Sólo gel Manual | Pippin Prep o gel Manual | Pippin Prep o gel Manual |
| 7 | La concentración de cebador de PCR | 25 mM | 25 mM | 10 M durante 14 ciclos; 25 m para 18 ciclos |
| Número de ciclos de PCR | 18 | 18 | 14-18 | |
Tabla 6:. Protocolo modificaciones paso para la entrada de las cantidades de materiales que van desde 5 hasta 50 ng varios pasos durante todo el protocolo necesario modificar las cantidades de reactivos utilizados para generar bibliotecas de calidad con diferentes cantidades de materiales de partida. Los cambios en las cantidades de reactivos clave se incluyen aquí. Ajuste tampón y los volúmenes de agua en las reacciones en consecuencia.
| Chr | Base | Hebra | Cobertura | freqC | freqT |
| chr1 | 10564 | R | 366 | 85.52 | 14.48 |
| chr1 | 10571 | F | 423 | 91.25 | 8.75 |
| chr1 | 10542 | F | 432 | 91.2 | 8.8 |
| chr1 | 10563 | F | 429 | 94.64 | 5.36 |
| chr1 | 10572 | R | 366 | 96.99 | 3.01 |
| chr1 | 10590 | R | 370 | 88.11 | 11.89 |
| chr1 | 10526 | R | 350 | 92 | 8 |
| chr1 | 10543 | R | 368 | 92.93 | 7.07 |
| chr1 | 10525 | F | 433 | 91.92 | 8.08 |
| chr1 | 10497 | F | 435 | 88.74 | 11.26 |
Tabla 7: Datos ERRBS representativos. Después de la alineación de datos y determinación de metilación de citosina, se obtiene un par de bases de datos Para cada CpG cubierto, el protocolo de alineación como se describe determinará la genómica coordenadas (columnas: chr = cromosoma, Base y Strand)., La tasa de cobertura del locus específico (Cobertura ), y la tasa de detección de citosina frente timidina como porcentaje (freqC y freqT respectivamente).
| Número de bibliotecas ERRBS por carril | Número medio de lecturas alineado de forma única | El número medio de CpG cubierto | Cobertura por CpG media |
| 1 | 152231184 ± 13189678 | 3.183.594 ± 713.547 | 85 ± 16 |
| 2 | 77680837 ± 7657058 | 2674823± 153494 | 49 ± 9 |
| 3 | 49938156 ± 2436865 | 2552186 ± - 76624 | 39 ± 2 |
| 4 | 34457208 ± 4441686 | 1.814.461 ± 144.339 | 28 ± 4 |
Tabla 8:. Parámetros representativos de la secuenciación de las bibliotecas ERRBS individuales y multiplexados Se muestra datos por línea de 51-ciclo se ejecuta una sola lectura de secuenciación: media y la desviación estándar de alineado de forma única lee, número de CpG cubierto y cobertura por sitio CpG obtenido a partir de la secuenciación de una sola bibliotecas ERRBS por carril (n = 100), dos bibliotecas ERRBS por carril (n = 128), tres ERRBS bibliotecas por carril (n = 11), y cuatro ERRBS bibliotecas por carril (n = 11).
Discusión
Los datos de resolución rendimientos pares de bases del protocolo presentado de metilación de citosina en regiones genómicas biológicamente relevantes. El protocolo escrito como está optimizado para 50 ng de material de partida, sin embargo, puede ser adaptada para manejar una amplia gama de material de entrada (5 ng o más) 26. Esto requerirá ajustes de algunos de los pasos del protocolo como se ve en la Tabla 6. Las bibliotecas ERRBS son susceptibles de secuenciación final emparejado y aún más la cobertura genómico también se puede lograr mediante secuenciación lee más de 51 ciclos. Secuenciación multiplexada ofrecerá un protocolo de menor costo por muestra, sin embargo, esto se traducirá en una cobertura reducida por sitio CpG representado en los datos (Figura 5 y Tabla 8), y no producir suficiente profundidad de cobertura para realizar análisis que requieren una alta cobertura por CpG sitio (por ejemplo, como se describe por Landan et al. 33). Por último, este protocolo (o cualquier protoco basada en bisulfitol) no puede distinguir entre metil-citosina y-hidroximetil citosina 46,47. Sin embargo, los datos generados se pueden integrar con otro protocolo de resultados 48,49 para delinear las diferentes modificaciones, y otras modificaciones de citosina recientemente reportaron 50, en caso de ser de su interés.
Aparecerán bibliotecas de alta calidad, como se muestra en la Figura 3A-C, y una vez combinado para la secuenciación produce una traza como se muestra en la Figura 3G (trazo rojo) que representa contribuciones molares iguales de ambas fracciones de la biblioteca. Insuficiencia preparación de la biblioteca puede resultar de cualquier etapa durante el procedimiento. Si se procesa ADN degradado que se traducirá en las bibliotecas que no están enriquecidas en fragmentos MspI y por lo tanto en la cobertura bajo CpG utilizando los parámetros de secuenciación descritos en este protocolo. Si una enzima no es funcional o inadvertidamente excluidos de una de las reacciones, el protocolo no dió la biblioteca esperado. Si el rea ligaduracción es ineficiente, adaptadores están en una concentración mayor de lo esperado, y / o la concentración de cebadores usado es un reactivo limitante para los pasos de amplificación final, se puede producir el fracaso de la biblioteca. El exceso de adaptadores (visto como picos en ~ 150 pb en los resultados del bioanalizador; Figura 3D-F) en la biblioteca también interferirá con la secuenciación debido al agrupamiento indiscriminado de la biblioteca y el exceso de adaptadores. Mientras que tales una biblioteca puede secuenciar aparentemente normalmente, una porción significativa de la lee será secuencias meramente adaptador. Si se observan exceso de adaptadores en una biblioteca, lo mejor es repetir la preparación de la biblioteca si el material está disponible utilizando material de entrada óptima al adaptador proporciones cuantitativas. Finalmente, para asegurar la amplificación por PCR eficiente de las bibliotecas, las fracciones de la biblioteca inferiores y superiores se mantienen como muestras separadas a lo largo de la conversión de bisulfito y etapas de enriquecimiento de PCR. De no hacerlo, se obtiene la eficacia diferencial de amplificación durante la PCR reacción de fracciones superiores e inferiores (como se ve en la Figura 3G traza azul) y el potencial de representación desigual de la respectiva loci genómico cubierto en cada fracción de la biblioteca durante la secuenciación. El usuario puede optar por incluir un paso de PCR cuantitativa inmediatamente después de la conversión de bisulfito para su posterior titulación de ciclos de PCR óptimos necesarios para amplificar las bibliotecas se generan.
ERRBS protocolo de preparación de biblioteca tiene varios pasos clave en el que se recomiendan reactivos específicos. En la etapa de reparación de extremo, el uso de una mezcla de dNTP de cuatro nucleótidos permite extremo reparación de cualquiera de los productos que no contienen el voladizo CG, tales como las resultantes de MspI actividad estrella enzimática y los fragmentos de ADN cortados presentes en la muestra de ADN original. Esto se traduce en una mejor representación CpG en los resultados. En la etapa de ligación es fundamental para utilizar una ligasa alta concentración (2.000.000 unidades / ml) y los adaptadores metilados para asegurar que el Reacti ligaduraen es eficiente y que la conversión de bisulfito no influye en las secuencias esenciales para la alineación de datos precisa de adaptador. En la etapa de PCR, usando una polimerasa capaz de amplificar fragmentos de ADN ricas en GC tratado con bisulfito es necesario para alta especificidad. Por último, para asegurar la eliminación del exceso de adaptadores y cebadores, purificación SPRI talón (por ejemplo: Agencourt AMPure XP) se recomienda en lugar de ensayos basados en columnas para la ligadura y aislamientos de productos de PCR.
Con el fin de generar datos de alta calidad, es importante para asegurar la conversión de bisulfito eficiente. El control presentado ofrece al usuario la capacidad de determinar la eficiencia de conversión antes de la secuenciación. Como alternativa, un ADN no humanos tales como ADN lambda se puede utilizar como un control interno (pico-in). Debido a las diferencias en las especies, este tipo de control puede incluirse directamente en la secuenciación de aguas abajo (por ejemplo, tal como se utiliza por Yu, et al. 34). Sin embargo, si el punto de I-ins utilizado, no puede ser utilizado para determinar la eficiencia de conversión antes de la secuenciación de la biblioteca amplificada a menos única e independientemente secuenciado antes de la secuenciación biblioteca. Los tipos de conversión determinados se basan en el estado de metilación en sitios no CpG. Esto puede no ser apropiado para el uso en el contexto de alta metilación de citosina en contexto no CpG (por ejemplo células madre embrionarias) y muestras paralelas o otros medios de evaluación de la eficiencia de conversión puede ser utilizado para este propósito.
Hay algunas advertencias a la dirección que son exclusivos de la secuenciación de las bibliotecas ERRBS. Las tres primeras bases de las fracciones de la biblioteca secuenciados son casi uniformemente no aleatoria debido a la sitio de corte reconocimiento MspI (C ^ CGG; véase la Figura 4B, C). Esto resulta en la posibilidad de pérdida de datos importantes debido a la baja calidad lee resultante de una mala localización grupo a pesar de la aparente alta densidad racimo durante la secuenciación. Para superar esta barrera,incluir una biblioteca de alta complejidad en un carril independiente (control PhiX u otro tipo de biblioteca) como un carril de control dedicado. Bibliotecas de alta complejidad tienen extremos que contienen una representación equilibrada de A, C, T y G en los primeros cuatro bases secuenciadas. Carriles de control adecuados incluyen bibliotecas tales como ARN-ss, chip-ss, la secuenciación del genoma, o un control ofrecido por el fabricante de la máquina de secuenciación (por ejemplo v3 control PhiX). Cuando designado como un carril de control para la respectiva ejecución de secuenciación, puede servir como base para la generación de la matriz que se utiliza durante los primeros cuatro bases de secuenciación para detectar posiciones de racimo. La mayor calidad lee capturado elevará la cobertura media por CpG sitio por 5,2 (n = 4). Alternativamente, esta dificultad técnica también se puede superar utilizando un enfoque de secuenciación oscuro como se ha descrito previamente 23. Otros criterios de secuenciación siguen procedimientos operativos estándar por los protocolos del fabricante. Por último, la cobertura por CpG ccalzas para el análisis de datos será guiada por el usuario y, en parte, por las cuestiones biológicas de interés. Umbral de cobertura de 10x proporciona un enfoque de análisis de alta cobertura, sin embargo este umbral se puede bajar debería que ser de interés.
Una discusión completa de análisis de datos ERRBS está más allá del alcance de este artículo, sin embargo, diferencialmente citosinas metiladas y regiones pueden determinarse utilizando herramientas de código abierto 31,51-53. Consideraciones de análisis adicionales y enfoques han sido bien descritos 54,55, y se anima al lector a buscar en la literatura para herramientas más adecuadas para el análisis planificado.
En comparación con otros métodos publicados, ERRBS ofrece un protocolo de cuatro días que, cuando se lleva a cabo ya que los rendimientos descritos altas tasas de reproducibilidad. Se ha validado en comparación con el estándar de oro MassARRAY EpiTYPER 26 años, es rentable para datos de alta cobertura, y es adaptable a diversos materiales de entradacantidades (favorables para el procesamiento de muestras clínicas y otros tipos de células de baja frecuencia) y enfoques de secuenciación. Se ofrece una resolución de pares de bases en loci biológicamente relevante y se puede utilizar en los análisis de integración con otras técnicas de perfilado factor de transcripción de todo el genoma de unión, remodelación de la cromatina, marcas epigenéticas y otras modificaciones de citosina de interés. El uso de datos ERRBS en tales estudios puede contribuir a un enfoque molecular amplia y permitir para alta análisis dimensionales en el estudio de modelos biológicos y enfermedades humanas.
Divulgaciones
Los autores no tienen conflictos de intereses a revelar.
Agradecimientos
We thank all the authors of the original ERRBS report. We thank Mame Fall for technical assistance. We acknowledge the Weill Cornell Medical College Epigenomics Core for technical services and assistance. The work was supported by a Sass Foundation Judah Folkman Fellowship, an NCI K08CA169055 and ASH-AMFDP12005 to FGB, NIH R01HG006798 and R01NS076465, funding from the Irma T. Hirschl and Monique Weill-Caulier Charitable Trusts and STARR Consortium (I7-A765) to CEM, and an LLS SCORE grant (7006-13) to AMM.
Materiales
| Name | Company | Catalog Number | Comments |
| MspI | New England Biolabs | R0106M | 100,000 units/ml |
| NEBuffer 2 | New England Biolabs | B7002S | Reaction buffer for MspI enzyme; protocol step 1.2 |
| Phenol solution | Sigma-Aldrich | P4557 | Equilibrated with 10 mM Tris HCl, pH 8.0; see safety and handling instructions at http://www.sigmaaldrich.com/catalog/product/sigma/p4557 |
| Chloroform | Sigma-Aldrich | C2432 | See safety and handling instructions at http://www.sigmaaldrich.com/catalog/product/sial/c2432 |
| Glycogen | Sigma-Aldrich | G1767 | 19-22 mg/ml |
| NaOAc | Sigma-Aldrich | S7899 | 3 M, pH 5.2 |
| Ethanol | Sigma-Aldrich | E7023 | 200 proof, for molecular biology |
| Buffer EB | Qiagen | 19086 | 10 mM Tris-Cl, pH 8.5 |
| tris(hydroxymethyl)aminomethane (Tris) | Sigma-Aldrich | T1503 | prepare a 1 M, pH 8.5 solution |
| Tris- Ethylenediaminetetraacetic acid (TE) | Sigma-Aldrich | T9285 | Dilute to 1x buffer solution per manufacturer's recommendations |
| T4 DNA Ligase Reaction Buffer | New England Biolabs | B0202S | 10x concentration |
| Deoxynucleotide triphosphate (dNTP) Solution Mix | New England Biolabs | N0447L | 10 mM each nucleotide |
| T4 DNA Polymerase | New England Biolabs | M0203L | 3,000 units/ml |
| DNA Polymerase I, Large (Klenow) Fragment | New England Biolabs | M0210L | 5,000 units/ml |
| T4 Polynucleotide Kinase | New England Biolabs | M0201L | 10,000 units/ml |
| QIAquick PCR Purification Kit | Qiagen | 28104 | Used for DNA product purification in protocol step 2.3 |
| 2'-deoxyadenosine 5'-triphosphate (dATP) | Promega | U1201 | 100 mM |
| Klenow Fragment (3'→5' exo-) | New England Biolabs | M0212L | 5,000 units/ml |
| MinElute PCR Purification Kit | Qiagen | 28004 | Used for DNA product purification in protocol step 3.3 |
| T4 DNA Ligase | New England Biolabs | M0202M | 2,000,000 units/ml |
| Methylation Adapter Oligo Kit | Illumina | ME-100-0010 | |
| Agencourt AMPure XP | Beckman Coulter | A63881 | Used in protocol sections that implement magnetic bead purification steps (steps 4.3 and 8.2). Equilibrate to room temperature before use. |
| Pippin Prep Gel Cassettes, 2% Agarose, dye-free | Sage Science | CDF2010 | with internal standards |
| Certified Low Range Ultra Agarose | Bio-Rad | 161-3106 | |
| Tris-Borate-EDTA (TBE) buffer | Sigma-Aldrich | T4415 | |
| Ethidium bromide solution | Sigma-Aldrich | E1510 | 10 mg/ml |
| 50 bp DNA Ladder | NEB | N3236S | |
| 100 bp DNA Ladder | NEB | N3231S | |
| Gel Loading Dye, Orange (6x) | NEB | B7022S | |
| Scalpel Blade No. 11 | Fisher Scientific | 3120030 | |
| QIAquick Gel Extraction Kit | Qiagen | 28704 | |
| EZ DNA Methylation Kit | Zymo Research | D5001 | Used in protocol step 6.2 |
| EZ DNA Methylation-Lightning Kit | Zymo Research | D5030 | Alternative for step 6.2 |
| Universal Methylated Human DNA Standard | Zymo Research | D5011 | Used as bisulfite conversion control |
| FastStart High Fidelity PCR System | Roche | 03553426001 | |
| Qubit dsDNA High Sensitivity Assay Kit | Life Technologies | Q32854 | A fluorescence-based DNA quantitation assay; used in protocol steps 1.1, 9.1 and 10.1 |
| DynaMag-2 Magnet | Life Technologies | 12321D | |
| High Sensitivity DNA Kit | Agilent Technologies | 5067-4626 | |
| 2100 Bioanalyzer | Agilent Technologies | ||
| PhiX Control v3 | Illumina | FC-110-3001 | |
| HiSeq 2500 | Illumina | ||
| Pippin Prep | Sage Science | ||
| Qubit 2.0 Fluorometer | Life Technologies | Q32872 | |
| TruSeq SR Cluster Kit v3-cBot-HS | Illumina | GD-401-3001 | |
| TruSeq SBS Kit v3-HS | Illumina | FC-401-3002 | |
| TruSeq RNA Sample prep | Illumina | RS-122-2001 | Barcoded adapters used for multiplexing libraries; See Supplemental file for multiplexing protocol. |
| Microcentrifuge | |||
| Vortex Mixer | |||
| Dry Block Heater | |||
| Thermal Cycler | |||
| Water Bath | |||
| Gel electrophoresis system | |||
| Electrophoresis power supply | |||
| Gel doc | |||
| UV or blue light transilluminator |
Referencias
- Jones, P. A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 13 (7), 484-492 (2012).
- Barlow, D. P. Genomic imprinting: a mammalian epigenetic discovery model. Annual Review Of Genetics. 45, 379-403 (2011).
- Thiagarajan, R. D., Morey, R., Laurent, L. C. The epigenome in pluripotency and differentiation. Epigenomics. 6 (1), 121-137 (2014).
- Reik, W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 447 (7143), 425-432 (2007).
- Hartnett, L., Egan, L. J. Inflammation, DNA methylation and colitis-associated cancer. Carcinogenesis. 33 (4), 723-731 (2012).
- Smith, Z. D., Meissner, A. DNA methylation: roles in mammalian development. Nat Rev Genet. 14 (3), 204-220 (2013).
- Li, E., Bestor, T. H., Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 69 (6), 915-926 (1992).
- Okano, M., Bell, D. W., Haber, D. A., Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 99 (3), 247-257 (1999).
- Feinberg, A. P. Phenotypic plasticity and the epigenetics of human disease. Nature. 447 (7143), 433-440 (2007).
- Bock, C. Epigenetic biomarker development. Epigenomics. 1 (1), 99-110 (2009).
- Laird, P. W. The power and the promise of DNA methylation markers. Nat Rev Cancer. 3 (4), 253-266 (2003).
- How Kit, A., Nielsen, H. M., Tost, J. DNA methylation based biomarkers: practical considerations and applications. Biochimie. 94 (11), 2314-2337 (2012).
- Mikeska, T., Bock, C., Do, H., Dobrovic, A. DNA methylation biomarkers in cancer: progress towards clinical implementation. Expert Review Of Molecular Diagnostics. 12 (5), 473-487 (2012).
- Gyparaki, M. T., Basdra, E. K., Papavassiliou, A. G. DNA methylation biomarkers as diagnostic and prognostic tools in colorectal cancer. Journal of Molecular Medicine. 91 (11), 1249-1256 (2013).
- Figueroa, M. E., et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 17 (1), 13-27 (2010).
- Heyn, H., Mendez-Gonzalez, J., Esteller, M. Epigenetic profiling joins personalized cancer medicine. Expert review of Molecular Diagnostics. 13 (5), 473-479 (2013).
- Kulis, M., Esteller, M. DNA methylation and cancer. Advances in Genetics. 70, 27-56 (2010).
- Xiong, Z., Laird, P. W. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 25 (12), 2532-2534 (1997).
- Meissner, A., et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 33 (18), 5868-5877 (2005).
- Gu, H., et al. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc. 6 (4), 468-481 (2011).
- Bock, C., et al. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol. 28 (10), 1106-1114 (2010).
- Harris, R. A., et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol. 28 (10), 1097-1105 (2010).
- Boyle, P., et al. Gel-free multiplexed reduced representation bisulfite sequencing for large-scale DNA methylation profiling. Genome Biol. 13 (10), R92(2012).
- Chatterjee, A., Rodger, E. J., Stockwell, P. A., Weeks, R. J., Morison, I. M. Technical considerations for reduced representation bisulfite sequencing with multiplexed libraries. Journal of Biomedicine & Biotechnology. 2012, 741542(2012).
- Lee, Y. K., et al. Improved reduced representation bisulfite sequencing for epigenomic profiling of clinical samples. Biological Procedures Online. 16 (1), 1(2014).
- Akalin, A., et al. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet. 8 (6), e1002781(2012).
- Hatzi, K., et al. A Hybrid Mechanism of Action for BCL6 in B Cells Defined by Formation of Functionally Distinct Complexes at Enhancers and Promoters. Cell Reports. 4 (3), 578-588 (2013).
- Will, B., et al. Satb1 regulates the self-renewal of hematopoietic stem cells by promoting quiescence and repressing differentiation commitment. Nature Immunology. 14 (5), 437-445 (2013).
- Lu, C., et al. Induction of sarcomas by mutant IDH2. Genes Dev. 27 (18), 1986-1998 (2013).
- Kumar, R., et al. AID stabilizes stem-cell phenotype by removing epigenetic memory of pluripotency genes. Nature. 500 (7460), 89-92 (2013).
- Li, S., et al. An optimized algorithm for detecting and annotating regional differential methylation. BMC Bioinformatics. 14, Suppl 5. S10(2013).
- Patterson, K., Molloy, L., Qu, W., Clark, S. DNA methylation: bisulphite modification and analysis. Journal of Visualized Experiments. (56), 3170(2011).
- Landan, G., et al. Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat Genet. 44 (11), 1207-1214 (2012).
- Yu, M., et al. Tet-assisted bisulfite sequencing of 5-hydroxymethylcytosine. Nat Protoc. 7 (12), 2159-2170 (2012).
- Goecks, J., Nekrutenko, A., Taylor, J., Galaxy, T. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 11 (8), R86(2010).
- Dorff, K. C., et al. GobyWeb: simplified management and analysis of gene expression and DNA methylation sequencing data. PLoS One. 8 (7), e69666(2013).
- Roehr, J. T., Dodt, M., Ahmed, R., Dieterich, C. Flexbar − flexible barcode and adapter processing for next-generation sequencing platforms. MDPI Biology. 1 (3), 895-905 (2012).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal, North America. 17 (1), 10-12 (2011).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (15), 2114-2120 (2014).
- Needleman, S. B., Wunsch, C. D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 48 (3), 443-453 (1970).
- Krueger, F., Andrews, S. R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 27 (11), 1571-1572 (2011).
- Li, H., et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25 (16), 2078-2079 (2009).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Res. 12 (6), 996-1006 (2002).
- Thorvaldsdottir, H., Robinson, J. T., Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in Bioinformatics. 14 (2), 178-192 (2013).
- Team, R. C. R. A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria. ISBN 3-900051-07-0, http://www.R-project.org (2012).
- Nestor, C., Ruzov, A., Meehan, R., Dunican, D. Enzymatic approaches and bisulfite sequencing cannot distinguish between 5-methylcytosine and 5-hydroxymethylcytosine in DNA. BioTechniques. 48 (4), 317-319 (2010).
- Huang, Y., et al. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS One. 5 (1), e8888(2010).
- Yu, M., et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 149 (6), 1368-1380 (2012).
- Song, C. X., et al. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell. 153 (3), 678-691 (2013).
- Ito, S., et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 333 (6047), 1300-1303 (2011).
- Akalin, A., et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 13 (10), R87-1186 (2012).
- Stockwell, P. A., Chatterjee, A., Rodger, E. J., Morison, I. M. DMAP: Differential Methylation Analysis Package for RRBS and WGBS data. Bioinformatics. 30 (13), 1814-1822 (2014).
- Sun, D., et al. MOABS: model based analysis of bisulfite sequencing data. Genome Biol. 15 (2), R38(2014).
- Bock, C. Analysing and interpreting DNA methylation data. Nat Rev Genet. 13 (10), 705-719 (2012).
- Rivera, C. M., Ren, B. Mapping human epigenomes. Cell. 155 (1), 39-55 (2013).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados