
Method Article
Enhanced réduit Représentation bisulfite séquençage pour l'évaluation de méthylation de l'ADN à la résolution de paires de base
* Ces auteurs ont contribué à parts égales
Dans cet article
Résumé
Enhanced Reduced Representation Bisulfite Sequencing is a method for the preparation of sequencing libraries for DNA methylation analysis based on restriction enzyme digestion combined with cytosine bisulfite conversion. This protocol requires 50 ng of starting material and yields base pair resolution data at GC-rich genomic regions.
Résumé
cartographie du réseau de méthylation de l'ADN est fortement étudié dans les tissus normaux et malades. Une variété de procédés ont été mis en place pour interroger les schémas de méthylation de cytosine dans des cellules. Représentation réduite du séquençage du génome de bisulfite ensemble a été conçu pour détecter des modèles de résolution de méthylation de cytosine quantitatives de paires de bases à des loci génomiques riche en GC. Ceci est réalisé en combinant l'utilisation d'une enzyme de restriction suivie de la conversion au bisulfite. Représentation accrue réduit Bisulfite Sequencing (ERRBS) augmente le locus génomique d'intérêt biologique couvert et a été utilisé pour profiler méthylation de cytosine dans l'ADN d'humain, de souris et d'autres organismes. ERRBS amorce avec une enzyme de restriction digestion de l'ADN pour générer des fragments de bas poids moléculaire pour une utilisation dans la préparation bibliothèque. Ces fragments sont soumis à la construction de la bibliothèque standard pour le séquençage de prochaine génération. Conversion au bisulfite de cytosines non méthylés avant la finale amplificatià l'étape permet la résolution de base quantitative des niveaux de méthylation cytosine dans des loci génomiques couverte. Le protocole peut être effectuée dans les quatre jours. Malgré une faible complexité dans les trois premières bases séquencées, bibliothèques ERRBS fournissent des données de haute qualité en utilisant une voie de commande de séquencement désigné. analyse de la cartographie et de la bio-informatique est alors effectuée et fournit des données qui peut être facilement intégré avec une variété de plates-formes de l'ensemble du génome. ERRBS peut utiliser de petites quantités de matières entrantes qui rend possible de traiter des échantillons cliniques humains et applicable dans une gamme d'applications de recherche. La vidéo produite démontre étapes critiques du protocole de ERRBS.
Introduction
Méthylation de l'ADN à la cytosine (5-méthylcytosine) est une marque épigénétique critique dans les cellules de mammifères pour une variété de processus biologiques, y compris mais non limité à l'empreinte, l'inactivation du chromosome X, le développement, et la régulation de l'expression génique 1-8. L'étude des modèles de méthylation d'ADN dans les troubles malignes et d'autres a déterminé les habitudes spécifiques de maladies et contribué à la compréhension de la pathogenèse de la maladie et de découvertes de biomarqueurs potentiels 9-17. Il existe de nombreux protocoles qui interrogent l'épigénome de statut de méthylation d'ADN. Ceux-ci peuvent être divisées en base d'affinité, sur la base-enzyme de restriction, et des essais à base de conversion-bisulfite qui utilisent des puces à ADN ou les plates-formes en aval séquençage. En outre, il ya quelques protocoles qui comblent ces catégories générales, y compris, mais sans s'y limiter, l'analyse combinée de restriction bisulfite 18 et réduit Représentation bisulfite séquençage (ORR 19).
RRBS a été initialement décrit par Meissner et al. 19,20. Le protocole introduit une étape d'enrichir régions génomiques riches en GC suivie par séquençage bisulfite, qui a abouti à des données quantitatives de la résolution paires de bases qui est rentable 21,22. Les régions riches en GC sont ciblés par le MspI (C ^ CGG) enzyme de restriction, et la cytosine méthylation est résolu par conversion au bisulfite de cytosines (désamination des cytosines non modifiés à l'uracile), suivie de la réaction en chaîne par polymérase (PCR). RRBS couvert la majorité des promoteurs de gènes et îlots CpG dans une fraction du séquençage requis pour un génome entier; cependant RRBS avaient une couverture limitée des rives de CpG et d'autres régions intergéniques de pertinence biologique. Plusieurs groupes ont publié des mises à jour des protocoles ORR depuis le rapport original améliorer la méthodologie et la couverture résultante de ces régions génomiques 23-25. Représentation accrue réduit bisulfite Sequdes diffi- (ERRBS) comprend des modifications de préparation bibliothèque et un alignement des données alternatif approche 26 par rapport à RRBS. ERRBS a entraîné une augmentation du nombre de CpG représentées dans les données générées et augmentation de la couverture de toutes les régions génomiques interrogé 26. Cette méthode a été utilisée pour résoudre des modèles de méthylation d'ADN dans patient humain et d'autres spécimens d'animaux de 26 à 30.
Le protocole de ERRBS décrit offre des détails sur toutes les mesures nécessaires pour l'achèvement et des données a été généré en utilisant l'ADN humain représentant (échantillons ont été obtenus à partir signalés précédemment, dépersonnalisées échantillons de patients 31 et CD34 + os échantillon de moelle d'un donneur humain normal). Le protocole comprend un processus de sélection du format automatisé, ce qui réduit le temps de traitement par l'échantillon et permet une précision accrue dans la sélection de la taille de bibliothèque. Le protocole combine une série de techniques de biologie moléculaire établies. ADN de haut poids moléculaire est digéré avecvec une enzyme de restriction de méthylation insensible (MspI) suivi d'ici la fin de réparation, A-tailing, et la ligature d'adaptateurs méthylés. Choix de la taille des fragments riches en GC est suivie par conversion au bisulfite et d'amplification par PCR avant le séquençage. Conversion au bisulfite a été décrit précédemment 32 et un examen détaillé de l'analyse des données et des applications est au-delà de la portée de ce document, cependant recommandations et des références sont inclus pour l'utilisation des lecteurs. Le protocole peut être effectuée sur quatre jours et se prête à la petite entrée (50 ng ou moins) des montants significatifs. Le protocole fournit des données décrits à forte couverture par site CpG non seulement suffisante pour site de méthylation et la région déterminations différentielles mais également pour la détection du polymorphisme épigénétique comme décrit par Landan, et al. 33.
Protocole
Toutes les procédures exécutées sont approuvés par l'École de médecine institutionnel de protection des animaux et l'utilisation Comité Université de l'Indiana et suivent Institut national de lignes directrices de la santé.
1. Technique chirurgicale
- Maintenir une technique aseptique lors de cette procédure en utilisant des gants stériles, des instruments et un champ opératoire stérile selon les directives du NIH 25. Stériliser les outils avant de commencer la chirurgie par eux autoclavage (voir le tableau des réactifs spécifiques / Équipement pour la liste complète). Utiliser un stérilisateur à billes de verre pour stériliser les outils pendant l'opération.
2. Anesthésie et préparation
- Anesthésier la souris dans une boîte d'anesthésie avec un mélange de 0,9 L / min d'oxygène et 2,5% d'isoflurane utilisant un système de vaporisateur isoflurane vétérinaire. Assurez-vous que la souris ne répond pas à des changements dans la position du corps avant de le retirer de la boîte.
- Appliquer une pommade ophtalmique à l'mouLes yeux de soi pour les protéger du dessèchement.
- Mettez le débit de gaz dans la zone du cône de nez. Placez la souris carrément sur son côté gauche sur un bloc couverte chauffée avec un tampon chirurgical et papier absorbant banc avec son nez et la bouche à l'intérieur du cône. Surveiller en permanence le rythme respiratoire et le taux de la souris et ajuster les niveaux isoflurane selon les besoins (entre 2,5 à 3% d'isoflurane) pour maintenir un niveau adéquat d'anesthésie, et utiliser le pincement de l'orteil réflexe pour confirmer la sédation totale.
3. Approche chirurgicale
- Aligner et concentrer le stéréoscope avec le champ opératoire. Ajuster le cône de nez et la bande vers le bas de sorte qu'il est positionné le long du bord du champ visuel.
- Avec la souris couché sur le côté gauche, la bande du bord de l'oreille droite à le cône de nez, exposant la zone située derrière l'oreille où l'incision sera faite. Assurez-vous que la veine auriculaire postérieure se déplace horizontalement à travers l'oreille. Notez que le placement correct de til animale et enregistrement de l'oreille sont cruciales afin de trouver rapidement le nerf facial.
- Mouiller la fourrure sur et derrière l'oreille avec 70% d'éthanol et de se raser le site chirurgical aide d'un rasoir ou un scalpel lame. Prémouillage la fourrure facilite le rasage à cet endroit anatomique.
- Nettoyer la peau avec une solution d'iode, telles que le lavage chirurgical Betadine (7,5% de povidone-iode), suivie de 70% d'éthanol. Répéter ce nettoyage deux fois plus pour désinfecter soigneusement la zone.
- Pour déterminer où faire l'incision, oligo la veine auriculaire postérieure de l'oreille caudale vers la zone postérieure de la protubérance de l'oreille. Avec des ciseaux de printemps, faire une incision de 4 mm 2 - 3 mm en arrière à la protubérance.
- Disséquer par la graisse sous-cutanée et le fascia utilisant dissection. Evitez de couper direct avec les ciseaux, car les vaisseaux sanguins ou les tissus musculaires peuvent être facilement endommagés.
- En cas de saignement, appliquer une pression sur le site chirurgical avec un coton-tige stérilependant au moins 30 sec. Si la perte de liquide importante se produit, injecter la souris par voie intraperitoneale avec 0,5 ml de solution saline stérile à 0,9% en utilisant une aiguille G 25 ou 27.
- Utilisez plusieurs sites clés, le nerf spinal, canal de l'oreille, et antérieure digastrique (décrit ci-dessous), pour localiser le nerf facial. Disséquer autour de ces sites jusqu'à ce que les branches du nerf facial sont visualisés. Le nerf apparaît comme une structure solide blanc importante quand il se est révélé et une couche de fascia adhère à des structures sous-jacentes.
- Trouver le nerf spinal, qui se déplace de la partie caudale du crâne pour innerver le muscle trapèze, une fois que la graisse sous-cutanée et le fascia ont été disséqués. Le nerf facial est profonde du nerf spinal.
- Trouvez le conduit auditif cartilagineux qui ressemble blanc nacré et peut être vu rostrale du nerf facial.
- Trouver le ventre musculaire du muscle digastrique antérieure qui se trouve sur le dessus et caudal du nerf facial.
- Lorsque les principales branches du nerf facial sont visualisées, les tracer le dos pour trouver leur origine du foramen stylomastoïdien. En utilisant une pince fine pointe de Dumont # 5/45 pour maintenir le site chirurgical ouvert, avancer les conseils printemps ciseaux suivants le chemin de nerf, puis déplacez la pince le dos pour garder la zone nouvellement ouverte avancée.
- Visualisez le tronc du nerf facial avec le zygomatique, buccale et branches mandibulaires marginaux à ce point.
REMARQUE: La branche temporelle sera trouvé plus près des foramen. Les branches nerveuses mandibulaires marginaux dans ses parties supérieure et inférieure de plus près à la mâchoire, ainsi ces branches nerveuses ne seront pas visibles à ce niveau.- Si l'exécution d'une transection du nerf, de stabiliser le nerf doucement avec les pinces fines de pointe et de couper le nerf avec les ciseaux à ressort. Éviter d'appliquer trop de traction du nerf avec la pince pour empêcher avulsing le nerf partir du tronc cérébral. Pousserles souches de distance les uns des autres, ou couper et enlever une partie du nerf distale pour se assurer qu'aucun reconnexion peut se produire.
- Si l'exécution d'une lésion par écrasement, utiliser Dumont # 5/45 pince pour comprimer le nerf pendant 30 secondes en utilisant une pression constante pour rompre tous les axones, puis répétez cette cohue à un second angle perpendiculaire à la première place à l'écrasement. Eviter d'appliquer des quantités variables de pression pendant la cohue de 30 secondes, sinon la blessure ne sera pas uniforme entre les animaux.
4. clôture et récupération
- Repositionner la graisse et les muscles sur les structures sous-jacentes.
- Rapprocher les bords de l'incision et de fermer la plaie en utilisant un clip de la plaie de 7,5 mm. Sutures ou de la colle est aussi acceptable pour la fermeture des plaies. Analgésiques post-opératoires peuvent être fournis à ce moment.
- Retirer le ruban de l'oreille de la souris. Éteignez le flux de l'isoflurane et laissez la souris pour respirer l'oxygène pur pendant 30 sec à 1 min. Place la souris dans une cage vide sans litière pour remettre de l'anesthésie.
- Lorsque la souris est récupéré, examiner son comportement pour des signes de confirmation de la paralysie faciale. Les moustaches seront paralysés et inclinés vers la joue, le nez sera dévié, et l'œil ne clignote pas en réponse à une bouffée d'air.
- Animaux Maison conjointement après la chirurgie si elles sont des femmes. Évitez héberge des souris conjointement parce qu'ils sont plus agressifs et ont tendance à retirer de force les clips de la plaie de leur compagnon de cage, ce qui conduit à l'infection. Fournir analgésiques post-opératoires en ce moment, si nécessaire.
- Surveiller les souris une fois par jour pendant plusieurs jours après l'opération afin de se assurer qu'aucune infection ou d'autres complications se produisent après l'opération. Retirer des agrafes 7-10 jours après la chirurgie se ils ne sont pas tombés sur leur propre.
- Appliquer pommade ophtalmique lubrifiante à l'œil touchée par jour pour prévenir les complications de la cornée, soit jusqu'à ce que le réflexe de clignement des yeux est rerecouverts ou jusqu'à l'euthanasie.
Résultats
Figure 1 donne un aperçu des ERRBS, soulignant les étapes clés, qui sont expliquées à travers le protocole décrit. ERRBS bibliothèques ont été préparés en utilisant l'ADN d'entrée de 50 ng.
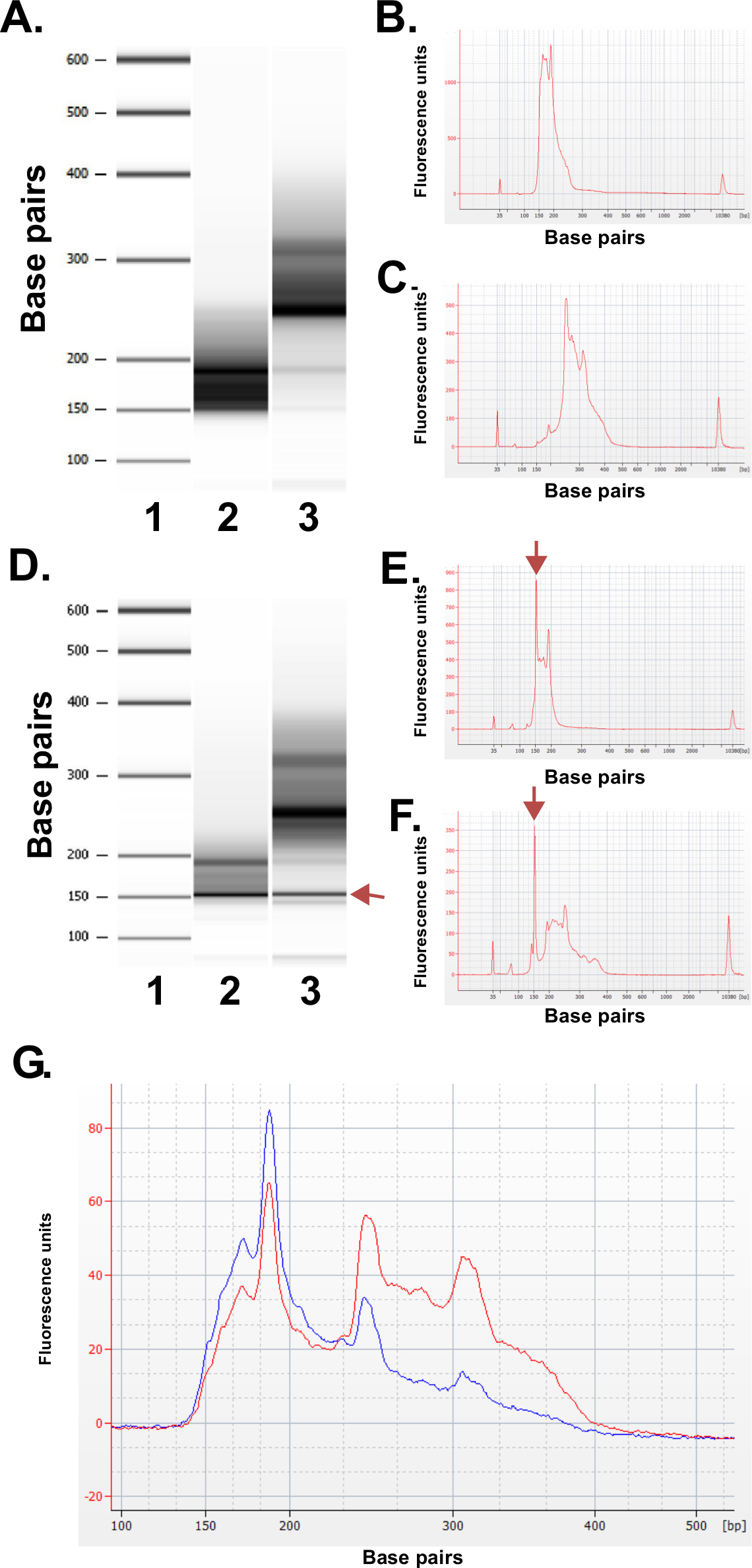
Évaluer la qualité des bibliothèques préparés. Bibliothèque production donne régulièrement tailles fraction de 150-250 pb et de 250 à 400 pb (figure 3A-C). De légères différences dans les distributions de taille de la bibliothèque entre les échantillons sont attendus. On notera que dans les deux fractions bibliothèque inférieures et supérieures sont là tailles d'ADN très intenses, indicatifs de l'enrichissement d'une séquence particulière. Les résultats de la digestion Mspl dans l'enrichissement d'une famille de l'ADN répétitif séquences présentes dans le génome humain à 190 pb, 250 pb et 310 pb dans les bibliothèques de ERRBS. Ces trois répétitions représentent une signature caractéristique d'une bibliothèque ERRBS 20 (voir les figures 3A-C et 3G). Bibliothèques représentatifs ont été séquencées sur une séquence de prochaine générationr en utilisant une seule fin lit. Lors du chargement à la concentration de la bibliothèque recommandée sur un séquenceur Illumina HiSeq 2500, la densité de la grappe de 500.000-700.000 par mm 2 sont attendus. A cette densité de clustering, 81,6% ± 3,14% (n = 81) des grappes filtre passe (figure 4A). En raison de la fin de faible complexité des inserts de la bibliothèque (MspI site de reconnaissance: C ^ CGG), valeurs d'intensité et les scores de qualité enregistrés au cours du séquençage sont très variables dans les trois premières bases (figure 4B-C), cependant, si une voie de contrôle indépendant est inclus (voir la discussion), 85% des bases aura scores de qualité de 30 ou plus (valeurs Q30; Figure 4D).
l'alignement des données et de détermination de la méthylation de la cytosine comme décrit dans les données de résolution rendements de protocole paires de bases (tableau 7). Pour le génome humain, un 51-seul cycle lire terme de séquençage d'une bibliothèque ERRBS sur une voie d'un HiSeq 2500 en mode haute de sortie régulièreLy génère 153 194 882 ± 12.918.302 totale lit que, après filtrage de la qualité et l'adaptateur coupe rendements 152231183 ± 13.189.678 lit pour l'entrée dans le pipeline d'analyse. L'efficacité de la cartographie moyenne pour une bibliothèque de ERRBS est généralement 62,95% ± 5,92% avec une représentation de 3.183.594 ± 713 547 CpG avec une couverture minimum par CpG de 10x et d'une couverture moyenne par CpG de 84,94 ± 16,29 (n = 100).
Le protocole de ERRBS se prête à multiplexage (voir le fichier complémentaire 1: Protocole d'adaptation pour le séquençage multiplexé). Les données de séquençage représentant exécute est résumé dans la figure 5 Les données de pistes de séquençage multiplexés (51 seul cycle lire terme de séquençage;. N = 128 pour les deux bibliothèques par voie; n = 11 pour trois bibliothèques par voie; n = 11 pour quatre bibliothèques par voie) ont été comparés à un séquençage pleine voie d'une bibliothèque de ERRBS (51-cycle unique lire pistes de séquençage; n = 100) ainsi que le sous-échantillonnage une seule voie à SIMULATe 50%, 33% et 25% du lit par voie (2, 3, et 4 échantillons de multiplexage par voie respectivement; n = 3). Comme le nombre de lectures par échantillon diminue avec le facteur de multiplexage, le nombre de CpG couvertes à une couverture minimale de 10x et la couverture par CpG diminue ainsi (figure 5 et tableau 8). Taux moyen de conversion de sites non-CpG attendus sont 99,85% ± 0,04% (n = 400). des taux inférieurs à 99% de conversion peuvent indiquer moins de conversion au bisulfite optimale qui peut entraîner des taux élevés de niveaux de méthylation faux.
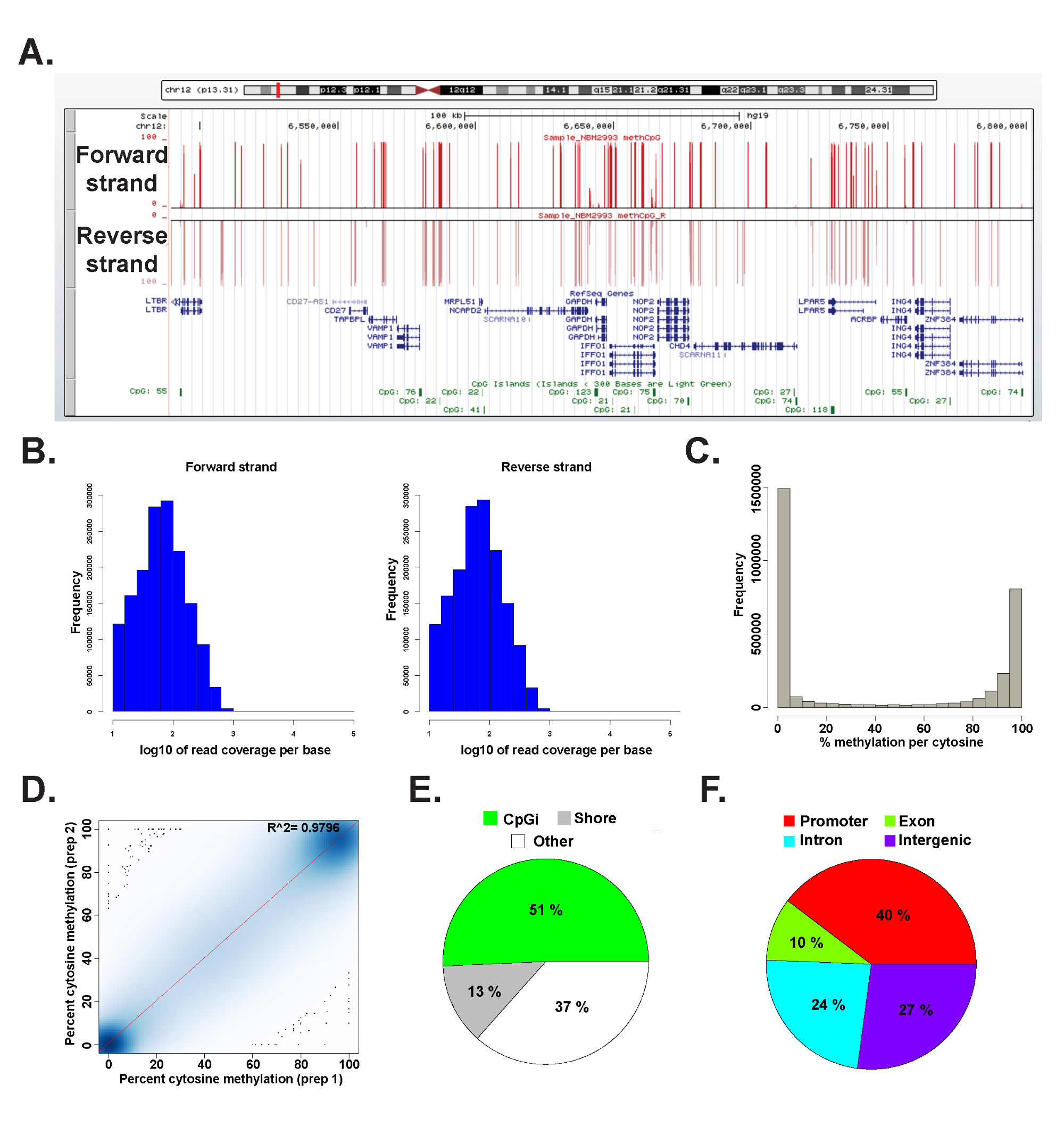
Les données d'une bibliothèque ERRBS préparé à partir d'un ADN génomique humain représentant a été analysée dans R 2.15.2 45 utilisant le paquet methylKit 26 (voir fichier de code supplémentaire 1 pour les détails de la commande). Les données peuvent être visualisées dans les navigateurs de génome couramment utilisés (figure 6A). Les données de méthylation de cytosine est également dérivée de deux brins (figure 6B) et se étend tout lespectre de niveaux de méthylation de cytosine potentiels (Figure 6C). Analyse des répétitions techniques d'une concordance élevée des rendements représentatifs d'échantillons d'ADN humains entre les résultats de données (Figure 6D) et couvre CpG dans un large éventail de loci génomiques (Figure 6E et F et comme décrit précédemment 26). Alors que des répliques techniques donneront haute R 2 valeurs (supérieure à 97%), des répliques biologique apportera des deux valeurs allant 0,92 au 0,96 26, et la comparaison de différents types de cellules humaines donnera R 2 valeurs inférieures à 0,86 (données non présentées) R.

Figure 1: Diagramme des étapes du protocole ERRBS. Graphique représente étapes, qui peuvent être complétés dans une journée de travail traditionnelle. * Indique un point de pause potentiel (suivent immédiatement ligature ing nettoyage et avant la sélection de la taille, le protocole étape 5) à laquelle les échantillons peut être congelé à -20 ° C avant de procéder à la durée du protocole.

Figure 2: Taille protocole de sélection. (A) Capture d'écran de paramètres utilisés dans le protocole ERRBS Pippin Prep (voir la section 5.1.2 du protocole - 5.1.6): (1) Sélectionnez le type de cassette. (2) Sélectionnez standard à utiliser. (3) Sélectionnez le mode de collecte pour chaque voie. (4) Entrez les gammes collection pb. (5) Enregistrer le protocole (B) Les étapes de l'extraction de gel manuel utilisé dans la section de protocole: 5.2. (1) des échelles de gel visualisées. (2) Tailles Marqué pour la sélection de taille en utilisant une lame de rasoir. (3) l'image d'échantillons excisés (fraction inférieure: 150-250 pb et de la fraction supérieure: 250-400 pb)."> Se il vous plaît cliquer ici pour voir une version plus grande de cette figure.

Figure 3: Les résultats de contrôle de qualité pour les bibliothèques ERRBS représentatifs préparés à partir d'échantillons d'ADN humain en utilisant une machine de bioanalyseur. (A) Gel d'image comme montrant une échelle standard (1), la fraction bibliothèque inférieur (135-240 fraction de pb de Pippin Prep); 2) et la fraction bibliothèque élevée (240 à 410 pb de la fraction de Pépin de préparation); . 3) (B) Bioanalyzer électrophérogramme de la fraction inférieure bibliothèque attendu (C) Bioanalyzer de électrophérogramme de la fraction bibliothèque supérieure attendue D -.. F) données représentatives d'une bibliothèque de mauvaise qualité prep. image de gel-like (D) de l'échelle standard (1), faible fraction bibliothèque (2) et la fraction bibliothèque supérieure (3). La bande à 150 pb marqué avec un arrow indique des quantités excessives de l'adaptateur. Électrophorégramme de la partie inférieure (E) et les fractions supérieures bibliothèque (F) avec les pics d'adaptateurs en excès à 150 pb (marqués par des flèches). (G) bioanalyseur électrophérogramme d'une bibliothèque ERRBS mis en commun pour le séquençage. Trace rouge représente une bibliothèque commun de haute qualité avec une représentation égale des fractions supérieures et inférieures. Bleu trace représente une bibliothèque groupé ne suffit pas pour le séquençage en raison d'une absence de représentation égale des fractions supérieures et inférieures. Se il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}

Figure 4: Le séquençage graphiques pour un représentant ERRBS 51 cycle séquençage seule lire terme sur un séquenceur HiSeq 2500 à haut rendementMode. (A) densités de cluster (K / mm 2 = 1 000 grappes par millimètre carré; bleu). Et les densités de cluster passant filtre (vert) dans deux voies avec des bibliothèques de ERRBS (B) intensités typiques vus dans les 30 premiers cycles dans une ruelle avec une bibliothèque ERRBS. Remarque la signature CGG de MspI digestion dans les intensités des trois premiers cycles. (C) Pourcentage de bases avec un score de qualité de 30 ou plus (%> Q30) pour chaque cycle dans une voie de ERRBS. (D) répartition du score de qualité pour tous les cycles dans une voie de ERRBS. Bleu = moins de Q30, Vert = supérieur ou égal à Q30. Dans cette voie, 84,7% des bases avaient des scores de qualité de 30 ou plus.

Figure 5: Séquençage de résultats de sortie. Box parcelles de données expérimentales de l'échantillon multiplexés et unique par séquençage de voie ru ns (affichés comme des carrés verts) et de données obtenues par le sous-échantillonnage simulé du séquençage des séries de trois ERRBS bibliothèques (affiché comme boîtes bleues; échantillonnés cinq fois pour chaque cycle de séquençage) à partir de 51 cycle séquençage seule lire exécute Le facteur de multiplexage correspond à. le nombre de bibliothèques ERRBS séquencé par voie. Une voie ou ensemble = 100% de lectures et représente les données d'une bibliothèque de ERRBS unique par voie; 2 = 50% des voies et représente les données de deux ERRBS bibliothèques par voie; 3 = 33% d'une voie et représente les données de trois ERRBS bibliothèques par voie; et, 4 = 25% d'une voie et représente des données de quatre ERRBS bibliothèques par voie. (A) Les chiffres de lecture, ou le nombre de séquences analysées, par facteur de multiplexage. (B) Le nombre de CpG est couvert par les données de séquençage par multiplexage facteur. (C) La couverture moyenne par CpG par facteur multiplexage._blank "> Se il vous plaît cliquer ici pour voir une version plus grande de cette figure.

Figure 6: Les données représentatives de une bibliothèque de ERRBS préparé à partir de l'ADN génomique humain (A) de l'Université de Californie, Santa Cruz (UCSC) navigateur de génome 43 image de données représentatives de une voie de séquençage de ERRBS.. La barre d'échelle de l'axe y représente 0-100% méthylation à chaque cytosine couvert avec un minimum de 10x. La piste personnalisé haut représente le brin avant et la piste personnalisé inférieur représente le brin inverse. Montré est chr12:.. 6,489,523-6,802,422 (hg19) inclusivement des gènes RefSeq et îlots CpG dans cette région génomique histogrammes (B) de distribution de couverture CpG long avant et inverser brins dans un CD34 + de la moelle osseuse échantillon représentatif (C) histogramme de distributiondes niveaux de méthylation CpG long des deux brins dans un CD34 + de la moelle osseuse échantillon représentatif. (D) courbe de corrélation entre les niveaux de méthylation CpG d'une réplique technique représentant d'un échantillon d'ADN humain. (E) Diagramme illustrant les proportions de CpG abordés dans ERRBS qui annoté îlots CpG (vert clair), CpG rives (gris) et d'autres régions (blanc) dans un échantillon représentatif préparé à partir de l'ADN génomique humain. (F) Diagramme illustrant les proportions de CpG abordés dans ERRBS qui annotés aux promoteurs de gènes (rouge ), les exons (vert), les introns (bleus) et des régions intergéniques (violets). Se il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
| Réactif | Volume | Commentaire |
| 10x ADN ligase T4 Reaction Buffer | 10 ul | |
| Triphosphate Deoxynucleotide (dNTP) Solution Mix | 4 ul | mélanger de 10 mM de chaque nucléotide |
| T4 DNA Polymerase | 5 ul | 3000 unités / ml |
| ADN polymerase I Large (Klenow) Fragment | 1 ul | 5000 unités / ml |
| T4 Polynucleotide Kinase | 5 ul | 10 000 unités / ml |
| L'eau DNase | 45 ul |
Tableau 1: Fin. Réactifs de la réaction de réparation noms et les quantités de réactifs utilisés dans la réaction de réparation d'extrémité (protocole de l'étape 2.1).
| Réactif | Volume | Commentaire |
| Un tampon de réaction 10X | 5 ul | par exemple, deux NEBuffer |
| 1 mM 2'-désoxyadénosine 5'-triphosphate (dATP) | 10 ul | |
| Fragment de Klenow (3 '→ 5' exo-) | 3 ul | 5000 unités / ml |
Tableau 2:. A-tailing réactifs de réaction noms et les quantités de réactifs utilisés dans la réaction de A-tailing (protocole étape 3.1).
| Réactif | Volume | Commentaire |
| 15 uM adaptateurs recuits dans l'eau DNase | 3 ul | PE adaptateur 1,0 et 2,0 PE adaptateur; voir le tableau 4 pour les séquences et référence |
| 10x ADN ligase T4 Reaction Buffer | 581; l | |
| T4 DNA Ligase | 1 ul | 2.000.000 unités / ml |
| L'eau DNase | 31 pi |
Tableau 3: Adapter ligation réactifs de réaction noms et les quantités de réactifs utilisés dans la réaction de ligature adaptateur (protocole de l'étape 4.2)..

Tableau 4:. Oligos utilisés dans le protocole de ERRBS Liste des oligonucléotides utilisés dans le protocole de ERRBS dans la réaction de ligature (protocole de l'étape 4) et les étapes d'amplification par PCR (protocole étape 7).
| Réactif | Volume | Commentaire |
| 10x FastStart High Fidelity Reaction Buffer wie 18 mM de chlorure de magnésium | 20 ul | |
| 10 mM dNTP Mix Solution | 5 ul | |
| 25 uM PE amorce de PCR 1,0 | 4 ul | Voir le tableau 4 |
| 25 uM PE amorce de PCR 2,0 | 4 ul | Voir le tableau 4 |
| FastStart High Fidelity Enzyme | 2 ul | 5 unités / ul FastStart Taq DNA Polymerase |
| L'eau DNase | 125 ul |
Tableau 5: PCR réactifs de réaction noms et les quantités de réactifs utilisés dans la réaction d'amplification par PCR (protocole de l'étape 7.1)..
| Protocole étape | Réactif / détail de protocole | quantité d'ADN d'entrée | ||
| 5-10 ng | 25 ng | 50 ng | ||
| 1 | MspI enzyme | 1 ul | 2 ul | 2 ul |
| Volume de réaction MspI digest | 50 | 100 | 100 | |
| 4 | Adaptateurs en réaction de ligature | 1 ul | 2 ul | 3 ul |
| le volume réactionnel de ligature | 20 ul | 25 ul | 50 ul | |
| 5 | protocole de sélection Taille | Gel Manuel uniquement | Pippin Prep ou de gel d'emploi | Pippin Prep ou de gel d'emploi |
| 7 | Concentration de l'amorce PCR | 25 pM | 25 pM | 10 uM pendant 14 cycles; 25 uM pendant 18 cycles |
| Nombre de cycles de PCR | 18 | 18 | 14-18 | |
Tableau 6: Protocole modifications. Étape pour quantités de matériaux d'entrée allant de 5 à 50 ng Plusieurs étapes à travers le protocole exigent la modification des quantités de réactifs utilisés pour générer des bibliothèques de haute qualité provenant de diverses quantités de matières premières. Les modifications apportées à des quantités de réactifs clés sont inclus ici. Réglez tampon et les volumes d'eau dans les réactions en conséquence.
| Chr | Base | Brin | Couverture | freqC | freqT |
| CHR1 | 10564 | R | 366 | 85,52 | 14,48 |
| CHR1 | 10571 | Fa | 423 | 91,25 | 8,75 |
| CHR1 | 10542 | Fa | 432 | 91,2 | 8,8 |
| CHR1 | 10563 | Fa | 429 | 94,64 | 5,36 |
| CHR1 | 10572 | R | 366 | 96,99 | 3.01 |
| CHR1 | 10590 | R | 370 | 88,11 | 11,89 |
| CHR1 | 10526 | R | 350 | 92 | 8 |
| CHR1 | 10543 | R | 368 | 92,93 | 7,07 |
| CHR1 | 10525 | Fa | 433 | 91,92 | 8,08 |
| CHR1 | 10497 | Fa | 435 | 88,74 | 11,26 |
Nombre de bibliothèques ERRBS par voie Nombre moyen de alignée unique lit Le nombre moyen des CpG couvert Couverture par CpG moyenne 1 152 231 184 ± 13.189.678 3183594 ± 713,547 85 ± 16 2 77680837 ± 7.657.058 2674823± 153 494 49 ± 9 3 49938156 ± 2.436.865 2552186 ± - 76624 39 ± 2 4 34457208 ± 4.441.686 1814461 ± 144,339 28 ± 4
Tableau 8:. Paramètres représentatifs de séquençage bibliothèques ERRBS simples et multiplexés Montré est données par voie de 51-cycle unique lire pistes de séquençage: moyenne et les écarts-types de alignée unique lectures, le nombre de CpG couvert et la couverture par site CpG obtenu à partir de séquençage unique bibliothèques ERRBS par voie (n = 100), deux ERRBS bibliothèques par voie (n = 128), trois ERRBS bibliothèques par voie (n = 11), et quatre ERRBS bibliothèques par voie (n = 11).
Discussion
Les données de résolution rendements paires de bases protocole présenté de méthylation de la cytosine à des régions génomiques biologiquement pertinents. Le protocole comme écrit est optimisé pour 50 ng de produit de départ, cependant, il peut être adapté pour traiter une gamme de matériel d'entrée (5 ng ou plus) 26. Cela nécessitera des ajustements de certaines des étapes de protocole comme on le voit dans le tableau 6. Les bibliothèques de ERRBS se prêtent à un séquençage d'extrémité et couplé en outre une couverture génomique peut également être réalisé par séquençage lectures plus de 51 cycles. Séquençage multiplexé offrira un protocole à moindre coût par échantillon, cependant, cela se traduira par une couverture réduite par site CpG représenté dans les données (Figure 5 et le Tableau 8), et ne cédera pas une profondeur suffisante de la couverture d'effectuer des analyses qui nécessitent une couverture élevée par CpG place (par exemple comme décrit par Landan et al. 33). Enfin, ce protocole (ou tout Protoco base bisulfite-l) ne peut pas distinguer entre méthyl-cytosine et hydroxyméthyle-cytosine 46,47. Toutefois, les données générées peuvent être intégrés à d'autres résultats protocole 48,49 pour délimiter les différentes modifications, et d'autres modifications de cytosine récemment rapporté 50, devraient-ils être d'intérêt.
Bibliothèques de haute qualité apparaîtra comme le montre la figure 3A-C, et une fois mis en commun pour le séquençage donne une trace comme le montre la figure 3G (trace rouge) représentant contributions molaires égales des deux fractions de la bibliothèque. Bibliothèque échec de préparation peut résulter de ne importe quelle étape pendant la procédure. Si l'ADN dégradé est traitée elle se traduira dans les bibliothèques qui ne sont pas enrichis en fragments Mspl et donc de la faible couverture CpG en utilisant les paramètres de séquençage décrites dans ce protocole. Si une enzyme est non fonctionnelle ou exclu par inadvertance de l'une des réactions, le protocole donné la bibliothèque attendu. Si le rea de ligaturection est inefficace, les adaptateurs sont à une concentration plus élevée que prévu, et / ou la concentration des amorces utilisé est un réactif limitant pour les étapes finales d'amplification, l'échec de la bibliothèque peut se produire. Adaptateurs excès (considéré comme un des pics à ~ 150 pb dans les résultats de bioanalyse; Figure 3D-F) à la bibliothèque sera également interférer avec le séquençage due au regroupement aveugle de la bibliothèque et adaptateurs excès. Si une telle bibliothèque peut séquencer apparemment normalement, une partie importante du lit sera séquences simplement l'adaptateur. Si des adaptateurs en excès sont observées dans une bibliothèque, il est préférable de répéter la préparation bibliothèque si le matériel est disponible en utilisant du matériel d'entrée optimal à l'adaptateur rapports quantitatifs. Enfin, pour assurer une amplification par PCR efficace des bibliothèques, les fractions bibliothèque inférieurs et supérieurs sont maintenus comme des échantillons distincts dans l'ensemble de la conversion au bisulfite et PCR étapes d'enrichissement. Ne pas le faire donne l'efficacité différentielle de l'amplification au cours de la PCR réaction de fractions supérieures et inférieures (comme on le voit sur la figure 3G trace bleue) et le potentiel de la représentation inégale des loci génomiques respectif couverte dans chaque fraction bibliothèque au cours du séquençage. L'utilisateur peut choisir d'inclure une étape de PCR quantitative immédiatement après la conversion au bisulfite de titrage en outre des cycles de PCR optimales nécessaires pour amplifier les bibliothèques générées.
ERRBS protocole de préparation de la bibliothèque comporte plusieurs étapes clés dans lesquels les réactifs spécifiques sont recommandées. Lors de l'étape de fin de réparation, l'utilisation d'un mélange de dNTP à quatre nucleotides permet fin de réparation de tous produits qui ne contiennent pas le surplomb CG, tels que ceux résultant de MspI activité enzymatique en étoile et des fragments d'ADN cisaillé présents dans l'échantillon d'ADN d'origine. Il en résulte une meilleure représentation CpG dans les résultats. A l'étape de ligature, il est essentiel d'utiliser une ligase forte concentration (2.000.000 unités / ml) et méthylés adaptateurs pour se assurer que la Reacti de ligationOn est efficace et que la conversion au bisulfite ne influence pas les séquences d'adaptation essentielles pour l'alignement de données précises. A l'étape PCR, en utilisant une polymérase capable d'amplifier des fragments d'ADN riches en GC traité au bisulfite est nécessaire pour une grande spécificité. Enfin, pour assurer l'élimination des adaptateurs et les amorces en excès, SPRI cordon purification (par exemple: Agencourt AMPure XP) est recommandé plutôt que de tests basés sur des colonnes pour ligature et isolements de produits PCR.
Afin de générer des données de haute qualité, il est important pour assurer une conversion au bisulfite efficace. Le contrôle présenté offre à l'utilisateur la possibilité de déterminer l'efficacité de conversion avant le séquençage. En variante, un ADN non humain tel que lambda ADN peut être utilisé en tant que contrôle interne (spike-in). En raison des différences entre les espèces, ce type de commande peut être directement inclus dans le séquençage en aval (par exemple, tel qu'il est utilisé par Yu, et al. 34). Cependant, si le pic i-ins utilisé, il ne peut être utilisé pour déterminer l'efficacité de conversion avant le séquençage bibliothèque amplifiée à moins unique et séquence avant le séquençage bibliothèque de façon indépendante. Les taux de conversion déterminés sont basés sur l'état de méthylation des sites CpG non. Cela peut ne pas être approprié pour une utilisation dans le contexte de forte méthylation de la cytosine dans le contexte non CpG (par exemple des cellules souches embryonnaires) et des échantillons parallèles ou d'autres moyens d'évaluation de l'efficacité de conversion peuvent être utilisés à cette fin.
Il ya quelques mises en garde à l'adresse qui sont uniques à l'enchaînement des bibliothèques ERRBS. Les trois premières bases des fractions de la bibliothèque séquencés sont presque uniformément non aléatoire en raison du site de coupe de reconnaissance MspI (C ^ CGG; voir la figure 4B, C). Il en résulte le risque de perte de données importantes en raison de la faible qualité lectures résultant d'une mauvaise localisation de cluster en dépit de la densité de cluster haute apparente au cours du séquençage. Pour surmonter cet obstacle,inclure une bibliothèque de grande complexité dans une voie indépendante (contrôle PhiX ou un autre type de bibliothèque) comme une voie de contrôle dédié. Bibliothèques de haute complexité ont des extrémités contenant une représentation équilibrée des A, C, T et G dans les quatre premières bases séquencées. Voies de contrôle appropriés comprennent les bibliothèques telles que l'ARN-Seq, ChIP-seq, le séquençage du génome entier, ou un contrôle offert par le fabricant de la machine de séquençage (par exemple v3 contrôle PhiX). Lorsque désignée comme une voie de commande pour la course respective de séquençage, il peut servir de base pour la génération de la matrice qui est utilisée au cours des quatre premières bases de séquençage pour détecter des positions de la grappe. La qualité supérieure lectures capturé augmentera la couverture moyenne par site CpG de 5,2 (n = 4). Alternativement, cette difficulté technique peut également être surmonté en utilisant une approche de séquençage sombre comme décrit précédemment 23. Autres critères de séquençage suivent des procédures normalisées d'exploitation par les protocoles du fabricant. Enfin, la couverture par CpG chosen pour l'analyse des données sera guidé par l'utilisateur et en partie par les questions biologiques d'intérêt. 10x seuil de couverture offre une approche d'analyse de couverture élevé, mais ce seuil peut être abaissé devrait-il être d'intérêt.
Une discussion complète de l'analyse de données ERRBS est au-delà du champ d'application de cet article, cependant, différemment cytosines méthylés et régions peuvent être déterminées en utilisant des outils open source 31,51-53. Considérations et des approches d'analyse supplémentaires ont été bien décrit 54,55, et le lecteur est encouragé à rechercher la littérature pour les outils les plus appropriés pour l'analyse prévue.
Comparé à d'autres méthodes publiées, ERRBS propose un protocole de quatre jours qui, lorsqu'il est effectué alors que les rendements décrits taux élevés de reproductibilité. Il a été validé par rapport à l'étalon-or MassARRAY EpiTYPER 26, est rentable pour des données de haute couverture, et est adaptable pour différents matériaux d'entrée(montants favorables pour le traitement de l'échantillon clinique et d'autres types cellulaires de basse fréquence) et des approches de séquençage. Il offre une résolution paires de bases à des loci biologiquement pertinente et peut être utilisé dans les analyses d'intégration avec d'autres techniques de profilage du génome entier facteur de transcription contraignant, remodelage de la chromatine, marques épigénétiques et d'autres modifications cytosine d'intérêt. L'utilisation des données ERRBS dans de telles études peut contribuer à une approche moléculaire complète et permettre grande dimension analyses dans l'étude de modèles biologiques et la maladie humaine.
Déclarations de divulgation
Les auteurs ne ont aucun conflit d'intérêt à divulguer.
Remerciements
We thank all the authors of the original ERRBS report. We thank Mame Fall for technical assistance. We acknowledge the Weill Cornell Medical College Epigenomics Core for technical services and assistance. The work was supported by a Sass Foundation Judah Folkman Fellowship, an NCI K08CA169055 and ASH-AMFDP12005 to FGB, NIH R01HG006798 and R01NS076465, funding from the Irma T. Hirschl and Monique Weill-Caulier Charitable Trusts and STARR Consortium (I7-A765) to CEM, and an LLS SCORE grant (7006-13) to AMM.
matériels
| Name | Company | Catalog Number | Comments |
| MspI | New England Biolabs | R0106M | 100,000 units/ml |
| NEBuffer 2 | New England Biolabs | B7002S | Reaction buffer for MspI enzyme; protocol step 1.2 |
| Phenol solution | Sigma-Aldrich | P4557 | Equilibrated with 10 mM Tris HCl, pH 8.0; see safety and handling instructions at http://www.sigmaaldrich.com/catalog/product/sigma/p4557 |
| Chloroform | Sigma-Aldrich | C2432 | See safety and handling instructions at http://www.sigmaaldrich.com/catalog/product/sial/c2432 |
| Glycogen | Sigma-Aldrich | G1767 | 19-22 mg/ml |
| NaOAc | Sigma-Aldrich | S7899 | 3 M, pH 5.2 |
| Ethanol | Sigma-Aldrich | E7023 | 200 proof, for molecular biology |
| Buffer EB | Qiagen | 19086 | 10 mM Tris-Cl, pH 8.5 |
| tris(hydroxymethyl)aminomethane (Tris) | Sigma-Aldrich | T1503 | prepare a 1 M, pH 8.5 solution |
| Tris- Ethylenediaminetetraacetic acid (TE) | Sigma-Aldrich | T9285 | Dilute to 1x buffer solution per manufacturer's recommendations |
| T4 DNA Ligase Reaction Buffer | New England Biolabs | B0202S | 10x concentration |
| Deoxynucleotide triphosphate (dNTP) Solution Mix | New England Biolabs | N0447L | 10 mM each nucleotide |
| T4 DNA Polymerase | New England Biolabs | M0203L | 3,000 units/ml |
| DNA Polymerase I, Large (Klenow) Fragment | New England Biolabs | M0210L | 5,000 units/ml |
| T4 Polynucleotide Kinase | New England Biolabs | M0201L | 10,000 units/ml |
| QIAquick PCR Purification Kit | Qiagen | 28104 | Used for DNA product purification in protocol step 2.3 |
| 2'-deoxyadenosine 5'-triphosphate (dATP) | Promega | U1201 | 100 mM |
| Klenow Fragment (3'→5' exo-) | New England Biolabs | M0212L | 5,000 units/ml |
| MinElute PCR Purification Kit | Qiagen | 28004 | Used for DNA product purification in protocol step 3.3 |
| T4 DNA Ligase | New England Biolabs | M0202M | 2,000,000 units/ml |
| Methylation Adapter Oligo Kit | Illumina | ME-100-0010 | |
| Agencourt AMPure XP | Beckman Coulter | A63881 | Used in protocol sections that implement magnetic bead purification steps (steps 4.3 and 8.2). Equilibrate to room temperature before use. |
| Pippin Prep Gel Cassettes, 2% Agarose, dye-free | Sage Science | CDF2010 | with internal standards |
| Certified Low Range Ultra Agarose | Bio-Rad | 161-3106 | |
| Tris-Borate-EDTA (TBE) buffer | Sigma-Aldrich | T4415 | |
| Ethidium bromide solution | Sigma-Aldrich | E1510 | 10 mg/ml |
| 50 bp DNA Ladder | NEB | N3236S | |
| 100 bp DNA Ladder | NEB | N3231S | |
| Gel Loading Dye, Orange (6x) | NEB | B7022S | |
| Scalpel Blade No. 11 | Fisher Scientific | 3120030 | |
| QIAquick Gel Extraction Kit | Qiagen | 28704 | |
| EZ DNA Methylation Kit | Zymo Research | D5001 | Used in protocol step 6.2 |
| EZ DNA Methylation-Lightning Kit | Zymo Research | D5030 | Alternative for step 6.2 |
| Universal Methylated Human DNA Standard | Zymo Research | D5011 | Used as bisulfite conversion control |
| FastStart High Fidelity PCR System | Roche | 03553426001 | |
| Qubit dsDNA High Sensitivity Assay Kit | Life Technologies | Q32854 | A fluorescence-based DNA quantitation assay; used in protocol steps 1.1, 9.1 and 10.1 |
| DynaMag-2 Magnet | Life Technologies | 12321D | |
| High Sensitivity DNA Kit | Agilent Technologies | 5067-4626 | |
| 2100 Bioanalyzer | Agilent Technologies | ||
| PhiX Control v3 | Illumina | FC-110-3001 | |
| HiSeq 2500 | Illumina | ||
| Pippin Prep | Sage Science | ||
| Qubit 2.0 Fluorometer | Life Technologies | Q32872 | |
| TruSeq SR Cluster Kit v3-cBot-HS | Illumina | GD-401-3001 | |
| TruSeq SBS Kit v3-HS | Illumina | FC-401-3002 | |
| TruSeq RNA Sample prep | Illumina | RS-122-2001 | Barcoded adapters used for multiplexing libraries; See Supplemental file for multiplexing protocol. |
| Microcentrifuge | |||
| Vortex Mixer | |||
| Dry Block Heater | |||
| Thermal Cycler | |||
| Water Bath | |||
| Gel electrophoresis system | |||
| Electrophoresis power supply | |||
| Gel doc | |||
| UV or blue light transilluminator |
Références
- Jones, P. A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 13 (7), 484-492 (2012).
- Barlow, D. P. Genomic imprinting: a mammalian epigenetic discovery model. Annual Review Of Genetics. 45, 379-403 (2011).
- Thiagarajan, R. D., Morey, R., Laurent, L. C. The epigenome in pluripotency and differentiation. Epigenomics. 6 (1), 121-137 (2014).
- Reik, W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 447 (7143), 425-432 (2007).
- Hartnett, L., Egan, L. J. Inflammation, DNA methylation and colitis-associated cancer. Carcinogenesis. 33 (4), 723-731 (2012).
- Smith, Z. D., Meissner, A. DNA methylation: roles in mammalian development. Nat Rev Genet. 14 (3), 204-220 (2013).
- Li, E., Bestor, T. H., Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 69 (6), 915-926 (1992).
- Okano, M., Bell, D. W., Haber, D. A., Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 99 (3), 247-257 (1999).
- Feinberg, A. P. Phenotypic plasticity and the epigenetics of human disease. Nature. 447 (7143), 433-440 (2007).
- Bock, C. Epigenetic biomarker development. Epigenomics. 1 (1), 99-110 (2009).
- Laird, P. W. The power and the promise of DNA methylation markers. Nat Rev Cancer. 3 (4), 253-266 (2003).
- How Kit, A., Nielsen, H. M., Tost, J. DNA methylation based biomarkers: practical considerations and applications. Biochimie. 94 (11), 2314-2337 (2012).
- Mikeska, T., Bock, C., Do, H., Dobrovic, A. DNA methylation biomarkers in cancer: progress towards clinical implementation. Expert Review Of Molecular Diagnostics. 12 (5), 473-487 (2012).
- Gyparaki, M. T., Basdra, E. K., Papavassiliou, A. G. DNA methylation biomarkers as diagnostic and prognostic tools in colorectal cancer. Journal of Molecular Medicine. 91 (11), 1249-1256 (2013).
- Figueroa, M. E., et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 17 (1), 13-27 (2010).
- Heyn, H., Mendez-Gonzalez, J., Esteller, M. Epigenetic profiling joins personalized cancer medicine. Expert review of Molecular Diagnostics. 13 (5), 473-479 (2013).
- Kulis, M., Esteller, M. DNA methylation and cancer. Advances in Genetics. 70, 27-56 (2010).
- Xiong, Z., Laird, P. W. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 25 (12), 2532-2534 (1997).
- Meissner, A., et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 33 (18), 5868-5877 (2005).
- Gu, H., et al. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc. 6 (4), 468-481 (2011).
- Bock, C., et al. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol. 28 (10), 1106-1114 (2010).
- Harris, R. A., et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol. 28 (10), 1097-1105 (2010).
- Boyle, P., et al. Gel-free multiplexed reduced representation bisulfite sequencing for large-scale DNA methylation profiling. Genome Biol. 13 (10), R92(2012).
- Chatterjee, A., Rodger, E. J., Stockwell, P. A., Weeks, R. J., Morison, I. M. Technical considerations for reduced representation bisulfite sequencing with multiplexed libraries. Journal of Biomedicine & Biotechnology. 2012, 741542(2012).
- Lee, Y. K., et al. Improved reduced representation bisulfite sequencing for epigenomic profiling of clinical samples. Biological Procedures Online. 16 (1), 1(2014).
- Akalin, A., et al. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet. 8 (6), e1002781(2012).
- Hatzi, K., et al. A Hybrid Mechanism of Action for BCL6 in B Cells Defined by Formation of Functionally Distinct Complexes at Enhancers and Promoters. Cell Reports. 4 (3), 578-588 (2013).
- Will, B., et al. Satb1 regulates the self-renewal of hematopoietic stem cells by promoting quiescence and repressing differentiation commitment. Nature Immunology. 14 (5), 437-445 (2013).
- Lu, C., et al. Induction of sarcomas by mutant IDH2. Genes Dev. 27 (18), 1986-1998 (2013).
- Kumar, R., et al. AID stabilizes stem-cell phenotype by removing epigenetic memory of pluripotency genes. Nature. 500 (7460), 89-92 (2013).
- Li, S., et al. An optimized algorithm for detecting and annotating regional differential methylation. BMC Bioinformatics. 14, Suppl 5. S10(2013).
- Patterson, K., Molloy, L., Qu, W., Clark, S. DNA methylation: bisulphite modification and analysis. Journal of Visualized Experiments. (56), 3170(2011).
- Landan, G., et al. Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat Genet. 44 (11), 1207-1214 (2012).
- Yu, M., et al. Tet-assisted bisulfite sequencing of 5-hydroxymethylcytosine. Nat Protoc. 7 (12), 2159-2170 (2012).
- Goecks, J., Nekrutenko, A., Taylor, J., Galaxy, T. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 11 (8), R86(2010).
- Dorff, K. C., et al. GobyWeb: simplified management and analysis of gene expression and DNA methylation sequencing data. PLoS One. 8 (7), e69666(2013).
- Roehr, J. T., Dodt, M., Ahmed, R., Dieterich, C. Flexbar − flexible barcode and adapter processing for next-generation sequencing platforms. MDPI Biology. 1 (3), 895-905 (2012).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal, North America. 17 (1), 10-12 (2011).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (15), 2114-2120 (2014).
- Needleman, S. B., Wunsch, C. D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 48 (3), 443-453 (1970).
- Krueger, F., Andrews, S. R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 27 (11), 1571-1572 (2011).
- Li, H., et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25 (16), 2078-2079 (2009).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Res. 12 (6), 996-1006 (2002).
- Thorvaldsdottir, H., Robinson, J. T., Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in Bioinformatics. 14 (2), 178-192 (2013).
- Team, R. C. R. A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria. ISBN 3-900051-07-0, http://www.R-project.org (2012).
- Nestor, C., Ruzov, A., Meehan, R., Dunican, D. Enzymatic approaches and bisulfite sequencing cannot distinguish between 5-methylcytosine and 5-hydroxymethylcytosine in DNA. BioTechniques. 48 (4), 317-319 (2010).
- Huang, Y., et al. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS One. 5 (1), e8888(2010).
- Yu, M., et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 149 (6), 1368-1380 (2012).
- Song, C. X., et al. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell. 153 (3), 678-691 (2013).
- Ito, S., et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 333 (6047), 1300-1303 (2011).
- Akalin, A., et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 13 (10), R87-1186 (2012).
- Stockwell, P. A., Chatterjee, A., Rodger, E. J., Morison, I. M. DMAP: Differential Methylation Analysis Package for RRBS and WGBS data. Bioinformatics. 30 (13), 1814-1822 (2014).
- Sun, D., et al. MOABS: model based analysis of bisulfite sequencing data. Genome Biol. 15 (2), R38(2014).
- Bock, C. Analysing and interpreting DNA methylation data. Nat Rev Genet. 13 (10), 705-719 (2012).
- Rivera, C. M., Ren, B. Mapping human epigenomes. Cell. 155 (1), 39-55 (2013).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.