
Method Article
자료 쌍 해상도에서 DNA 메틸화의 평가에 대한 표현 바이 설 파이트 시퀀싱을 감소 향상
* 이 저자들은 동등하게 기여했습니다
요약
Enhanced Reduced Representation Bisulfite Sequencing is a method for the preparation of sequencing libraries for DNA methylation analysis based on restriction enzyme digestion combined with cytosine bisulfite conversion. This protocol requires 50 ng of starting material and yields base pair resolution data at GC-rich genomic regions.
초록
DNA 메틸화 패턴 매핑은 크게 정상 및 병에 걸린 조직에서 연구되고있다. 다양한 방법이 세포에서 시토신 메틸화 패턴을 심문하기 위해 설립되었다. 전체 게놈 서열의 중아 감소 표현 GC 풍부한 게놈 유전자좌 양적 염기쌍 해상도 시토신 메틸화 패턴을 검출하기 위해 개발되었다. 이 바이 설 파이트 변환이어서 제한 효소의 조합을 사용함으로써 달성된다. 향상된 감소 표현 바이 설 파이트 시퀀싱 (ERRBS)가 적용되는 생물학적으로 관련 게놈 궤적을 증가시키고 인간, 마우스 및 다른 생물의 DNA에서 시토신 메틸화을 프로파일 링하는 데 사용되었습니다. ERRBS 라이브러리 제조에 사용하기위한 저 분자량의 단편을 생성하는 DNA의 제한 효소 절단함으로써 시작된다. 이 조각은 차세대 시퀀싱을위한 표준 라이브러리 건설을 실시한다. 최종 amplificati 이전에 비 메틸화 된 시토신의 중아 변환단계에 덮여 게놈 유전자 좌에서 시토신 메틸화 수준의 양적 기본 해상도 수 있습니다. 이 프로토콜은 네 일 이내에 완료 할 수 있습니다. 지정된 시퀀싱 제어 레인 사용시 서열화 처음 세 염기 낮은 복잡성에도 불구 ERRBS 라이브러리는 고품질의 데이터를 얻었다. 매핑 및 생물 정보학 분석 수행된다 수율 데이터 용이 게놈 넓은 다양한 플랫폼과 통합 될 수있다. ERRBS는 가능한 연구 어플리케이션의 범위에서 인간 임상 샘플 및 해당 사항을 처리 할 수있게 작은 입력 재료 수량을 이용할 수있다. 생성 된 비디오는 ERRBS 프로토콜의 중요한 단계를 보여줍니다.
서문
시토신 (5- 메틸 시토신)에서 DNA 메틸화는 생물학적 과정의 다양한 포유 동물 세포에서 중요한 포함하지만 임프린트, X 염색체 불 활성화, 개발 및 유전자 발현 조절 1-8에 한정되지 후생 유전 학적 마크이다. 악성 질환 및 기타 질환에서의 DNA 메틸화 패턴의 연구는 질병 특정 패턴을 결정하고 질병의 발병 기전과 잠재적 인 바이오 마커 발견 9-17의 이해에 기여하고있다. DNA 메틸화 상태를 가지로 구분을 심문하는 프로토콜이있다. 이들은 선호도 기반으로 분할 될 수 있고, 제한 효소 기반 및 하류 마이크로 어레이 또는 시퀀싱 플랫폼을 활용 중아 변환 - 기반 분석법. 또한, 다음과 같은 일반적인 범주에 포함되지만, 결합 바이 설 파이트 제한 분석 (18)에 한정 표현 바이 설 파이트 시퀀싱 (RRBS 19) 감소하지 다리를 몇 가지 프로토콜이있다.
RRBS는 원래 마이스너 등. (19, 20)에 의해 설명되었다. 이 프로토콜은 비용 (21, 22) 효과가 정량적 염기쌍 해상도 데이터 결과 바이 설 파이트 시퀀싱, 다음 GC 풍부한 게놈 영역을 풍부하게하는 단계를 소개했다. GC 풍부 영역들은 MspI (C ^ CGG) 제한 효소에 의해 대상으로하고 시토신 메틸화는 중합 효소 연쇄 반응 (PCR) 증폭에 이어 시토신의 중아 변환 (우라실 변성 시토신의 탈 아미 노화)에 의해 해결된다. RRBS 유전자 프로모터 및 전체 게놈에 필요한 순서의 일부에서의 CpG 섬의 대부분을 커버; 그러나 RRBS는 CpG 디 해안과 생물학적 관련성의 다른 유전자 간 지역의 제한 범위를했다. 몇몇 그룹은 이러한 게놈 지역 23 ~ 25의 방법론과 결과 범위를 개선 원래의 보고서 이후 RRBS 프로토콜을 업데이트 게시 한. 향상된 감소 표현 바이 설 파이트 Sequencing (ERRBS)는 RRBS에 비해 라이브러리 준비 수정 및 대체 데이터 정렬 방식 (26)를 포함한다. ERRBS는 생성 된 데이터로 표현 CpGs 더 많은 수의 결과와 모든 게놈 영역의 범위가 26 심문 증가했다. 이 방법은 인간 및 기타 동물 환자 검체 26-30에서 DNA 메틸화 패턴을 확인하는 데 사용되어왔다.
ERRBS 프로토콜 완료 대표적인 인간 DNA를 이용하여 생성 된 데이터에 필요한 모든 단계에서 이벤트 정보를 기재 (샘플은 이전에보고 된, 디 식별 환자 샘플 (31)로부터 얻어지고, 정상적인 인간 공여자로부터의 CD34 + 골수 샘플 하였다). 프로토콜은 샘플 당 처리 시간을 감소시키고, 라이브러리 크기 선택의 정확도 향상을 가능하게하는 자동 사이즈 선택 프로세스를 포함한다. 프로토콜은 수립 분자 생물학 기법을 결합한 계열. 고 분자량의 DNA를 소화 w최종 수리, A-미행하고, 메틸화 된 어댑터의 결찰 다음에 메틸화를 구분하지 않는 제한 효소 (MspI) i 번째. GC 풍부한 단편의 사이즈 선택은 바이 설 파이트 변환 및 시퀀싱 전에 PCR 증폭 이어진다. 중아 변환 이전에 있었다 (32) 기술과 데이터 분석 및 응용 프로그램의 상세한 검토를 권고하고 참조가 독자를위한 부분 포함되어 있지만이 문서의 범위를 벗어했다. 이 프로토콜은 사일을 통해 수행과 작은 입력 (50 NG 이하) 재료 양의 의무가 될 수있다. 차동 메틸화 사이트 및 영역에 대한 결정뿐만 아니라 후생 유전 학적 다형성 검출뿐만 아니라 충분한 된 CpG 사이트 당에 따르면 높은 수율로 설명 데이터로서 프로토콜, 등. 33 랜댄 기재된 바와 같이.
프로토콜
실행 모든 절차는 의학 기관 동물 관리 및 사용위원회의 인디애나 대학의 승인을 건강 지침의 국립 연구소 따릅니다.
1. 수술 기법
- 멸균 장갑, 악기, 그리고 NIH 가이드 라인 (25)에 따라 멸균 수술 필드를 사용하여이 절차를 수행하는 동안 무균 기술을 유지한다. 를 오토 클레이브에 의해 수술을 시작하기 전에 도구를 소독 (전체 목록은 특정 시약 / 장비의 표 참조). 작업 중에 도구를 소독 유리 구슬 살균기를 사용합니다.
2. 마취 및 준비
- 수의학 이소 플루 란 기화기 시스템을 이용하여 0.9 L / 분의 산소 및 2.5 %의 이소 플루 란 혼합물로 마취 상자에 마우스를 마취. 마우스가 상자에서 제거하기 전에 몸의 위치 변화에 응답하지 않습니다 있는지 확인하십시오.
- 양해 각서 (MOU)에 안과 연고를 적용그 자체의 눈이 마르지을 보호 할 수 있습니다.
- 코 콘에있는 상자에서 가스 흐름을 전환합니다. 콘 내부의 코와 입으로 수술 패드와 흡수 벤치 종이로 덮여 가열 패드의 왼쪽에 정면으로 마우스를 놓습니다. 지속적으로 마우스의 호흡 리듬과 속도를 모니터링하고 필요에 따라 이소 플루 란 레벨을 조정 - 마취의 적절한 수준을 유지하기 위해 (2.5 사이의 3 % 이소 플루 란), 총 진정 작용을 확인하기 위해 발가락 핀치 반사를 사용합니다.
3. 수술 적 접근
- 맞추고 수술 필드 입체경을 초점을 맞 춥니 다. 노즈콘을 조정하고이 시야의 가장자리에 위치하도록 아래로하는 테이프.
- 마우스의 왼쪽에 누워, 절개 될 것이다 귀 뒤 영역을 노출 노즈콘을 우측 귀의 가장자리를 테이프. 후방 귀의 정맥 귀 가로로 이동하는 확인하십시오. 참고 t의 정확한 위치그 동물 귀의 테이핑 신속 안면 신경을 찾기 위해 매우 중요하다.
- 에 70 % 에탄올 귀 뒤에 모피 적시고 면도기 또는 메스 블레이드를 사용하여 수술 부위를 면도. 사전 습윤 모피하는이 해부학 적 위치에 쉽게 면도 있습니다.
- 70 % 에탄올 다음과 같은 베타 딘 수술 스크럽 (7.5 % 포비돈 요오드)와 같은 요오드 용액,로 피부를 청소합니다. 철저하게 지역을 소독이 청소를 두 번 더 반복합니다.
- , 절개를 귀 돌기에 지역의 후방에 꼬리 쪽 귀에서 후방 귀의 정맥을 추적해서 결정해야합니다. 돌기에 3mm 후방 - 봄 가위를 사용하여 4mm의 절개 (2)를 확인합니다.
- 무딘 절개를 이용하여 피하 지방 및 근막을 절개하다. 혈관 또는 근육 조직이 쉽게 손상 될 수 있기 때문에 가위로 직접 절단하지 마십시오.
- 출혈이 발생하는 경우, 멸균 면봉으로 수술 부위에 압력을 적용적어도 30 초. 유체 상당한 손실이 발생하면, 최대 25 또는 제 27 G 바늘을 사용하여 멸균 0.9 % 식염수 용액 0.5 ml를 복강 내 주사를 마우스.
- 안면 신경의 위치를, 몇 가지 주요 랜드 마크, 척추 부신경, 외이도를 사용하여 (아래에 설명) digastric 근육을 전방. 안면 신경의 가지가 가시화 될 때까지 이러한 랜드 마크 주위를 해부하다. 이 공개 될 때 신경은 크게 백색 고체 구조로 표시됩니다 및 근막 층이 기본 구조를 준수합니다.
- 피하 지방과 근막이 해부 된 이후, 승모근의 근육에 분포하는 두개골의 꼬리 부분에서 이동 척추 액세서리 신경을 찾을 수 있습니다. 안면 신경은 척추 부신경에 깊은이다.
- 진주 흰색 보이는 안면 신경에 주동이 볼 수있는 연골 귀 운하를 찾을 수 있습니다.
- 의 상단과 C에 놓여 전방 digastric 근육의 근육 배꼽 찾기안면 신경에 audal.
- 안면 신경의 가지 주요 가시화되면 stylomastoid 난원에서 기원을 찾을 등쪽 그들을 추적. 개방형 수술 부위를 보유하고있는 신경의 경로를 따라 봄 가위 팁을 발전 연 다음, 새로 고급 영역을 유지하기 위해 등쪽 집게를 이동 뾰족한 뒤몽 포셉 # 45분의 5를 사용.
- 이 시점에서 광대뼈, 구강과 안면 신경의 트렁크, 변연 하악 가지를 시각화.
주 : 시간 분기 난원에 가까운 발견 될 것이다. 상부 및 하부에 한계 하악 신경 가지에 가까운 턱, 따라서 그 신경 지점이 수준에서 표시되지 않습니다.- 신경 절개를 수행하는 경우, 미세 팁 포셉으로 부드럽게 신경을 안정시키고 스프링 가위로 신경을 잘라. 뇌간의 신경을 avulsing 방지하기 위해 집게와 신경에 너무 많은 견인을 적용하지 마십시오. 푸시멀리 서로, 또는 절단 및 말초 신경의 일부를 제거에서 젊고 아름다운 여자가 재 연결이 발생하지 않도록합니다.
- 호감 부상을 수행하는 경우, 모든 축삭을 절단 첫 번째 호감 사이트에 수직 인 제 2 각도로이 호감을 반복 일정한 압력을 사용하여 30 초 동안 신경을 압축 뒤몽 #에게 45분의 5 집게를 사용합니다. 그렇지 않으면 부상 동물 사이에 일관성이 없습니다, 30 초 호감 동안 압력의 변수 금액을 적용하지 마십시오.
4. 결산 및 복구
- 기본 구조를 통해 지방과 근육 위치를 조정합니다.
- 절개의 가장자리를 대략적인 7.5 mm 상처 클립을 사용하여 상처를 닫는다. 봉합 또는 접착제는 또한 상처 폐쇄에 대한 허용됩니다. 수술후 진통제는이 시점에 제공 될 수있다.
- 마우스의 귀에서 테이프를 제거합니다. 이소 플루 란 흐름을 끄고 마우스가 1 분 30 초 동안 순수한 산소를 흡입 할 수 있습니다. PL마취에서 회복 할 수없는 침구 빈 케이지에서 마우스 에이스.
- 마우스가 복구 될 때, 안면 마비의 확증 흔적이 동작을 검사합니다. 수염이 마비 다시 뺨을 향해 각도됩니다, 코가 이탈되고, 눈은 공기의 퍼프에 대한 응답으로 깜박하지 않습니다.
- 주택의 동물 공동 수술 후 그들은 여성의 경우. 그들은 더 공격적 강제 감염에 이르게 자신의 cagemate의 상처 클립을 제거하는 경향이 있기 때문에 공동으로 남성 쥐를 수용하지 마십시오. 필요한 경우,이 시점에서 수술후 진통제를 제공한다.
- 어떤 감염 또는 기타 합병증은 수술 후 발생하지 않도록 수술 후 며칠 동안 하루에 한 번 마우스를 모니터링합니다. 그들은 자신에 타락하지 않은 경우 수술 10 일 - 상처 클립 (7)를 제거합니다.
- 눈 깜빡임이 재 할 때까지, 매일 각막 합병증을 방지하기 위해 영향을받는 눈에 눈 연고를 윤활 적용적용 또는 안락사까지.
결과
그림 1은 기술 된 프로토콜을 통해 설명 주요 단계를 강조 ERRBS의 개요를 제공합니다. ERRBS 라이브러리는 50 NG 입력 DNA를 사용하여 제조 하였다.
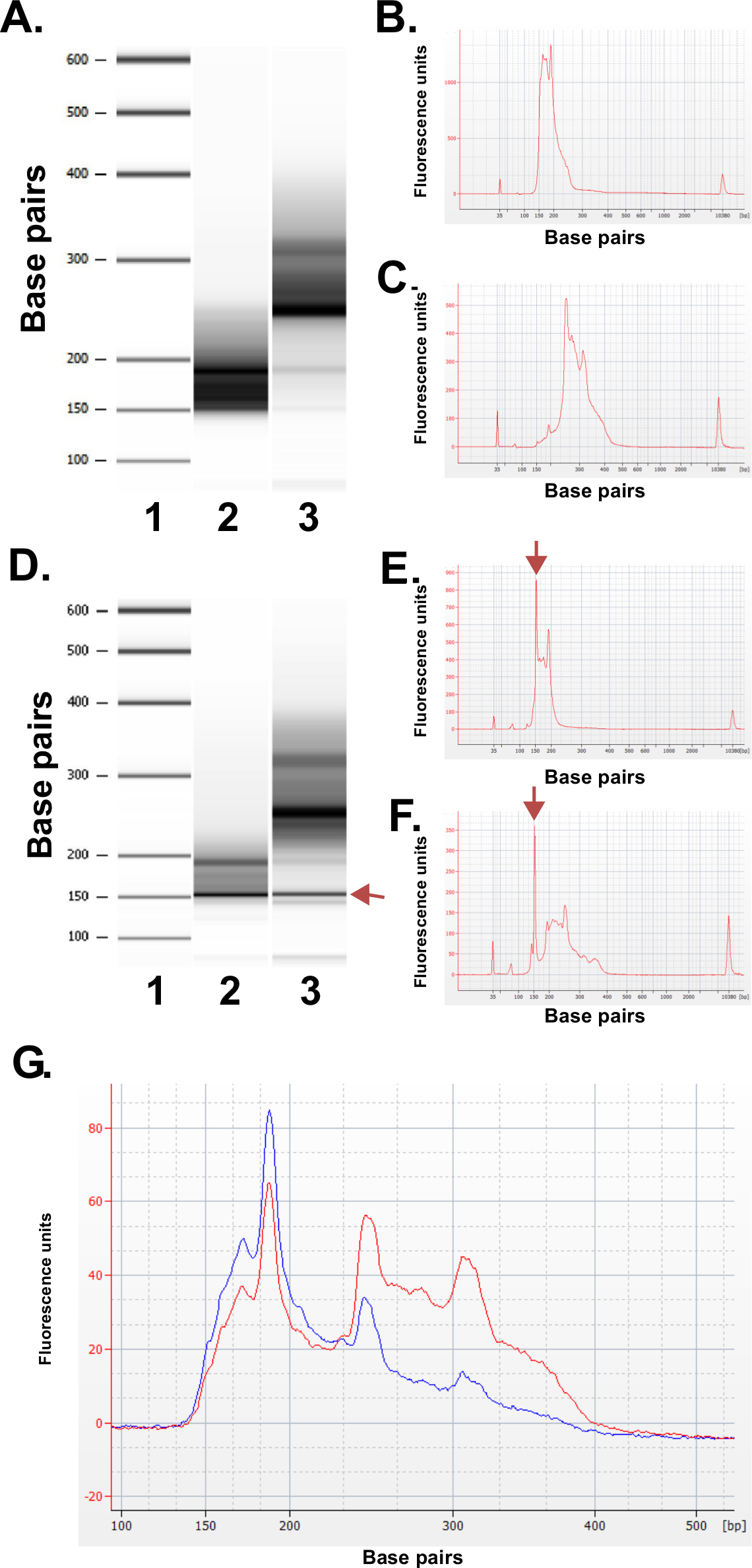
준비 라이브러리의 품질을 평가합니다. 라이브러리 제작 일상적 BP 150-250 250-400 염기쌍 (도 3A-C)의 분획 크기를 산출한다. 샘플 사이 라이브러리 크기 분포에 약간의 차이가 예상된다. 특정 시퀀스의 농축을 나타내는 매우 강렬 DNA 크기, 거기 모두 낮은 높은 라이브러리 분수 모릅니다. 반복적 인 DNA의 가족의 농축에 MspI 소화 결과 190 BP, 250 BP와 ERRBS 라이브러리의 310 bp의에서 인간 게놈에 존재하는 시퀀스. 이 세 가지가 반복 (그림 3A-C 및 3G 참조) ERRBS 라이브러리 (20)의 특성 서명을 나타냅니다. 대표적인 라이브러리는 차세대 서열에 서열 분석 하였다R 사용하여 단일 엔드 읽습니다. 일루미나 HiSeq 2500 시퀀서에 권장되는 라이브러리 농도에서로드 할 때, mm 2 당 500,000-700,000의 클러스터 밀도가 예상된다. 클러스터의이 클러스터링 밀도, 81.6 % ± 3.14 % (N = 81)에서 필터 (그림 4A)를 전달합니다. 인해 라이브러리 삽입물의 낮은 복잡도의 단부 (MspI 인식 부위 : C ^ CGG)에 독립 제어 레인 경우 시퀀싱 동안 기록 강도 값 및 품질 점수는, 그러나, 처음 세 개의 염기 (도 4B-C)에서 매우 가변적 (설명 참조)에 포함되어, 염기의 85 % 이상 30의 품질 점수를 가질 것이다 (Q30 값도 4D 참조).
데이터 정렬 및 프로토콜의 수율 염기쌍 해상도 데이터에 설명 시토신 메틸화 판정 (표 7). 인간 게놈의 경우, 높은 출력 모드에서 HiSeq 2500의 한 차선에 ERRBS 라이브러리의 51주기 단일 읽기 시퀀싱 실행 일반LY는 153,194,882 ± 12,918,302 총 품질 필터링 및 어댑터 수율을 트리밍 한 후 152,231,183 ± 13189678가 분석 파이프 라인에 입력을 읽 읽고 생성합니다. ERRBS 라이브러리에 대한 평균 매핑 효율은 10 배의 CpG 당 최소 범위와 84.94 ± 16.29 (N = 100)의의 CpG 당 평균 적용 범위 일반적으로 62.95 % 3,183,594 ± 713547 CpGs의 표현과 함께 % 5.92 ±이다.
ERRBS 프로토콜 (: 멀티 플렉스 시퀀싱 프로토콜 적응 보충 파일 1 참조) 다중 의무입니다. (11)에 대한 네 개의 라이브러리를 N =;. 대표 시퀀싱 데이터는 다중화 된 순서 실행 (51 사이클 단일 읽기 시퀀싱 실행에서 그림 5 데이터에 요약되어 실행되며 N = 레인 당 두 개의 라이브러리 128; n은 레인 당 11 세를위한 라이브러리 N = 100)과 시뮬 단일 차선을 다운 샘플링 (51 사이클 하나의 읽기 순서 실행 ERRBS 라이브러리의 전체 차선 염기 서열과 비교했다) 레인 당즉 50 %, 33 % 및 25 %는 레인 당 판독한다 (2, 3, 4 레인 당 샘플 각각 다중화; N = 3). 샘플 근처의 번호가 판독 멀티플렉싱 인자 감소 된 바와 같이, 10 배의 커버리지 및 최소의 CpG 당에 따르면 덮여 CpGs의 수뿐만 아니라 (도 5 및 표 8) 감소한다. 예상되는 비의 CpG 사이트의 평균 전환율은 0.04 % (N = 400) ± 99.85 %이다. 99 %보다 낮은 전환율은 false 메틸화 수준의 높은 요금이 발생할 수 있습니다 최적의 중아 변환보다 나타낼 수 있습니다.
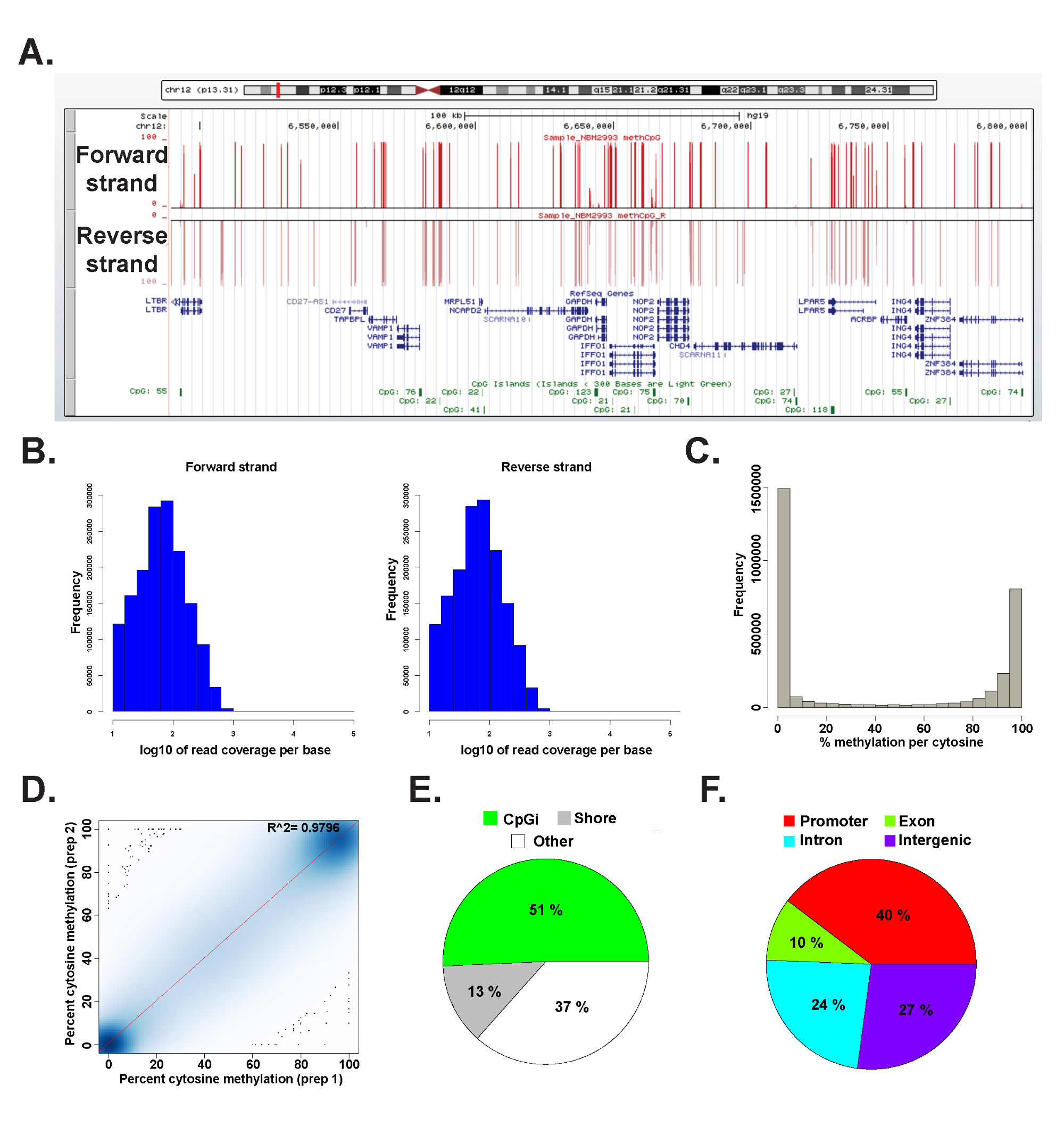
대표적인 인간 게놈 DNA에서 준비 ERRBS 라이브러리에서 데이터는 methylKit 패키지 (26) (명령 세부 사항에 대한 보충 코드 파일 1 참조)를 사용하여 R 2.15.2 (45)에서 분석 하였다. 데이터는 일반적으로 사용되는 게놈 브라우저 (도 6a)에서 가시화 될 수있다. 시토신 메틸화 데이터는 모두 동일 가닥 (도 6B)로부터 유도되는 전체 범위잠재적 인 시토신 메틸화 수준 (그림 6C)의 스펙트럼. 데이터 결과 (그림 6D)과 게놈 유전자 좌의 광범위한 스펙트럼에 CpGs 커버 사이의 대표적인 인간의 DNA 샘플 수율 높은 일치의 기술 복제물의 분석 (같은 그림 (e) 및 F 및 이전에 26 설명). 기술적 복제본 높은 R 2 값 (97 % 이상)을 수득되지만, 생물학적 레플리카 R를 0.92에서 0.96 (26)에 이르기까지, 0.86 (데이터 미기재) 2보다 낮은 값 R을 산출 할 것이다 다른 인간 세포 유형을 비교 2 값을 산출 할 것이다.

그림 1 : ERRBS 프로토콜 단계의 흐름 차트. 차트 전통적인 작업 일에 완료 할 수있는 단계를 나타냅니다. * 잠재적 일시 정지 지점을 나타냅니다 (즉시 수행 보내고 결찰은 정리하고 크기 선택, 프로토콜의 5 단계) 전에하는 샘플은 프로토콜의 지속 시간을 진행하기 전에 -20 ° C에서 동결 할 수 있습니다.

그림 2 : 사이즈 선택 프로토콜입니다. (A) ERRBS 사과의 일종 준비 프로토콜에서 사용되는 설정의 스크린 샷 (프로토콜 5.1.2 절 참조 - 5.1.6) : (1) 선택 카세트 타입입니다. (2) 선택 기준은 사용한다. (3) 각 레인에 대한 컬렉션 모드를 선택합니다. (4) 컬렉션 혈압 범위를 입력합니다. (5) 프로토콜을 저장 프로토콜 5.2 절에 사용되는 수동 겔 추출 (B) 단계 :. (1) 가시화 젤 사다리. (2) 면도날을 사용하여 사이즈 선택에 대한 크기를 표시됨. 절제 샘플 (: 150 ~ 250 bp의 높은 비율 : 250-400 bp의 낮은 부분) (3) 이미지.">이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.

그림 3 : bioanalyzer 기계를 사용하여 인간의 DNA 샘플로부터 제조 된 대표 ERRBS 라이브러리에 대한 품질 관리 결과. (A) 표준 사다리 (1)를 보여주는 젤 같은 이미지, 낮은 라이브러리 분획 (피핀 초등학교에서 135-240 bp의 분율) 2) 높은 라이브러리 분율 (피핀 초등학교에서 240-410 bp의 분율) . 예상보다 낮은 라이브러리 부분의 3) (B) Bioanalyzer의 electropherogram (C) 예상보다 높은 라이브러리 부분의 Bioanalyzer의 electropherogram D -.. F) 품질이 낮은 라이브러리 준비에서 대표 데이터. 표준 래더 (1)의 겔형 화상 (D), 저급 라이브러리 분획 (2)과 높은 분획 라이브러리 (3). 150 BP에서 밴드는 arro 표시w는 어댑터의 과도한 양을 나타냅니다. 아래의 Electropherogram (E) 및 (화살표로 표시) 150 BP의 과잉 어댑터 봉우리와 높은 라이브러리 분수 (F). 시퀀싱 풀링 ERRBS 라이브러리 (G) Bioanalyzer의 electropherogram. 레드 추적 높고 낮은 분수의 동일한 표현과 높은 품질의 풀 라이브러리를 나타냅니다. 블루 추적 인해 높고 낮은 분수의 동일한 표현의 부족으로 순서에 적합하지 풀링 된 라이브러리를 나타냅니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 4 : 대표에 대한 차트를 시퀀싱은 높은 출력의 HiSeq 2500 시퀀서 (51) 사이클 하나의 읽기 순서 실행 ERRBS모드. (A) 클러스터 밀도 (밀리미터 당 2 = 1000 클러스터의 제곱 K / mm, 파란색).과 ERRBS 라이브러리와 두 개의 차선 필터 (녹색)을 통과 클러스터 밀도 (B)와 차선의 첫 번째 30 회에서 볼 전형적인 강도 ERRBS 라이브러리. 대한 최초의 3주기의 강도에 MspI 소화의 CGG 서명을합니다. 하나 ERRBS 차선의 각 사이클 30 이상의 품질 평가 점수 (%> Q30)와 기지의 (C) 비율입니다. (D) 품질 점수 분포 하나 ERRBS 레인의 모든 사이클. 블루 = Q30, 녹색 미만 = 이하 또는 Q30에 같음. 이 레인에서, 염기의 84.7 %가 30 이상 품질 평가 점수를했다.

그림 5 : 출력 결과를 시퀀싱. 레인 순서 RU 당 다중 및 단일 샘플에서 실험 데이터의 박스 플롯 NS (녹색 상자로 표시)과 세 ERRBS 라이브러리의 시퀀싱 실행에서 시뮬레이션 다운 샘플링에 의해 도출 된 데이터 (파란색 상자로 표시 각 시퀀싱 실행에 대한 다섯 번을 샘플링). 하나의 읽기 순서가 실행 51주기가 다중 요인에 대응에서 ERRBS 라이브러리의 수는 레인 당 시퀀싱. 1 = 전체 차선 또는 100 읽고 레인 당 하나의 ERRBS 라이브러리에서 데이터를 나타냅니다의 %; 레인 2 = 50 %와 레인 당 두 ERRBS 라이브러리에서 데이터를 나타냅니다; 차선의 3 = 33 %와 레인 당 세 ERRBS 라이브러리에서 데이터를 나타냅니다; 그리고, 레인 4 = 25 %와 레인 당 네 ERRBS 라이브러리의 데이터를 나타낸다. (A) 멀티플렉싱 인자 당 판독 횟수, 또는 분석 서열 번호. (B) 다중화 당 시퀀싱 데이터에 포함 된 CpG의 수가 인자. (C) 다중 요소마다의 CpG 당 평균 범위."_blank>이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.

그림 6 : 인간 게놈 DNA에서 준비 ERRBS 라이브러리에서 대표 데이터 캘리포니아 (A) 대학, 산타 크루즈 (UCSC) 게놈 브라우저 ERRBS 시퀀싱 차선 대표 데이터의 43 이미지.. y 축 스케일 바는 10 배의 최소 덮여 각 시토신의 0 ~ 100 % 메틸화를 나타냅니다. 상위 사용자 정의 트랙은 앞으로 가닥을 나타내고 낮은 사용자 정의 트랙은 반대 가닥을 나타냅니다. . 표시가 CHR12입니다. 앞으로 함께의 CpG 범위의 (B) 분포 히스토그램 6,489,523-6,802,422 (hg19)이 게놈 영역 내에서 나온 RefSeq 유전자와의 CpG 섬의 포괄적이고 대표적인 인간의 CD34 + 골수 샘플에서 가닥을 역 (C) 유통 히스토그램대표적인 인간의 CD34 + 골수 샘플의 두 가닥을 따라의 CpG 메틸화 수준의. 인간의 DNA 샘플의 대표 기술 복제본의 CpG 메틸화 수준의 (D)의 상관 관계 플롯. (E) 원형 차트는 ERRBS 덮여 CpGs의 비율을 설명하는 인간의 게놈 DNA로부터 제조 된 대표 샘플에서의 CpG 섬 (밝은 녹색)의 CpG 해안 (회색) 및 기타 지역 (흰색)에 주석. (F) 파이 차트는 유전자 프로모터에 주석 ERRBS 덮여 CpGs의 비율 (빨간색을 설명 ), 엑손 (녹색), 인트론 (파란색)과 유전자 간 영역 (보라색). 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}
| 시약 | 체적 | 논평 |
| 10 배 T4 DNA 리가 제 반응 완충액 | 10 μL | |
| 옥시 뉴클레오티드 인산 (의 dNTP) 솔루션 믹스 | 4 μL | 각 염기의 10 mm의 혼합 |
| T4 DNA 중합 효소 | 5 μL | 3000 단위 / ㎖ |
| DNA 중합 효소 I 대형 (클레 나우) 조각 | 1 μL | 5,000 단위 / ㎖ |
| T4 폴리 뉴클레오티드 키나제 | 5 μL | 10,000 단위 / ㎖ |
| DNase의없는 물 | 45 μL |
표 1 : 종료. 수리 반응 시약 시약 명 및 수량 단부 수리 반응 (프로토콜 단계 2.1)에 사용되는.
| 시약 | 체적 | 논평 |
| 10 배 반응 완충액 | 5 μL | 예를 들어, NEBuffer 2 |
| 1 mM의 2'- 데 옥시 아데노신 5'- 삼인산 (으로 dATP) | 10 μL | |
| 클레 나우 단편 (3 '→ 5'엑소) | 3 μL | 5,000 단위 / ㎖ |
표 2 :. A-미행 반응 시약 시약 이름과 수량은 A-미행 반응 (프로토콜 3.1 단계)에 사용됩니다.
| 시약 | 체적 | 논평 |
| DNase의없는 물에 15 μm의 어닐링 어댑터 | 3 μL | PE 어댑터 1.0 PE 어댑터 2.0; 시퀀스 및 기준은 표 4 참조 |
| 10 배의 T4 DNA 리가 제 반응 완충액 | (5)(81), L | |
| T4 DNA 리가 | 1 μL | 200 단위 / ㎖ |
| DNase의없는 물 | 31 μL |
표 3 : 어댑터 연결 반응 시약 시약 명 및 어댑터 연결 반응 (프로토콜 단계 4.2)에서 사용되는 양..

표 4 :. 연결 반응 (프로토콜 4 단계)과 PCR 증폭 단계 (프로토콜 7 단계)에서 ERRBS 프로토콜에서 사용 올리고의 ERRBS 프로토콜에 사용 올리고 목록.
| 시약 | 체적 | 논평 |
| 10 배는 FastStart 고 충실도 반응 버퍼 위스콘신18 mM의 염화 마그네슘 번째 | 20 μL | |
| 10 mM의 dNTP의 솔루션 믹스 | 5 μL | |
| 25 μM PCR PE 프라이머 1.0 | 4 μL | 표 4 참조 |
| 25 μM PCR PE 프라이머 2.0 | 4 μL | 표 4 참조 |
| 는 FastStart 고 충실도 효소 | 2 μL | 5 단위 / μL는 FastStart의 Taq DNA 중합 효소 |
| DNase의없는 물 | 125 μL |
표 5 : PCR 반응 시약 시약 명 및 PCR 증폭 반응 (프로토콜 단계 7.1)에서 사용되는 양..
| 프로토콜 단계 | 시약 / 프로토콜 세부 사항 | 입력 DNA 금액 | ||
| 5-10 NG | 25 NG | 50 NG | ||
| (1) | MspI 효소 | 1 μL | 2 μL | 2 μL |
| MspI 다이제스트 반응 부피 | (50) | (100) | (100) | |
| 4 | 연결 반응에 어댑터 | 1 μL | 2 μL | 3 μL |
| 라이 게이션 반응 부피 | 20 μL | 25 μL | 50 μL | |
| (5) | 사이즈 선택 프로토콜 | 수동 젤 만 | 피핀 준비 또는 수동 젤 | 피핀 준비 또는 수동 젤 |
| 7 | PCR 프라이머 농도 | 25 μM | 25 μM | 14주기 10 μM; 18주기 25 μM |
| PCR 횟수 | (18) | (18) | 14-18 | |
표 6 :. 5-50 NG에 이르기까지 입력 자료 수량 프로토콜 단계 수정 프로토콜에 걸쳐 여러 단계의 출발 물질의 다양한 양에서 높은 품질의 라이브러리를 생성하는 데 사용되는 시약 수량의 수정을 필요로한다. 키 시약 수량의 변경이 여기에 포함된다. 그에 따라 반응에서 버퍼와 물 볼륨을 조정합니다.
| 대하 | 베이스 | 스트랜드 | 적용 범위 | freqC | freqT |
| chr1 | 10564 | R | (366) | 85.52 | 14.48 |
| chr1 | 10571 | F | (423) | 91.25 | 8.75 |
| chr1 | 10542 | F | (432) | 91.2 | 8.8 |
| chr1 | 10563 | F | (429) | 94.64 | 5.36 |
| chr1 | 10572 | R | (366) | 96.99 | 3.01 |
| chr1 | 10590 | R | (370) | 88.11 | 11.89 |
| chr1 | 10526 | R | (350) | (92) | 8 |
| chr1 | 10543 | R | (368) | 92.93 | 7.07 |
| chr1 | 10525 | F | (433) | 91.92 | 8.08 |
| chr1 | 10497 | F | (435) | 88.74 | 11.26 |
도표 7 : 대표 ERRBS 데이터. 데이터 정렬 시토신 메틸화 판정 후에 염기쌍 데이터가 얻어진다 각각의 CpG 덮여 들어 바와 같이 배향 프로토콜 게놈 좌표 (컬럼 : CHR = 염색체, Base와 스트랜드)을 결정한다. 특정 유전자좌의 커버리지 레이트 (커버리지 )와 퍼센트 (freqC 및 freqT으로 감지 시토신 대 티미 딘의 비율을 각각).
| 레인 당 ERRBS 라이브러리의 수 | 유일하게 정렬 된 읽기의 수를 의미 | CpGs의 숫자가 덮여 평균 | 의 CpG 당 커버리지를 의미 |
| (1) | 152231184 ± 13189678 | 3,183,594 ± 713547 | 85 ± 16 |
| 이 | 77,680,837 ± 7,657,058 | 2674823153494을 ± | 49 ± 9 |
| 3 | 49,938,156 ± 2,436,865 | 2,552,186 ± - 76624 | 39 ± 2 |
| 4 | 34,457,208 ± 4,441,686 | 1,814,461 ± 144339 | 28 ± 4 |
도표 8 :. 단일 염기 서열에서 얻은의 CpG 사이트 당 평균 고유 정렬의 표준 편차, CpGs의 수는 포함 읽기 및 적용 : 단일 및 다중 ERRBS 라이브러리를 시퀀싱 대표 매개 변수 표시됨 51주기 단일 읽기 시퀀싱 실행에서 레인 당 데이터입니다 레인 당 ERRBS 라이브러리 (N = 100), 레인 당 두 ERRBS 라이브러리 (N = 128), 레인 (N = 11)에 세 ERRBS 라이브러리, 레인 (N = 11)에 네 ERRBS 라이브러리.
토론
생물학적으로 관련 게놈 영역에서 시토신 메틸화의 프로토콜을 제시 수익률의 염기쌍 해상도 데이터. 출발 물질 50 NG 최적화되어 작성된 프로토콜은, 그러나, 입력 범위의 재료 (5 NG 이상) (26)을 처리하도록 적응 될 수있다. 표 6에서 볼 수 있듯이이 프로토콜의 일부 단계의 조정을 필요로 할 것이다. ERRBS 라이브러리는 시퀀싱에 의해 달성 될 수있는 한 쌍의 단부 및 상기 게놈 시퀀싱 따르면 의무가있는 더 긴 51 회 이상의 판독한다. 샘플 당 낮은 비용 프로토콜을 제안 할 것이다 다중화 순서는, 그러나, 이것은 데이터 (도 5 및 표 8)에 표현 된 CpG 사이트 당 감소 따르면 초래할 것이며, 당 높은 커버리지를 요구 분석을 수행하기에 따르면 충분한 깊이를 산출하지 않을 (랜댄 등. (33)에 의해 설명 된 바와 같이 예 :)의 CpG 사이트. 마지막으로,이 프로토콜 (또는 중아 기반 protoco난) 메틸 시토신과 하이드 록시 메틸 시토신 (46, 47) 사이에 구별 할 수 없습니다. 그러나, 다른 프로토콜로 통합 될 수있는 데이터를 생성 다른 변형을 묘사하는 48,49 결과, 시토신 및 다른 변형들이 최근 관심이어야 50을보고했다.
그림 3 세대 (빨간색 추적) 모두 라이브러리 분수에서 동일한 몰 기여를 나타내는과 같이 그림 3A-C에 표시하고, 한 번 시퀀싱 풀 추적을 산출로 높은 품질의 라이브러리가 표시됩니다. 도서관 준비 실패는 절차를 수행하는 동안 모든 단계에서 발생할 수 있습니다. DNA 분해 처리되어 있으면이 프로토콜에 기재된 순서 변수를 이용하여 낮은 커버리지의 CpG 따라서 MspI 단편 및 농축되지 않은 라이브러리 초래할 것이다. 효소가 비 기능적 또는 부주의 한 반응에서 제외되면, 프로토콜은 예상 라이브러리를 산출하지 않는다. 결찰 것이란 경우ction의 비효율적이며, 어댑터는 예상보다 더 높은 농도로, 그리고 / 또는 사용 된 프라이머 농도가, 최종 증폭 단계에 대한 제한적인 시약이고, 라이브러리 오류가 발생할 수있다. 때문에 라이브러리와 과잉 어댑터 모두의 무차별 적 클러스터링 또한 시퀀싱을 방해 라이브러리 (도 3D-F bioanalyzer 결과에 ~ 150 BP에서 피크로 본) 초과 어댑터. 이러한 라이브러리 명백하게 정상적으로 시퀀스 수 있지만, 상당 부분은 단지 어댑터 서열 것이다 읽는다. 과량 어댑터 라이브러리에서 관찰되는 경우, 재료는 수량 비율 어댑 최적 입력 재료를 이용 가능한 경우 라이브러리 제조를 반복하는 것이 최상이다. 마지막으로, 라이브러리의 PCR 증폭 효율을 보장하기 위해, 상기 하부 및 높은 라이브러리 분획물 변환 비설 파이트 및 PCR 농축 단계에 걸쳐 별도의 샘플로 유지된다. 그렇게하지 않으면 P 동안 증폭 차동 효율을 산출CR의 (그림 3G 파란색 추적에서 볼 수 있듯이) 높고 낮은 분수의 반응 및 시퀀싱 동안 각 라이브러리 부분에 언급 한 각각의 게놈 유전자 좌의 불평등 한 표현의 가능성. 사용자는 라이브러리가 생성되는 증폭하기 위해 필요한 최적의 PCR 사이클의 추가적인 적정 중아 변환 후 즉시 정량적 PCR 공정을 포함하는 것을 선택할 수있다.
ERRBS 도서관 준비 프로토콜은 특정 시약을 권장하는 몇 가지 주요 단계가 있습니다. 최종 수리 공정에서, 네 개의 뉴클레오티드의 dNTP 혼합의 사용은 MspI 효소 등급 활성 및 원래 DNA 샘플에 존재하는 전단 된 DNA 단편으로부터 얻어진 것과 CG 오버행을 함유하지 않는 임의의 제품의 최종 수리를 허용한다. 이 결과에서 개선의 CpG 표현 초래한다. 결찰 단계에서 그것은 보장 고농도 리가 제 (200 단위 / ml) 및 메틸화 어댑터를 사용하는 것이 중요하다는 결찰를 Reacti에 효율적이고 중아 변환은 정확한 데이터의 위치 맞춤을위한 필수적인 어댑터 서열에 영향을주지 않는다는 것을. PCR 단계에서 바이 설 파이트 처리 GC가 풍부한 DNA 단편을 증폭 할 수있는 중합 효소를 사용하여 높은 특이성에 대한 필요가있다. 마지막으로, 과량의 어댑터 및 프라이머의 제거를 보장하기 위해, (예 : Agencourt AMPure XP) SPRI 비드 정제 결찰 및 PCR 생성물 아이솔레이션 용 칼럼 기재 분석법 아닌 권장된다.
고품질의 데이터를 생성하기 위해서는, 바이 설 파이트 변환 효율을 보장하는 것이 중요하다. 표시 제어부는 유저에게 시퀀싱 전에 변환 효율을 결정하는 능력을 제공한다. 대안으로, 람다 DNA와 같은 비 - 인간 DNA는 내부 제어 (스파이크)에서 사용될 수있다. 인해 종의 차이에 따라, 이런 종류의 제어가 직접 시퀀싱 하류에 포함될 수있다 (예를 들면 유 의해 사용되는 바와 같이, 등. 34). 그러나, 스파이크의 난 경우이용 S, 이는 고유 증폭 독립적 라이브러리 시퀀싱 전에 서열화 않는 라이브러리 시퀀싱 이전에 변환 효율을 결정하기 위해 사용될 수 없다. 결정 전환율은 비의 CpG 사이트에서 메틸화 상태를 기준으로합니다. 이것은 (예를 들어 배아 줄기 세포) 이외의 CpG 컨텍스트와 평행 샘플 또는 이러한 목적을 위해 이용 될 수 변환 효율을 평가하는 다른 수단이 높은 사이토 신 메틸화의 맥락에서 사용하기에 적합하지 않을 수있다.
ERRBS 라이브러리의 순서에 고유 한 주소로 몇 가지주의해야 할 점이있다. 서열 라이브러리 분획 처음 세 염기 인해 MspI 인식 컷 사이트를 거의 균일하게 비 - 랜덤 (C ^ CGG도 4B, C 참조). 때문에 낮은 품질에 중요한 데이터 손실의 가능성이 결과는 순서 동안 명백한 높은 클러스터 밀도에도 불구하고 가난한 클러스터 현지화로 인한 읽습니다. 이 장벽을 극복하기 위해,전용 제어 차선으로 독립적 인 레인 (PhiX 제어 또는 다른 라이브러리 유형)에서 높은 복잡성 라이브러리를 포함한다. 높은 복잡성 라이브러리는 서열 처음 네 기지에서 A, C, T와 G의 균형 표현을 포함한 끝이있다. 적절한 제어 차선은 RNA-SEQ, 칩 서열, 전체 게놈 시퀀싱, 또는 시퀀싱 장비 제조업체 (예 : PhiX 제어 V3)에 의해 제공되는 제어와 같은 라이브러리를 포함한다. 각각의 시퀀싱 런 제어 차선으로 지정되면, 클러스터 위치를 검출하는 순서의 최초의 DNA 염기 중에 이용되는 행렬 생성을위한 기초로서 기능 할 수있다. 높은 품질로 된 CpG 사이트 당 평균 범위를 올릴 캡처 읽기 5.2 (N = 4). 대안 적으로, 기술적 인 어려움이 또한 앞서 설명한 바와 같이 23 어두운 시퀀싱 방법을 사용하여 극복 될 수있다. 다른 염기 서열 기준은 제조사의 프로토콜에 따라 표준 운영 절차를 따르십시오. CpG 디 C 당 마지막 따르면데이터 분석을위한 호센 관심 생물학적 질문하여 사용자에 의해 부분적으로 유도 될 것이다. 10 배의 커버리지는 높은 임계 범위 분석 방법 그러나이 임계 값은 그 관심이어야 낮아질 수를 제공한다.
ERRBS 데이터 분석의 전체 논의는하지만, 차별적 메틸화 된 시토신와 지역은 오픈 소스 도구 31,51-53를 사용하여 결정할 수있다,이 문서의 범위를 벗어납니다. 추가 고려 사항 및 분석 방법은 54, 55을 잘 설명되어 있고, 리더는 계획된 분석에 가장 적합한 도구 문헌을 검색 할 것을 권장한다.
게시 된 다른 방법에 비해, ERRBS 재현성의 설명 수익률 높은 속도로 수행 나흘 간 프로토콜을 제공합니다. 그것은 MassARRAY EpiTYPER 26 일 표준 금에 비교 검증되었습니다, 비용 효율적인 커버력이 높은 데이터이며, 다양한 입력 자료에 대한 적응력시퀀싱 방법 (낮은 주파수의 임상 샘플 처리 및 다른 세포 유형에 유리한) 양. 그것은 게놈 전체의 전사 조절 인자 결합, 크로 마틴 리모델링, 후생 유전 학적 마크와 관심의 다른 시토신 수정을 프로파일 링 다른 기술과 분석 생물학적으로 관련 유전자좌에서 염기쌍 해상도를 제공하며, 통합 사용할 수 있습니다. 이러한 연구에서 ERRBS 데이터 사용은 포괄적 인 분자 접근에 기여 차원 생물학적 모델과 인간 질병의 연구에서 분석에 대한 높은 허용 할 수 있습니다.
공개
저자는 공개 할 관심의 충돌이 없습니다.
감사의 말
We thank all the authors of the original ERRBS report. We thank Mame Fall for technical assistance. We acknowledge the Weill Cornell Medical College Epigenomics Core for technical services and assistance. The work was supported by a Sass Foundation Judah Folkman Fellowship, an NCI K08CA169055 and ASH-AMFDP12005 to FGB, NIH R01HG006798 and R01NS076465, funding from the Irma T. Hirschl and Monique Weill-Caulier Charitable Trusts and STARR Consortium (I7-A765) to CEM, and an LLS SCORE grant (7006-13) to AMM.
자료
| Name | Company | Catalog Number | Comments |
| MspI | New England Biolabs | R0106M | 100,000 units/ml |
| NEBuffer 2 | New England Biolabs | B7002S | Reaction buffer for MspI enzyme; protocol step 1.2 |
| Phenol solution | Sigma-Aldrich | P4557 | Equilibrated with 10 mM Tris HCl, pH 8.0; see safety and handling instructions at http://www.sigmaaldrich.com/catalog/product/sigma/p4557 |
| Chloroform | Sigma-Aldrich | C2432 | See safety and handling instructions at http://www.sigmaaldrich.com/catalog/product/sial/c2432 |
| Glycogen | Sigma-Aldrich | G1767 | 19-22 mg/ml |
| NaOAc | Sigma-Aldrich | S7899 | 3 M, pH 5.2 |
| Ethanol | Sigma-Aldrich | E7023 | 200 proof, for molecular biology |
| Buffer EB | Qiagen | 19086 | 10 mM Tris-Cl, pH 8.5 |
| tris(hydroxymethyl)aminomethane (Tris) | Sigma-Aldrich | T1503 | prepare a 1 M, pH 8.5 solution |
| Tris- Ethylenediaminetetraacetic acid (TE) | Sigma-Aldrich | T9285 | Dilute to 1x buffer solution per manufacturer's recommendations |
| T4 DNA Ligase Reaction Buffer | New England Biolabs | B0202S | 10x concentration |
| Deoxynucleotide triphosphate (dNTP) Solution Mix | New England Biolabs | N0447L | 10 mM each nucleotide |
| T4 DNA Polymerase | New England Biolabs | M0203L | 3,000 units/ml |
| DNA Polymerase I, Large (Klenow) Fragment | New England Biolabs | M0210L | 5,000 units/ml |
| T4 Polynucleotide Kinase | New England Biolabs | M0201L | 10,000 units/ml |
| QIAquick PCR Purification Kit | Qiagen | 28104 | Used for DNA product purification in protocol step 2.3 |
| 2'-deoxyadenosine 5'-triphosphate (dATP) | Promega | U1201 | 100 mM |
| Klenow Fragment (3'→5' exo-) | New England Biolabs | M0212L | 5,000 units/ml |
| MinElute PCR Purification Kit | Qiagen | 28004 | Used for DNA product purification in protocol step 3.3 |
| T4 DNA Ligase | New England Biolabs | M0202M | 2,000,000 units/ml |
| Methylation Adapter Oligo Kit | Illumina | ME-100-0010 | |
| Agencourt AMPure XP | Beckman Coulter | A63881 | Used in protocol sections that implement magnetic bead purification steps (steps 4.3 and 8.2). Equilibrate to room temperature before use. |
| Pippin Prep Gel Cassettes, 2% Agarose, dye-free | Sage Science | CDF2010 | with internal standards |
| Certified Low Range Ultra Agarose | Bio-Rad | 161-3106 | |
| Tris-Borate-EDTA (TBE) buffer | Sigma-Aldrich | T4415 | |
| Ethidium bromide solution | Sigma-Aldrich | E1510 | 10 mg/ml |
| 50 bp DNA Ladder | NEB | N3236S | |
| 100 bp DNA Ladder | NEB | N3231S | |
| Gel Loading Dye, Orange (6x) | NEB | B7022S | |
| Scalpel Blade No. 11 | Fisher Scientific | 3120030 | |
| QIAquick Gel Extraction Kit | Qiagen | 28704 | |
| EZ DNA Methylation Kit | Zymo Research | D5001 | Used in protocol step 6.2 |
| EZ DNA Methylation-Lightning Kit | Zymo Research | D5030 | Alternative for step 6.2 |
| Universal Methylated Human DNA Standard | Zymo Research | D5011 | Used as bisulfite conversion control |
| FastStart High Fidelity PCR System | Roche | 03553426001 | |
| Qubit dsDNA High Sensitivity Assay Kit | Life Technologies | Q32854 | A fluorescence-based DNA quantitation assay; used in protocol steps 1.1, 9.1 and 10.1 |
| DynaMag-2 Magnet | Life Technologies | 12321D | |
| High Sensitivity DNA Kit | Agilent Technologies | 5067-4626 | |
| 2100 Bioanalyzer | Agilent Technologies | ||
| PhiX Control v3 | Illumina | FC-110-3001 | |
| HiSeq 2500 | Illumina | ||
| Pippin Prep | Sage Science | ||
| Qubit 2.0 Fluorometer | Life Technologies | Q32872 | |
| TruSeq SR Cluster Kit v3-cBot-HS | Illumina | GD-401-3001 | |
| TruSeq SBS Kit v3-HS | Illumina | FC-401-3002 | |
| TruSeq RNA Sample prep | Illumina | RS-122-2001 | Barcoded adapters used for multiplexing libraries; See Supplemental file for multiplexing protocol. |
| Microcentrifuge | |||
| Vortex Mixer | |||
| Dry Block Heater | |||
| Thermal Cycler | |||
| Water Bath | |||
| Gel electrophoresis system | |||
| Electrophoresis power supply | |||
| Gel doc | |||
| UV or blue light transilluminator |
참고문헌
- Jones, P. A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 13 (7), 484-492 (2012).
- Barlow, D. P. Genomic imprinting: a mammalian epigenetic discovery model. Annual Review Of Genetics. 45, 379-403 (2011).
- Thiagarajan, R. D., Morey, R., Laurent, L. C. The epigenome in pluripotency and differentiation. Epigenomics. 6 (1), 121-137 (2014).
- Reik, W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 447 (7143), 425-432 (2007).
- Hartnett, L., Egan, L. J. Inflammation, DNA methylation and colitis-associated cancer. Carcinogenesis. 33 (4), 723-731 (2012).
- Smith, Z. D., Meissner, A. DNA methylation: roles in mammalian development. Nat Rev Genet. 14 (3), 204-220 (2013).
- Li, E., Bestor, T. H., Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 69 (6), 915-926 (1992).
- Okano, M., Bell, D. W., Haber, D. A., Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 99 (3), 247-257 (1999).
- Feinberg, A. P. Phenotypic plasticity and the epigenetics of human disease. Nature. 447 (7143), 433-440 (2007).
- Bock, C. Epigenetic biomarker development. Epigenomics. 1 (1), 99-110 (2009).
- Laird, P. W. The power and the promise of DNA methylation markers. Nat Rev Cancer. 3 (4), 253-266 (2003).
- How Kit, A., Nielsen, H. M., Tost, J. DNA methylation based biomarkers: practical considerations and applications. Biochimie. 94 (11), 2314-2337 (2012).
- Mikeska, T., Bock, C., Do, H., Dobrovic, A. DNA methylation biomarkers in cancer: progress towards clinical implementation. Expert Review Of Molecular Diagnostics. 12 (5), 473-487 (2012).
- Gyparaki, M. T., Basdra, E. K., Papavassiliou, A. G. DNA methylation biomarkers as diagnostic and prognostic tools in colorectal cancer. Journal of Molecular Medicine. 91 (11), 1249-1256 (2013).
- Figueroa, M. E., et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 17 (1), 13-27 (2010).
- Heyn, H., Mendez-Gonzalez, J., Esteller, M. Epigenetic profiling joins personalized cancer medicine. Expert review of Molecular Diagnostics. 13 (5), 473-479 (2013).
- Kulis, M., Esteller, M. DNA methylation and cancer. Advances in Genetics. 70, 27-56 (2010).
- Xiong, Z., Laird, P. W. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 25 (12), 2532-2534 (1997).
- Meissner, A., et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 33 (18), 5868-5877 (2005).
- Gu, H., et al. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc. 6 (4), 468-481 (2011).
- Bock, C., et al. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol. 28 (10), 1106-1114 (2010).
- Harris, R. A., et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol. 28 (10), 1097-1105 (2010).
- Boyle, P., et al. Gel-free multiplexed reduced representation bisulfite sequencing for large-scale DNA methylation profiling. Genome Biol. 13 (10), R92(2012).
- Chatterjee, A., Rodger, E. J., Stockwell, P. A., Weeks, R. J., Morison, I. M. Technical considerations for reduced representation bisulfite sequencing with multiplexed libraries. Journal of Biomedicine & Biotechnology. 2012, 741542(2012).
- Lee, Y. K., et al. Improved reduced representation bisulfite sequencing for epigenomic profiling of clinical samples. Biological Procedures Online. 16 (1), 1(2014).
- Akalin, A., et al. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet. 8 (6), e1002781(2012).
- Hatzi, K., et al. A Hybrid Mechanism of Action for BCL6 in B Cells Defined by Formation of Functionally Distinct Complexes at Enhancers and Promoters. Cell Reports. 4 (3), 578-588 (2013).
- Will, B., et al. Satb1 regulates the self-renewal of hematopoietic stem cells by promoting quiescence and repressing differentiation commitment. Nature Immunology. 14 (5), 437-445 (2013).
- Lu, C., et al. Induction of sarcomas by mutant IDH2. Genes Dev. 27 (18), 1986-1998 (2013).
- Kumar, R., et al. AID stabilizes stem-cell phenotype by removing epigenetic memory of pluripotency genes. Nature. 500 (7460), 89-92 (2013).
- Li, S., et al. An optimized algorithm for detecting and annotating regional differential methylation. BMC Bioinformatics. 14, Suppl 5. S10(2013).
- Patterson, K., Molloy, L., Qu, W., Clark, S. DNA methylation: bisulphite modification and analysis. Journal of Visualized Experiments. (56), 3170(2011).
- Landan, G., et al. Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat Genet. 44 (11), 1207-1214 (2012).
- Yu, M., et al. Tet-assisted bisulfite sequencing of 5-hydroxymethylcytosine. Nat Protoc. 7 (12), 2159-2170 (2012).
- Goecks, J., Nekrutenko, A., Taylor, J., Galaxy, T. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 11 (8), R86(2010).
- Dorff, K. C., et al. GobyWeb: simplified management and analysis of gene expression and DNA methylation sequencing data. PLoS One. 8 (7), e69666(2013).
- Roehr, J. T., Dodt, M., Ahmed, R., Dieterich, C. Flexbar − flexible barcode and adapter processing for next-generation sequencing platforms. MDPI Biology. 1 (3), 895-905 (2012).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal, North America. 17 (1), 10-12 (2011).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (15), 2114-2120 (2014).
- Needleman, S. B., Wunsch, C. D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 48 (3), 443-453 (1970).
- Krueger, F., Andrews, S. R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 27 (11), 1571-1572 (2011).
- Li, H., et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25 (16), 2078-2079 (2009).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Res. 12 (6), 996-1006 (2002).
- Thorvaldsdottir, H., Robinson, J. T., Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in Bioinformatics. 14 (2), 178-192 (2013).
- Team, R. C. R. A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria. ISBN 3-900051-07-0, http://www.R-project.org (2012).
- Nestor, C., Ruzov, A., Meehan, R., Dunican, D. Enzymatic approaches and bisulfite sequencing cannot distinguish between 5-methylcytosine and 5-hydroxymethylcytosine in DNA. BioTechniques. 48 (4), 317-319 (2010).
- Huang, Y., et al. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS One. 5 (1), e8888(2010).
- Yu, M., et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 149 (6), 1368-1380 (2012).
- Song, C. X., et al. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell. 153 (3), 678-691 (2013).
- Ito, S., et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 333 (6047), 1300-1303 (2011).
- Akalin, A., et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 13 (10), R87-1186 (2012).
- Stockwell, P. A., Chatterjee, A., Rodger, E. J., Morison, I. M. DMAP: Differential Methylation Analysis Package for RRBS and WGBS data. Bioinformatics. 30 (13), 1814-1822 (2014).
- Sun, D., et al. MOABS: model based analysis of bisulfite sequencing data. Genome Biol. 15 (2), R38(2014).
- Bock, C. Analysing and interpreting DNA methylation data. Nat Rev Genet. 13 (10), 705-719 (2012).
- Rivera, C. M., Ren, B. Mapping human epigenomes. Cell. 155 (1), 39-55 (2013).
재인쇄 및 허가
JoVE'article의 텍스트 или 그림을 다시 사용하시려면 허가 살펴보기
허가 살펴보기This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. 판권 소유