
Method Article
Verbesserte Reduzierte Darstellung Bisulfit-Sequenzierung für die Bewertung der DNA-Methylierung bei Basenpaar Auflösung
* Diese Autoren haben gleichermaßen beigetragen
In diesem Artikel
Zusammenfassung
Enhanced Reduced Representation Bisulfite Sequencing is a method for the preparation of sequencing libraries for DNA methylation analysis based on restriction enzyme digestion combined with cytosine bisulfite conversion. This protocol requires 50 ng of starting material and yields base pair resolution data at GC-rich genomic regions.
Zusammenfassung
DNA-Methylierungsmuster Mapping ist stark in normalen und erkrankten Geweben untersucht. Eine Vielzahl von Methoden wurden etabliert, um die Cytosin-Methylierungsmustern in Zellen abzufragen. Reduzierte Darstellung des gesamten Genoms Bisulfit-Sequenzierung wurde entwickelt, um quantitative Basenpaar Auflösung Cytosin-Methylierungsmuster bei GC-reichen genomischen Loci zu erkennen. Dies wird durch die Kombination der Verwendung von einem Restriktionsenzym, gefolgt von Bisulfitumwandlung bewerkstelligt. Verbesserte Reduzierte Darstellung Bisulfit-Sequenzierung (ERRBS) erhöht die biologisch relevanten genomischen Loci bedeckt und wurde genutzt, um Cytosin-Methylierung in DNA aus Mensch, Maus und anderen Organismen zu profilieren. ERRBS initiiert mit dem Restriktionsenzym Verdauung von DNA, um niedermolekulare Bruchstücke für die Verwendung in der Bibliothek Vorbereitung erzeugen. Diese Fragmente werden zu Standardbibliothek Bau der nächsten Generation Sequenzierung unterzogen. Bisulfit-Umwandlung von nicht methylierten Cytosine vor der endgültigen amplificatiauf Schritt ermöglicht die quantitative Basisauflösung von Cytosin-Methylierung Niveaus in gedeckten genomischen Loci. Das Protokoll kann innerhalb von vier Tagen abgeschlossen sein. Trotz geringer Komplexität in den ersten drei Basen sequenziert, ERRBS Bibliotheken liefern hohe Datenqualität bei Verwendung eines vorgesehenen Ablaufsteuerung Spur. Mapping und Bioinformatik-Analyse wird dann durchgeführt und liefert Ergebnisse, die leicht mit einer Vielzahl von genomweite Plattformen integriert werden kann. ERRBS können kleine Mengen der Vorleistungsgüter nutzen, so dass es möglich ist, den menschlichen klinischen Proben und anwendbar in einer Reihe von Forschungsanwendungen verarbeiten. Das produzierte Video zeigt kritischen Schritte des ERRBS Protokoll.
Einleitung
DNA Methylierung an Cytosin (5-Methylcytosin) eine epigenetische Filter kritisch in Säugerzellen für eine Vielzahl von biologischen Prozessen, einschließlich aber nicht beschränkt auf Prägen, X-Chromosom Inaktivierung, die Entwicklung und die Regulation der Genexpression 1-8 beschränkt. Die Untersuchung der DNA-Methylierungsmuster in malignen und anderen Erkrankungen wurde krankheitsspezifische Muster bestimmt und dazu beigetragen, das Verständnis der Pathogenese der Krankheit und potentielle Biomarker Entdeckungen 9-17. Es gibt viele Protokolle, die das Epigenom für DNA-Methylierungs-Status abfragen. Diese können in der Affinitäts-geteilt werden, Restriktionsenzymbasis und Bisulfitumwandlung basierten Assays, Mikroarray oder Sequenzierplattformen abwärts zu nutzen. Darüber hinaus gibt es einige Protokolle, die diese allgemeinen Kategorien, einschließlich, aber nicht beschränkt auf, Kombinierte Bisulfit Restriktionsanalyse 18 begrenzt und reduzierte Darstellung Bisulfit-Sequenzierung (RRBS 19) zu überbrücken.
RRBS wurde ursprünglich von Meissner et al. 19,20 beschrieben. Das Protokoll wurde ein Schritt zur GC-reiche genomischen Regionen, gefolgt von Bisulfit-Sequenzierung, die quantitativen Basenpaarauflösung Daten geführt, die kostengünstig ist 21,22 bereichern. Die GC-reiche Regionen werden durch das MspI (C ^ CGG) Restriktionsenzym gezielte und Cytosin Methylierung durch Bisulfit-Umwandlung von Cytosinen (Deaminierung von unmodifizierten Cytosine in Uracil), gefolgt von Polymerasekettenreaktion (PCR) gelöst. RRBS bedeckt den Großteil der Genpromotoren und CpG-Inseln in einem Bruchteil der für einen gesamten Genoms erforderliche Reihenfolge; jedoch RRBS nur eine begrenzte Verbreitung von CpG-Ufer und anderen intergenen Regionen biologische Relevanz. Mehrere Gruppen haben seit dem ursprünglichen Bericht, der von der Methode und der daraus resultierenden Abdeckung dieser genomischen Regionen 23-25 verbessern veröffentlicht aktualisierte RRBS Protokolle. Verbesserte Reduzierte Darstellung Bisulfite Sequflussen (ERRBS) umfasst Bibliothek Vorbereitung Änderungen und eine alternative Datenabgleich Ansatz 26 im Vergleich zu RRBS. ERRBS führte zu einer höheren Anzahl an CpGs in den erzeugten Daten dargestellt wird und eine erhöhte Abdeckung aller genomischen Regionen 26 abgefragt. Dieses Verfahren wurde verwendet, um DNA-Methylierungsmuster in menschlichen Patienten und anderen tierischen Proben 26-30 lösen.
Die ERRBS Protokoll beschrieben Angebote Details über alle Schritte für den Abschluss und die Daten wurden unter Verwendung repräsentativer menschlicher DNA erzeugt benötigt (Proben wurden von zuvor berichteten deidentifizierte Patientenproben 31 erhalten und ein CD34 + Knochenmarksprobe von einem normalen menschlichen Spender). Das Protokoll umfasst eine automatische Größenauswahlprozesses, der die Bearbeitungszeit pro Probe reduziert und ermöglicht eine erhöhte Genauigkeit bei der Bibliotheksgrße Auswahl. Das Protokoll kombiniert eine Reihe etablierter molekularbiologischer Techniken. Hochmolekulare DNA wird w verdautit einem Methylierungsunempfindlichen Restriktionsenzym (MspI), gefolgt von End-Reparatur, A-Tailing, und Ligation von methylierter Adapter. Größe Auswahl der GC-reiche Fragmente durch Bisulfit-Konvertierung und PCR-Amplifikation vor der Sequenzierung gefolgt. Bisulfitumwandlung zuvor beschrieben 32 und detaillierte Überprüfung der Datenanalyse und Anwendungen würde den Rahmen dieses Artikels jedoch Empfehlungen und Referenzen sind für die Leser Einsatz enthalten gewesen. Das Protokoll kann über vier Tage durchgeführt werden und ist offen für kleine Eingangs (50 ng oder weniger) Materialmengen. Das Protokoll, wie beschrieben, liefert Ergebnisse mit hoher Deckkraft pro CpG-reichen nicht nur für Differenzmethylierungsstelle und der Region Bestimmungen, sondern auch für epigenetische Polymorphismus Erkennung durch Landan, et al. 33 beschrieben.
Protokoll
Alle Geschäfte durchgeführt werden von der Indiana University School of Medicine Institutional Animal Care und Verwenden Committee genehmigt und befolgt, National Institute of Health Richtlinien.
1. Operationstechnik
- Pflegen aseptische Technik bei diesem Verfahren, indem sie sterile Handschuhe, Instrumente, und ein steriles Operationsfeld nach NIH-Richtlinien 25. Sterilisieren Instrumente vor Beginn der Operation durch Autoklavieren ihnen (siehe Tabelle der spezifischen Reagenzien / Ausrüstung für die komplette Liste). Verwenden Sie eine Glasperlen Sterilisator Werkzeuge während der Operation zu sterilisieren.
2. Anästhesie und Vorbereitung
- Anesthetize die Maus in einem Anästhesie-Box mit einer Mischung aus 0,9 l / min Sauerstoff und 2,5% Isofluran mit einem veterinär Isofluran Verdampfersystem. Stellen Sie sicher, dass die Maus nicht zu Veränderungen in der Körperposition, bevor es dem Auspacken zu reagieren.
- Bewerben Augensalbe auf die mouse in die Augen, um sie vor dem Austrocknen zu schützen.
- Schalten Sie den Gasstrom aus der Box an den Nasenkegel. Platzieren Sie die Maus direkt auf seiner linken Seite auf einer erhitzten Unterlage mit einem Operationsfeld und absorbierende Papierbank mit der Nase und den Mund innerhalb des Kegels bedeckt. Die Maus Atemrhythmus und bewerten fortlaufend zu überwachen und anzupassen Isofluran Ebenen nach Bedarf (zwischen 2,5 bis 3% Isofluran), ein angemessenes Niveau der Narkose zu halten und verwenden die Zehe Prise Reflex auf insgesamt Sedierung bestätigen.
3. Chirurgische Vorgehensweise
- Ausrichten und konzentrieren das Stereoskop mit dem OP-Feld. Passen Sie den Nasenkegel und kleben Sie es nach unten, so dass es am Rand des Gesichtsfeldes positioniert ist.
- Mit der Maus, der auf der linken Seite, kleben Sie den Rand des rechten Ohr auf die Bugnase, Freilegung der Bereich hinter dem Ohr, wo der Schnitt gemacht werden. Stellen Sie sicher, dass die hintere Ohrvene fährt horizontal über das Ohr. Man beachte, dass die korrekte Platzierung ter Tier- und Taping des Ohres sind von entscheidender Bedeutung, um die Gesichtsnerven schnell zu finden.
- Befeuchten Sie das Fell auf und hinter dem Ohr mit 70% Ethanol und rasieren die Operationsstelle mit einer Rasierklinge oder Skalpell. Vorbenetzung das Fell macht Rasieren leichter in dieser anatomischen Lage.
- Reinigen der Haut mit einer Jodlösung, wie Betadine chirurgischen Scheuer (7,5% Povidon-Iod), gefolgt von 70% Ethanol. Wiederholen Sie diese Reinigung noch zweimal, um den Bereich gründlich zu desinfizieren.
- Um festzustellen, wo der Schnitt zu machen, verfolgen Sie die hinteren Ohrvene aus dem Ohr nach kaudal auf den Bereich hinter dem Ohr Vorsprung. Mit Federschere, einen 4 mm Schnitt 2-3 mm hinter dem Vorsprung.
- Präparieren durch das subkutane Fett und Bindegewebe mit stumpf. Vermeiden Sie direkte Schneiden mit der Schere, weil die Blutgefäße oder Muskelgewebe leicht beschädigt werden könnte.
- Wenn die Blutung auf, Druck auf die Operationsstelle mit einem sterilen Wattestäbchenfür mindestens 30 sec. Wenn bedeutende Flüssigkeitsverlust auftritt, injizieren die Maus intraperitoneal mit bis zu 0,5 ml steriler 0,9% iger Kochsalzlösung unter Verwendung einer 25 oder 27 G-Nadel.
- Verwenden Sie mehrere wichtige Sehenswürdigkeiten, die N. accessorius, Gehörgang, und vorderen M. digastricus (siehe unten), um die Gesichtsnerven zu lokalisieren. Sezieren um diese Landmarken, bis die Zweige des Gesichtsnervs visualisiert werden. Der Nerv wird als bedeutender solid weiß Struktur angezeigt, wenn es aufgedeckt wird und eine Schicht aus Faszie haftet sie an die zugrunde liegenden Strukturen.
- Finden Sie die N. accessorius, die aus dem kaudalen Teil des Schädels reist, um die Trapezmuskel innervieren, sobald das subkutane Fett und Bindegewebe wurden seziert. Der Gesichtsnerv ist tief mit dem N. accessorius.
- Finden Sie die knorpeligen Gehörgang, die strahlend weißen aussieht und rostral des Gesichtsnerven zu sehen.
- Finden Sie die Muskelbauch des vorderen M. digastricus, der oben auf und c liegtaudal des Gesichtsnervs.
- Wenn die Hauptzweige des Gesichtsnervs visualisiert, Spuren sie dorsal ihrer Herkunft aus den Foramen stylomastoideum finden. Mit feiner Spitze Dumont Pinzetten # 5/45 um die Operationsstelle offen zu halten, zu fördern die Federscherenspitzen nach dem Pfad des Nervs, und bewegen Sie die Zange dorsal um den neu erweiterten Bereich offen zu halten.
- Visualisieren Sie die Stamm des Facialis mit dem Jochbein, bukkale, und Randkieferzweige an dieser Stelle.
HINWEIS: Die zeitliche Niederlassung wird näher an das Foramen finden. Die Randkiefernervenäste in die oberen und unteren Teil näher an der Backe, damit diese Nervenäste nicht auf dieser Ebene sichtbar sein.- Bei Durchführung einer Nervendurchtrennung, Stabilisierung des Nerven vorsichtig mit der feinen Spitze Pinzette und schneiden Sie die Nerven mit den Feder Schere. Vermeiden Sie die Anwendung zu viel Traktion auf die Nerven mit der Zange avulsing den Nerv aus dem Hirnstamm zu verhindern. Drückendie Stümpfe voneinander entfernt oder ausgeschnitten und ein Abschnitt des distalen Nerv zu entfernen, um sicherzustellen, dass keine Wiederverbindung auftreten kann.
- Bei Durchführung einer Quetschverletzungen, verwenden Dumont # 5/45 Pinzette, um den Nerv für 30 Sekunden mit konstantem Druck, alle Axone trennen, wiederholen Sie diesen Andrang in einem zweiten Winkel senkrecht zur ersten Quetschstelle zu komprimieren. Vermeiden Sie die Anwendung unterschiedliche Mengen an Druck während der 30 Sekunden zu vernichten, da sonst die Verletzung inkonsistent zwischen den Tieren sein.
4. Schließen und Wiederherstellung
- Positionieren Sie das Fett und die Muskeln in den darunterliegenden Strukturen.
- Näherungsweise die Kanten des Einschnitts und schließen Sie die Wunde mit einem 7,5 mm Wundklammer. Nahtmaterial oder Klebstoff sind auch für den Wundverschluss akzeptabel. Postoperative Schmerz kann zu diesem Zeitpunkt zur Verfügung gestellt werden.
- Entfernen Sie das Band von Ohr die Maus. Schalten Sie die Isofluran Fluss und damit die Maus, um reinen Sauerstoff für 30 Sekunden zu atmen, um 1 Min. PlAss der Maus in einem leeren Käfig ohne Einstreu aus der Narkose zu erholen.
- Wenn die Maus wieder, verfolgen Sie sein Verhalten für bestätigende Anzeichen von Gesichtslähmung. Die Schnurrhaare werden gelähmt und abgewinkelt zurück in Richtung der Wange sein, wird die Nase abgewichen werden, und das Auge wird nicht als Reaktion auf einen Luftstoß blinken.
- Haustieren gemeinsam nach der Operation, wenn sie weiblich sind. Vermeiden männlichen Mäusen gemeinsam, weil sie sind aggressiver und neigen dazu, Gewalt zu entfernen Wundklammern ihres cagemate, die Infektion führt. Bereitzustellen postoperativen Schmerz zu dieser Zeit, falls erforderlich.
- Überwachen die Mäuse einmal am Tag über mehrere Tage nach der Operation, um sicherzustellen, dass keine Infektion oder andere Komplikation tritt postoperativ. Entfernen Wundklammern 7-10 Tage nach der Operation, wenn sie nicht auf eigene Faust zurückgegangen.
- Bewerben Schmieraugensalbe in das betroffene Auge täglich Hornhautkomplikationen zu vermeiden, sei es, bis das Auge Blinkreflex ist neuabgedeckt oder bis Euthanasie.
Ergebnisse
Abbildung 1 gibt einen Überblick über ERRBS, wobei die wesentlichen Schritte, die im gesamten beschriebenen Protokoll erläutert werden. ERRBS Bibliotheken wurden mit 50 ng Eingabe-DNA hergestellt.
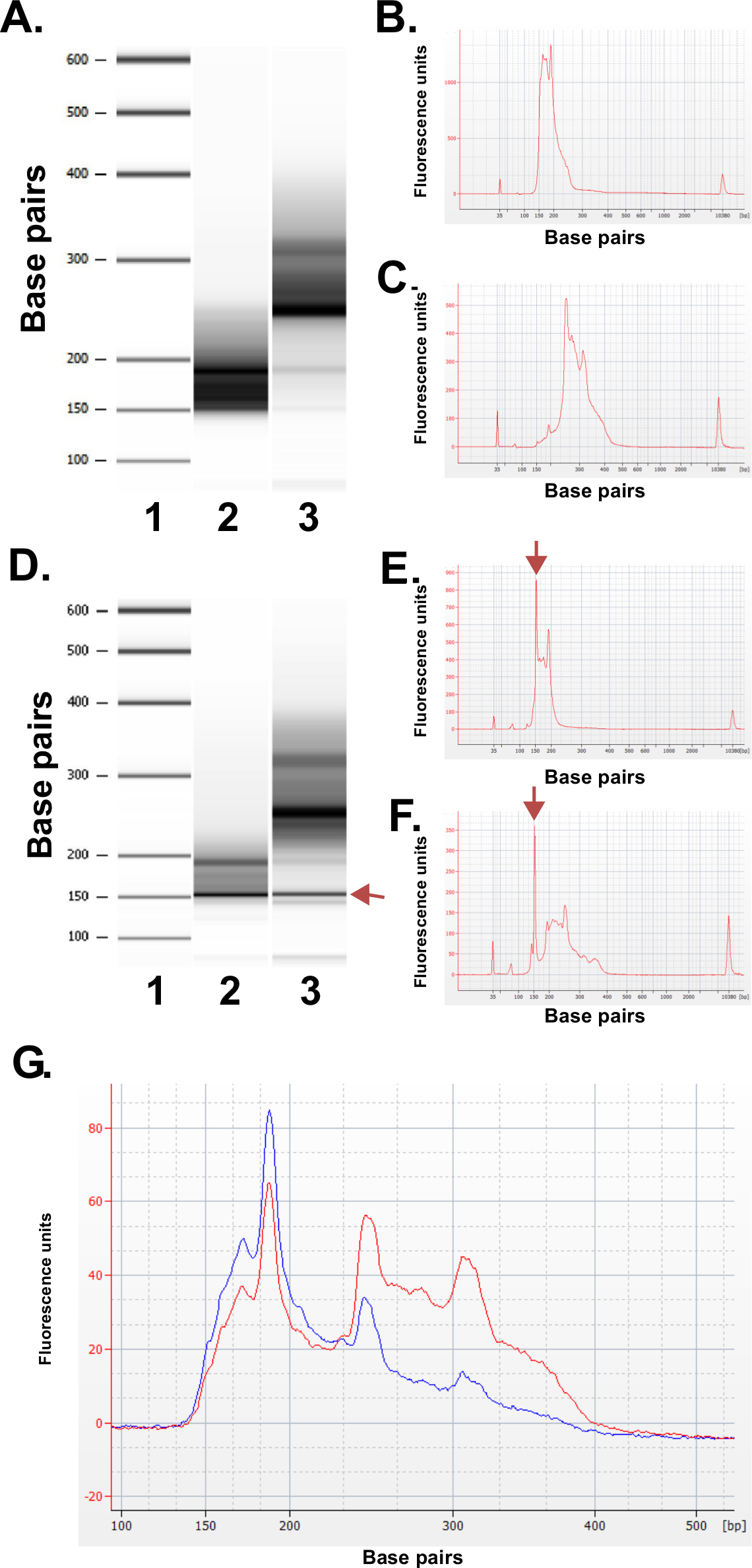
Bewerten Sie die Qualität der Bibliotheken erstellt. Bibliothek Produktion routine ergibt Fraktionsgrößen von 150 bis 250 bp und 250 bis 400 bp (3A-C). Leichte Unterschiede in der Bibliothek Größenverteilungen zwischen den Proben zu erwarten sind. Man beachte, dass in den beiden unteren und oberen Bankfraktionen gibt es sehr intensive DNA Größen anzeigt Anreicherung einer bestimmten Sequenz. MspI-Verdau führt zur Anreicherung einer Familie repetitiver DNA-Sequenzen im menschlichen Genom zu 190 bp, 250 bp und 310 bp in den ERRBS Bibliotheken vorhanden. Diese drei Repeats repräsentieren eine charakteristische Signatur eines ERRBS Bibliothek 20 (siehe Figuren 3A-C und 3G). Repräsentative Bibliotheken wurden auf einer nächsten Generation Sequenz sequenziertr mit Single-Ende liest. Achten Sie beim Einlegen auf die empfohlene Konzentration Bibliothek auf einem Illumina HiSeq 2500 Sequenzer werden Cluster Dichten 500.000-700.000 pro mm 2 zu erwarten. An dieser Clustering Dichte 81,6% ± 3,14% (n = 81) der Cluster Tiefpaßfilter (4A). Aufgrund der geringen Komplexität Ende der Bibliothek Einsätze (MspI Erkennungsstelle: C ^ CGG), sind Intensitätswerte und Qualitätswerte während der Sequenzierung aufgezeichnet sehr variabel in den ersten drei Basen (4B-C), jedoch, wenn eine unabhängige Kontrollspur ist im Preis inbegriffen (siehe Diskussion), werden 85% der Basen Qualitätswerte von 30 oder höher (Q30-Werte; 4D).
Datenabgleich und Cytosin-Methylierung Bestimmung wie in den Protokoll Erträge Basenpaar auflösende Daten beschrieben (Tabelle 7). Für das menschliche Genom, ein 51-Zyklus ein- gelesen Sequenzierungslauf einer ERRBS Bibliothek in einer Spur eines HiSeq 2500 in Hochleistungs Modus regelmäßigely erzeugt 153.194.882 ± 12.918.302 Gesamt liest, dass nach dem Qualitätsfilter und Adapter Trimmen Erträge 152.231.183 ± 13.189.678 liest für die Eingabe in die Analyse-Pipeline. Durchschnittliche Mapping Effizienz für eine ERRBS Bibliothek ist in der Regel 62,95% ± 5,92% mit Darstellung 3.183.594 ± 713.547 CpGs mit einer Mindestdeckungssumme pro CpG von 10x und einem durchschnittlichen Verbrauch pro CpG von 84,94 ± 16,29 (n = 100).
Die ERRBS Protokoll ist offen für Multiplexing (siehe Zusatzdatei 1: Protokollanpassung für Multiplex-Sequenzierung). . Die Daten aus repräsentativen Sequenzierungsläufe ist in Abbildung 5 Daten aus Multiplex-Sequenzierungsdurchläufe (51-Zyklus ein- gelesen Sequenzierungslauf zusammengefasst; n = 128 für zwei Bibliotheken pro Bahn; n = 11 für drei Bibliotheken pro Bahn; n = 11 für vier Bibliotheken pro Spur) waren im Vergleich zu einer vollständigen Spur Sequenzierung eines ERRBS Bibliothek (51 Zyklus Einzel lesen Sequenzierläufe; n = 100) als auch Downsampling eine einzige Spur zu SimulatE 50%, 33% und 25% der liest pro Bahn (2, 3, und 4 Sample Multiplexing pro Bahn sind; n = 3). Da die Anzahl der Lesevorgänge pro Probe mit dem Multiplexfaktor abnimmt, nimmt die Anzahl der CpGs bei einer Deckung von mindestens 10-fach und die Abdeckung pro CpG ebenso abgedeckt (Figur 5 und Tabelle 8). Die mittlere Umwandlungsraten von nicht-CpG-Stellen zu erwarten sind 99,85% ± 0,04% (n = 400). Conversion-Raten niedriger als 99% kann darauf hindeuten, nicht optimal Bisulfitumwandlung, die in hohen Raten der falschen Methylierung Niveaus führen kann.
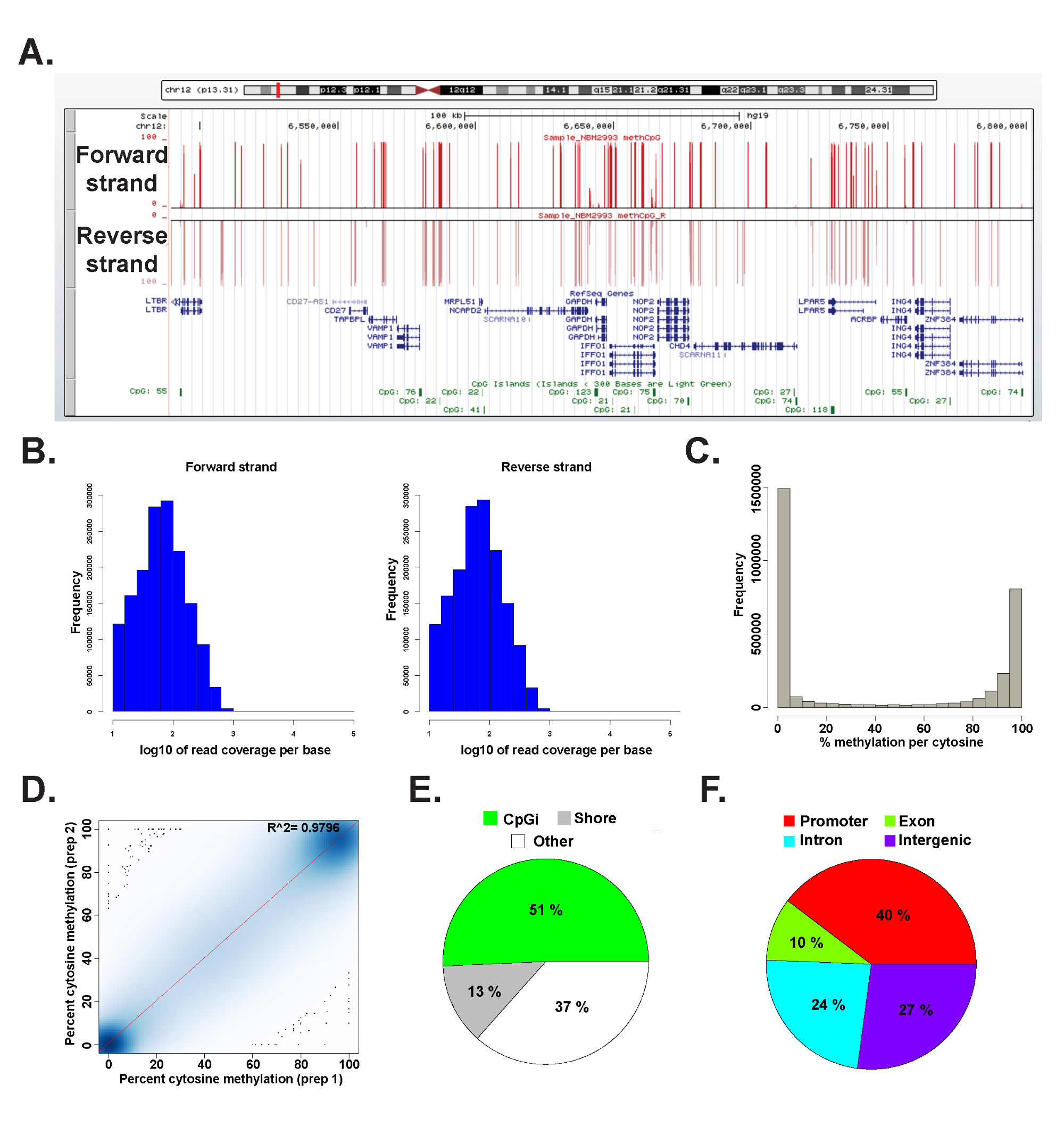
Daten aus einer ERRBS Bibliothek aus einem Vertreter der Personal genomischer DNA wurde in R 2.15.2 45 mit dem methylKit Paket 26 (siehe Zusatzcode-Datei 1 für Befehls Details) analysiert. Die Daten können in üblicherweise verwendeten Genom Browsern (6A) visualisiert werden. Die Cytosinmethylierung Daten gleichmäßig von beiden Strängen (6B) abgeleitet und reicht die gesamteSpektrum potentieller Cytosinmethylierung Ebenen (6C). Analyse technische Replikate aus einer repräsentativen menschlichen DNA-Probe ergibt große Übereinstimmung zwischen den Datenergebnissen (6D) und deckt CpGs in einem breiten Spektrum von genomischen Loci (6E und F wie vorher beschrieben 26). Während technische Replikate 2-Werte (über 97%) zu erhalten hohe R, werden biologische Repliken R 2 Werten zwischen 0,92-0,96 26 und den Vergleich verschiedener Zelltypen des Menschen wird R 2 Werte unter 0,86 (Daten nicht gezeigt) zu erhalten erhalten.

Abbildung 1: Flussdiagramm des ERRBS Protokollschritte. Diagramm stellt Schritte, die in einem traditionellen Arbeitstages abgeschlossen werden kann. * Kennzeichnet eine mögliche Pause Punkt (unmittelbar folgen ing Ligierung bereinigen und bevor die Größe Auswahl Protokoll Schritt 5), an dem Proben können bei -20 ° C vor der Dauer Protokoll ausgehend eingefroren werden.

Abbildung 2: Größenauswahl Protokoll. (A) Screenshot der Einstellungen im ERRBS Pippin Prep-Protokoll verwendet wird (siehe Protokoll 5.1.2 - 5.1.6): (1) Wählen Sie die Kassettentyp. (2) Wählen Sie Standard verwendet werden soll. (3) Wählen Sie den Erfassungsmodus für jede Spur. (4) Geben Sie die Sammlung bp reicht. (5) Speichern Sie das Protokoll (B) Stufen des Hand Gelextraktion in Abschnitt 5.2-Protokoll verwendet:. (1) Visualisierte Gel Leitern. (2) bezeichnete Größen zur Größenauswahl mit einer Rasierklinge. (3) Bild von herausgeschnittenen Proben (niedrigere Bruchteil: 150 bis 250 bp und höheren Anteil: 250 bis 400 bp)."> Bitte klicken Sie hier, um eine größere Version dieses Bild anzuzeigen.

Abbildung 3: Qualitätskontrolle Ergebnisse für repräsentative ERRBS Bibliotheken von menschlichen DNA-Proben mit einem Bioanalyzer Maschine hergestellt. (A) Gel-ähnliche Bild, das eine Standard-Leiter (1), Unter Bibliothek Fraktion (135-240 bp Fraktion von Pippin Prep); 2) und die höhere Bankfraktion (240 bis 410 bp-Fraktion aus Pippin Prep); . 3) (B) Bioanalyzer Elektropherogramm der erwarteten niedrigeren Bankfraktion (C) Bioanalyzer Elektropherogramm der erwarteten höheren Bankfraktion D -.. F) Repräsentative Daten von schlechter Qualität Bibliothek prep. Gelartigen Bildes (D) der Standardleiter (1), unteren Bankfraktion (2) und die höhere Bankfraktion (3). Die Bande bei 150 bp mit einem arro markiertw gibt große Mengen an Adapter. Elektropherogramm der unteren (E) und höhere Bankfraktionen (F) mit den überschüssigen Adapter Peaks bei 150 bp (mit Pfeilen markiert). (G) Bioanalyzer Elektropherogramm einer gepoolten ERRBS Bibliothek zur Sequenzierung. Rote Kurve stellt eine hohe Qualität gebündelt Bibliothek paritätisch mit höherer und niedrigerer Fraktionen. Blaue Kurve stellt eine gepoolte Bibliothek für die Sequenzierung nicht ausreichend aufgrund mangel der gleichberechtigten Vertretung der höheren und niederen Fraktionen. Bitte klicken Sie hier, um eine größere Version dieses Bild anzuzeigen.
{kind=link}

Abbildung 4: Sequenzierung Charts für eine repräsentative ERRBS 51 Zyklus Einzel lesen Sequenzierungsdurchlauf auf einem HiSeq 2500 Sequenzer in hoher LeistungModus. (A) Cluster-Dichten (K / mm 2 = 1.000 Cluster pro Millimeter im Quadrat, blau). Und Cluster-Dichten vorbei Filter (grün) in zwei Fahrspuren mit ERRBS Bibliotheken (B) Typische Intensitäten in den ersten 30 Zyklen in einer Gasse mit einem gesehen ERRBS Bibliothek. Beachten Sie die CGG Unterschrift MspI Verdauung in den Intensitäten der ersten drei Zyklen. (C) Prozentsatz der Basen mit einem Qualitätsfaktor von 30 oder höher (%> Q30) für jeden Zyklus in einem ERRBS Spur. (D) Qualitäts Partituren für alle Zyklen in einem ERRBS Spur. Blau = weniger als Q30, Grün = größer als oder gleich zu Q30. In dieser Gasse, 84,7% der Basen hatten Qualitätswerte von 30 oder höher.

Abbildung 5: Die Sequenzierung Ausgabeergebnisse. Box-Plots von experimentellen Daten aus Multiplex und einzigen Probe pro Bahn Sequenzierung ru ns (als grüne Kästchen dargestellt) und der Daten durch simulierte Downsampling von Sequenzierungsläufen von drei ERRBS Bibliotheken abgeleitet wurde (als blaue Kästchen dargestellt, fünf Mal pro Sequenzierungslauf abgetastet) ab 51 Zyklus Einzel lesen Sequenzierung läuft Die Multiplexfaktor entspricht. pro Bahn die Zahl der ERRBS Bibliotheken sequenziert. 1 = gesamte Fahrspur oder 100% der liest und stellt Daten aus einer einzigen Bibliothek ERRBS pro Bahn; 2 = 50% der Spur und stellt Daten aus zwei ERRBS Bibliotheken pro Bahn; 3 = 33% einer Spur und stellt Daten aus drei ERRBS Bibliotheken pro Bahn; und 4 = 25% einer Spur und stellt Daten von vier ERRBS Bibliotheken pro Bahn. (A) Die Lese zählt, oder die Anzahl der Sequenzen analysiert, pro Multiplexfaktor. (B) Die Anzahl der CpG die durch die Sequenzdaten pro Multiplexen abgedeckt Faktor. (C) Der mittlere Verbrauch pro CpG pro Multiplexfaktor._blank "> Bitte klicken Sie hier, um eine größere Version dieses Bild anzuzeigen.

Abbildung 6: Repräsentative Daten aus einer ERRBS Bibliothek aus humaner genomischer DNA, die (A) University of California, Santa Cruz (UCSC) Genom-Browser 43 Bild von repräsentativen Daten aus einer ERRBS Sequenzierung Spur.. Der Maßstab der y-Achse stellt 0-100% Methylierung an jedem Cytosin mit einem Minimum von 10-fach abgedeckt. Die Platte Brauch Spur stellt die Vorwärtsstrang und die untere benutzerdefinierte Spur stellt den Rückwärtsstrang. Dargestellt ist chr12:.. 6,489,523-6,802,422 (HG19) inklusive RefSeq Gene und CpG-Inseln innerhalb dieser genomischen Region (B) Verteilung Histogramme von CpG Abdeckung entlang Vorwärts- und Rückwärts Stränge in einer repräsentativen humanen CD34 + Knochenmarksprobe (C) Verteilung Histogrammvon CpG-Methylierung Ebenen entlang der beiden Stränge in einer repräsentativen humanen CD34 + Knochenmarksprobe. (D) Correlation Grundstück von CpG-Methylierung Niveaus aus einer repräsentativen technischen Nachbildung eines menschlichen DNA-Probe. (E) Kreisdiagramm, welches die Anteile von CpGs in ERRBS bedeckt die auf CpG-Inseln (hellgrün), CpG-Ufer (grau) und anderen Regionen (weiß) in einer repräsentativen Stichprobe von menschlicher genomischer DNA vorbereitet kommentiert. (F) Kreisdiagramm, welches die Anteile von CpGs in ERRBS abgedeckt, die Gen-Promotoren kommentierten (rot ), die Exons (grün), Introns (blau) und Intergenregionen (lila). Bitte klicken Sie hier, um eine größere Version dieses Bild anzuzeigen.
{kind=link}
| Reagens | Volumen | Kommentar |
| 10x T4 DNA Ligase-Reaktionspuffer | 10 & mgr; | |
| Desoxynucleotidtriphosphat (dNTP) Lösung Mix | 4 ul | Mischen von 10 mM jedes Nukleotid |
| T4 DNA-Polymerase | 5 & mgr; | 3.000 Einheiten / ml |
| DNA-Polymerase I Large (Klenow) Fragment | 1 & mgr; | 5.000 Einheiten / ml |
| T4 Polynucleotide Kinase | 5 & mgr; | 10.000 Einheiten / ml |
| DNase-freiem Wasser | 45 & mgr; |
Tabelle 1:. End Reparaturreaktion Reagenzien Reagenz Namen und Mengen in dem Ende Reparaturreaktion (Protokollschritt 2.1) verwendet.
| Reagens | Volumen | Kommentar |
| 10x Reaktionspuffer | 5 & mgr; | B. NEBuffer 2 |
| 1 mM 2'-Desoxyadenosin-5'-triphosphat (dATP) | 10 & mgr; | |
| Klenow-Fragment (3 '→ 5'-Exo-) | 3 ul | 5.000 Einheiten / ml |
Tabelle 2:. A-Tailing-Reaktion Reagenzien Reagenz Namen und Mengen in der A-Tailing-Reaktion (Protokoll Schritt 3.1) verwendet.
| Reagens | Volumen | Kommentar |
| 15 uM geglüht Adapter in DNase-freiem Wasser | 3 ul | PE-Adapter 1,0 und PE-Adapter 2.0; siehe Tabelle 4 Sequenzen und Referenz |
| 10x T4 DNA Ligase-Reaktionspuffer | 581; l | |
| T4 DNA Ligase | 1 & mgr; | 2.000.000 Einheiten / ml |
| DNase-freiem Wasser | 31 & mgr; |
Tabelle 3: Adapter Ligationsreaktion Reagenzien Reagenz Namen und im Adapter Ligationsreaktion (Protokoll Schritt 4.2) verwendeten Mengen..

Tabelle 4:. Oligos im ERRBS Protokoll Liste der Oligos im gesamten ERRBS Protokoll in der Ligationsreaktion (Protokoll Schritt 4) und PCR-Amplifikation Schritte (Protokoll Schritt 7) verwendet.
| Reagens | Volumen | Kommentar |
| 10x Faststart High Fidelity Reaktionspuffer with 18 mM Magnesiumchlorid | 20 & mgr; | |
| 10 mM dNTP Mix-Lösung | 5 & mgr; | |
| 25 uM Primer PCR PE 1.0 | 4 ul | Siehe Tabelle 4 |
| 25 uM Primer PCR PE 2.0 | 4 ul | Siehe Tabelle 4 |
| Faststart High Fidelity Enzyme | 2 & mgr; | 5 Einheiten / ul Faststart Taq DNA Polymerase |
| DNase-freiem Wasser | 125 ul |
Tabelle 5: PCR Reagenzien Reagenz Namen und in der PCR-Amplifikationsreaktion (Protokoll Schritt 7.1) verwendeten Mengen..
| Protokoll Schritt | Reagenz / Protokoll Detail | Eingangs DNA Menge | ||
| 5-10 ng | 25 ng | 50 ng | ||
| 1 | MspI Enzym | 1 & mgr; | 2 & mgr; | 2 & mgr; |
| MspI Digest Reaktionsvolumen | 50 | 100 | 100 | |
| 4 | Adapter in Ligationsreaktion | 1 & mgr; | 2 & mgr; | 3 ul |
| Die Ligationsreaktionsvolumen | 20 & mgr; | 25 & mgr; | 50 & mgr; | |
| 5 | Größenauswahl Protokoll | Nur Hand Gel | Pippin Prep oder manuelle Gel | Pippin Prep oder manuelle Gel |
| 7 | PCR-Primer-Konzentration | 25 & mgr; M | 25 & mgr; M | 10 uM für 14 Zyklen; 25 uM für 18 Zyklen |
| Anzahl der PCR-Zyklen | 18 | 18 | 14-18 | |
Tabelle 6:. Schritt Protocol Modifikationen für Vormaterial Mengen im Bereich von 5 bis 50 ng mehrere Schritte in der gesamten Protokoll ist eine Änderung der verwendet werden, um qualitativ hochwertige Bibliotheken aus verschiedenen Mengen an Ausgangsmaterialien zu erzeugen Reagenzienmengen. Änderungen an Schlüsselverpackungsgrößen sind hier eingeschlossen. Passen Puffer und Wassermengen in Reaktionen entsprechend.
| Chr | Basis | Strang | Berichterstattung | freqC | freqts |
| chr1 | 10.564 | R | 366 | 85,52 | 14,48 |
| chr1 | 10.571 | F | 423 | 91,25 | 8,75 |
| chr1 | 10542 | F | 432 | 91,2 | 8.8 |
| chr1 | 10563 | F | 429 | 94,64 | 5.36 |
| chr1 | 10.572 | R | 366 | 96,99 | 3.01 |
| chr1 | 10.590 | R | 370 | 88,11 | 11,89 |
| chr1 | 10.526 | R | 350 | 92 | 8 |
| chr1 | 10.543 | R | 368 | 92,93 | 7,07 |
| chr1 | 10.525 | F | 433 | 91,92 | 8.08 |
| chr1 | 10497 | F | 435 | 88,74 | 11,26 |
Tabelle 7: Vertreter ERRBS Daten. Nach dem Datenabgleich und Cytosin-Methylierung Bestimmung, Basenpaar Daten erhalten für jedes CpG fällt, wird die Ausrichtung Protokoll wie beschrieben die genomische Koordinate (Spalten: chr = Chromosom, Basis- und Strand) zu bestimmen. Der Deckungsgrad der spezifischen Locus (Coverage ), und die Rate von Detektions Cytosin gegen Thymidin als Prozent (freqC und freqts beziehungsweise).
| Anzahl der ERRBS Bibliotheken pro Spur | Die mittlere Anzahl der einzigartig ausgerichtet liest | Die mittlere Anzahl der CpGs abgedeckt | Mittlere Deckung pro CpG |
| 1 | 152.231.184 ± 13.189.678 | 3.183.594 ± 713.547 | 85 ± 16 |
| 2 | 77.680.837 ± 7.657.058 | 2.674.823± 153.494 | 49 ± 9 |
| 3 | 49.938.156 ± 2.436.865 | 2.552.186 ± - 76.624 | 39 ± 2 |
| 4 | 34.457.208 ± 4.441.686 | 1.814.461 ± 144.339 | 28 ± 4 |
Tabelle 8:. Vertreter Parameter aus der Sequenzierung Einzel- und Multiplex ERRBS Bibliotheken Gezeigt wird Daten pro Spur von 51-Zyklus ein- gelesen Sequenzierläufe: Mittelwert und Standardabweichungen der einzigartig ausgerichtet liest, Anzahl der CpGs bedeckt und Deckung pro CpG-Sequenzierung von Einzel erhalten ERRBS Bibliotheken pro Spur (n = 100), zwei ERRBS Bibliotheken pro Spur (n = 128), drei ERRBS Bibliotheken pro Spur (n = 11) und vier ERRBS Bibliotheken pro Spur (n = 11).
Diskussion
Die vorgestellten Protokoll Erträge Basenpaar-Feindaten von Cytosin-Methylierung in biologisch relevanten genomischen Regionen. Das Protokoll geschrieben wird 50 ng des Ausgangsmaterials optimiert, kann es jedoch geeignet ist, einen Bereich von Eingangsmaterial (5 ng oder mehr) 26 handhaben. Dies wird Anpassungen einiger der Protokollschritte erforderlich, wie in Tabelle 6 zu sehen. Die ERRBS Bibliotheken zugänglich sind gepaart Ende Sequenzierung und weitere genomische Abdeckung kann auch durch Sequenzierung durchgeführt werden liest mehr als 51 Zyklen. Multiplex-Sequenzierung wird eine kostengünstigere Protokoll pro Probe anbieten, dies jedoch zu einer verringerten Verbrauch pro CpG-in den Daten (Abbildung 5 und Tabelle 8) vertreten führen und ausreichend Tiefe der Berichterstattung, um Analysen, die hohe Reichweite pro erfordern führen nicht nachgeben CpG-Stelle (zB durch Landan et al. 33 beschrieben). Schließlich dieses Protokoll (oder ein Bisulfit-basierten Protokolls inl) kann nicht zwischen Methyl-Cytosin und Hydroxymethyl-Cytosin 46,47 unterscheiden. Jedoch sind die erzeugten mit anderen Protokoll integriert werden Daten ergibt 48,49, um die verschiedenen Modifikationen zu beschreiben, und andere Modifikationen Cytosin kürzlich berichtet 50, sollten sie von Interesse sein.
Hochwertige Bibliotheken wird angezeigt, wie in 3A-C gezeigt, und einmal für die Sequenzierung zusammengefasst ergibt sich eine Spur wie in 3G (rote Linie), die gleiche molare Beiträge von beiden Bankfraktionen gezeigt. Bibliothek Vorbereitung Ausfall kann von jedem Schritt während des Verfahrens führen. Wenn degradierte DNA verarbeitet sie in Bibliotheken, die nicht in MspI Fragmente in niedrigen CpG Berichterstattung über die in diesem Protokoll beschriebenen Sequenzierung Parameter angereichert sind und somit zur Folge haben. Wenn ein Enzym nicht-funktionell ist oder unbeabsichtigt von einer der Reaktionen auszuschließen, wird das Protokoll nicht zu den gewünschten Bibliothek. Wenn der Ligation reaRésol ineffizient sind Adapter in einer höheren Konzentration als erwartet, und / oder die Primer-Konzentration verwendet wird, eine begrenzende Reagenz für die Endverstärkung Schritten können Bibliotheksfehler auftreten. Überschüssiges Adaptern (als Peaks bei ~ 150 bp Bioanalyzer Ergebnissen gesehen; Figur 3D-F) in der Bibliothek werden auch mit Sequenzierung stören aufgrund des wahllosen Clustering von der Bibliothek und überschüssiges Adapter. Während eine solche Bibliothek kann offensichtlich normal Sequenz, einen signifikanten Teil der Leseoperationen lediglich Adaptersequenzen sein. Wenn überschüssige Adapter werden in einer Bibliothek zu beobachten, ist es am besten, wiederholen Sie die Bibliothek, wenn Vorbereitung Material ist mit optimalen Ausgangsmaterial, um Mengenverhältnisse Adapter. Schließlich, um eine effiziente PCR-Amplifikation der Bibliotheken zu gewährleisten, werden die niedrigeren und höheren Bankfraktionen als getrennte Proben der gesamten Bisulfitumwandlung und PCR Anreicherungsschritten aufrechterhalten. Andernfalls ergibt Differential Effizienz der Amplifikation in der PCR-Reaktion von höheren und niederen Fraktionen (wie in 3G-blaue Spur zu sehen) und das Potential für ungleiche Repräsentation der jeweiligen genomischen Loci in jeder Bibliothek Anteil während der Sequenzierung bedeckt. Der Benutzer kann entscheiden, eine quantitative PCR-Schritt unmittelbar nach der Bisulfit-Konvertierung für die weitere Titration optimale PCR-Zyklen notwendig, um verstärkt die Bibliotheken erzeugt sind.
ERRBS Bibliothek Vorbereitung Protokoll hat mehrere Schritte, in denen spezifische Reagenzien empfohlen. Am Ende Reparaturschritt ermöglicht End-Reparatur aller Produkte der CG Überhang, wie die von MspI enzymatische Sterne-Aktivität und geschert DNA in der ursprünglichen DNA-Probe vorhandenen Fragmente ergibt, nicht enthalten, die Verwendung eines Vier-Nucleotid-dNTP-Mix. Dies führt zu einer verbesserten CpG Darstellung der Ergebnisse. An der Ligationsschritt ist es wichtig, eine hohe Konzentration Ligase (2.000.000 Einheiten / ml) und methylierten Adapter, um sicherzustellen, daß die Ligations Reactiauf effiziente und dass die Bisulfitumwandlung nicht den Adapter Voraussetzung für eine genaue Datenabgleich Sequenzen beeinflussen. Bei der PCR-Schritt unter Verwendung einer Polymerase zur Amplifikation Bisulphit-behandelten GC-reichen DNA-Fragmente ist für hohe Spezifität notwendig. Schließlich, um die Beseitigung von überschüssigem Adapter und Primer zu gewährleisten, SPRI Wulst Reinigung (zum Beispiel: Agen AMPure XP) wird vielmehr empfohlen, als Säule basierte Assays für Ligation und PCR Produkt Isolierungen.
Um Daten hoher Qualität zu erzeugen, ist es wichtig, um eine effiziente Bisulfitumwandlung gewährleisten. Das vorgestellte Regel bietet dem Benutzer die Möglichkeit, Umwandlungseffizienz vor der Sequenzierung zu bestimmen. Als Alternative kann eine nicht-menschliche DNA wie Lambda DNA als eine interne Kontrolle (Spike-in) verwendet werden. Aufgrund der Unterschiede in der Art, kann diese Art einer Steuerung direkt in Downstream-Sequenzierung enthalten sein (zB wie beschrieben von Yu verwendet, et al. 34). Allerdings, wenn der Spike-in is verwendet wird, kann es nicht verwendet werden, um Umwandlungseffizienz vor der Bibliothek-Sequenzierung zu bestimmen, es sei denn eindeutig verstärkt und unabhängig sequenziert vor Bibliothek Sequenzierung werden. Die festgestellten Umrechnungskurse auf der Methylierungsstatus bei nicht-CpG Seiten. Dies ist möglicherweise nicht für die Verwendung im Zusammenhang mit Hoch Cytosinmethylierung in nicht-CpG-Kontext (zum Beispiel embryonale Stammzellen) und parallel Proben oder andere Mittel zur Bewertung der Umwandlungseffizienz kann für diesen Zweck verwendet werden, geeignet sein.
Es gibt einige Einschränkungen an die Adresse, die einzigartig für die Sequenzierung ERRBS Bibliotheken sind. Die ersten drei Basen des Bankfraktionen sequenziert sind nahezu gleichförmig nicht-zufälligen aufgrund der MspI Erkennungsschnittstelle (C ^ CGG; siehe 4B, C). Daraus ergibt sich das Potenzial für erhebliche Datenverluste aufgrund von niedriger Qualität liest aus armen Cluster Lokalisierung trotz der offensichtlich hohen Clusterdichte während der Sequenzierung resultieren. Um dieses Hindernis zu überwinden,sind eine hohe Komplexität Bibliothek in einem unabhängigen Spur (PhiX Kontrolle oder andere Bibliothek Typ) als dedizierter Kontrollspur. Hohe Komplexität Bibliotheken haben Enden, die ein ausgewogenes Verhältnis von A, C, T und G in den ersten vier Basen sequenziert. Geeignete Kontrollspuren sind Bibliotheken wie RNA-seq, ChIP-seq, Sequenzierung ganzer Genome, oder eine Kontrolle durch die Sequenzierung Maschinenhersteller (zB PhiX Steuer v3) angeboten. Wenn sie als eine Kontrollspur für die jeweilige Sequenzierungslauf bezeichnet wird, kann es als Basis für die Matrix Generation, während der ersten vier Basen der Sequenzierung verwendet wird, um Clusteranordnungen detektieren dienen. Je höher die Qualität liest erfasst den mittleren Verbrauch pro CpG-durch zu erhöhen 5.2 (n = 4). Alternativ kann diese technische Schwierigkeit auch mit einem dunklen Sequenzierung Ansatz wie zuvor beschrieben 23 überwunden werden. Andere Sequenzierung Kriterien folgen Standard-Betriebsverfahren gemäß Herstellerprotokolle. Schließlich ist die Deckung pro CpG cHosen für die Datenanalyse wird durch den Benutzer und zum Teil von den biologischen Fragen von Interesse geführt werden. 10x Abdeckung Schwelle bietet eine hohe Abdeckungsanalyse Ansatz ist jedoch diese Schwelle gesenkt sollte, dass die von Interesse sein werden.
Eine vollständige Diskussion von ERRBS Datenanalyse geht über den Rahmen dieses Artikels jedoch differentiell methylierte Cytosine und Regionen können mit Open Source Tools 31,51-53 bestimmt werden. Zusätzliche Analyse Überlegungen und Ansätze sind gut beschrieben worden, 54,55, und der Leser wird aufgefordert, die Literatur für die Werkzeuge am besten geeignet, um die geplante Analyse zu suchen.
Im Vergleich zu anderen veröffentlichten Verfahren bietet ERRBS eine Vier-Tage-Protokoll, wenn wie beschrieben Erträge hohe Reproduzierbarkeit durchgeführt. Es hat sich im Vergleich zum Goldstandard Massarray EpiTYPER 26 validiert, ist kostengünstig für hohe Reichweite von Daten und ist für verschiedene Eingangsmaterial anpassbarMengen (günstig für klinische Probenverarbeitung und anderen Zelltypen der Niederfrequenz) und Sequenzierungsansätzen. Es bietet Basenpaar-Auflösung bei biologisch relevanten loci und können in integrativen-Analysen mit anderen Techniken Profilierung genomweiten Transkriptionsfaktor-Bindungs, Chromatin-Remodeling, epigenetische Markierungen und andere Cytosin Modifikationen von Interesse. ERRBS Daten Einsatz in solchen Studien können zu einem umfassenden molekularen Ansatz bei und ermöglichen eine hohe Dimensionsanalysen bei der Untersuchung von biologischen Modellen und Krankheit beim Menschen.
Offenlegungen
Die Autoren haben keine Interessenkonflikte offen zu legen.
Danksagungen
We thank all the authors of the original ERRBS report. We thank Mame Fall for technical assistance. We acknowledge the Weill Cornell Medical College Epigenomics Core for technical services and assistance. The work was supported by a Sass Foundation Judah Folkman Fellowship, an NCI K08CA169055 and ASH-AMFDP12005 to FGB, NIH R01HG006798 and R01NS076465, funding from the Irma T. Hirschl and Monique Weill-Caulier Charitable Trusts and STARR Consortium (I7-A765) to CEM, and an LLS SCORE grant (7006-13) to AMM.
Materialien
| Name | Company | Catalog Number | Comments |
| MspI | New England Biolabs | R0106M | 100,000 units/ml |
| NEBuffer 2 | New England Biolabs | B7002S | Reaction buffer for MspI enzyme; protocol step 1.2 |
| Phenol solution | Sigma-Aldrich | P4557 | Equilibrated with 10 mM Tris HCl, pH 8.0; see safety and handling instructions at http://www.sigmaaldrich.com/catalog/product/sigma/p4557 |
| Chloroform | Sigma-Aldrich | C2432 | See safety and handling instructions at http://www.sigmaaldrich.com/catalog/product/sial/c2432 |
| Glycogen | Sigma-Aldrich | G1767 | 19-22 mg/ml |
| NaOAc | Sigma-Aldrich | S7899 | 3 M, pH 5.2 |
| Ethanol | Sigma-Aldrich | E7023 | 200 proof, for molecular biology |
| Buffer EB | Qiagen | 19086 | 10 mM Tris-Cl, pH 8.5 |
| tris(hydroxymethyl)aminomethane (Tris) | Sigma-Aldrich | T1503 | prepare a 1 M, pH 8.5 solution |
| Tris- Ethylenediaminetetraacetic acid (TE) | Sigma-Aldrich | T9285 | Dilute to 1x buffer solution per manufacturer's recommendations |
| T4 DNA Ligase Reaction Buffer | New England Biolabs | B0202S | 10x concentration |
| Deoxynucleotide triphosphate (dNTP) Solution Mix | New England Biolabs | N0447L | 10 mM each nucleotide |
| T4 DNA Polymerase | New England Biolabs | M0203L | 3,000 units/ml |
| DNA Polymerase I, Large (Klenow) Fragment | New England Biolabs | M0210L | 5,000 units/ml |
| T4 Polynucleotide Kinase | New England Biolabs | M0201L | 10,000 units/ml |
| QIAquick PCR Purification Kit | Qiagen | 28104 | Used for DNA product purification in protocol step 2.3 |
| 2'-deoxyadenosine 5'-triphosphate (dATP) | Promega | U1201 | 100 mM |
| Klenow Fragment (3'→5' exo-) | New England Biolabs | M0212L | 5,000 units/ml |
| MinElute PCR Purification Kit | Qiagen | 28004 | Used for DNA product purification in protocol step 3.3 |
| T4 DNA Ligase | New England Biolabs | M0202M | 2,000,000 units/ml |
| Methylation Adapter Oligo Kit | Illumina | ME-100-0010 | |
| Agencourt AMPure XP | Beckman Coulter | A63881 | Used in protocol sections that implement magnetic bead purification steps (steps 4.3 and 8.2). Equilibrate to room temperature before use. |
| Pippin Prep Gel Cassettes, 2% Agarose, dye-free | Sage Science | CDF2010 | with internal standards |
| Certified Low Range Ultra Agarose | Bio-Rad | 161-3106 | |
| Tris-Borate-EDTA (TBE) buffer | Sigma-Aldrich | T4415 | |
| Ethidium bromide solution | Sigma-Aldrich | E1510 | 10 mg/ml |
| 50 bp DNA Ladder | NEB | N3236S | |
| 100 bp DNA Ladder | NEB | N3231S | |
| Gel Loading Dye, Orange (6x) | NEB | B7022S | |
| Scalpel Blade No. 11 | Fisher Scientific | 3120030 | |
| QIAquick Gel Extraction Kit | Qiagen | 28704 | |
| EZ DNA Methylation Kit | Zymo Research | D5001 | Used in protocol step 6.2 |
| EZ DNA Methylation-Lightning Kit | Zymo Research | D5030 | Alternative for step 6.2 |
| Universal Methylated Human DNA Standard | Zymo Research | D5011 | Used as bisulfite conversion control |
| FastStart High Fidelity PCR System | Roche | 03553426001 | |
| Qubit dsDNA High Sensitivity Assay Kit | Life Technologies | Q32854 | A fluorescence-based DNA quantitation assay; used in protocol steps 1.1, 9.1 and 10.1 |
| DynaMag-2 Magnet | Life Technologies | 12321D | |
| High Sensitivity DNA Kit | Agilent Technologies | 5067-4626 | |
| 2100 Bioanalyzer | Agilent Technologies | ||
| PhiX Control v3 | Illumina | FC-110-3001 | |
| HiSeq 2500 | Illumina | ||
| Pippin Prep | Sage Science | ||
| Qubit 2.0 Fluorometer | Life Technologies | Q32872 | |
| TruSeq SR Cluster Kit v3-cBot-HS | Illumina | GD-401-3001 | |
| TruSeq SBS Kit v3-HS | Illumina | FC-401-3002 | |
| TruSeq RNA Sample prep | Illumina | RS-122-2001 | Barcoded adapters used for multiplexing libraries; See Supplemental file for multiplexing protocol. |
| Microcentrifuge | |||
| Vortex Mixer | |||
| Dry Block Heater | |||
| Thermal Cycler | |||
| Water Bath | |||
| Gel electrophoresis system | |||
| Electrophoresis power supply | |||
| Gel doc | |||
| UV or blue light transilluminator |
Referenzen
- Jones, P. A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 13 (7), 484-492 (2012).
- Barlow, D. P. Genomic imprinting: a mammalian epigenetic discovery model. Annual Review Of Genetics. 45, 379-403 (2011).
- Thiagarajan, R. D., Morey, R., Laurent, L. C. The epigenome in pluripotency and differentiation. Epigenomics. 6 (1), 121-137 (2014).
- Reik, W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 447 (7143), 425-432 (2007).
- Hartnett, L., Egan, L. J. Inflammation, DNA methylation and colitis-associated cancer. Carcinogenesis. 33 (4), 723-731 (2012).
- Smith, Z. D., Meissner, A. DNA methylation: roles in mammalian development. Nat Rev Genet. 14 (3), 204-220 (2013).
- Li, E., Bestor, T. H., Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 69 (6), 915-926 (1992).
- Okano, M., Bell, D. W., Haber, D. A., Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 99 (3), 247-257 (1999).
- Feinberg, A. P. Phenotypic plasticity and the epigenetics of human disease. Nature. 447 (7143), 433-440 (2007).
- Bock, C. Epigenetic biomarker development. Epigenomics. 1 (1), 99-110 (2009).
- Laird, P. W. The power and the promise of DNA methylation markers. Nat Rev Cancer. 3 (4), 253-266 (2003).
- How Kit, A., Nielsen, H. M., Tost, J. DNA methylation based biomarkers: practical considerations and applications. Biochimie. 94 (11), 2314-2337 (2012).
- Mikeska, T., Bock, C., Do, H., Dobrovic, A. DNA methylation biomarkers in cancer: progress towards clinical implementation. Expert Review Of Molecular Diagnostics. 12 (5), 473-487 (2012).
- Gyparaki, M. T., Basdra, E. K., Papavassiliou, A. G. DNA methylation biomarkers as diagnostic and prognostic tools in colorectal cancer. Journal of Molecular Medicine. 91 (11), 1249-1256 (2013).
- Figueroa, M. E., et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 17 (1), 13-27 (2010).
- Heyn, H., Mendez-Gonzalez, J., Esteller, M. Epigenetic profiling joins personalized cancer medicine. Expert review of Molecular Diagnostics. 13 (5), 473-479 (2013).
- Kulis, M., Esteller, M. DNA methylation and cancer. Advances in Genetics. 70, 27-56 (2010).
- Xiong, Z., Laird, P. W. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 25 (12), 2532-2534 (1997).
- Meissner, A., et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 33 (18), 5868-5877 (2005).
- Gu, H., et al. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc. 6 (4), 468-481 (2011).
- Bock, C., et al. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol. 28 (10), 1106-1114 (2010).
- Harris, R. A., et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol. 28 (10), 1097-1105 (2010).
- Boyle, P., et al. Gel-free multiplexed reduced representation bisulfite sequencing for large-scale DNA methylation profiling. Genome Biol. 13 (10), R92 (2012).
- Chatterjee, A., Rodger, E. J., Stockwell, P. A., Weeks, R. J., Morison, I. M. Technical considerations for reduced representation bisulfite sequencing with multiplexed libraries. Journal of Biomedicine & Biotechnology. 2012, 741542 (2012).
- Lee, Y. K., et al. Improved reduced representation bisulfite sequencing for epigenomic profiling of clinical samples. Biological Procedures Online. 16 (1), 1 (2014).
- Akalin, A., et al. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet. 8 (6), e1002781 (2012).
- Hatzi, K., et al. A Hybrid Mechanism of Action for BCL6 in B Cells Defined by Formation of Functionally Distinct Complexes at Enhancers and Promoters. Cell Reports. 4 (3), 578-588 (2013).
- Will, B., et al. Satb1 regulates the self-renewal of hematopoietic stem cells by promoting quiescence and repressing differentiation commitment. Nature Immunology. 14 (5), 437-445 (2013).
- Lu, C., et al. Induction of sarcomas by mutant IDH2. Genes Dev. 27 (18), 1986-1998 (2013).
- Kumar, R., et al. AID stabilizes stem-cell phenotype by removing epigenetic memory of pluripotency genes. Nature. 500 (7460), 89-92 (2013).
- Li, S., et al. An optimized algorithm for detecting and annotating regional differential methylation. BMC Bioinformatics. 14, S10 (2013).
- Patterson, K., Molloy, L., Qu, W., Clark, S. DNA methylation: bisulphite modification and analysis. Journal of Visualized Experiments. (56), 3170 (2011).
- Landan, G., et al. Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat Genet. 44 (11), 1207-1214 (2012).
- Yu, M., et al. Tet-assisted bisulfite sequencing of 5-hydroxymethylcytosine. Nat Protoc. 7 (12), 2159-2170 (2012).
- Goecks, J., Nekrutenko, A., Taylor, J., Galaxy, T. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 11 (8), R86 (2010).
- Dorff, K. C., et al. GobyWeb: simplified management and analysis of gene expression and DNA methylation sequencing data. PLoS One. 8 (7), e69666 (2013).
- Roehr, J. T., Dodt, M., Ahmed, R., Dieterich, C. Flexbar − flexible barcode and adapter processing for next-generation sequencing platforms. MDPI Biology. 1 (3), 895-905 (2012).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal, North America. 17 (1), 10-12 (2011).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (15), 2114-2120 (2014).
- Needleman, S. B., Wunsch, C. D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 48 (3), 443-453 (1970).
- Krueger, F., Andrews, S. R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 27 (11), 1571-1572 (2011).
- Li, H., et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25 (16), 2078-2079 (2009).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Res. 12 (6), 996-1006 (2002).
- Thorvaldsdottir, H., Robinson, J. T., Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in Bioinformatics. 14 (2), 178-192 (2013).
- Team, R. C. R. A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria. ISBN 3-900051-07-0, (2012).
- Nestor, C., Ruzov, A., Meehan, R., Dunican, D. Enzymatic approaches and bisulfite sequencing cannot distinguish between 5-methylcytosine and 5-hydroxymethylcytosine in DNA. BioTechniques. 48 (4), 317-319 (2010).
- Huang, Y., et al. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS One. 5 (1), e8888 (2010).
- Yu, M., et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 149 (6), 1368-1380 (2012).
- Song, C. X., et al. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell. 153 (3), 678-691 (2013).
- Ito, S., et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 333 (6047), 1300-1303 (2011).
- Akalin, A., et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 13 (10), R87-1186 (2012).
- Stockwell, P. A., Chatterjee, A., Rodger, E. J., Morison, I. M. DMAP: Differential Methylation Analysis Package for RRBS and WGBS data. Bioinformatics. 30 (13), 1814-1822 (2014).
- Sun, D., et al. MOABS: model based analysis of bisulfite sequencing data. Genome Biol. 15 (2), R38 (2014).
- Bock, C. Analysing and interpreting DNA methylation data. Nat Rev Genet. 13 (10), 705-719 (2012).
- Rivera, C. M., Ren, B. Mapping human epigenomes. Cell. 155 (1), 39-55 (2013).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten