
Method Article
Misa cuantitativa espectrometría de perfiles de los proteomas de células del cáncer derivados de líquidos y tumores sólidos
En este artículo
Resumen
In-depth analyses of cancer cell proteomes facilitate identification of novel drug targets and diagnostic biomarkers. We describe an experimental workflow for quantitative analysis of (phospho-)proteomes in cancer cell subpopulations derived from liquid and solid tumors. This is achieved by combining cellular enrichment strategies with quantitative Super-SILAC-based mass spectrometry.
Resumen
El análisis en profundidad de los proteomas celulares de cáncer son necesarias para dilucidar mecanismos patológicos oncogénicos, así como para identificar los posibles objetivos farmacológicos y biomarcadores de diagnóstico. Sin embargo, los métodos para la caracterización proteómica cuantitativa de los tumores derivados del paciente y, en particular, sus subpoblaciones celulares se carece en gran medida. Aquí se describe un montaje experimental que permite el análisis cuantitativo de proteomas de subpoblaciones de células de cáncer derivadas de tumores, ya sea líquida o sólida. Esto se logra mediante la combinación de estrategias de enriquecimiento celulares con espectrometría de masas basado-Super-SILAC cuantitativa seguido de análisis de datos bioinformáticas. Para enriquecer subgrupos celulares específicas, tumores líquidos están primero immunophenotyped por citometría de flujo seguido por FACS de clasificación; para tumores sólidos, láser de microdisección de captura se utiliza para purificar subpoblaciones celulares específicas. En un segundo paso, las proteínas se extraen de las células purificadas y posteriormente combinarse con un tumor-específica,SILAC marcado estándar espiga-en que permite la cuantificación de proteínas. La mezcla de proteína resultante se somete a electroforesis en gel o filtro Aided Preparación de la muestra (FASP) seguido por la digestión con tripsina. Finalmente, los péptidos trípticos se analizaron utilizando un espectrómetro de masas cuadrupolo-orbitrap híbrido, y los datos obtenidos se procesaron con paquetes de software de bioinformática, incluyendo MaxQuant. Por medio del flujo de trabajo que aquí se presenta, hasta 8.000 proteínas pueden ser identificados y cuantificados en las muestras de los pacientes derivados, y los perfiles de expresión de proteínas resultantes pueden ser comparados entre los pacientes para identificar firmas proteómicos de diagnóstico o los posibles objetivos farmacológicos.
Introducción
Proteómica basada en la espectrometría de masas ha surgido y ahora es una disciplina ampliamente utilizado en la biología celular y la investigación biomédica traslacional. Los avances técnicos en la materia han hecho posible estudiar procesos celulares complejos en líneas celulares y modelos animales, como miles de proteínas pueden ser identificados en un solo experimento de espectrometría de masas 1,2. Se han realizado progresos similares para el análisis de muchas modificaciones postraduccionales tales como fosforilación o ubiquitinación, aunque esto usualmente requiere flujos de trabajo a medida para enriquecimiento y análisis de datos para cada tipo de 2,3 modificación. Por otra parte, la implicación de las estrategias de etiquetado químicas y metabólicas, incluyendo SILAC, permite la cuantificación relativa precisa de proteínas y PTM, haciendo de este método particularmente atractivo para el descubrimiento de procesos celulares básicos, biomarcadores diagnósticos y dianas de fármacos potenciales en el cáncer humano 4.
Sin embargo,varios desafíos tienen que ser superadas con respecto al análisis proteómico de los cánceres humanos primarios 5. En primer lugar, las muestras de cáncer humanos a menudo muestran un alto grado de heterogeneidad celular que es debido a la presencia de diversos tipos de células que pertenecen a la microambiente del tumor, incluyendo células inmunes y fibroblastos. En segundo lugar, la evolución clonal conduce a la diversidad genética dentro de los tumores a sí mismos, resultando en la existencia de varias subpoblaciones celulares con propiedades funcionales distintas. Se espera De acuerdo con el concepto actual de unas pocas células madre del cáncer son las fuerzas impulsoras detrás del desarrollo y progresión del cáncer, el análisis proteómico de dichas subpoblaciones celulares (funcionalmente muy relevantes) a ser de gran importancia para una mejor comprensión de los mecanismos oncogénicos relevante para la aplicación clínica 6. En tercer lugar, los perfiles de expresión de proteínas cuantitativos de grandes conjuntos de muestras se requieren a menudo para la identificación de BioMar clínica robustakers; esto no se puede lograr por medio del etiquetado químico tal como iTRAQ 7, mientras que las estrategias metabólicas etiquetado - basándose, como lo hacen, sobre la proliferación celular 4 - son igualmente inaplicable. En cuarto lugar, la mayoría de las muestras de tumores sólidos disponibles son fijados con formalina, lo que complica el análisis del proteoma de espectrometría de masas debido a la formación de reticulaciones de proteínas 8. Por último, la mayoría de los flujos de trabajo existentes proteómica requieren cantidades significativas de procesamiento de la muestra y adquisición de datos, haciendo que el análisis de los números de las muestras pertinentes para la investigación clínica difícil, y llamando a nuevo flujo de trabajo paradigmas 9,10.
Para hacer frente a estos obstáculos, hemos desarrollado un sistema experimental que combina ya sea celular por citometría de flujo de la clasificación 11 o láser captura microdissection 12,13 para el enriquecimiento celular con una estrategia de Super-SILAC 14 para introducir una norma interna global para el análisis integral de espectrometría de masas. Al utilizar el método descrito aquí, es posible cuantificar hasta 8000 proteínas en muestras de tumores humanos individuales derivados de cualquiera de los cánceres de líquidos o sólidos.
Protocolo
Todos los experimentos con tejidos o muestras de sangre humana deben ser aprobados por un comité de ética y llevaron a cabo de acuerdo con las directrices que figuran en el voto ética.
1. Celular Enriquecimiento
- Tumores líquidos / activada por fluorescencia de células.
- Aislar las células mononucleares de la médula ósea o muestra de sangre mediante la realización de lisis de los eritrocitos con cloruro de amonio o por Ficoll-centrifugación en gradiente de densidad.
- Para la lisis de eritrocitos, combine 1 ml de muestra con 4 ml de solución de cloruro de amonio. Incubar la solución en hielo durante 5 a 10 min. Añadir 40 ml de tampón fosfato salino (PBS) + 2% de suero de ternera fetal (FCS) y centrifugar durante 5 min a 400 xg y 4 ° C.
- Para Ficoll centrifugación en gradiente, capa de 10 ml de la muestra en 15 ml de Ficoll-Hypaque para formar 2 capas distintas (tenga cuidado de no molestar a las capas). Centrifugar a 400 xg durante 30 min a TA (15 - 25 ° C) con cuidado y cosechar el mononuclearlas células que se acumulan en la interfase.
- Centrifugar las células y tomarlas en PBS + 2% FCS. Centrifugar a 400 xg durante 5 minutos y tomar en 1 ml de PBS + 2% FCS. Tomar una alícuota como control negativo y una alícuota para cada colorante fluorescente utilizado en la mancha final.
- Incubar con anticuerpos marcados con fluorescencia útiles para aislar la población de células leucémicas en la dilución de acuerdo con las instrucciones del fabricante en hielo durante 30 min. También manchar las alícuotas utilizadas para la compensación con cada anticuerpo individual. Lavar dos veces con PBS + 2% FCS.
NOTA: Los anticuerpos utilizados fueron anti-ratón humano contra CD117-PE (clon 95C3), CD34-FITC (clon 8G12) y CD33-PE (Clone P67.6). - Las células de filtro a través de filtro de células de 35 micras, lo ideal sería que utilizan tubos con filtro de células integradas en la tapa.
- Añadir tinción de viabilidad celular (por ejemplo, 7-AAD). Ordenar células utilizando un clasificador celular apropiado 11 (Figura 2). Recoger las células en Isc Modificado de Dulbecco de OVE Medio (IMDM) que contiene 10% de FCS.
- Aislar las células mononucleares de la médula ósea o muestra de sangre mediante la realización de lisis de los eritrocitos con cloruro de amonio o por Ficoll-centrifugación en gradiente de densidad.
- Los tumores sólidos / Laser captura microdissection.
- Para los experimentos descritos, se utilizaron muestras de FFPE de espécimen de cáncer de pulmón. Para este tejido de cáncer de pulmón propósito se fijó inmediatamente en 4% de formalina tamponada después de la resección quirúrgica y trozos de unos 3 x 2 x 1 cm, donde se incluyeron en parafina para su posterior análisis.
- Cortar secciones de 5 a 10 micras de espesor de la (FFPE) muestra fijado en formol e parafina embebido con un microtomo. Mount secciones en las diapositivas de membrana con película cubierto y seco a 37 ° C durante 1 hora. Desparafinar y rehidratar las secciones montadas por incubación sucesiva en xileno, etanol absoluto, 70% y agua, cada uno durante 1 min.
- Teñir las secciones con hematoxilina durante 20 segundos y luego enjuague con agua del grifo. Recoger la población celular de interés mediante el uso de un sistema de microdisección por láser de captura (véase también 12).
- Tumores Líquido.
- Centrifugar la suspensión celular de la etapa 1.1.5 a 400 xg, 4 ° C durante 4 min y descartar el sobrenadante. Lavar dos veces con 500 l de PBS frío y centrifugación durante 5 min a 400 xg y 4 ° C
- Añadir 40 l de tampón de lisis por 10 6 células y se incuba durante 15 min en hielo (10 5 células (aprox. 10 g de proteína total) debe ser el número de células mínimo). Centrifugar el lisado a 14.000 xg, 4 ° C durante 10 min y transferir el sobrenadante (lisado celular aclarado) a un nuevo tubo de reacción. Deseche la pastilla.
- Tumores sólidos.
- Añadir 60 l de tampón de lisis de tejido al tejido microdissected y se incuba durante 15 min en hielo, recoger el fluido por centrifugación corta y transferir la suspensión a un nuevo tubo de reacción. Sonicar el lisado en hielo durante 3 min.
- Añadir 15 l de 20% de dodecilsulfato de sodio (SDS) a un llegar a una final SDS concentration de 4%. Incubar el tejido microdissected a 99 ° C en un bloque de calentamiento durante 1 hora y se agitó a 600 rpm. Centrifugar el lisado a 16.000 xg, 18 ° C durante 10 min y transferir el sobrenadante a un nuevo tubo.
3. Establecimiento de un SILAC Punto-en Cuantificación Standard (Super-SILAC Standard)
NOTA: El estándar de cuantificación consiste en una mezcla de proteínas SILAC marcado derivada 4-6 líneas de células que coinciden con el tipo de tumor de interés. Para lograr el máximo solapamiento entre el proteoma de referencia marcado con SILAC y el proteoma derivado de tumor, un análisis de componentes principales tiene que ser realizado antes se seleccionan líneas de células para el estándar de cuantificación 14.
- Análisis de componentes principales (PCA).
- Para determinar los patrones de expresión de proteínas de líneas celulares, cultivan cerca de diez diferentes líneas de células en medios de cultivo celular apropiado. Lisar las células como se describeen el paso 2.1. Preparar lisados celulares por espectrometría de masas como se describe en el paso 6.1.
- Analizar el patrón de expresión de proteínas de cada línea celular en una alta resolución, espectrómetro de masas cromatografía líquida acoplada (nanoLC-MS / MS).
- Analizar los datos brutos resultantes utilizando un MaxQuant libre de las mercancías y llevar a cabo un análisis de componentes principales con el software asociado, como Perseo 16,17.
- Seleccione 4-6 líneas celulares que muestran la mayor diversidad entre sus perfiles de proteínas de expresión y los utilizan para generar la norma espiga en Super-SILAC.
- SILAC etiquetado y verificación de etiquetas.
- Cultivar las líneas celulares seleccionadas durante al menos cinco ciclos celulares en un medio adecuado de cultivo celular SILAC, en el que la arginina y la lisina se marcan con isótopos estables de carbono y nitrógeno (medio SILAC) 4.
- Lisar las células, como se describe en el paso 3.1 y preparar lisados celulares por espectrometría de masas como se describe en el paso 6.1.Medir la eficacia de incorporación de etiquetado SILAC por nanoLC-MS / MS. Analizar los datos de MS primas resultantes con MaxQuant y determinar la eficiencia de SILAC etiquetado. Esto se logra mediante el recuento del número de péptidos identificados en su etiqueta (pesado) y sus formas endógenas (luz), y el cálculo de la relación (pesada) / (+ luz pesada). Etiquetado de eficiencia debe exceder el 98%.
- Combinación de proteomas SILAC y validación.
- Cultivar y ampliar las líneas celulares seleccionadas de acuerdo a las instrucciones del fabricante en medio SILAC apropiado. Lisar las células, como se describe en el paso 2.1 para tumores líquidos o como se describe en el paso 3.2 para tumores sólidos y determinar la concentración de proteínas de cada lisado celular.
- Mezcle cantidades de proteínas equimolares de cada línea celular y dividir la mezcla en alícuotas. Snap-congelar las alícuotas y se almacena a -80 ° C hasta su medición. Para un experimento que necesita 20 - 50 g de estándar Super-SILAC. Tenga cuidado al preparar el stanDard en exceso como cambiar el estándar dentro de una serie de experimentos debe ser evitado.
4. Medición de la concentración de proteínas y Spike-in
- Tumores Líquido.
- Usando el ensayo de cuantificación de proteínas determinar las concentraciones de proteína de los respectivos lisados celulares y el estándar Super-SILAC.
- Mezcle cantidades iguales de lisado celular aclarado y el estándar Super-SILAC y, posteriormente, añadir sulfato de dodecil (LDS) tampón de litio (25% del volumen de la muestra) y el agente reductor (10% del volumen de la muestra).
- Calentar la solución resultante en un bloque de calentamiento a 72 ° C durante 10 min. Opcionalmente, almacenar las proteínas desnaturalizadas resultantes a -80 ° C.
- Tumores sólidos.
- Para la medición de la concentración de proteínas en un lector de placas, mezclar un estándar de albúmina de suero bovino (BSA) series solución de dilución, el lisado y el estándar Super-SILAC apropiado con una proteína disponible comercialmentessay en una placa de 96 pocillos y agitar durante 1 min, se incuba durante el tiempo indicado y medir absorbancia según lo indicado por el fabricante.
NOTA: La alta concentración de SDS y ditiotreitol (DTT) en el lisado es un problema para la mayoría de los ensayos disponibles para la determinación de la concentración de proteína. - Mezcle cantidades iguales de lisado clarificado y el estándar Super-SILAC con 200 l de urea en la unidad de filtro y se centrifuga a 14.000 xg durante 30 min a 20 ° C. No utilice más de 50 l de lisado clarificado y el estándar Super-SILAC. Evitar temperaturas por debajo de 15 ° C, por lo que la urea no cristaliza a cabo.
- Para la medición de la concentración de proteínas en un lector de placas, mezclar un estándar de albúmina de suero bovino (BSA) series solución de dilución, el lisado y el estándar Super-SILAC apropiado con una proteína disponible comercialmentessay en una placa de 96 pocillos y agitar durante 1 min, se incuba durante el tiempo indicado y medir absorbancia según lo indicado por el fabricante.
5. Separación de muestra y Proteína Recopilación
- Tumores Líquido.
- Separadas 30 - 100 g de proteína total por carril en un 4 - Gel SDS-PAGE gradiente de 12%. Proteínas se tiñen con Coomassie azul O / N. Retire el exceso de tinción de Coomassie de dos lavados posteriores con agua.
- Cortar cada carril del gel y dividirlaen 23 rodajas de igual tamaño, independientemente del patrón de tinción de gel. Procesar las rodajas de gel por separado, cada uno en una en 0,6 ml de polipropileno vial.
- Lavar las rodajas de gel con agua y metanol / agua (50:50, v / v), a reducir con DTT 10 mM por incubación durante 30 min a 56 ° C. Se alquila el rodajas de gel con 55 mM yodoacetamida (IAA) mediante la incubación a 60 minutos a temperatura ambiente en la oscuridad.
- Entre los pasos de manipulación de muestras-, lavar rebanadas con acetonitrilo durante 15 min y seco en un SpeedVac para eliminar el exceso de disolvente y mejorar la absorción de la solución de reactivo.
- Realizar corte de proteasa rehidratando cortes de gel secas con la cantidad mínima de solución de tripsina porcina (12,5 ng / l en bicarbonato de amonio acuoso M 0.025) durante 16 horas a 37 ° C.
- Añadir agua 10 l de corte de gel e incubar durante 15 minutos a 37 ° C. Añadir 80 l de acetonitrilo y se incuba durante 15 minutos a 37 ° C. Centrifugar a 15.800 xg, durante 1 minuto. Recoger el sobrenadante y guárdelo en un separadas 0,6 mltubo.
- Añadir 65 l de 5% de ácido fórmico solución, vórtice y se incuba durante 15 minutos a 37 ° C. Añadir 65 l de acetonitrilo y se incuba durante 15 minutos a 37 ° C. Centrifugar a 15.800 xg durante 1 min. Recoger el sobrenadante y añadir al sobrenadante de la etapa anterior. Evaporar el sobrenadante combinado a sequedad en un concentrador de vacío.
- Tumores sólidos.
- Para eliminar SDS de lisado tejido utilice el siguiente protocolo FASP, también descrito como en la referencia 15.
- Después de la primera centrifugación se describe en el paso 5.2.3 añadir otros 200 l de 8 M urea en el filtro y, además, se centrifuga a 14.000 xg durante 20 min a 20 ° C. Desechar el filtrado de paso de flujo.
- Añadir 100 l de IAA y mezclar en un termomezclador a 600 rpm durante 1 min. Incubar el filtro durante 20 min a 20 ° C en la oscuridad. Centrifugar el filtro a 14.000 xg durante 10 min a 20 ° C.
- Añadir 100 l de urea al filtro y CENtrifuge a 14.000 xg, durante 15 min a 20 ° C. Repita este paso una vez más.
- Añadir 100 l de NH 4 HCO 3 al filtro y se centrifuga a 14.000 xg durante 10 min a 20 ° C. Repita este paso dos veces.
- Añadir 40 l NH 4 HCO 3 + 1 l (= 0,4 g) tripsina y mezclar en termomezclador a 20 ° C durante 600 rpm, 1 min. Incubar el filtro de O / N en una cámara húmeda a 37 ° C. Transferir el filtro de nuevos tubos de recolección.
- Centrifugar el filtro a 14.000 xg durante 10 min a 20 ° C. Añadir 50 l de NH 4 HCO 3 y centrifugar el filtro a 14.000 g durante 10 minutos a 20 ° C. Guarde los péptidos resultantes a -20 ° C hasta que la medición de espectrometría de masas.
- Para la medición de la concentración de péptido, 50 l de dispensar el flujo a través resultante y una serie de diluciones apropiadas de triptófano ordenada en una placa de 96 pocillos. Medir la fluorescencia de triptófano. Convertir la concentración resultante detriptófano a la concentración de péptido, tal como 0,1 g de triptófano corresponde a la proteína de 9 mg.
Análisis 6. Cromatografía líquida y espectrometría de masas
- Se vuelve a disolver péptidos en tampón de carga de 30 l durante 5 min en un baño de sonicación. Haz girar en una centrífuga a 15.800 xg durante 1 min y una pipeta la solución clara en un vial de MS muestreador automático.
- Inyectar 5 l de muestra por análisis utilizando el inyector automático del sistema nanoLC-MS / MS. Se concentra y péptidos desalar en línea en una fase inversa C18 pre-columna (0,15 mm ID x 20 mm con 5 micras de tamaño de poro de material C18) montado ya sea en una configuración de columna de ventilación o una configuración de precolumna de conmutación.
- Péptidos separados en una fase inversa C18 microcolumna (0,075 mm ID x 200 mm auto-lleno de 3 micras o menor de poro material de tamaño en una columna C18 de nanopulverización auto-pack como PicoFrit, usando un gradiente de 90 min de 5> 35% de acetonitrilo vs . ácido fórmico acuoso al 0,1% a 300 nl /min. Eluyente de transferencia en un cuadrupolo híbrido / espectrómetro de masas orbitrap a través de una fuente de iones de nanopulverización.
- Analizar péptidos usando un método Top15 datos dependientes de adquisición (EM m / z gama 350 -. 1600, resolución de destino 70,000 FWHM, AGC objetivo 1 x 10 6, tiempo de llenado máximo 60 ms MS / MS inicia masa 100, resolución de destino 17,500 FWHM, AGC objetivo 2 x 10 5, el tiempo de llenado máximo 60 ms MS / MS umbral del 3 x 10 4, incluir estados de carga. 2 - 5, Normalized Collision Energía NCE 25%, dinámicas de exclusión de 15 segundos).
Análisis 7. Datos
- Para el análisis de los datos de uso del software libre disposición MaxQuant 16. Un protocolo detallado para el análisis bioinformático de datos se describe en 16,17. Para el análisis, combinar archivos de datos brutos de todos los cortes de un carril de SDS-PAGE en un solo experimento 17.
Resultados
Perfiles proteómicos de tumores líquidos y sólidos de los pacientes es un enfoque prometedor para el descubrimiento de nuevos biomarcadores de diagnóstico y de predicción. Sin embargo, el procedimiento de preparación de muestras y el análisis de espectrometría de masas son un reto, debido a la complejidad de las muestras y la necesidad de cuantificación de proteínas precisa en grandes conjuntos de muestras. El procedimiento experimental descrito anteriormente comienza con el aislamiento de células de interés, ya sea por fluorescencia de clasificación de células activadas por láser o de captura de microdisección.
Para este propósito, las células de aspirados de médula ósea derivadas del paciente o muestras de sangre se tiñeron con anticuerpos marcados con fluorescencia contra marcadores de superficie definidas de interés antes de la clasificación basada en FACS y la lisis celular.
Para enriquecer subconjuntos celulares de las secciones de tejidos, los primero tienen que ser montados en portaobjetos apropiadas para permitir la captura de microdisección por láser. Para este propósito montar tque las secciones de tejido en los portaobjetos de membrana correspondientes y utilizar un láser captura microdissector según las instrucciones del fabricante. Selección de las áreas y las células de interés requiere un conocimiento adecuado sobre el histomorfología de tejido sano y neoplásica. Las células extraídas se transfieren entonces a un tubo de recogida adecuado. Los experimentos iniciales se deben realizar con el fin de definir la cantidad de tejido necesaria para la extracción de cantidades suficientes de proteína (mínimo de proteína total de 10 mg) por cada nuevo tejido de interés. Cada muestra microdissected se trata con 60 l de tampón de lisis del tejido. Para la lisis eficiente, las células de muestras de tejido tienen que ser sonicó durante 3 min y después de la adición de 15 l de muestras de SDS se incuban en un termomezclador a 99 ° C durante 1 hora para eliminar las reticulaciones de proteínas inducidos por formalina. La centrifugación finalmente facilitará la recogida del lisado celular despejado.
Para cada enfermedad de interés (líquido y tumores sólidos) una norma adecuada cuantificación Super-SILAC tiene que ser establecidos 14. Una norma adecuada debe representar más de 90% de las proteínas expresadas en las muestras de interés. Para la preparación de un estándar de cuantificación, las líneas celulares relacionadas con el tipo de cáncer de interés primero deben ser analizados por espectrometría de masas para determinar sus perfiles de expresión de proteínas y análisis de componentes principales, posteriormente, se deben realizar. 4 - 6 líneas de células diferentes con patrones de expresión diferencial de proteínas deben ser elegidos y SILAC etiquetados con arginina "pesado" y lisina. Etiquetado de eficiencia debe ser revisado por nanoLC-MS / MS y debe ser superior al 98%. Posteriormente, las líneas celulares deben ser lisadas en la misma forma que la muestra respectiva derivada de paciente y, posteriormente, cantidades equimolares de proteína de cada muestra y el estándar de espiga-en SILAC se deben mezclar. Un paso crítico para la cuantificación de proteínas exacta de las muestras clínicas ointerés f es la mezcla exacta de las cantidades de proteínas equimolares de los derivados de las muestras de los pacientes y el estándar de cuantificación espiga en SILAC. Para determinar la concentración de proteína de lisados derivados de células clasificadas, se pueden utilizar varios ensayos de cuantificación de proteína. Para lisados celulares a partir de células derivadas de tejido se necesita un ensayo que puede hacer frente a altas concentraciones de SDS y DTT en el lisado. Las concentraciones de proteína de las muestras derivadas del paciente y el estándar de espiga-en SILAC deben medirse cada vez antes de la combinación con las respectivas muestras clínicas con el fin de asegurar la mezcla correcta, que es crucial en la fabricación de incluso cientos de muestras comparables.
Después de la mezcla de cantidades iguales de espiga-en muestras estándar y de lisado, obtenidos a partir de tumores líquidos se mezclan con LDS, se calienta en un termomezclador a 72 ° C durante 10 min y posteriormente sometidas a 1D-PAGE y en gel digestión de los separados proteínas con tripsina. Muestras obtenidas deel tejido se libera de SDS y se digirió con tripsina utilizando el enfoque FASP según lo establecido por Wisniewski et al. 15
Los péptidos trípticos obtenidos se pueden cuantificar midiendo la fluorescencia de triptófano, que es de especial interés para las muestras de tejido como la cantidad de péptidos liberados del filtro difiere entre 15% y 75% en comparación con las cantidades de proteína cargados inicialmente en el filtro. Para este propósito se mide la fluorescencia de triptófano. Comparación con una serie de dilución de triptófano apropiado permite la cuantificación de péptidos, como 1,1 g de triptófano corresponde a aproximadamente 100 g de péptido. Si es necesario, las muestras derivadas especialmente del procesamiento FASP pueden ser pre-purificado en cualquiera de consejos comerciales o caseras etapas llenas de (RP-C18) Material de fase inversa C18 antes de cargar en el sistema nanoLC / MS / MS 18.
Análisis de espectrometría de masas se lleva a cabo en un alto-Resolución, alta sensibilidad del sistema de espectrometría de masas. En resumen, las muestras de péptidos se desalaron y preconcentrado en una precolumna RP-C18, y separaron en una columna analítica RP-C18 acoplado directamente al espectrómetro de masas. Para lograr suficiente profundidad de análisis, empleamos ya sea una combinación de SDS-PAGE de proteínas fraccionamiento previo con 40 min gradientes de RP-C18 (por tumores líquidos) o inyecciones individuales con largas 2 - 3 hr gradientes de RP-C18 (por tumores sólidos procesados por FASP ). MS espectros se adquirieron a una resolución de 70.000 FWHM o mejor, para permitir la cuantificación exacta de pares de SILAC por integración de perfiles de picos cromatográficos. Para la identificación de proteínas, un método de adquisición de datos dependiente de Top15 se utiliza para generar un gran número de péptido espectros MS / MS para el péptido y la identificación de proteínas.
A partir de los datos brutos resultantes, la identificación de proteínas y la cuantificación se consiguen mediante la búsqueda de base de datos con el software MaxQuant contra un UniProt Knowledgebase Proteoma Humano completa base de datos de secuencia 17. En el software MaxQuant (versión actual 1.5.0.25) péptidos se identificaron a partir de la adquirida espectros MS / MS por el péptido fragmento de coincidencia contra espectros derivados in silico de la base de datos de secuencias de proteínas. Al mismo tiempo, precursor de iones perfiles isotópicos se extraen alrededor de sus tiempos de retención de cromatografía, y sus áreas de los picos integrados se utiliza para la cuantificación relativa de la luz: pares de péptidos pesados generados por SILAC etiquetado. Identidades de péptidos y las intensidades relativas se asignan a las propiedades de las proteínas correspondientes. Software Perseo (versión actual 1.5.0.15) se utiliza para realizar una evaluación adicional estadística aguas abajo de los resultados del procesamiento MaxQuant, incluyendo de muestra a muestra comparaciones, PCA y la agrupación jerárquica.
Usando la preparación experimental descrito hemos identificado y cuantificado hasta 8000 proteínas a partir de tan poco como 30 g de proteína totalderivado de tumores líquidos.
Hasta 2.500 proteínas a partir de muestras de tumores sólidos pueden ser identificados y cuantificados en un enfoque proteómico escopeta con un gradiente LC de sólo 2 horas, lo que permite el análisis de cientos de muestras clínicas en un tiempo relativamente corto.

Figura 1. Flujo de trabajo experimental. Los pasos principales de enriquecimiento celular-subconjunto, el aislamiento de proteínas, Spike-en de la norma cuantificación y análisis por espectrometría de masas se muestran para los tumores sólidos y líquidos. Haga clic aquí para ver una versión más grande de esta figura.
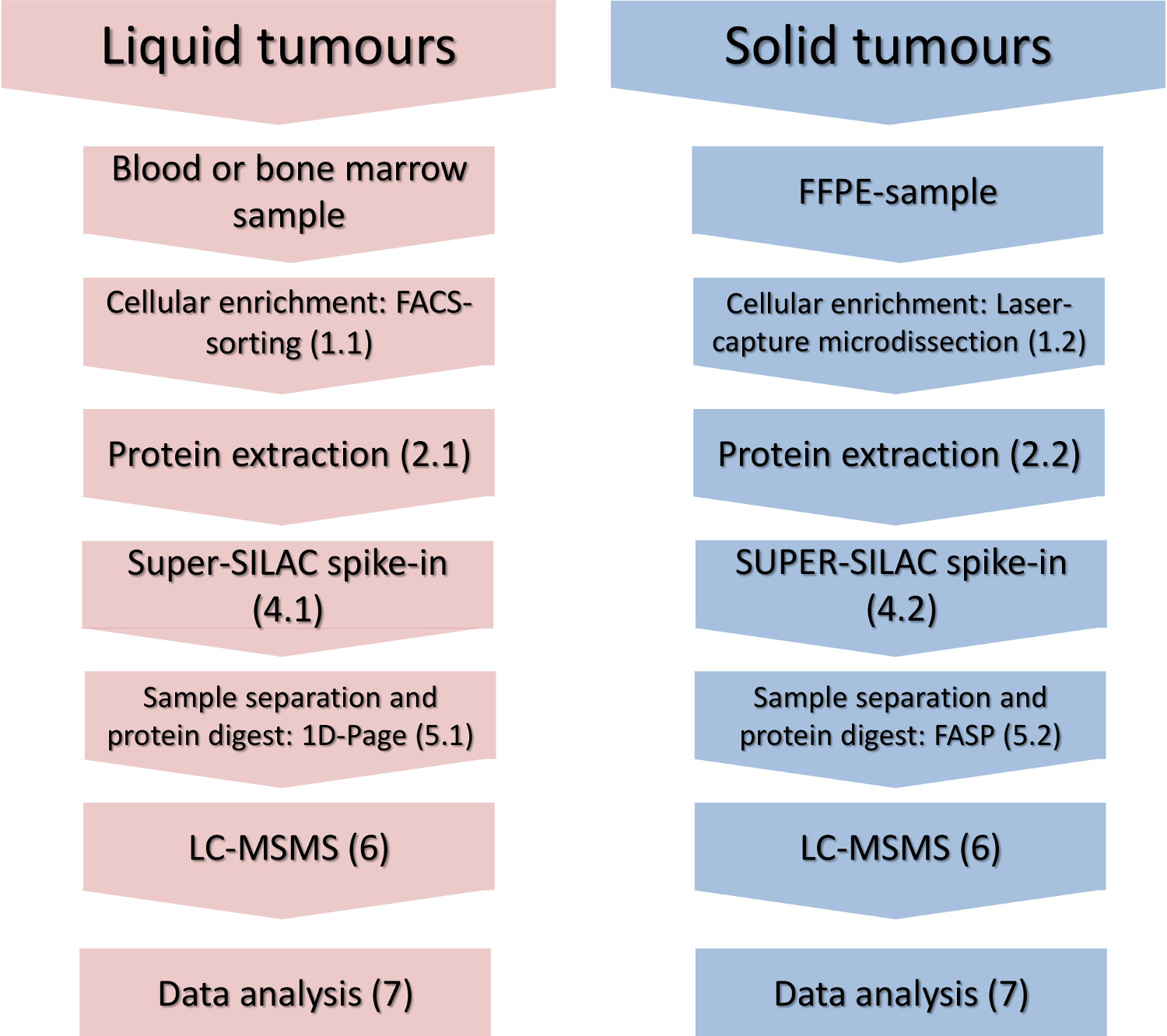{kind=link}

Figura 2. celular strategi enriquecimientoES. FACS clasificación y LCM. (A) estrategia de gating ilustrativa que representa gating en una población de células leucémicas CD117-manchado adquirida por un clasificador de células. (B) microdisección de captura por láser de tejido del tumor sólido. Las secciones de tejido de 5 a 10 micras de espesor fueron montados en portaobjetos cubiertos de membrana de película antes de la tinción. Región de los intereses fue seleccionada manualmente para la captura microdissection láser. Se muestran secciones antes de microdisección con la región de interés marcado en amarillo y después de microdisección. Una barra de escala de 50 micras se incluye en la figura. Por favor, haga clic aquí para ver una versión más grande de esta figura.
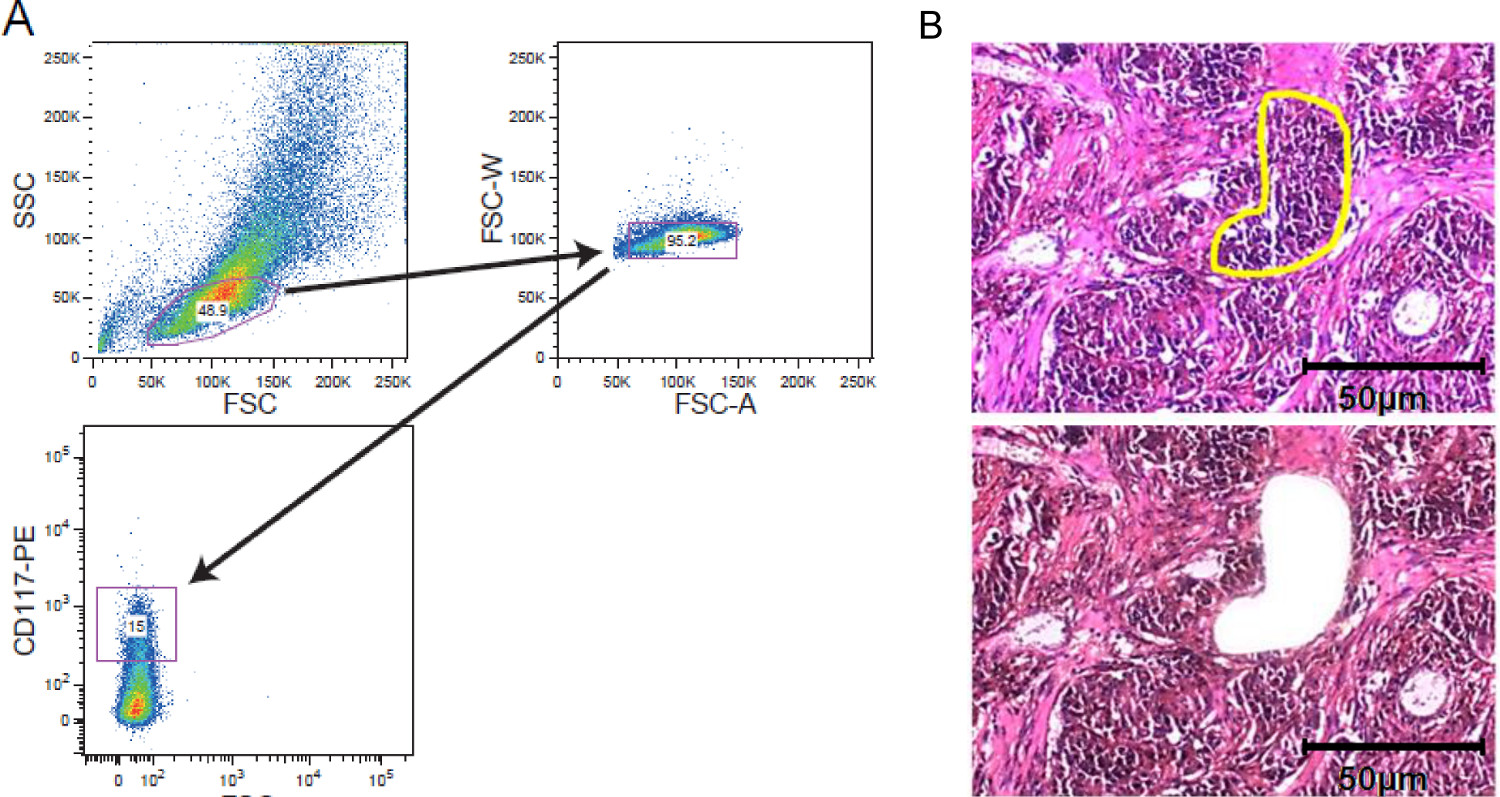{kind=link}

Figura 3. El análisis de agrupamiento de los proteomas de células de cáncer humano. Análisis de agrupamiento no supervisado de prperfiles de expresión Otein de tumores sólidos y líquidos se realizó a través de la plataforma computacional Perseo. AC - Adenocarcinoma, SCC - Carcinoma de células escamosas, SCC-Met - metástasis de carcinoma de células escamosas de cáncer de cabeza y cuello, R - técnica de reproducir.
Discusión
Se necesita Misa perfiles de espectrometría de proteomas de células de cáncer derivados de pacientes para el descubrimiento de nuevos biomarcadores de diagnóstico / predictivos, así como para dar una mejor comprensión de la biología de las células del cáncer, lo que podría a su vez conducir a la identificación de nuevas dianas farmacológicas potenciales. Sin embargo, este tipo de análisis de espectrometría de masas son muy difíciles, sobre todo porque varios temas pre-analíticos tienen que ser resueltos, si uno va a obtener resultados robustos y biológicamente relevantes.
El flujo de trabajo experimental descrito aquí permite la caracterización proteómica cuantitativa de proteomas derivados de subpoblaciones celulares de tumores tanto líquidos como sólidos. Se necesita el enriquecimiento inicial de las células tumorales ya sea por FACS basada en células sortingor microdisección para evitar la contaminación por las células del microambiente del tumor. Además, estas técnicas permiten una para aislar subpoblaciones celulares de interés. Estudios de células biológicas recientes han demoniotrado que ciertas subpoblaciones celulares tienen propiedades iniciadoras del tumor y por tanto son de gran relevancia para la patogénesis del cáncer 19,20. Como espectrometría de masas se ha vuelto más sensible en los últimos años, los análisis proteómicos cuantitativos son factibles para las pequeñas cantidades de proteína que se pueden derivar de unos pocos miles de células, por lo que es posible para centrarse en poblaciones de células funcionalmente relevantes.
La puesta a punto que aquí se presenta se puede utilizar para identificar y validar nuevos biomarcadores de diagnóstico en muestras FFPE. Por lo tanto, se compromete a bea herramienta útil para la mejora de los diagnósticos clínicos, como hasta la fecha todavía hay una falta de biomarcadores moleculares en número y calidad adecuadas para muchos tipos de cáncer. Ejemplos importantes de diagnósticos diferenciales difíciles, para las que se carece de biomarcadores, son la discriminación entre el cáncer de pulmón primario de las metástasis en el pulmón, carcinoma colangiocelular intrapancreática y adenocarcinoma de páncreas, así como difierenciación de neurofibroma tumores benignos de los nervios periféricos de la vaina altamente malignos. Además, nosotros y otros han demostrado que la elucidación cuantitativa de firmas proteómicos puede ser útil en el estudio de la biología de células de cáncer en general, y por revelar biomarcadores predictivos de respuesta terapéutica en pacientes con cáncer 21.
Dos inconvenientes actuales del método presentado aquí son el requisito para el proceso extenso manual de las muestras y la demanda de nanoLC-MS / MS tiempo de adquisición. Mientras que el primero se pueden abordar moviendo la preparación de muestras para por ejemplo, formatos de 96 pocillos y utilizando el procesamiento de robot, éste requerirá un cambio en la estrategia de adquisición de espectrometría de masas. Una vez subconjuntos de proteínas objetivo se han identificado que puede estar asociada con, por ejemplo, la clasificación de tumores, prevemos el diseño de métodos de espectrometría de masas específicas que proporcionan lecturas cuantitativas para estos subconjuntos con un esfuerzo de separación muy reducida, ypor lo tanto, con un tiempo de adquisición correspondientemente reducida. Si el tiempo de adquisición requerido requerido podría reducirse 24-36 hr (tumores líquidos) o 3 hr (tumores sólidos) a, por ejemplo, 1 hr mediante espectrometría de masas y una simple separación unidimensional de péptidos, a continuación, la ganancia resultante en el rendimiento dirigido podría utilizarse para aumentar significativamente el número de repeticiones biológicos y técnicos examinados, con las mejoras correspondientes en la importancia de los resultados de cuantificación. Enfoques de espectrometría de masas específicos han demostrado ser una herramienta adecuada para la verificación de la proteína de biomarcadores-candidatos asociados con el cáncer 22, y se han desarrollado a un punto en el que se muestran prometedores para la validación o incluso como una herramienta potencial para el uso clínico rutinario 23 , 24.
Divulgaciones
Los autores no tienen ningún conflicto de intereses u otros temas a revelar.
Agradecimientos
The authors thank Uwe Plessmann, Monika Raabe und Silvia Münch for technical support.
Materiales
| Name | Company | Catalog Number | Comments |
| 660 nm Kit | Thermo scientific | 22662 | |
| Cell culture medium depleted of arginine and lysine | Thermo Scientific | 88421 | |
| Coomassie Brilliant Blue R-250 staining solution | Bio Rad | 161-0436 | |
| Dialyzed fetal calf serum (FCS) | PAA | A15-107 | |
| Diffuser caps for microdissection | MMI | 50202 | |
| FACS-sorter | BD | FACSAria III | |
| Ionic Detergent Compatibility Reagent | Thermo scientific | 22663 | |
| Laser-capture microdissector | MMI | cell cut plus | |
| LDS buffer | Life Technologies | NP0009 | |
| Membrane slides for microdissection | MMI | 50103 | |
| Microcon YM-30 | Millipore | MRCF0R030 | |
| NuPAGE 4-12% Bis-Tris Mini Gels | Life Technologies | NP0335PK2 | |
| Picofrit Self-Pack Columns | New Objective | PF360-75-15-N-5 | Mass Spectrometry Column/Emitter |
| Reducing agent | Life Technologies | NP0007 | |
| Reprosil-Pur LC/MS/MS Column stationary phase | Dr. Maisch | 120 C18-AQ, 3 µm | |
| Reprosil-Pur LC/MS/MS Precolumn stationary phase | Dr. Maisch | 120 C18-AQ, 5 µm | |
| SILAC-labeled arginine | Eurisotop | CLM-2265-H-0.1 | |
| SILAC-labeled lysine | Eurisotop | DLM-2640-0.25 | |
| Trypsin, NB Sequencing Grade | Serva | 3728301 | for in-gel digests |
| Trypsin, Sequencing Grade | Promega | V5111 | for in-solution digests |
| Buffer and solutions | |||
| Cell lysis buffer: 150 mM NaCl, 50 mM Tris/HCl pH 7.8, 5 mM NaF, 0.5% NP40, 0.1% laurylmaltoside, Roche complete protease inhibitor, 1 mM Na3VO4 | |||
| Tissue lysis buffer: 100 mM Tris/HCl pH 7.8, 0.1 M DTT | |||
| Urea: 8 M urea in 0.1 M Tris-HCl, pH 8.5 | for FASP-protocoll | ||
| IAA: 0.05 M iodoacetamide, 8 M urea, 0.1 M Tris-HCl, pH 8.5 | for FASP-protocoll | ||
| 0.05 M NH4HCO3 | |||
| 10 mM dithiothreitol (DTT) in 0.1 M ammonium bicarbonate | for in-gel digest | ||
| 55 mM iodoacetamide (IAA) in 0.1 mM ammonium bicarbonate | for in-gel digest | ||
| 5% aqueous formic acid. |
Referencias
- Walther, T. C., Mann, M. Mass spectrometry-based proteomics in cell biology. J Cell Biol. 190 (4), 491-500 (2010).
- Lenz, C., Urlaub, H. Separation methodology to improve proteome coverage depth. Expert Rev Proteomics. 11 (4), 409-414 (2014).
- Olsen, J. V., Mann, M. Status of large-scale analysis of post-translational modifications by mass spectrometry. Mol Cell Proteomics. 12 (12), 3444-3452 (2013).
- Ong, S. E., et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 1 (5), 376-386 (2002).
- Jimenez, C. R., Verheul, H. M. Mass spectrometry-based proteomics: from cancer biology to protein biomarkers, drug targets, and clinical applications. Am Soc Clin Oncol Educ Book. , e504-e510 (2014).
- Tang, D. G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 22 (3), 457-472 (2012).
- Evans, C., et al. An insight into iTRAQ: where do we stand now. Anal Bioanal Chem. 404 (4), 1011-1027 (2012).
- Ostasiewicz, P., Zielinska, D. F., Mann, M., Wisniewski, J. R. Proteome, phosphoproteome, and N-glycoproteome are quantitatively preserved in formalin-fixed paraffin-embedded tissue and analyzable by high-resolution mass spectrometry. J Proteome Res. 9 (7), 3688-3700 (2010).
- Malmström, J., Picotti, P., Aebersold, R. Perspectives of targeted mass spectrometry for protein biomarker verification. Curr Opin Chem Biol. 13 (5-6), 518-525 (2009).
- Gillet, L. C., et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 11 (6), (2012).
- Basu, S., Campbell, H. M., Dittel, B. N., Ray, A. Purification of specific cell population by fluorescence activated cell sorting (FACS). J Vis Exp. 41, (2010).
- Edwards, R. A. Laser capture microdissection of mammalian tissue. J Vis Exp. 8, 309 (2007).
- Liu, N. Q., et al. Proteomics pipeline for biomarker discovery of laser capture microdissected breast cancer tissue. J Mammary Gland Biol Neoplasia. 17 (2), 155-164 (2012).
- Geiger, T., et al. Use of stable isotope labeling by amino acids in cell culture as a spike-in standard in quantitative proteomics. Nat Protoc. 6 (2), 147-157 (2011).
- Wisniewski, J. R. Proteomic sample preparation from formalin fixed and paraffin embedded tissue. J Vis Exp. (79), (2013).
- Cox, J., Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 26 (12), 1367-1372 (2008).
- Cox, J., et al. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat Protoc. 4 (5), 698-705 (2009).
- Rappsilber, J., Mann, M., Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2 (8), 1896-1906 (2007).
- Sarvi, S., et al. CD133+ cancer stem-like cells in small cell lung cancer are highly tumorigenic and chemoresistant but sensitive to a novel neuropeptide antagonist. Cancer Res. 74 (5), 1554-1565 (2014).
- Shlush, L. I., et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 506 (7488), 328-333 (2014).
- Schaab, C., et al. Global phosphoproteome analysis of human bone marrow reveals predictive phosphorylation markers for the treatment of acute myeloid leukemia with quizartinib. Leukemia. 28 (3), 716-719 (2014).
- Hüttenhain, R., et al. Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Sci Transl Med. 4 (142), 142ra94 (2012).
- Burgess, M. W., et al. Simplified and efficient quantification of low-abundance proteins at very high multiplex via targeted mass spectrometry. Mol Cell Proteomics. 13 (4), 1137-1149 (2014).
- Boja, E. S., et al. Analytical Validation Considerations of Multiplex Mass Spectrometry-based Proteomic Platforms for Measuring Protein Biomarkers. J Proteome Res. , (2014).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados