
Method Article
Quantitative massenspektrometrische Analyse von Krebs-Zellen-Proteome Abgeleitet von Liquid und soliden Tumoren
In diesem Artikel
Zusammenfassung
In-depth analyses of cancer cell proteomes facilitate identification of novel drug targets and diagnostic biomarkers. We describe an experimental workflow for quantitative analysis of (phospho-)proteomes in cancer cell subpopulations derived from liquid and solid tumors. This is achieved by combining cellular enrichment strategies with quantitative Super-SILAC-based mass spectrometry.
Zusammenfassung
In eingehenden Analysen der Krebszelle Proteome sind erforderlich, um onkogene Pathomechanismen aufzuklären, aber auch, um potenzielle Zielmoleküle und diagnostische Biomarker zu identifizieren. Jedoch sind Verfahren zum quantitativen Proteom-Charakterisierung von Patienten stammende Tumoren und insbesondere ihre zellulären Subpopulationen weitgehend. Hier beschreiben wir eine Versuchsanordnung, die quantitative Analyse der Proteome von Krebs Zell-Subpopulationen von entweder flüssigen oder festen Tumoren abgeleitet werden können. Dies wird durch die Kombination von Zellanreicherungsstrategien mit quantitativen Super SILAC basierte Massenspektrometrie gefolgt von bioinformatischen Datenanalyse erreicht. Um bestimmte zelluläre Untergruppen bereichern werden flüssige Tumoren zunächst durch Fließ immunophenotyped Zytometrie durch FACS-Sortierung gefolgt; bei soliden Tumoren wird laser Mikrodissektion verwendet, um bestimmte Zellpopulationen zu reinigen. In einem zweiten Schritt werden die Proteine aus den gereinigten Zellen extrahiert und anschließend mit einem tumorspezifischen kombiniert,Spike-In-Standard, der Proteinquantifizierung ermöglicht SILAC-markiert. Das resultierende Protein Mischung wird entweder Gelelektrophorese oder Filter Aided Probenvorbereitung (FASP), gefolgt von tryptischen Verdau unterzogen. Schließlich werden tryptischen Peptide mit einem Hybrid-Quadrupol-Orbitrap Massenspektrometer analysiert, und die erhaltenen Daten werden mit Bioinformatik-Software-Suiten, einschließlich MaxQuant verarbeitet. Mittels der hier dargestellten Arbeitsablauf bis zu 8.000 Proteinen identifiziert werden können und in einem Patienten stammenden Proben quantifiziert werden, und die resultierenden Proteinexpressionsprofile kann bei Patienten zu Diagnose proteomic Signaturen oder mögliche Wirkstoffziele zu identifizieren, verglichen werden.
Einleitung
Die Massenspektrometrie-basierte Proteomik entstanden und ist heute eine weit verbreitete Disziplin in der Zellbiologie und translationalen biomedizinischen Forschung. Der technische Fortschritt in diesem Gebiet haben es möglich, komplexe zelluläre Prozesse in Zelllinien und Tiermodellen zu untersuchen gemacht, da Tausende von Proteinen in einer einzigen massenspektrometrischen Experiment 1,2 identifiziert werden. Ähnliche Fortschritte bei der Analyse für viele posttranslationale Modifikationen wie Phosphorylierung oder Ubiquitinierung gemacht worden, obwohl dies erfordert in der Regel maßgeschneiderte Abläufe für die Anreicherung und die Datenanalyse für jede Art von Modifikation 2,3. Darüber hinaus ist die Beteiligung von chemischen und metabolischen Markierung Strategien, einschließlich SILAC, ermöglicht eine genaue relative Quantifizierung von Proteinen und PTMs, so dass dieses Verfahren besonders für die Entdeckung der grundlegenden zellulären Prozesse, diagnostische Biomarker und potenzielle Angriffspunkte für Arzneimittel in menschlichen Krebs 4 attraktiv.
Jedochmehrere Herausforderungen müssen hinsichtlich Proteom-Analyse von primären humanen Krebs 5 überwunden werden. Zunächst zeigen menschliche Krebsproben oft einen hohen Grad an zellulärer Heterogenität, die durch die Anwesenheit verschiedener Zellentypen in der Tumor-Mikroumgebung, einschließlich Immunzellen und Fibroblasten gehör. Zweitens führt klonalen Evolution die genetische Vielfalt innerhalb der Tumoren selbst, was in der Existenz mehrerer Zellpopulationen mit unterschiedlichen funktionellen Eigenschaften. Nach dem derzeitigen Konzept der wenigen Krebsstammzellen als die treibenden Kräfte bei der Krebsentstehung und Progression, Proteomanalyse eines solchen (funktional höchst relevant) Zellpopulationen wird erwartet, dass von großer Bedeutung für ein besseres Verständnis der onkogenen Mechanismen für die klinische Anwendung relevant sein 6. Drittens werden quantitative Proteinexpressionsprofile von großen Sätzen von Proben oft zur Identifizierung robuste klinische Biomar erforderlichekers; Dies kann nicht durch chemische Markierung wie iTRAQ 7 durchgeführt werden, während die metabolische Markierung Strategien - die sich, wie sie es tun, auf die zelluläre Proliferation 4 - sind ebenfalls nicht anwendbar. Viertens sind die meisten verfügbaren festen Tumorproben in Formalin fixierten, die massenspektrometrische Proteomanalyse aufgrund der Bildung von Proteinquervernetzungen 8 verkompliziert. Schließlich sind die meisten bestehenden Proteomik Workflows erfordern erhebliche Mengen an Probenverarbeitung und Datenerfassung, wodurch die Analyse der Probennummern für die klinische Forschung relevanten schwierig und fordern neue Workflow-Paradigmen 9,10.
Um diese Hindernisse zu adressieren wir einen Versuchsaufbau, die entweder durchflusszytometrische Zell kombiniert Sortier 11 oder Laser-Mikrodissektion 12,13 für die Zellanreicherung mit einem Super SILAC 14 Strategie, ein globales interner Standard für eine umfassende massenspektrometrische Analyse einzuführen entwickelt. Durch die Verwendung der hier beschriebenen Verfahren ist es möglich, bis 8.000 Proteine in einzelnen menschlichen Tumorproben von entweder flüssigen oder festen Krebsarten abgeleitet quantifizieren up.
Protokoll
Alle Experimente an menschlichen Gewebe- oder Blutproben muss von einer Ethikkommission nach beliebigen Vorgaben in der Ethik-Votum gegeben genehmigt und durchgeführt werden.
1. Cellular Enrichment
- Flüssige Tumoren / Fluoreszenz-aktivierte Zellsortierung.
- Mononukleäre Zellen aus dem Knochenmark oder Blut Wasserprobe durch Erythrozytenlyse mit Ammoniumchlorid oder durch Ficoll-Dichtegradientenzentrifugation.
- Für Erythrozytenlyse, kombinieren 1 ml der Probe mit 4 ml Ammoniumchloridlösung. Inkubieren der Lösung auf Eis für 5 - 10 min. 40 ml phosphatgepufferte Salzlösung (PBS) + 2% fötales Kälberserum (FCS) und Zentrifuge 5 min bei 400 × g und 4 ° C.
- Für Ficollgradientenzentrifugation, Schicht 10 ml Probe auf 15 ml Ficoll-Hypaque bis 2 unterschiedliche Schichten bilden (achten Sie darauf, um die Schichten zu stören). Zentrifugation bei 400 g für 30 min bei RT (15-25 ° C) sorgfältig und ernten den einkernigenZellen, die in der Interphase gesammelt.
- Spin down die Zellen und nehmen sie in PBS + 2% FCS. Zentrifuge bei 400 xg für 5 min und nehme in 1 ml PBS + 2% FCS. Nehmen Sie eine aliquote Menge als negative Kontrolle und ein Aliquot für jeden Fluoreszenzfarbstoff in der letzten Flecken verwendet.
- Inkubation mit fluoreszenzmarkierten Antikörpern nützlich, um die Leukämiezellpopulation in Verdünnung nach Herstellerangaben auf Eis für 30 min zu isolieren. Färben Sie auch die für die Entschädigung mit jedem Antikörper einzeln Aliquots. Zweimal waschen mit PBS + 2% FCS.
HINWEIS: Die verwendeten Antikörper waren Anti-Mensch gegen CD117-PE (Klon 95C3), CD34-FITC (Klon 8G12) und CD33-PE (Clone P67.6). - Filterzellen durch 35 um Zellsieb, idealerweise unter Verwendung von Hülsen mit Zellsieb in Kappe integriert.
- Fügen Sie die Lebensfähigkeit der Zellen Fleck (zB 7-AAD). Sortierzellen unter Verwendung eines geeigneten Zellsortierer 11 (Abbildung 2). Sammeln Zellen in Isc Ove modifiziertem Dulbecco-Medium (IMDM) mit 10% FCS.
- Mononukleäre Zellen aus dem Knochenmark oder Blut Wasserprobe durch Erythrozytenlyse mit Ammoniumchlorid oder durch Ficoll-Dichtegradientenzentrifugation.
- Solide Tumoren / Laser-Mikrodissektion.
- Aus den beschriebenen Versuchen wurden FFPE-Proben von Lungenkrebs Probe verwendet. Zu diesem Zweck Lungenkrebsgewebe wurde sofort in 4% gepuffertem Formalin nach chirurgischer Resektion und Stücke von etwa 3 x 2 x 1 cm befestigt, wo in Paraffin für die weitere Analyse eingebettet.
- Schneiden Abschnitte 5-10 & mgr; m Dicke aus der Formalin-fixierten und in Paraffin eingebetteten (FFPE) Probe mit einem Mikrotom. Berg Abschnitte auf Folie bespannt Membran Rutschen und trocken bei 37 ° C für 1 Stunde. Entparaffinieren und rehydratisieren die Einbaufelder durch aufeinanderfolgende Inkubation in Xylol, absolutem Ethanol, 70% und Wasser für jeweils 1 min.
- Färben die Schnitte mit Hämatoxylin für 20 Sekunden und dann mit Leitungswasser spülen. Sammeln Sie die Zellpopulation mit einem Laser-Mikrodissektion System (siehe auch 12).
- Flüssigkeitstumoren.
- Zentrifugieren Sie die Zellsuspension aus Schritt 1.1.5 bei 400 × g, 4 ° C für 4 Minuten und den Überstand verwerfen. Zweimaliges Waschen mit 500 ul kaltem PBS und Zentrifugation für 5 min bei 400 × g und 4 ° C
- In 40 ul Lysepuffer pro 10 6 Zellen und Inkubation für 15 min auf Eis (10 5 Zellen (ca. 10 ug Gesamtprotein) sollte die minimale Anzahl von Zellen sein). Zentrifugieren Sie das Lysat bei 14.000 × g, 4 ° C für 10 min und den Überstand (gelöscht Zelllysat) in ein neues Reaktionsgefäß. Entsorgen Sie das Pellet.
- Solide Tumoren.
- In 60 ul Gewebe Lysepuffer zu der mikrodissezierten Gewebe und Inkubation für 15 Minuten auf Eis, sammeln die Flüssigkeit durch kurze Zentrifugation und übertragen Sie die Suspension auf ein neues Reaktionsgefäß. Beschallen das Lysat auf Eis für 3 min.
- Werden 15 ul 20% Natriumdodecylsulfat (SDS), um ein endgültiges SDS concentration von 4%. Inkubieren microdissected Gewebe bei 99 ° C in einem Heizblock für 1 h und bei 600 Upm gerührt. Zentrifugieren Sie das Lysat bei 16.000 × g, 18 ° C für 10 min und den Überstand in ein neues Röhrchen.
3. Einrichtung eines SILAC Spike-In Quantifizierung Standard (Super SILAC Standard)
HINWEIS: Die Quantifizierung Standard besteht aus einem SILAC-markierten Proteinmischung aus 4 abgeleitet - 6 Zelllinien, die das Tumor-Typ von Interesse entsprechen. Um die maximale Überlappung zwischen dem SILAC-markierten Referenz Proteom und Tumor-abgeleitete Proteom zu erreichen, muss ein Hauptkomponentenanalyse durchgeführt, bevor Zelllinien werden für die Quantifizierungsstandard 14 ausgewählt werden.
- Hauptkomponentenanalyse (PCA).
- Um die Proteinexpressionsmuster von Zelllinien zu bestimmen, zu kultivieren etwa zehn verschiedenen Zelllinien in geeigneten Zellkulturmedien. Lyse der Zellen, wie beschrieben,in Schritt 2.1. Bereiten Zellysaten für die Massenspektrometrie, wie in Schritt 6.1 beschrieben.
- Analyse der Proteinexpressionsmuster von jeder Zelllinie auf einem hochauflösenden Flüssigchromatographie-gekoppelte Massenspektrometer (nanoLCs-MS / MS).
- Analysieren Sie die resultierenden Rohdaten mit einem kostenlosen ware MaxQuant und führen Sie eine Hauptkomponentenanalyse mit Hilfe der zugehörigen Software, wie Perseus 16,17.
- Wählen Sie 4-6 Zelllinien, die die größte Vielfalt zwischen ihren Protein-Expressionsprofile zeigen, und verwenden Sie sie, um das Super SILAC Spike-In Standard zu generieren.
- SILAC-Kennzeichnung und Beschriftung Prüfung.
- Kultivieren der ausgewählten Zellinien mindestens fünf Zellzyklen in entsprechende SILAC Zellkulturmedium, in dem Arginin und Lysin sind mit stabilen Isotope von Kohlenstoff und Stickstoff (SILAC Medium) 4 gekennzeichnet.
- Lyse der Zellen, wie in Schritt 3.1 beschrieben, und bereiten Zelllysaten für die Massenspektrometrie, wie in Schritt 6.1 beschrieben.Messen Sie die Einbaueffizienz von SILAC Markierung durch nanoLCs-MS / MS. Analysieren Sie die resultierende Roh MS Daten mit MaxQuant und bestimmen die Effizienz der SILAC-Kennzeichnung. Dies wird durch Zählen der Anzahl von Peptiden in ihren beschriftet (schwer) identifiziert und ihre endogene Formen (Licht) und Berechnung des Verhältnisses (schwere) / (schwere + Licht) erreicht. Markierungseffizienz sollte 98% nicht überschreiten.
- Kombination von SILAC Proteome und Validierung.
- Kultivieren und nach Herstelleranweisungen im entsprechenden SILAC Medium erweitern ausgewählten Zelllinien. Lyse der Zellen, wie in Schritt 2.1 für flüssige Tumoren beschrieben oder wie in Schritt 3.2 bei soliden Tumoren beschrieben und festzustellen, Proteinkonzentration für jeden Zelllysat.
- Mischen äquimolarer Proteinmengen von jeder Zelllinie und teilen Sie die Mischung in aliquoten. Snap-Einfrieren der Teilmengen und bei -80 ° C bis zur Messung. Bei einem Experiment benötigen Sie 20-50 ug Super SILAC Standard. Achten Sie auf die stan vorbereitendard im Überschuss als Änderung des Standard innerhalb einer Reihe von Versuchen zu vermeiden ist.
4. Messung der Proteinkonzentration und Spike-in
- Flüssigkeitstumoren.
- Mit Proteinquantifizierung Assay bestimmen die Proteinkonzentrationen der jeweiligen Zelllysaten und der Super-SILAC Standard.
- Gleiche Mengen der geklärten Zelllysat und der Super-SILAC Standard und anschliessend hinzufügen Lithiumdodecylsulfat (LDS) Puffer (25% des Volumens der Probe) und Reduktionsmittel (10% des Volumens der Probe).
- Erhitzen der erhaltenen Lösung in einem Heizblock bei 72 ° C für 10 min. Wahlweise speichern die resultierende denaturierten Proteinen bei -80 ° C.
- Solide Tumoren.
- Zur Proteinkonzentrationsmessung auf einem Plattenleser, zu mischen einen Standard Rinderserumalbumin (BSA) Lösung Verdünnungsreihe, das Lysat und das entsprechende Super SILAC mäßig mit einem im Handel erhältlichen Protein assay in einer 96-Well-Platte, und schütteln Sie für 1 min inkubieren angegebenen Zeit und Messung der Extinktion, wie vom Hersteller angegeben.
HINWEIS: Die hohe Konzentration von SDS und Dithiothreit (DTT) in dem Lysat ist ein Problem für die meisten verfügbaren Assays zur Bestimmung der Proteinkonzentration. - Gleiche Mengen des geklärten Lysats und dem Super SILAC Standard mit 200 ul von Harnstoff in der Filtereinheit und Zentrifugieren bei 14.000 xg für 30 min bei 20 ° C. Verwenden Sie nicht mehr als 50 & mgr; l des geklärten Lysats und die Super SILAC Standard. Zu vermeiden sind Temperaturen unter 15 ° C, so dass der Harnstoff nicht auskristallisiert.
- Zur Proteinkonzentrationsmessung auf einem Plattenleser, zu mischen einen Standard Rinderserumalbumin (BSA) Lösung Verdünnungsreihe, das Lysat und das entsprechende Super SILAC mäßig mit einem im Handel erhältlichen Protein assay in einer 96-Well-Platte, und schütteln Sie für 1 min inkubieren angegebenen Zeit und Messung der Extinktion, wie vom Hersteller angegeben.
5. Probentrennung und Protein Digest
- Flüssigkeitstumoren.
- Separate 30-100 & mgr; g Gesamtprotein pro Spur auf einem 4-12% Gradienten-SDS-PAGE-Gel. Stain Proteine mit Coomassie-Blau O / N. Überschüssiges Coomassie-Färbung von zwei aufeinanderfolgenden Waschvorgängen mit Wasser.
- Schneiden Sie jede Spur von dem Gel und teilen siein 23 gleich große Scheiben unabhängig von dem Muster der Gelfärbung. Verarbeiten Sie die Gel-Scheiben getrennt, die jeweils in eine in 0,6 ml Polypropylen-Röhrchen.
- Mit Wasser und Methanol / Wasser (50:50, v / v), mit 10 mM DTT reduzieren durch Inkubation für 30 min bei 56 ° C waschen Gelscheiben. Alkylieren die Gelscheiben mit 55 mM Iodacetamid (IAA) durch Inkubation bei 60 min bei RT im Dunkeln.
- Zwischen Probe-Handhabungsschritte, waschen Scheiben mit Acetonitril für 15 min und trocken in einer SpeedVac um überschüssiges Lösungsmittel zu entfernen und verbessern die Aufnahme von Reagenzienlösung.
- Führen Protease Spaltung durch Rehydratisieren getrocknete Gel Scheiben mit der minimalen Menge von Schweine-Trypsin-Lösung (12,5 ng / ul in 0,025 M wässrige Ammoniumhydrogencarbonatlösung) für 16 h bei 37 ° C.
- In 10 ul Wasser Gelstück und Inkubation für 15 min bei 37 ° C. In 80 ul Acetonitril und Inkubation für 15 min bei 37 ° C. Zentrifugieren bei 15.800 · g, 1 min. Sammeln Stand und lagern in einem separaten 0,6 mlRohr.
- Hinzufügen 65 ul 5% igen Ameisensäurelösung, Vortex und inkubiere für 15 min bei 37 ° C. In 65 ul Acetonitril und Inkubation für 15 min bei 37 ° C. Zentrifuge bei 15.800 · g für 1 min. Die überstehende Flüssigkeit und zum Überstand hinzu aus dem vorherigen Schritt. Dampfe das kombinierte Überstand zur Trockene im Vakuum-Konzentrator.
- Solide Tumoren.
- SDS entfernen Gewebelysat verwenden Sie die folgende FASP Protokoll, auch in Bezug 15 beschrieben.
- Nach dem ersten Schritt 5.2.3 beschrieben Zentrifugation fügen weitere 200 & mgr; l 8 M Harnstoff auf dem Filter und weitere Zentrifuge bei 14.000 xg für 20 min bei 20 ° C. Entsorgen Sie die Durchfluss Filtrat.
- 100 l IAA und mischen in einem Thermomixer bei 600 UpM für 1 Minute. Die Filter für 20 min Inkubation bei 20 ° C im Dunkeln. Zentrifugieren Sie den Filter bei 14.000 g für 10 min bei 20 ° C.
- Füge 100 & mgr; l von Harnstoff zu dem Filter und CENZentrifuge bei 14.000 × g für 15 min bei 20 ° C. Wiederholen Sie diesen Schritt noch einmal.
- 100 l NH 4 HCO 3 auf dem Filter und Zentrifuge bei 14.000 g für 10 min bei 20 ° C. Wiederholen Sie diesen Schritt zweimal.
- Man fügt 40 & mgr; l NH 4 HCO 3 + 1 & mgr; l (= 0,4 & mgr; g) Trypsin und mischen in Thermomixer bei 20 ° C für 600 min, 1 min. Inkubieren des Filters O / N in einem feuchten Kammer bei 37 ° C. Übertragen Sie die Filter, um neue Sammelröhrchen.
- Zentrifugieren Sie den Filter bei 14.000 g für 10 min bei 20 ° C. Zugeben von 50 ul NH 4 HCO 3 und der Filterzentrifuge bei 14.000 g für 10 min bei 20 ° C. Speichern der resultierenden Peptide bei -20 ° C bis massenspektrometrische Messung.
- Peptidkonzentrationsmessung verzichten 50 ul der resultierenden Durchfluss und eine Reihe von geeigneten Verdünnungen von reinem Tryptophan in einer 96-Well-Platte. Tryptophan-Fluoreszenz zu messen. Konvertieren Sie die resultierende KonzentrationTryptophan Peptidkonzentration, wie 0,1 ug Tryptophan entspricht 9 ug Protein.
6. Flüssigkeitschromatographie und massenspektrometrischer Analyse
- Abgeblasen, Peptiden in 30 & mgr; l Beladungspuffer für 5 Minuten in einem Ultraschallbad. Spin down in einer Zentrifuge bei 15.800 · g für 1 min und pipettieren Sie die klare Lösung in eine MS Autosamplergefäß.
- Injizieren 5 ul Probe pro Analyse mit dem automatischen Probennehmer nanoLCs-MS / MS-System. Konzentrieren und Entsalzen von Peptiden Online auf einer Umkehrphasen-C18-Vorsäule (0,15 mm ID x 20 mm mit 5 um Porengröße C18-Material) entweder in einer belüfteten Säule Setup oder eine Vorsäule Schaltaufbau montiert.
- Separate Peptide auf einer Umkehrphasen-C18-Mikrosäule (0,075 mm ID x 200 mm Selbst gepackt mit 3 um oder kleiner Porengrße C18 Material in einer selbst Packung Nanospray Spalte wie PicoFrit Verwendung eines 90 min-Gradienten von 5> 35% Acetonitril vs . 0,1% wässriger Ameisensäure in 300 nl /Minute Überlaufmittel in einen Hybrid-Quadrupol / Orbitrap Massenspektrometer über eine Nanospray-Ionenquelle.
- Analysieren Peptide mit einem Top15-Daten abhängigen Erwerbsmethode (MS m / z-Bereich 350 -. 1600, Auflösung Ziel 70.000 FWHM, AGC Ziel 1 x 10 6, max Füllzeit 60 msec MS / MS beginnen Masse 100, Auflösung Ziel 17.500 FWHM, AGC Ziel 2 × 10 5, max Füllzeit 60 msec MS / MS-Schwelle 3 x 10 4, sind Ladungszustände. 2 - 5, normalisierten Kollisionsenergie NCE 25%, dynamischen Ausschluss 15 sec).
7. Datenanalyse
- Für die Datenanalyse mit dem frei verfügbaren MaxQuant 16 Software. Ein detailliertes Protokoll für Bioinformatik-Datenanalyse wird in 16,17 beschrieben. Zur Analyse kombinieren Rohdaten von allen Scheiben aus einem SDS-PAGE-Spur in einem einzigen Experiment 17.
Ergebnisse
Proteomanalyse von flüssigen und festen Tumoren von Patienten ist ein vielversprechender Ansatz für die Entdeckung neuer diagnostischer und prädiktiver Biomarker. Die Probenvorbereitung und die massenspektrometrische Analyse gibt jedoch eine Herausforderung, sie aufgrund der Komplexität der Proben und die Notwendigkeit für eine genaue Quantifizierung von Proteinen in großen Mengen von Proben. Die oben beschriebenen experimentellen Prozedur beginnt mit der Isolierung von Zellen von Interesse, entweder durch fluoreszenzaktivierte Zellsortierung oder durch Laser-Mikrodissektion.
Hierzu werden die Zellen aus einem Patienten stamm Knochenmarkabsaugungen oder Blutproben mit fluoreszenzmarkierten Antikörpern gegen definierte Oberflächenmarker von Interesse vor dem FACS-basierte Sortierung und Zelllyse gefärbt.
Um zelluläre Untergruppen von Gewebeschnitten zu bereichern, müssen diese erst auf entsprechende Objektträger aufgebracht werden, um Laser-Mikrodissektion ermöglichen. Hierzu Halterung ter Gewebeschnitte auf geeignete Membran gleitet und mit einem Laser-Capture-MicroDissector nach den Anweisungen des Herstellers. Die Auswahl der Bereiche und Zellen von Interesse erfordert angemessene Kenntnisse über die Histomorphologie von gesunden und Tumorgewebe. Die extrahierten Zellen werden dann in einem geeigneten Sammelröhrchen überführt. Anfängliche Experimente sollte, um die Menge an Gewebe zur Extraktion von ausreichenden Mengen an Protein (mindestens 10 & mgr; g Gesamtprotein) für jeden neuen Gewebe von Interesse notwendig definiert durchgeführt werden. Jede microdissected Probe wird mit 60 ul Gewebe Lysepuffer behandelt. Für eine effiziente Lyse haben Zellen von Gewebeproben für 3 min, und nach der Zugabe von 15 ul SDS-Proben werden in einem Thermomixer bei 99 ° C für 1 h inkubiert, um die Formalin-induzierten Protein-Quervernetzungen zu entfernen beschallt werden. Die Zentrifugation wird endlich erleichtern die Sammlung der geklärten Zelllysat.
Für jede Krankheit interest (flüssigen und festen Tumoren) eine geeignete Super SILAC Quantifizierungsstandard ist zu bilden, 14 werden. Eine entsprechende Norm sollte mehr als 90% der Proteine in den Proben von Interesse zum Ausdruck zu vertreten. Zur Herstellung einer Quantifizierungsstandard sollte Zellinien zur Krebstyp von Interesse bezogenen ersten durch Massenspektrometrie analysiert werden, um ihren Proteinexpressionsprofile festzulegen und anschließend eine Hauptkomponentenanalyse durchgeführt werden soll. 4-6 verschiedenen Zelllinien mit Differenzproteinexpressionsmuster zu wählen und mit "schweren" Arginin und Lysin SILAC-markiert. Markierungseffizienz sollte nanoLCs-MS / MS überprüft werden und sollte mehr als 98% betragen. Danach sollten die Zelllinien in der gleichen Weise wie die entsprechenden Patienten stammenden Probe, und anschließend sollte äquimolarer Proteinmengen von jeder Probe und dem SILAC spike in Standard gemischt werden, lysiert werden. Ein entscheidender Schritt zur genauen Quantifizierung von Proteinen von klinischen Proben of Interesse ist die genaue Mischung äquimolarer Proteinmengen von Patienten stammende Proben und die SILAC spike in Quantifizierungsstandard. Um die Proteinkonzentration der Lysate von sortierten Zellen gewonnen werden, können verschiedene Proteinquantifizierung Assays verwendet werden. Für Zellysaten aus Gewebe stammenden Zellen ein Assay benötigt, die mit hohen Konzentrationen an SDS und DTT in dem Lysat umgehen kann. Proteinkonzentrationen der Patienten stammenden Proben und die SILAC spike in Standard sollte jedes Mal vor dem Kombinieren mit den jeweiligen klinischen Proben, um eine korrekte Mischung, die von entscheidender Bedeutung, sogar Hunderte von Proben vergleichbar ist sicherzustellen gemessen werden.
Nachdem die Mischung aus gleichen Mengen von spike in Standard und Lysat, Proben von Flüssigkeitstumoren mit HLT vermischt, in einem Thermomixer erhitzt, bei 72 ° C für 10 min gewonnen und anschließend in 1D-PAGE unterzogen und In-Gel-Verdauung des abgetrennten Proteine mit Trypsin. Proben aus erhaltenGewebe aus SDS mit der FASP Ansatz wie von Wisniewski et al fest 15 befreit und mit Trypsin verdaut.
Die tryptischen Peptiden erhalten durch Messen Tryptophan-Fluoreszenz, die von für die Gewebeproben besonderem Interesse ist, wie die Menge der Peptide, die von dem Filter gelöst unterscheidet zwischen 15% und 75% im Vergleich zu den Proteinmengen zunächst auf den Filter geladen quantifizieren. Dazu messen wir die Fluoreszenz von Tryptophan. Der Vergleich mit einer geeigneten Tryptophan Verdünnungsreihe ermöglicht die Quantifizierung von Peptiden, wie 1,1 ug Tryptophan entspricht etwa 100 ug Peptid. Falls erforderlich, Proben vor allem aus FASP Verarbeitung abgeleitet werden auf kommerziellen oder hausgemachte Bühne Tipps vor dem Laden auf den nanoLC / MS / MS-System 18 mit Umkehrphasen-C18 (RP-C18) Material verpackt vorgereinigt werden.
Massenspektrometrische Analyse basiert auf einem hohen geführt-Auflösung, hoher Empfindlichkeit Massenspektrometriesystem. Kurz gesagt, werden Peptidproben entsalzt und auf einer RP-C18 Vorsäule vorkonzentriert und auf einer RP-C18 analytischen Säule direkt an das Massenspektrometer gekoppelt getrennt. Um eine ausreichende Tiefe der Analyse zu erzielen, wir verwenden entweder eine Kombination aus SDS-PAGE Protein Vorfraktionierung mit 40 min RP-C18-Gradienten (für flüssige Tumoren) oder Einzelinjektionen mit langen 2 - 3 h RP-C18-Gradienten (soliden Tumoren durch FASP verarbeiteten ). MS-Spektren werden mit einer Auflösung von 70.000 FWHM oder besser erworben, um eine genaue Quantifizierung der SILAC Paare durch Integration der chromatographischen Peakprofile ermöglichen. Zur Proteinidentifizierung wird eine Top15 datenabhängigen Erfassungsverfahren verwendet, um eine große Anzahl von Peptid MS / MS-Spektren für Peptid- und Proteinidentifizierung zu erzeugen.
Aus den resultierenden Rohdaten werden Protein Identifizierung und Quantifizierung von Datenbank erreicht Suche mit MaxQuant Software gegen ein UniProt Knowledgebase Menschen komplette Proteom-Sequenzdatenbank 17. In MaxQuant Software (aktuelle Version 1.5.0.25) Peptide aus dem erworbenen MS / MS-Spektren von Peptidfragment-Abgleich mit Spektren in silico aus der Proteinsequenz-Datenbank abgeleitet identifiziert. Zur gleichen Zeit, sind Vorläuferion Isotopenprofile um ihre chromatographischen Retentionszeiten extrahiert und ihre integrierten Peakflächen werden zur relativen Quantifizierung des verwendeten Lichts: Schwer von SILAC Kennzeichnung erzeugt Peptidpaare. Peptid Identitäten und relativen Intensitäten werden dann zu den entsprechenden Proteineigenschaften versehen. Dann wird Perseus Software (aktuelle Version 1.5.0.15) verwendet werden, um weiter stromabwärts statistische Auswertung der MaxQuant Verarbeitungsergebnisse, einschließlich Probe zu Probe-Vergleiche, PCA und hierarchisches Clustering durchzuführen.
Mit dem Versuchsaufbau beschrieben haben wir identifiziert und quantifiziert bis 8.000 Proteine von weniger als 30 & mgr; g Gesamtproteinvon flüssigen Tumoren abgeleitet.
Bis 2.500 Proteine aus festen Tumorproben können identifiziert und in einer Schrotflinte Proteomanalyse mit einem LC-Gradienten von nur 2 Stunden quantifiziert, so dass die Analyse von Hunderten von klinischen Proben in einer relativ kurzen Zeit.

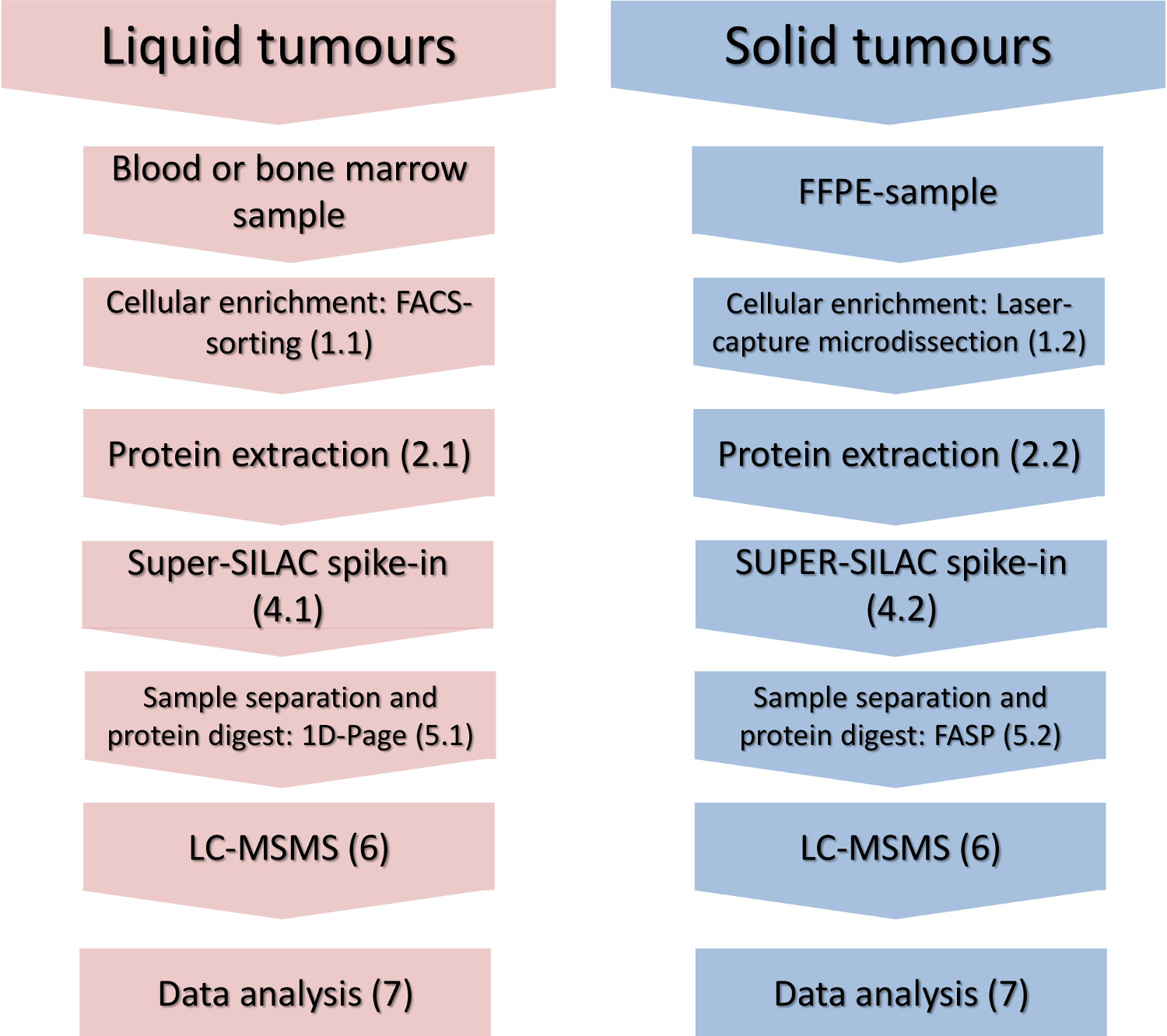
Abbildung 1. Experimentelle Arbeitsablauf. Die wichtigsten Schritte der zellulären-Teilmenge Bereicherung, Proteinisolierung, Spike-In des Quantifizierungsstandards und massenspektrometrische Analyse sind für flüssige und feste Tumoren gezeigt. Bitte klicken Sie hier, um eine größere Version dieses Bild anzuzeigen.
{kind=link}

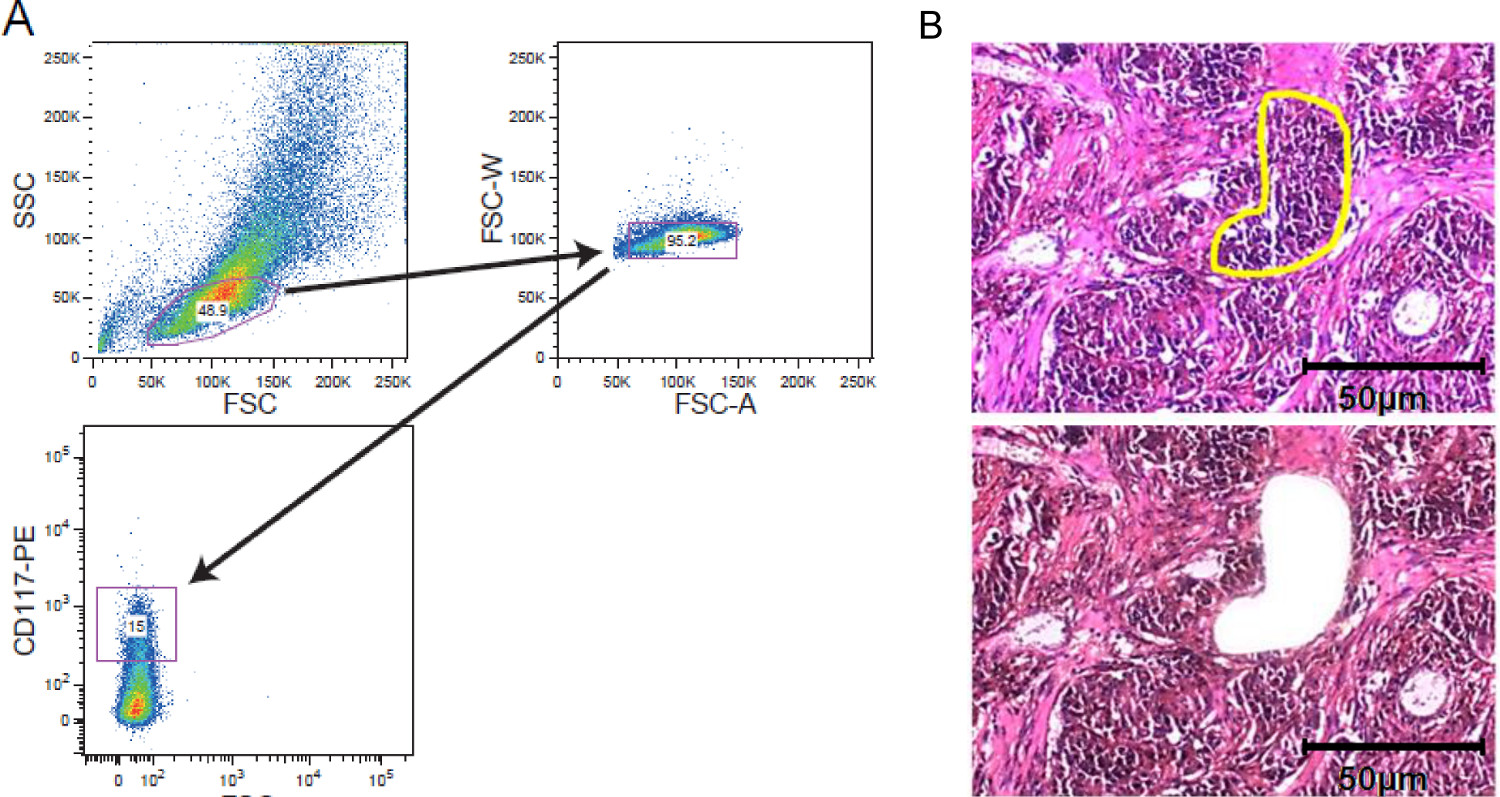
Abbildung 2. Cellular Bereicherung strategischn. FACS-Sortierung und LCM. (A) Beispielhafte Gating-Strategie darstellt Gating auf einem CD117-gefärbten Leukämiezellpopulation durch einen Zellsortierer erworben. (B) Laser Mikrodissektion von solidem Tumorgewebe. Gewebeschnitte von 5 bis 10 um Dicke wurden auf Film beschichteter Membran Folien vor der Färbung gelagert. Region von Interesse wurde manuell für die Laser Mikrodissektion ausgewählt. Gezeigt werden Abschnitte vor Mikrodissektion mit dem interessierenden Bereich gelb markiert und nach Mikrodissektion. Ein Maßstab von 50 & mgr; m ist in der Abbildung enthalten. Bitte klicken Sie hier, um eine größere Version dieses Bild anzuzeigen.
{kind=link}

Abbildung 3. Cluster-Analyse der menschlichen Krebszell Proteome. Unbeaufsichtigte Clusteranalyse protein Expressionsprofile von flüssigen und festen Tumoren wurde unter Verwendung der Rechenplattform Perseus. AC - Adenokarzinom, SCC - Plattenepithelkarzinom, SCC-Met - Plattenepithelkarzinom Metastasen von Kopf- und Halskrebs, R - Technische Replikation.
Diskussion
Massenspektrometrische Analyse von Patienten stammende Krebszell Proteome wird für die Entdeckung neuer diagnostischer / prädiktive Biomarker benötigt, als auch zu einem besseren Verständnis von Krebs-Zellbiologie, geben, die vielleicht wiederum zu der Identifizierung von neuen potenziellen Wirkstoff-Targets. Allerdings sind solche massenspektrometrische Analysen sehr anspruchsvoll, vor allem, weil verschiedene präanalytische Probleme müssen gelöst werden, wenn man sich um robuste und biologisch relevante Ergebnisse zu erhalten.
Die hier beschriebenen experimentellen Arbeitsablauf ermöglicht quantitative Proteomik Charakterisierung von Proteomen von zellulären Subpopulationen von flüssigen und festen Tumoren abgeleitet. Die anfängliche Anreicherung von Tumorzellen entweder durch FACS-basierte Zell sortingor Mikrodissektion benötigt wird, um eine Verunreinigung durch Zellen der Tumormikroumgebung zu vermeiden. Darüber hinaus sind diese Techniken erlauben es, zelluläre Subpopulationen von Interesse zu isolieren. Neueste zellbiologische Studien haben Dämonstrated, dass bestimmte Zellpopulationen haben tumorauslösende Eigenschaften und sind somit für die Krebs Pathogenese 19,20 höchst relevant. Massenspektrometrie sensibler geworden in den letzten Jahren sind quantitative proteomische Analyse denkbar, dass die geringen Mengen an Protein, das von einigen tausend Zellen abgeleitet werden können, was es ermöglicht, den Schwerpunkt auf die funktionsrelevanten Zellpopulationen.
Die Einrichtung hier vorgestellten kann verwendet werden, um in FFPE-Proben Identifizierung und Validierung neuartiger diagnostischer Biomarker werden. Daher verspricht sie nützliches Werkzeug für die Verbesserung der klinischen Diagnostik BEA, als bisher gibt es noch ein Mangel an molekulare Biomarker in ausreichender Zahl und Qualität für viele Krebsarten. Wichtige Beispiele für schwierige Differentialdiagnosen, für die Biomarker fehlen, sind die Unterscheidung zwischen primären Lungenkrebs von Metastasen in der Lunge, intrapankreatischen cholangiozelluläres Karzinom und Adenokarzinom des Pankreas sowie unterscheidenzierung von gutartigen Neurofibrom von hoch malignen peripheren Nervenscheidentumoren. Darüber hinaus haben wir und andere haben gezeigt, dass quantitative Aufklärung der Proteom-Signaturen können bei der Untersuchung von Krebszellen Biologie im Allgemeinen und für die Entdeckung prädiktiver Biomarker für therapeutische Reaktion bei Krebspatienten 21 Jahre alt sein.
Zwei aktuelle Nachteile der hier vorgestellten Verfahren sind die Voraussetzung für umfangreiche manuelle Probenaufbereitung und die Nachfrage am nanoLC-MS / MS Erfassungszeit. Während erstere können durch Bewegen der Probenvorbereitung bis zum Beispiel 96-Well-Format und mit Roboter gewandt werden, wird dieser eine Änderung der massenspektrometrischen Akquisitionsstrategie benötigen. Nach Teilmengen von Zielproteine identifiziert, die mit zB Tumorklassifikation in Verbindung gebracht werden können, sehen wir die Gestaltung von gezielten Massenspektrometrie Methoden, die quantitative Ablesungen für diese Teilmengen mit einem stark reduzierten Trennaufwand zu schaffen, unddaher mit einem entsprechend reduzierten Akquisitionszeit. Falls die benötigte Erfassungszeit benötigt konnte von 24 reduziert werden - 36 h (flüssige Tumoren) oder 3 Stunden (solide Tumoren), um zB, 1 h durch gezielte Massenspektrometrie und eine einfache eindimensionale Trennung von Peptiden, wird der daraus resultierende Gewinn im Durchsatz könnte verwendet werden, um wesentlich die Anzahl von biologischen und technischen Wiederholungen geprüft zu erhöhen, mit entsprechenden Verbesserungen in der Bedeutung der Quantifizierungsergebnisse werden. Gezielte massenspektrometrische Ansätze wurden bereits gezeigt, dass sie ein geeignetes Instrument für die Überprüfung der Krebs assoziiertes Protein-Biomarker-Kandidaten 22 zu sein, und sind an einem Punkt, wo sie Versprechen für die Validierung oder sogar als mögliches Instrument für die klinische Routineanwendung 23 zeigen entwickelt , 24.
Offenlegungen
Die Autoren haben keinen Interessenkonflikt oder andere Fragen offen zu legen.
Danksagungen
The authors thank Uwe Plessmann, Monika Raabe und Silvia Münch for technical support.
Materialien
| Name | Company | Catalog Number | Comments |
| 660 nm Kit | Thermo scientific | 22662 | |
| Cell culture medium depleted of arginine and lysine | Thermo Scientific | 88421 | |
| Coomassie Brilliant Blue R-250 staining solution | Bio Rad | 161-0436 | |
| Dialyzed fetal calf serum (FCS) | PAA | A15-107 | |
| Diffuser caps for microdissection | MMI | 50202 | |
| FACS-sorter | BD | FACSAria III | |
| Ionic Detergent Compatibility Reagent | Thermo scientific | 22663 | |
| Laser-capture microdissector | MMI | cell cut plus | |
| LDS buffer | Life Technologies | NP0009 | |
| Membrane slides for microdissection | MMI | 50103 | |
| Microcon YM-30 | Millipore | MRCF0R030 | |
| NuPAGE 4-12% Bis-Tris Mini Gels | Life Technologies | NP0335PK2 | |
| Picofrit Self-Pack Columns | New Objective | PF360-75-15-N-5 | Mass Spectrometry Column/Emitter |
| Reducing agent | Life Technologies | NP0007 | |
| Reprosil-Pur LC/MS/MS Column stationary phase | Dr. Maisch | 120 C18-AQ, 3 µm | |
| Reprosil-Pur LC/MS/MS Precolumn stationary phase | Dr. Maisch | 120 C18-AQ, 5 µm | |
| SILAC-labeled arginine | Eurisotop | CLM-2265-H-0.1 | |
| SILAC-labeled lysine | Eurisotop | DLM-2640-0.25 | |
| Trypsin, NB Sequencing Grade | Serva | 3728301 | for in-gel digests |
| Trypsin, Sequencing Grade | Promega | V5111 | for in-solution digests |
| Buffer and solutions | |||
| Cell lysis buffer: 150 mM NaCl, 50 mM Tris/HCl pH 7.8, 5 mM NaF, 0.5% NP40, 0.1% laurylmaltoside, Roche complete protease inhibitor, 1 mM Na3VO4 | |||
| Tissue lysis buffer: 100 mM Tris/HCl pH 7.8, 0.1 M DTT | |||
| Urea: 8 M urea in 0.1 M Tris-HCl, pH 8.5 | for FASP-protocoll | ||
| IAA: 0.05 M iodoacetamide, 8 M urea, 0.1 M Tris-HCl, pH 8.5 | for FASP-protocoll | ||
| 0.05 M NH4HCO3 | |||
| 10 mM dithiothreitol (DTT) in 0.1 M ammonium bicarbonate | for in-gel digest | ||
| 55 mM iodoacetamide (IAA) in 0.1 mM ammonium bicarbonate | for in-gel digest | ||
| 5% aqueous formic acid. |
Referenzen
- Walther, T. C., Mann, M. Mass spectrometry-based proteomics in cell biology. J Cell Biol. 190 (4), 491-500 (2010).
- Lenz, C., Urlaub, H. Separation methodology to improve proteome coverage depth. Expert Rev Proteomics. 11 (4), 409-414 (2014).
- Olsen, J. V., Mann, M. Status of large-scale analysis of post-translational modifications by mass spectrometry. Mol Cell Proteomics. 12 (12), 3444-3452 (2013).
- Ong, S. E., et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 1 (5), 376-386 (2002).
- Jimenez, C. R., Verheul, H. M. Mass spectrometry-based proteomics: from cancer biology to protein biomarkers, drug targets, and clinical applications. Am Soc Clin Oncol Educ Book. , e504-e510 (2014).
- Tang, D. G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 22 (3), 457-472 (2012).
- Evans, C., et al. An insight into iTRAQ: where do we stand now. Anal Bioanal Chem. 404 (4), 1011-1027 (2012).
- Ostasiewicz, P., Zielinska, D. F., Mann, M., Wisniewski, J. R. Proteome, phosphoproteome, and N-glycoproteome are quantitatively preserved in formalin-fixed paraffin-embedded tissue and analyzable by high-resolution mass spectrometry. J Proteome Res. 9 (7), 3688-3700 (2010).
- Malmström, J., Picotti, P., Aebersold, R. Perspectives of targeted mass spectrometry for protein biomarker verification. Curr Opin Chem Biol. 13 (5-6), 518-525 (2009).
- Gillet, L. C., et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 11 (6), (2012).
- Basu, S., Campbell, H. M., Dittel, B. N., Ray, A. Purification of specific cell population by fluorescence activated cell sorting (FACS). J Vis Exp. 41, (2010).
- Edwards, R. A. Laser capture microdissection of mammalian tissue. J Vis Exp. 8, 309 (2007).
- Liu, N. Q., et al. Proteomics pipeline for biomarker discovery of laser capture microdissected breast cancer tissue. J Mammary Gland Biol Neoplasia. 17 (2), 155-164 (2012).
- Geiger, T., et al. Use of stable isotope labeling by amino acids in cell culture as a spike-in standard in quantitative proteomics. Nat Protoc. 6 (2), 147-157 (2011).
- Wisniewski, J. R. Proteomic sample preparation from formalin fixed and paraffin embedded tissue. J Vis Exp. (79), (2013).
- Cox, J., Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 26 (12), 1367-1372 (2008).
- Cox, J., et al. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat Protoc. 4 (5), 698-705 (2009).
- Rappsilber, J., Mann, M., Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2 (8), 1896-1906 (2007).
- Sarvi, S., et al. CD133+ cancer stem-like cells in small cell lung cancer are highly tumorigenic and chemoresistant but sensitive to a novel neuropeptide antagonist. Cancer Res. 74 (5), 1554-1565 (2014).
- Shlush, L. I., et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 506 (7488), 328-333 (2014).
- Schaab, C., et al. Global phosphoproteome analysis of human bone marrow reveals predictive phosphorylation markers for the treatment of acute myeloid leukemia with quizartinib. Leukemia. 28 (3), 716-719 (2014).
- Hüttenhain, R., et al. Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Sci Transl Med. 4 (142), 142ra94 (2012).
- Burgess, M. W., et al. Simplified and efficient quantification of low-abundance proteins at very high multiplex via targeted mass spectrometry. Mol Cell Proteomics. 13 (4), 1137-1149 (2014).
- Boja, E. S., et al. Analytical Validation Considerations of Multiplex Mass Spectrometry-based Proteomic Platforms for Measuring Protein Biomarkers. J Proteome Res. , (2014).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten