
Method Article
Количественный масс-спектрометрический Профилирование раковых клеток протеомов, полученные из жидких и твердых опухолей
В этой статье
Резюме
In-depth analyses of cancer cell proteomes facilitate identification of novel drug targets and diagnostic biomarkers. We describe an experimental workflow for quantitative analysis of (phospho-)proteomes in cancer cell subpopulations derived from liquid and solid tumors. This is achieved by combining cellular enrichment strategies with quantitative Super-SILAC-based mass spectrometry.
Аннотация
Углубленный анализ раковых клеток протеомов необходимы для выяснения онкогенные pathomechanisms, а также для выявления потенциальных лекарственных целей и диагностические биомаркеры. Тем не менее, методы количественного протеомики характеристик опухолей пациентов, полученных и в частности их клеточных субпопуляций в значительной степени не хватает. Здесь мы описываем экспериментальную настройку, что позволяет количественный анализ протеомов раковых клеток, полученных из субпопуляций жидкие или твердые опухоли. Это достигается путем объединения сотовых стратегии обогащения количественного Супер-SILAC основе масс-спектрометрии с последующим анализом данных биоинформатики. Чтобы обогатить специфических клеточных подмножеств, жидкие опухоли сначала immunophenotyped с помощью проточной цитометрии с последующим FACS сортировки; для солидных опухолей, лазерная захвата микродиссекции используется для очистки специфических клеточных субпопуляций. На втором этапе, белки извлекают из очищенных клеток, а затем в сочетании с опухоль-специфических,SILAC-меченых шип в стандарт, который позволяет белка количественное. Полученный белковый смесь подвергают гель-электрофореза либо или фильтр автоматизированного приготовлени образца (FASP) с последующим трипсином переваривания. Наконец, триптические пептиды анализировали с использованием гибридного квадрупольного масс-спектрометра Orbitrap, и полученные данные обрабатываются с биоинформационных программные пакеты, включая MaxQuant. С помощью процесса, представленного здесь, до 8000 белков могут быть идентифицированы и количественно в образцах пациента, полученных, и полученные профили экспрессии белка можно сравнить у пациентов с целью выявления диагностических протеомических подписи или потенциальных лекарственных целей.
Введение
Масс-спектрометрия на основе протеомики стала и теперь широко используется дисциплина в клеточной биологии и поступательного биомедицинских исследований. Технический прогресс в области дали возможность изучить сложные клеточные процессы в клеточных линий и животных моделях, как тысячи белков могут быть идентифицированы в одном масс-спектрометрического эксперимента 1,2. Аналогичный прогресс был достигнут для анализа многих пост-трансляционных модификаций, таких как фосфорилирование или убиквитинирования, хотя обычно для этого требуется на заказ рабочие процессы для обогащения и анализа данных для каждого типа модификации 2,3. Кроме того, участие химических и метаболических стратегий маркировки, в том числе SILAC, обеспечивает точное относительное количественное белков и PTMs, что делает этот метод особенно привлекателен для открытия основных клеточных процессов, диагностических биомаркеров и потенциальных мишеней для лекарственных средств в человеческий рака 4.
Тем не менее,несколько проблем, которую необходимо преодолеть в связи с протеомного анализа первичных злокачественных опухолей человека 5. Прежде всего, образцы рака человека часто показывают высокую степень клеточной гетерогенности, что обусловлено наличием различных типов клеток, принадлежащих к микроокружения опухоли, в том числе иммунных клеток и фибробластов. Во-вторых, клональный эволюция приводит к генетического разнообразия внутри самих опухолей, в результате существования нескольких клеточных субпопуляций с различными функциональными свойствами. В соответствии с нынешней концепции нескольких раковых стволовых клеток, находящихся движущих сил развития рака и прогрессии, протеомики анализа таких (функционально очень актуально) клеточных субпопуляций, как ожидается, иметь большое значение для лучшего понимания онкогенных механизмов, имеющих значение для клинического применения 6. В-третьих, количественные профили экспрессии белка больших наборов образцов часто требуются для идентификации надежной клинической BiomarKERS; это не может быть достигнуто путем химической маркировки, таких как iTRAQ 7, в то время как метаболические стратегии маркировки - полагаясь, как они это делают, на клеточной пролиферации 4 - также являются неприменимыми. В-четвертых, большинство доступных твердые образцы опухоли являются фиксированных формалином, что затрудняет массовое спектрометрического анализа протеома за счет образования белковых сшивок 8. Наконец, большинство существующих протеомики рабочие процессы требуют значительного количества обработки образцов и сбора данных, что делает анализ чисел выборки соответствующих клинических исследований сложно, и призывают к новым процессом парадигмы 9,10.
Для решения этих препятствий мы разработали экспериментальную установку, которая сочетает в себе либо потока цитометрии сортировки клеток 11 или лазер-захвата микродиссекции 12,13 сотовой обогащения с супер-SILAC 14 стратегии внедрения глобальной внутренний стандарт для комплексного анализа масс-спектрометрического, С помощью метода, описанного здесь, можно количественно до 8000 белков в отдельных образцах опухолей человека, полученных из жидкого или твердого рака.
протокол
Все эксперименты по ткани или образцы крови человека должно быть одобрено комитетом по этике и осуществляется в соответствии с любыми руководящими указаниями в голосовании по вопросам этики.
1. Сотовый Обогащение
- Жидкие опухоли / флуоресценции активирован сортировки клеток.
- Изолировать мононуклеарных клеток из костного мозга или образца крови, выполняя эритроцитов лизис с использованием хлорида аммония или Ficoll плотности центрифугированием в градиенте.
- Для эритроцитов лизиса, объединить 1 мл образца с 4 мл раствора хлорида аммония. Выдержите раствор на льду в течение 5 - 10 мин. Добавить 40 мл забуференного фосфатом физиологического раствора (PBS) + 2% фетальной телячьей сыворотки (FCS) и центрифуге в течение 5 мин при 400 х г и 4 ° С.
- Для Фиколла центрифугирования в градиенте, слоя 10 мл образца на 15 мл Ficoll-Hypaque форме 2 отдельных слоев (будьте осторожны, чтобы не нарушить слои). Центрифуга на 400 мкг в течение 30 мин при комнатной температуре (15 - 25 ° C), осторожно и урожай одноядерныеклетки, которые собирают в интерфазе.
- Спин вниз клетки и принять их в PBS + 2% FCS. Центрифуга на 400 мкг в течение 5 мин и принимать в 1 мл PBS + 2% FCS. Возьмите одну аликвоту в качестве отрицательного контроля и одну аликвоту для каждого флуоресцентного красителя в конечном пятно.
- Выдержите с флуоресцентно-меченного антитела, используемые для изоляции лейкозных клеток населения в разведении в соответствии с инструкциями изготовителя на льду в течение 30 мин. Также пятно аликвоты, используемые для компенсации с каждого антитела в отдельности. Мыть два раза с PBS + 2% FCS.
ПРИМЕЧАНИЕ: Эти антитела были использованы мыши против человеческого против CD117-PE (клон 95C3), CD34-FITC (Clone 8G12) и CD33-PE (Clone p67.6). - Фильтрующие ячейки через сито 35 мкм клетки, в идеале использовать трубы с ячейки фильтра интегрированы в кепке.
- Добавить жизнеспособности клеток пятно (например, 7-AAD). Сортировать клетки с помощью соответствующих сортировщик клеток 11 (рисунок 2). Сбор клеток в Isc OVE модифицированной Дульбекко Среда (IMDM), содержащей 10% FCS.
- Изолировать мононуклеарных клеток из костного мозга или образца крови, выполняя эритроцитов лизис с использованием хлорида аммония или Ficoll плотности центрифугированием в градиенте.
- Солидные опухоли / Лазерная захвата микродиссекции.
- Для описанных экспериментов были использованы образцы FFPE от образца рака легких. Для этого рака легких ткань сразу же фиксировали в 4% формалине с буфером, после хирургической резекции и кусочки примерно 3 х 2 х 1 см, где заливали в парафин для дальнейшего анализа.
- Вырезать разделы 5 - 10 толщиной мкм с формалином фиксированной парафин (FFPE) образца с помощью микротома. Mount разделы, посвященные кино, покрытых мембраной горками и сухим при 37 ° С в течение 1 ч. Deparaffinize и увлажняет смонтированные секции путем последовательного инкубации в ксилоле, абсолютного этанола, 70% воды и, каждый в течение 1 мин.
- Пятно секции с гематоксилином в течение 20 сек, а затем сполоснуть водой. Сбор клеточной популяции интереса с помощью системы микродиссекции лазерного захвата (также см 12).
- Жидкие опухоли.
- Центрифуга клеточной суспензии, полученной на стадии 1.1.5 при 400 х г, 4 ° С в течение 4 мин и отбросить супернатант. Дважды промывали 500 мкл холодного PBS и центрифугированием в течение 5 мин при 400 х г и 4 ° С
- Добавить 40 мкл буфера для лизиса на 10 6 клеток и инкубировать в течение 15 минут на льду (10 5 клеток (ок. 10 мкг общего белка) должны быть минимальное количество клеток). Центрифуга лизата при 14000 х г, 4 ° С в течение 10 мин и супернатант передавать очищается (сотовой) лизата в новую реакционную трубку. Откажитесь от гранул.
- Солидные опухоли.
- Добавить 60 мкл буфера для лизиса ткани в микродиссекции ткани и инкубируют в течение 15 мин на льду, собирают жидкость, короткого центрифугирования и передавать суспензии в новой реакционной трубы. Разрушать ультразвуком лизата на льду в течение 3 мин.
- Добавить 15 мкл 20% додецилсульфата натрия (SDS), чтобы достичь окончательного SDS сотрудничестваncentration 4%. Инкубируйте микродиссекции ткани на 99 ° С в нагревательном блоке в течение 1 часа и перемешивают при 600 оборотах в минуту. Центрифуга лизата при 16000 х г, 18 ° С в течение 10 мин и передавать супернатант в новую пробирку.
3. Создание SILAC шип-в Количественная стандарта (Super-SILAC Standard)
ПРИМЕЧАНИЕ: стандарт количественного состоит из смеси белков SILAC-меченого производного от 4 - 6 клеточных линий, которые соответствуют типу опухоли интерес. Для достижения максимального совпадения между SILAC меченного эталонного протеом и опухоли, полученных протеом, анализ главных компонентов должна быть выполнена перед выбраны клеточные линии для количественного стандарта 14.
- Анализ главных компонент (PCA).
- Чтобы определить паттерны экспрессии белка клеточных линий, развивать около десяти различных клеточных линий в соответствующей среде для культивирования клеток. Лизиса клеток, как описанов шаге 2.1. Подготовка клеточные лизаты для масс-спектрометрии, как описано в стадии 6,1.
- Анализ экспрессии белка образец каждой клеточной линии с высоким разрешением, жидкостная хроматография-масс-спектрометр в сочетании (nanoLC-МС / МС).
- Проанализируйте полученные данные, используя бесплатный вещевой MaxQuant и выполнить основную-компонентного анализа с использованием соответствующего программного обеспечения такой как Персей 16,17.
- Выберите 4 - 6 клеточных линий, которые показывают наибольшее разнообразие между их профилями белок экспрессии и использовать их для создания всплеска в стандарт Super-SILAC.
- SILAC маркировка и проверка этикетки.
- Развивайте выбранные клеточные линии, по крайней мере, пяти клеточных циклов в соответствующей среде культивирования SILAC клеток, в которых аргинин и лизин, меченных стабильными изотопами углерода и азота (SILAC среды) 4.
- Лизиса клеток, как описано в стадии 3,1 и подготовка клеточные лизаты для масс-спектрометрии, как описано в стадии 6,1.Измерьте эффективность включения SILAC маркировки по nanoLC-MS / MS. Анализ полученных исходных данных MS с MaxQuant и определения эффективности SILAC-маркировки. Это достигается путем подсчета числа пептидов, идентифицированных в их маркировкой (тяжелый) и их эндогенных форм (света), и расчета отношения (тяжелая) / (тяжелая + свет). Эффективность Маркировка должна превышать 98%.
- Сочетание SILAC протеомов и проверки.
- Развивайте и расширить отобранных клеточных линий в соответствии с инструкции производителя в соответствующих SILAC среды. Лизиса клеток, как описано в стадии 2,1 для жидких опухолей или как описано в стадии 3,2 для солидных опухолей и определить концентрацию белка для каждого клеточного лизата.
- Смешайте эквимолярные количества белка из каждой клеточной линии и разделить смесь в аликвоты. Snap-заморозить аликвоты и хранят при температуре -80 ° С до измерения. Для одного эксперимента вам нужно 20 - 50 мкг стандарта Super-SILAC. Позаботьтесь, чтобы подготовить стандарт в избытке, как изменения стандарта в рамках серии экспериментов следует избегать.
4. Измерение концентрации белка и Spike-в
- Жидкие опухоли.
- Использование белковых количественный анализ определения концентрации протеина соответствующих клеточных лизатов и стандарта Super-SILAC.
- Смешать в равных количествах очищенной сотовой лизата и стандарта Super-SILAC, а затем добавить литий додецилсульфата (LDS) буфер (25% от объема образца) и восстановитель (10% от объема образца).
- Тепло полученный раствор в нагревательном блоке при 72 ° С в течение 10 мин. По желанию, хранить в результате денатурированные белки при -80 ° С.
- Солидные опухоли.
- Для измерения концентрации белка на планшет-ридере, смешать стандартный бычьего сывороточного альбумина (БСА) разбавление раствора серии, лизат и соответствующий стандарт Супер-SILAC с коммерчески доступным белком Аssay в 96-луночного планшета и встряхивают в течение 1 мин, выдержать в течение указанного времени и измерения поглощения, как указано производителем.
ПРИМЕЧАНИЕ: высокая концентрация SDS и дитиотреитол (DTT) в лизат проблемой для большинства имеющихся анализов для определения концентрации белка. - Смешайте равные количества очищенного лизата и стандарта Супер-SILAC с 200 мкл мочевины в блоке фильтров и центрифуге при 14000 х г в течение 30 мин при 20 ° С. Не следует использовать более чем 50 мкл осветленной лизата и стандарт Super-SILAC. Избегайте температур ниже 15 ° C, так что мочевина не кристаллизуется из.
- Для измерения концентрации белка на планшет-ридере, смешать стандартный бычьего сывороточного альбумина (БСА) разбавление раствора серии, лизат и соответствующий стандарт Супер-SILAC с коммерчески доступным белком Аssay в 96-луночного планшета и встряхивают в течение 1 мин, выдержать в течение указанного времени и измерения поглощения, как указано производителем.
5. Пример Разделение и белка Дайджест
- Жидкие опухоли.
- Отдельный 30 - 100 мкг общего белка на полосу на четыре с - 12% градиентном геле SDS-PAGE. Пятно белки кумасси синим O / N. Удалите излишки Кумасси пятно на двух последующих промывок водой.
- Разрежьте каждый переулок из геля и разделить егов 23 одинакового размера кусочки, независимо от форм окрашивания геля. Процесс ломтики гель отдельно, каждый в в 0,6 мл полипропиленовую пробирку.
- Промыть ломтики гель водой и метанол / вода (50:50, об / об), снизить с 10 мМ DTT путем инкубации в течение 30 мин при 56 ° С. АЛКИЛАТ ломтики гель с 55 мм иодацетамида (IAA) путем инкубации при температуре 60 мин при комнатной температуре в темноте.
- Между шагов обработки образцов, мыть ломтики ацетонитрилом в течение 15 мин и сушат в SpeedVac, чтобы удалить избыток растворителя и улучшить поглощение раствора реагента.
- Выполнение расщепления протеазой путем регидратации сушеные кусочки геля с минимальным количеством свиного раствора трипсина (12,5 нг / мкл в 0,025 М водного бикарбоната аммония) в течение 16 ч при 37 ° С.
- Добавить 10 мкл воды, чтобы гель срез и инкубируют в течение 15 мин при 37 ° С. Добавить 80 мкл ацетонитрила и инкубировать в течение 15 мин при 37 ° С. Центрифуга на 15800 мкг, в течение 1 мин. Соберите супернатант и хранить в отдельных 0,6 млтруба.
- Добавить 65 мкл 5% раствора муравьиной кислоты, вихря и инкубировать в течение 15 мин при 37 ° С. Добавить 65 мкл ацетонитрила и инкубировать в течение 15 мин при 37 ° С. Центрифуга на 15800 мкг в течение 1 мин. Соберите супернатант и добавить его в надосадочной жидкости из предыдущего шага. Упаривают в сочетании супернатант досуха в вакууме концентратора.
- Солидные опухоли.
- Чтобы удалить SDS от ткани лизата с помощью следующей протокол FASP, также описанной в ссылочном 15.
- После первого центрифугирования, описанного в пункте 5.2.3 добавить еще 200 мкл 8 М мочевины на фильтре, а в дальнейшем центрифуге при 14000 х г в течение 20 мин при 20 ° С. Откажитесь от фильтрата проточный.
- Добавить 100 мкл IAA и смешать в термомиксере на 600 оборотов в минуту в течение 1 мин. Инкубируйте фильтр в течение 20 мин при 20 ° С в темноте. Центрифуга фильтр при 14000 х г в течение 10 мин при 20 ° С.
- Добавить 100 мкл мочевины к фильтру и CENtrifuge при 14000 х г в течение 15 мин при 20 ° С. Повторите этот шаг еще раз.
- Добавить 100 мкл NH 4 HCO 3 с фильтром и центрифуге при 14000 х г в течение 10 мин при 20 ° С. Повторите этот шаг дважды.
- Добавить 40 мкл NH 4 HCO 3 + 1 мкл (= 0,4 мкг) трипсина и перемешивают в термосмеситель при 20 ° С в течение 600 мин, 1 мин. Инкубируйте фильтр O / N в мокром камере при 37 ° С. Передача фильтр в новые пробирки для сбора.
- Центрифуга фильтр при 14000 х г в течение 10 мин при 20 ° С. Добавить 50 мкл NH 4 HCO 3 и центрифуги фильтра на 14000 г в течение 10 мин при 20 ° С. Хранить Полученные пептиды при температуре -20 ° С до измерения масс-спектрометрического.
- Для измерения концентрации пептида, обойтись 50 мкл полученного проточные и серии соответствующих разведений аккуратный триптофана в 96-луночный планшет. Измерьте флуоресценции триптофана. Преобразование полученного концентрациитриптофан концентрации пептида, как 0,1 мкг триптофана соответствует 9 мкг белка.
Анализ 6. жидкостной хроматографии и масс-спектрометрического
- Растворяться пептидов в 30 мкл буфера для нанесения в течение 5 мин в ультразвуковой обработки ванны. Спин вниз в центрифуге при 15800 х г в течение 1 мин и пипетки прозрачного раствора в качестве МС автоматического пробоотборника флаконе.
- Подайте 5 мкл образца на анализ, используя пробоотборник системы nanoLC-MS / MS. Концентрат и обессоливания пептидов онлайн на обращенной фазой C18 предварительно колонка (0,15 мм ID х 20 мм с размером пор C18 материала 5 мкм) установлен в любой вентилируемой установке колонки или установке переключения предколоночной.
- Отдельные пептиды на обращенной фазой C18, микроколонке (0,075 мм внутренний диаметр х 200 мм себя упакованы с 3 мкм или меньше размер пор C18 материала в колонне микросфер себя пакета, такие как PicoFrit, с использованием 90 мин градиент 5> 35% ацетонитрила против . 0,1% водной муравьиной кислоты в 300 нл /минимум Передача элюент в гибридных квадрупольного / Orbitrap масс-спектрометре с помощью источника ионов микросфер.
- Анализ пептидов с использованием данных зависимая Способ top15 приобретения (MS м / диапазон г 350 -. 1600, цель 70,000 FWHM разрешение, АРУ цель 1 х 10 6, максимальное время заполнения 60 мс MS / MS масс-старте 100, целевой разрешение 17,500 FWHM, AGC Задача 2 х 10 5, максимальное время заполнения 60 мс MS / MS порог 3 х 10 4, включают в себя зарядовых состояний 2 -. 5, нормированная энергии столкновения NCE 25%, динамический исключение 15 сек).
Анализ 7. Данные
- Для анализа данных с помощью свободно доступных MaxQuant 16 программного обеспечения. Подробный протокол биоинформатики анализа данных описана в 16,17. Для анализа объединить файлы исходных данных из всех кусочков из SDS-PAGE полосу в одном эксперименте 17.
Результаты
Протеомный профилирование жидких и твердых опухолей у пациентов является перспективным подходом для открытия новых диагностических и прогностических биомаркеров. Тем не менее, процедура подготовки проб и анализ масс-спектрометрический являются сложными, в связи со сложностью образцов и необходимости точного количественного белка в больших наборов образцов. Экспериментальная процедура, описанная выше начинается с выделения клеток, представляющих интерес, либо с помощью флуоресцентной сортировкой активированных клеток или лазерной захвата микродиссекции.
С этой целью клетки от пациента, полученных клетках костного мозга или образцы крови окрашивали флуоресцентно-меченного антитела против определенных поверхностных маркеров, представляющих интерес до FACS-сортировки на основе и лизиса клеток.
Для обогащения клеточных подмножеств из срезов тканей, они сначала должны быть установлены на соответствующих слайдов, чтобы обеспечить лазера захвата микродиссекции. С этой целью установить тон срезов ткани на соответствующих мембранных слайдов и использовать лазерный захвата microdissector в соответствии с инструкциями изготовителя. Выбор направления и клетки, представляющие интерес требует соответствующих знаний о histomorphology здорового и опухолевой ткани. Извлеченные клетки затем переносили в пробирку для сбора соответствующей. Первоначальные эксперименты должны быть выполнены, чтобы определить количество ткани, необходимое для извлечения достаточного количества белка (не менее 10 мкг общего белка) для каждой новой интересующей ткани. Каждый микродиссекции образец обрабатывают 60 мкл буфера для лизиса ткани. Для эффективного лизиса клетки из образцов ткани должны быть ультразвуком в течение 3 мин и после добавления 15 мкл образцов SDS инкубируют в термосмеситель при 99 ° С в течение 1 часа, чтобы удалить формалином, вызванной белковые сшивки. Центрифугирование, наконец, облегчить сбор очищенной сотовой лизата.
Для каждого заболевания Iпроценты (жидкие и твердые опухоли) пригодны Супер-SILAC стандарт количественного должна быть установлена 14. Соответствующий стандарт должна составлять более 90% белков, выраженных в образцах, представляющих интерес. Для получения количественного стандарта, клеточные линии, относящиеся к типу рака интереса первым должен быть проанализирован с помощью масс-спектрометрии с целью определения их профили экспрессии белка и затем анализ главных компонентов должна быть выполнена. 4 - 6 различных клеточных линий с дифференциалом белковых паттернов экспрессии должны быть выбраны и SILAC меченных с «тяжелой» аргинин и лизин. Эффективность маркировки должны быть проверены nanoLC-MS / MS и должна быть больше, чем 98%. После этого клеточные линии должны быть лизируют так же, как и соответствующего пациента, полученных образца и, впоследствии, эквимолярные количества белка каждого образца и шип в стандарте SILAC должны быть смешаны. Важным шагом для точного белка количественного клинических образцов оF интерес точной смесь эквимолярных количествах белка в образцах пациентов, полученных и SILAC шип в стандарте количественного определения. Для определения концентрации белка в лизатах, полученных из отсортированных клеток, различные белок количественного анализов могут быть использованы. Для сотовых лизатов из ткани клеток, полученных анализ необходим, которые могут иметь дело с высокими концентрациями SDS и DTT в лизата. Концентрации белка в образцах пациентов, полученных и Спайк в стандарте SILAC следует измерять каждый раз перед объединением с соответствующими клиническими образцами в целях обеспечения правильную смесь, которая имеет решающее значение в принятии даже сотни образцов сопоставимы.
После того как смесь равных количеств шип в стандартных и лизатов, образцов, полученных из жидких опухолей, смешивают с LDS, нагреваемых в термомиксер при 72 ° С в течение 10 мин и затем подвергают 1D-странице и в геле переваривания разделены белки с трипсином. Образцы получены изткани освобождаются от SDS и расщепляли трипсином, используя подход FASP в порядке, установленном Вишневский и др. 15
Триптические пептиды, полученные может быть определена количественно путем измерения флуоресценции триптофана, которое представляет особый интерес для образцов ткани, как количество пептидов, выпущенных из фильтра отличается от 15% до 75% по сравнению с белковыми количествах первоначально загруженных на фильтр. Для этого мы измеряем флуоресценции триптофана. Сравнение с соответствующим триптофана серии разведений позволяет для количественного определения пептидов, как 1,1 мкг триптофана соответствует примерно 100 мкг пептида. При необходимости образцы, полученные от переработки, особенно FASP может быть предварительно очищены от коммерческих или самодельных подсказок стадии упакованных с обращенной фазой-С18 (ОФ-С18) материала перед загрузкой в систему nanoLC / MS / MS 18.
Масс-спектрометрический анализ выполняется на высокой-Разрешение, высокая чувствительность системы масс-спектрометрии. Вкратце, образцы пептидов обессоливают и preconcentrated на предколонкой RP-C18, и разделены на аналитической колонке RP-C18 соедин ют непосредственно в масс-спектрометр. Для достижения достаточной глубины анализа, мы используем либо сочетание SDS-PAGE белка фракционирование с 40 мин RP-C18 градиентов (для жидких опухолей) или отдельных инъекций с длинными 2 - 3 ч RP-C18 градиентов (для твердых опухолей, обработанных FASP ). MS спектры приобретаются с разрешением 70000 FWHM или лучше, позволяют точно количественное SILAC пар путем интеграции хроматографического пика профилей. Для идентификации белков, способ сбора данных TOP15-зависимой используется для генерации большого количества пептида MS / MS спектры пептидов и идентификации белков.
Из полученных исходных данных, определение белка и количественное достигается за счет поиска в базе данных с MaxQuant программного обеспечения против UniProt Knowledgebasе человека базу данных Полный Proteome последовательность 17. В MaxQuant программного обеспечения (текущая версия 1.5.0.25) пептиды идентифицированы из полученного MS / MS спектры с пептидным фрагментом сопоставления против спектров, полученных в кремнии из базы данных белковых последовательностей. В то же время, предшественником иона изотопных профилей извлекаются вокруг их хроматографических времен удерживания, и их интегрированные площади пиков используются для относительного количественного света: тяжелых пар пептидных генерируемых SILAC маркировки. Пептидные идентичности и относительные интенсивности, то назначаются соответствующими свойствами белка. Персей программное обеспечение (текущая версия 1.5.0.15) используется для выполнения далее вниз по течению статистическую оценку результатов обработки MaxQuant, в том числе от образца к образцу сравнения, СПС и иерархической кластеризации.
Использование экспериментальной установки описано мы определили и количественно до 8000 белков от всего 30 мкг общего белкаполучают из жидких опухолей.
До 2500 белки из образцов твердых опухолей можно определить и количественно дробовика протеомики подхода с LC градиентом только два часа, что позволяет анализ сотен клинических образцов в течение относительно короткого времени.

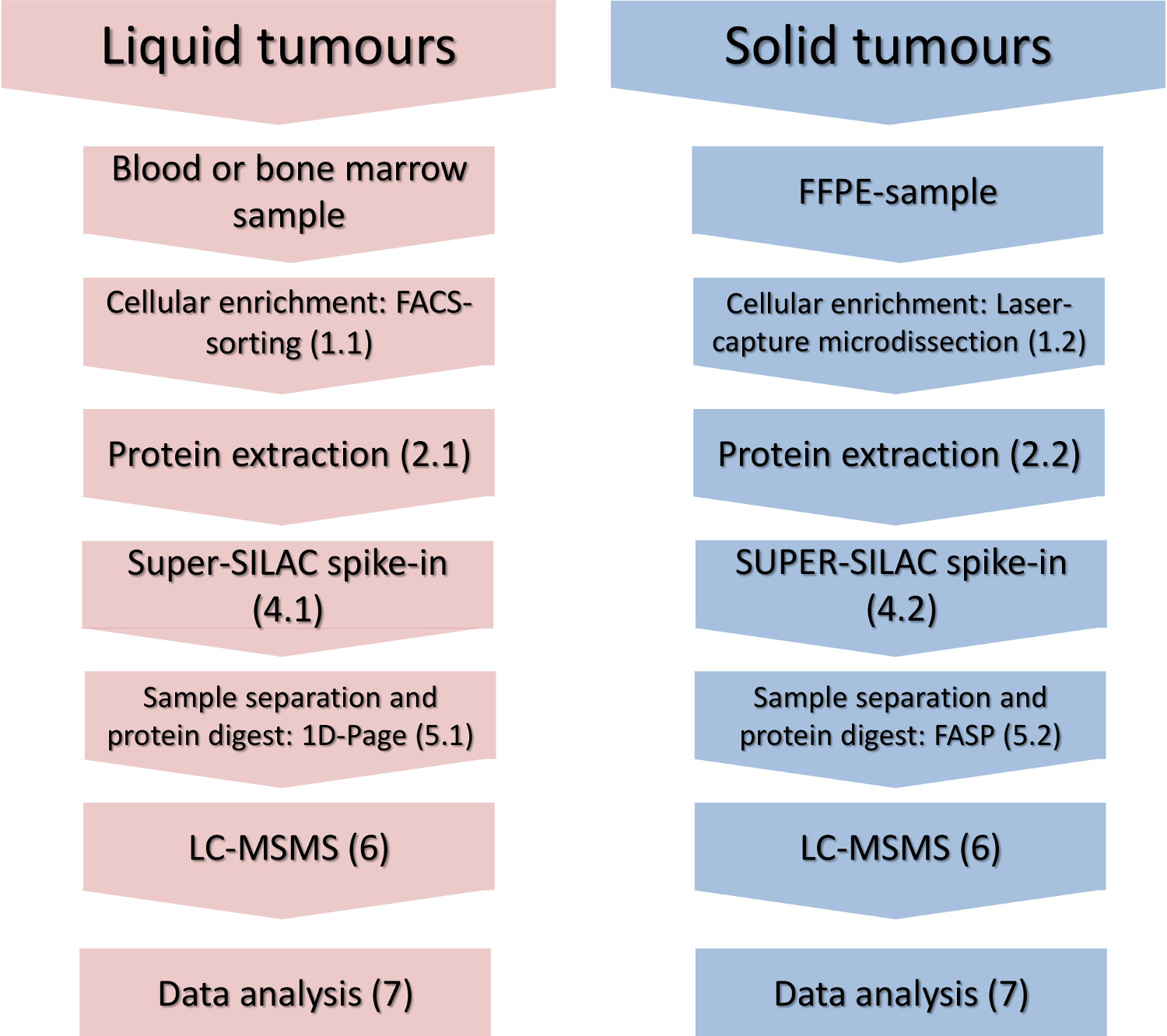
Рисунок 1. Экспериментальная процесса. Основные этапы обогащения сотовой подмножества, изоляции белка, шип-ин стандарта количественного и масс-спектрометрического анализа приведены для жидких и твердых опухолей. Пожалуйста, нажмите здесь, чтобы увидеть увеличенную версию этой цифры.
{kind=link}

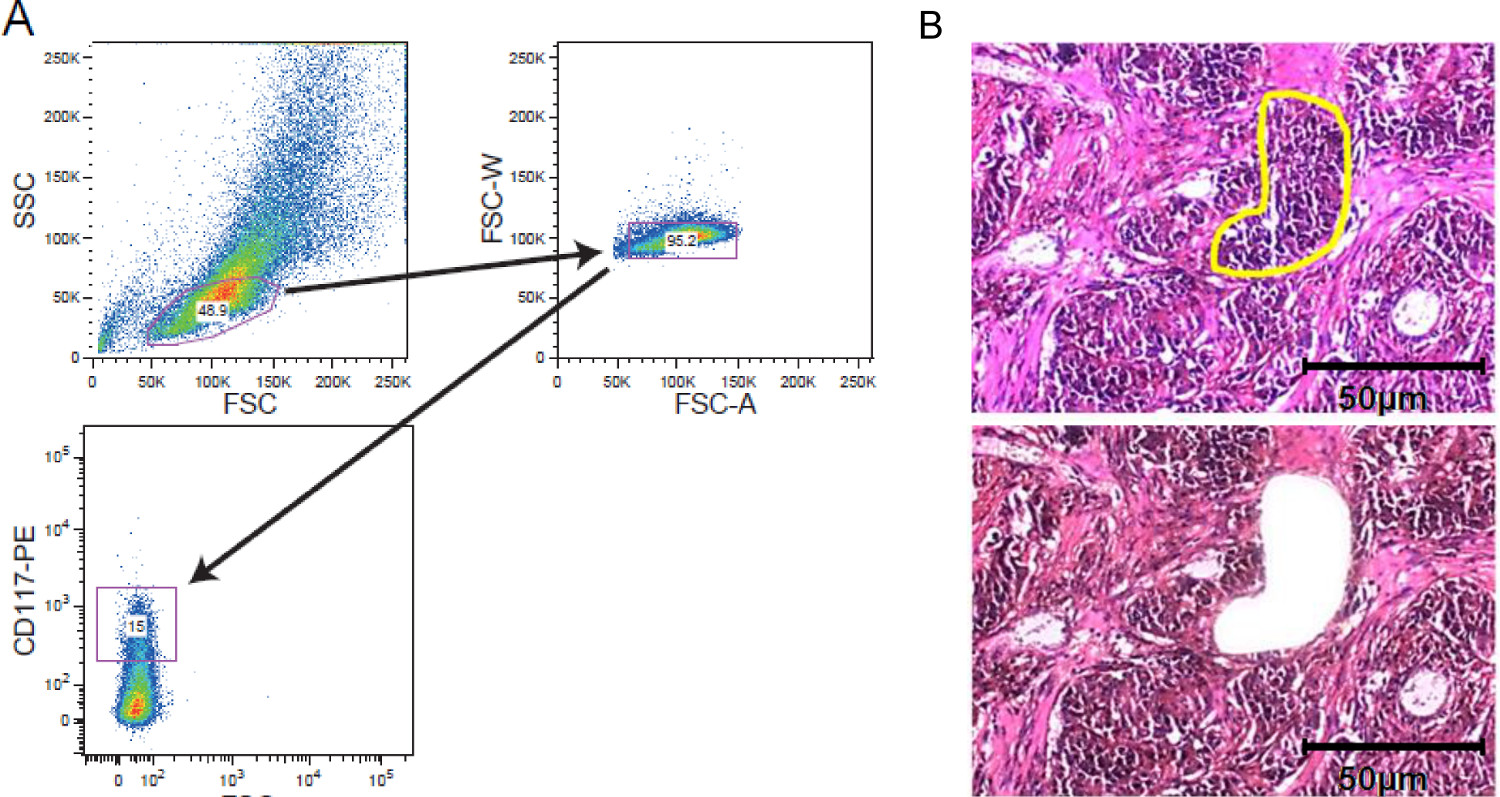
Рисунок 2. Сотовые стратегов обогащениеES. FACS сортировки и LCM. () Примерный стратегия стробирования с изображением стробирования на CD117 окрашенных лейкозных клеточной популяции, приобретенных клеточного сортера. (Б) Лазерная захватывающая микродиссекция твердого опухолевой ткани. Срезов ткани 5 - 10 толщиной мкм были установлены на пленке, покрытой мембраны слайдов перед окрашиванием. Регион интересов был выбран вручную для лазерной захвата микродиссекции. Показаны секции до микродиссекции с интересующей нас области помечаются желтым цветом и после микродиссекции. Масштабная линейка из 50 мкм входит в рисунке. Пожалуйста, нажмите здесь, чтобы увидеть увеличенную версию этой цифры.
{kind=link}

Рисунок 3. Кластерный анализ протеома раковых клеток человека. Неконтролируемая кластерный анализ PRotein профили экспрессии жидких и твердых опухолей проводили с использованием вычислительной платформы Персей. AC - Аденокарцинома, SCC - плоскоклеточный рак, SCC-Met - плоскоклеточный рак метастазы рака головы и шеи, R - техническая репликации.
Обсуждение
Масс-спектрометрическое профилирование пациентов, полученных протеомов раковых клеток необходим для открытия новых диагностических / прогностических биомаркеров, а также, чтобы дать более глубокое понимание рака клеточной биологии, который в свою очередь может привести к выявлению новых потенциальных мишеней для лекарственных средств. Тем не менее, такие масс-спектрометрии анализ весьма сложной, в частности потому, что различные преаналитического вопросы должны быть решены, если кто-то, чтобы получить надежные и биологически соответствующие результаты.
Экспериментальный рабочий описано здесь, дает возможность количественного протеомики характеристик протеомов, полученных из клеточных субпопуляций жидких и твердых опухолей. Начальное обогащение опухолевых клеток либо FACS на основе клеток-sortingor микродиссекции необходима, чтобы избежать загрязнения клетками микроокружения опухоли. Кроме того, эти методы позволяют выделить клеточные субпопуляции интерес. Недавние исследования клеточно-биологического есть демонstrated, что некоторые клеточные субпопуляции есть опухоль начала свойствами и, таким образом, весьма актуальны для патогенезе рака 19,20. Масс-спектрометрии, как стало более чувствительным в последние годы, количественные анализы протеомические являются выполнимыми для малых количеств белка, который может быть получен из нескольких тысяч клеток, что делает возможным, чтобы сосредоточить внимание на функционально соответствующие клеточных популяций.
Настройка, представленная здесь может быть использован для идентификации и проверки новых диагностических биомаркеров в образцах FFPE. Таким образом, он обещает BEA полезный инструмент для улучшения клинической диагностики, как на сегодняшний день по-прежнему отсутствие молекулярных биомаркеров в необходимом количестве и качестве для многих типов рака. Важные примеры сложных дифференциальных диагнозов, для которого биомаркеров отсутствуют, являются дискриминации между первичным раком легких с метастазами в легких, intrapancreatic холангиоцеллюлярный карциномы и аденокарциномы поджелудочной, а также отличаетсяПроизводная доброкачественной нейрофибромы от высокой злокачественных периферийных опухолей нервных оболочки. Более того, мы и другие показали, что количественное выяснение протеомическим подписей могут быть полезны при изучении биологии клеток рака в целом, так и для выявления прогностических биомаркеров терапевтического ответа у больных раком 21.
В настоящее время два недостатки метода, представленные здесь, требование для широкого ручной обработки образца и спрос на nanoLC-MS / MS время приобретения. В то время как первые могут быть решены путем перемещения подготовки пробы, например, форматирует 96-а и используя робота обработку, последний потребует изменения в средствах массовой стратегию приобретения спектрометрический. После того, как подмножества белков-мишеней были определены, которые могут быть связаны с, например, классификации опухолей, мы предусматриваем конструкцию целевых методов масс-спектрометрии, которые обеспечивают количественные показания для этих подмножеств с сильно пониженным усилием отрыва, иПоэтому с соответствующим сокращением времени приобретения. Если требуемое время приобретения требуется может быть сокращен с 24 - 36 ч (жидкие опухоли) или 3 ч (солидные опухоли), чтобы например, 1 час использование целевых масс-спектрометрии и простой одномерный разделение пептидов, то в результате усиления пропускной могут быть использованы, чтобы значительно увеличить количество биологических и технических повторов рассмотренных с соответствующими улучшения в значимости результатов количественного определения. Целевые подходы масс-спектрометрии уже было продемонстрировано, что подходящий инструмент для проверки раком белок, ассоциированный с биомаркеров-кандидатов 22, и были разработаны до точки, где они показывают обещание для проверки или даже в качестве потенциального инструмента для рутинного клинического использования 23 , 24.
Раскрытие информации
Авторы не имеют никакого конфликта интересов или других вопросов раскрывать.
Благодарности
The authors thank Uwe Plessmann, Monika Raabe und Silvia Münch for technical support.
Материалы
| Name | Company | Catalog Number | Comments |
| 660 nm Kit | Thermo scientific | 22662 | |
| Cell culture medium depleted of arginine and lysine | Thermo Scientific | 88421 | |
| Coomassie Brilliant Blue R-250 staining solution | Bio Rad | 161-0436 | |
| Dialyzed fetal calf serum (FCS) | PAA | A15-107 | |
| Diffuser caps for microdissection | MMI | 50202 | |
| FACS-sorter | BD | FACSAria III | |
| Ionic Detergent Compatibility Reagent | Thermo scientific | 22663 | |
| Laser-capture microdissector | MMI | cell cut plus | |
| LDS buffer | Life Technologies | NP0009 | |
| Membrane slides for microdissection | MMI | 50103 | |
| Microcon YM-30 | Millipore | MRCF0R030 | |
| NuPAGE 4-12% Bis-Tris Mini Gels | Life Technologies | NP0335PK2 | |
| Picofrit Self-Pack Columns | New Objective | PF360-75-15-N-5 | Mass Spectrometry Column/Emitter |
| Reducing agent | Life Technologies | NP0007 | |
| Reprosil-Pur LC/MS/MS Column stationary phase | Dr. Maisch | 120 C18-AQ, 3 µm | |
| Reprosil-Pur LC/MS/MS Precolumn stationary phase | Dr. Maisch | 120 C18-AQ, 5 µm | |
| SILAC-labeled arginine | Eurisotop | CLM-2265-H-0.1 | |
| SILAC-labeled lysine | Eurisotop | DLM-2640-0.25 | |
| Trypsin, NB Sequencing Grade | Serva | 3728301 | for in-gel digests |
| Trypsin, Sequencing Grade | Promega | V5111 | for in-solution digests |
| Buffer and solutions | |||
| Cell lysis buffer: 150 mM NaCl, 50 mM Tris/HCl pH 7.8, 5 mM NaF, 0.5% NP40, 0.1% laurylmaltoside, Roche complete protease inhibitor, 1 mM Na3VO4 | |||
| Tissue lysis buffer: 100 mM Tris/HCl pH 7.8, 0.1 M DTT | |||
| Urea: 8 M urea in 0.1 M Tris-HCl, pH 8.5 | for FASP-protocoll | ||
| IAA: 0.05 M iodoacetamide, 8 M urea, 0.1 M Tris-HCl, pH 8.5 | for FASP-protocoll | ||
| 0.05 M NH4HCO3 | |||
| 10 mM dithiothreitol (DTT) in 0.1 M ammonium bicarbonate | for in-gel digest | ||
| 55 mM iodoacetamide (IAA) in 0.1 mM ammonium bicarbonate | for in-gel digest | ||
| 5% aqueous formic acid. |
Ссылки
- Walther, T. C., Mann, M. Mass spectrometry-based proteomics in cell biology. J Cell Biol. 190 (4), 491-500 (2010).
- Lenz, C., Urlaub, H. Separation methodology to improve proteome coverage depth. Expert Rev Proteomics. 11 (4), 409-414 (2014).
- Olsen, J. V., Mann, M. Status of large-scale analysis of post-translational modifications by mass spectrometry. Mol Cell Proteomics. 12 (12), 3444-3452 (2013).
- Ong, S. E., et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 1 (5), 376-386 (2002).
- Jimenez, C. R., Verheul, H. M. Mass spectrometry-based proteomics: from cancer biology to protein biomarkers, drug targets, and clinical applications. Am Soc Clin Oncol Educ Book. , e504-e510 (2014).
- Tang, D. G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 22 (3), 457-472 (2012).
- Evans, C., et al. An insight into iTRAQ: where do we stand now. Anal Bioanal Chem. 404 (4), 1011-1027 (2012).
- Ostasiewicz, P., Zielinska, D. F., Mann, M., Wisniewski, J. R. Proteome, phosphoproteome, and N-glycoproteome are quantitatively preserved in formalin-fixed paraffin-embedded tissue and analyzable by high-resolution mass spectrometry. J Proteome Res. 9 (7), 3688-3700 (2010).
- Malmström, J., Picotti, P., Aebersold, R. Perspectives of targeted mass spectrometry for protein biomarker verification. Curr Opin Chem Biol. 13 (5-6), 518-525 (2009).
- Gillet, L. C., et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 11 (6), (2012).
- Basu, S., Campbell, H. M., Dittel, B. N., Ray, A. Purification of specific cell population by fluorescence activated cell sorting (FACS). J Vis Exp. 41, (2010).
- Edwards, R. A. Laser capture microdissection of mammalian tissue. J Vis Exp. 8, 309 (2007).
- Liu, N. Q., et al. Proteomics pipeline for biomarker discovery of laser capture microdissected breast cancer tissue. J Mammary Gland Biol Neoplasia. 17 (2), 155-164 (2012).
- Geiger, T., et al. Use of stable isotope labeling by amino acids in cell culture as a spike-in standard in quantitative proteomics. Nat Protoc. 6 (2), 147-157 (2011).
- Wisniewski, J. R. Proteomic sample preparation from formalin fixed and paraffin embedded tissue. J Vis Exp. (79), (2013).
- Cox, J., Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 26 (12), 1367-1372 (2008).
- Cox, J., et al. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat Protoc. 4 (5), 698-705 (2009).
- Rappsilber, J., Mann, M., Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2 (8), 1896-1906 (2007).
- Sarvi, S., et al. CD133+ cancer stem-like cells in small cell lung cancer are highly tumorigenic and chemoresistant but sensitive to a novel neuropeptide antagonist. Cancer Res. 74 (5), 1554-1565 (2014).
- Shlush, L. I., et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 506 (7488), 328-333 (2014).
- Schaab, C., et al. Global phosphoproteome analysis of human bone marrow reveals predictive phosphorylation markers for the treatment of acute myeloid leukemia with quizartinib. Leukemia. 28 (3), 716-719 (2014).
- Hüttenhain, R., et al. Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Sci Transl Med. 4 (142), 142ra94 (2012).
- Burgess, M. W., et al. Simplified and efficient quantification of low-abundance proteins at very high multiplex via targeted mass spectrometry. Mol Cell Proteomics. 13 (4), 1137-1149 (2014).
- Boja, E. S., et al. Analytical Validation Considerations of Multiplex Mass Spectrometry-based Proteomic Platforms for Measuring Protein Biomarkers. J Proteome Res. , (2014).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеThis article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены