
Method Article
Mass quantitativa spettrometrica Profiling di proteomi cancro-cellulari derivate da liquido e tumori solidi
In questo articolo
Riepilogo
In-depth analyses of cancer cell proteomes facilitate identification of novel drug targets and diagnostic biomarkers. We describe an experimental workflow for quantitative analysis of (phospho-)proteomes in cancer cell subpopulations derived from liquid and solid tumors. This is achieved by combining cellular enrichment strategies with quantitative Super-SILAC-based mass spectrometry.
Abstract
In analisi approfondite dei proteomi di cellule di cancro sono necessari per chiarire meccanismi patogenetici oncogeni, nonché di identificare potenziali bersagli farmacologici e biomarcatori diagnostici. Tuttavia, i metodi per la caratterizzazione proteomica quantitativa dei tumori di derivazione pazienti e in particolare le loro sottopopolazioni cellulari sono in gran parte mancano. Qui si descrive un set-up sperimentale che permette l'analisi quantitativa dei proteomi di sottopopolazioni di cellule tumorali derivate da tumori liquidi o solidi. Ciò si ottiene combinando strategie di arricchimento cellulari con spettrometria di massa quantitativa basata-Super-SILAC seguita da analisi bioinformatica dei dati. Per arricchire sottoinsiemi cellulari specifici, tumori liquidi vengono prima immunophenotyped mediante citometria di flusso seguita da FACS-cernita; per i tumori solidi, laser-capture microdissezione è usato per purificare sottopopolazioni cellulari specifici. In una seconda fase, le proteine vengono estratti dalle cellule purificate e successivamente agganciato un tumore-specifica,SILAC marcato standard di spike-in che consente di proteine quantificazione. La miscela proteica risultante viene sottoposta a uno elettroforesi su gel o Filtro Aided Preparazione del campione (FASP) seguita da digestione trittico. Infine, peptidi triptici vengono analizzati utilizzando un ibrido spettrometro di massa-Orbitrap, ei dati ottenuti sono trattati con suite di software bioinformatici compreso MaxQuant. Per mezzo del flusso di lavoro qui presentato, fino a 8.000 proteine possono essere identificati e quantificati in campioni derivati da pazienti, e le conseguenti profili di espressione proteica possono essere paragonati tra i pazienti per individuare le firme proteomica diagnostici o potenziali bersagli farmacologici.
Introduzione
Proteomica basata spettrometria di massa è emerso ed è ora una disciplina ampiamente utilizzato in biologia cellulare e della ricerca biomedica traslazionale. I progressi tecnici nel campo hanno permesso di studiare i processi cellulari complessi in linee cellulari e modelli animali, come migliaia di proteine possono essere identificati in un singolo esperimento spettrometria di massa 1,2. Sono stati compiuti progressi simile per l'analisi di numerose modifiche post-come la fosforilazione o ubiquitination, anche se questo richiede di solito i flussi di lavoro su misura per l'arricchimento e l'analisi dei dati per ogni tipo di modifica 2,3. Inoltre, il coinvolgimento delle strategie in materia di etichettatura chimiche e metaboliche, tra cui SILAC, consente accurata quantificazione relativa di proteine e PTM, rendendo questo metodo particolarmente interessante per la scoperta di processi cellulari fondamentali, biomarcatori diagnostici e potenziali bersagli farmacologici nei tumori umani 4.
Tuttavia,numerose sfide devono essere superate in materia di analisi proteomica delle principali tumori umani 5. Prima di tutto, i campioni tumorali umane spesso mostrano un elevato grado di eterogeneità cellulare che è dovuto alla presenza di vari tipi di cellule appartenenti al microambiente tumorale, comprese le cellule immunitarie e fibroblasti. In secondo luogo, l'evoluzione clonale porta alla diversità genetica all'interno dei tumori stessi, con conseguente l'esistenza di più sottopopolazioni cellulari con diverse proprietà funzionali. Secondo l'attuale concetto di poche cellule staminali tumorali di essere le forze trainanti dello sviluppo e progressione del cancro, l'analisi proteomica di tali (funzionalmente molto rilevanti) sottopopolazioni cellulari dovrebbe essere di grande importanza per una migliore comprensione dei meccanismi oncogenici rilevanti per l'applicazione clinica 6. In terzo luogo, i profili di espressione proteica quantitativi di grandi serie di campioni sono spesso necessarie per l'identificazione di robusta Biomar clinicakers; questo non può essere realizzato mediante l'etichettatura chimica quali iTRAQ 7, mentre le strategie etichettatura metabolici - basandosi, come fanno, sulla proliferazione cellulare 4 - sono altrettanto inapplicabile. In quarto luogo, la maggior parte dei campioni di tumore solidi disponibili sono fissati in formalina, che complica massa spettrometria proteoma a causa della formazione di legami crociati proteici 8. Infine, la maggior parte dei flussi di lavoro esistenti proteomica richiedono notevoli quantità di campione di trasformazione e di acquisizione dei dati, rendendo l'analisi del numero di campioni rilevanti per la ricerca clinica difficile, e chiedendo nuovo flusso di lavoro paradigmi 9,10.
Per affrontare questi ostacoli abbiamo sviluppato un apparato sperimentale che combina sia cella di flusso-citometria ordinamento 11 o laser-capture microdissezione 12,13 per l'arricchimento cellulare con una strategia Super-SILAC 14 per introdurre una norma interna globale per una completa analisi di spettrometria di massa. Utilizzando il metodo qui descritto, è possibile quantificare fino a 8.000 proteine nei campioni tumorali umane derivate da singoli tumori liquidi o solidi.
Protocollo
Tutti gli esperimenti su campioni di tessuto o di sangue umano devono essere approvati da un comitato etico e condotti secondo linee guida fornite nella votazione etica.
1. Cellular Enrichment
- Tumori liquidi / fluorescenza-attivato ordinamento cellulare.
- Isolare cellule mononucleari di midollo osseo o campione di sangue effettuando eritrociti lisi con cloruro di ammonio o Ficoll densità gradiente di centrifugazione.
- Per eritrociti lisi, unire 1 ml di campione con 4 ml di soluzione di cloruro di ammonio. Incubare la soluzione su ghiaccio per 5 - 10 min. Aggiungere 40 ml di tampone fosfato salino (PBS) di siero fetale bovino + 2% (FCS) e centrifugare per 5 min a 400 xg e 4 ° C.
- Per Ficoll centrifugazione in gradiente, strato di 10 ml di campione di 15 ml di Ficoll-Hypaque per formare 2 strati distinti (attenzione a non disturbare gli strati). Centrifugare a 400 xg per 30 min a temperatura ambiente (15 - 25 ° C) accuratamente e raccogliere il mononuclearecellule che raccolgono all'interfase.
- Spin giù le cellule e portarli in PBS + 2% FCS. Centrifugare a 400 xg per 5 minuti e prendere in 1 ml di PBS + 2% FCS. Prendere una frazione come controllo negativo e un'aliquota di ciascun colorante fluorescente utilizzato nella macchia finale.
- Incubare con anticorpi fluorescenza marcati utili per isolare la popolazione di cellule leucemiche in diluizione secondo le istruzioni del produttore su ghiaccio per 30 min. Macchiare anche le aliquote utilizzate per la compensazione con ogni anticorpo individualmente. Lavare due volte con PBS + 2% FCS.
NOTA: Gli anticorpi utilizzati erano topo anti-umana contro CD117-PE (Clone 95C3), CD34-FITC (Clone 8G12) e CD33-PE (Clone P67.6). - Celle filtranti attraverso 35 micron filtro cellule, preferibilmente utilizzando tubi con filtro cella integrati in cap.
- Aggiungi macchia vitalità cellulare (ad esempio, 7-AAD). Ordina cellule utilizzando un cell sorter adeguata 11 (Figura 2). Raccogliere le cellule a Isc Modified Dulbecco del ove Medium (IMDM) contenente il 10% FCS.
- Isolare cellule mononucleari di midollo osseo o campione di sangue effettuando eritrociti lisi con cloruro di ammonio o Ficoll densità gradiente di centrifugazione.
- Tumori solidi / laser-capture di microdissezione.
- Per gli esperimenti descritti, sono stati utilizzati campioni FFPE provenienti da campioni di cancro ai polmoni. Per questo tessuto tumore polmonare scopo è stato immediatamente fissato in 4% formalina tamponata dopo la resezione chirurgica e pezzi di circa 3 x 2 x 1 cm dove inclusi in paraffina per ulteriori analisi.
- Tagliare sezioni di 5-10 micron di spessore dal incluso in paraffina (FFPE) campione fissato in formalina con un microtomo. Sezioni Montare su vetrini membrana film coperto e asciutto, a 37 ° C per 1 ora. Deparaffinare e reidratare le sezioni montati mediante incubazione successive in xilene, etanolo assoluto, il 70% e di acqua, ciascuno per 1 min.
- Colorare le sezioni con Ematossilina per 20 secondi e poi sciacquare con acqua di rubinetto. Raccogliere la popolazione di cellule utilizzando un sistema microdissezione laser cattura (vedi anche 12).
- Tumori liquidi.
- Centrifugare la sospensione cellulare dal punto 1.1.5 a 400 xg, 4 ° C per 4 minuti e scartare il surnatante. Lavare due volte con 500 microlitri di PBS freddo e centrifugazione per 5 min a 400 xg e 4 ° C
- Aggiungere 40 microlitri tampone di lisi per 10 6 cellule e incubare per 15 min in ghiaccio (10 5 cellule (circa. 10 mg di proteine totali) dovrebbe essere il minimo numero di cellule). Centrifugare il lisato a 14000 xg, a 4 ° C per 10 min e trasferire il surnatante (lisato cellulare cancellato) in un tubo di reazione. Eliminare il pellet.
- Tumori solidi.
- Aggiungere 60 ml di tampone di lisi tessuto al tessuto microdissezione e incubare per 15 min in ghiaccio, raccogliere il fluido dal breve centrifugazione e trasferire la sospensione in un tubo di reazione. Sonicare il lisato in ghiaccio per 3 min.
- Aggiungere 15 ml di 20% sodio dodecil solfato (SDS) a raggiungere un finale SDS concentration del 4%. Incubare il tessuto microdissezione a 99 ° C su una piastra riscaldante per 1 ora e agitato a 600 rpm. Centrifugare il lisato a 16.000 xg, 18 ° C per 10 min e trasferire il surnatante in una nuova provetta.
3. Istituzione di un SILAC Spike-in Quantificazione standard (Super-SILAC Standard)
NOTA: Lo standard quantificazione è costituito da una miscela di proteine SILAC marcata derivata 4-6 cellule linee corrispondenti al tipo di tumore di interesse. Per ottenere la massima sovrapposizione tra il proteoma di riferimento SILAC marcato e il proteoma del tumore di derivazione, una analisi delle componenti principali deve essere eseguita prima di linee cellulari vengono selezionati per lo standard quantificazione 14.
- Analisi delle componenti principali (PCA).
- Per determinare i pattern di espressione proteica di linee cellulari, coltivare una decina di diverse linee cellulari in mezzi appropriati di coltura cellulare. Lyse le cellule come descrittoal punto 2.1. Preparare lisati cellulari per spettrometria di massa come descritto al punto 6.1.
- Analizzare il pattern di espressione proteica di ciascuna linea cellulare su una ad alta risoluzione, cromatografia liquida accoppiata spettrometro di massa (nanoLC-MS / MS).
- Analizzare i dati grezzi risultanti utilizzando un software MaxQuant gratuito ed effettuare un'analisi dei principali componenti utilizzando il software associato, come Perseo 16,17.
- Selezionare 4 - 6 linee di cellule che mostrano la più grande diversità tra i loro profili di espressione proteica e le usano per generare il livello picco-in Super-SILAC.
- SILAC-etichettatura e controllo etichetta.
- Coltivare le linee cellulari selezionate per almeno cinque cicli cellulari in appropriato terreno di coltura cellulare SILAC, in cui arginina e lisina sono etichettati con isotopi stabili di carbonio e azoto (media SILAC) 4.
- Lyse le cellule come descritto al punto 3.1 e preparare lisati cellulari per spettrometria di massa come descritto al punto 6.1.Misurare l'efficienza incorporazione di etichettatura SILAC da nanoLC-MS / MS. Analizzare i risultanti dati grezzi MS con MaxQuant e determinare l'efficienza di SILAC etichettatura. Ciò si ottiene contando il numero di peptidi identificati nella loro etichettati (pesante) e loro forme endogene (luce), e calcolando il rapporto (pesante) / (luce pesanti +). Efficienza etichettatura deve superare il 98%.
- Combinazione di proteomi SILAC e validazione.
- Coltivare e ampliare le linee di cellule selezionate in base alle istruzioni produce circa il corretto medio SILAC. Lisare le cellule come descritto al punto 2.1 per tumori liquidi o come descritto al punto 3.2 per tumori solidi e determinare la concentrazione di proteine per ogni lisato cellulare.
- Mix di proteine equimolari quantità di ciascuna linea cellulare e dividere il composto in aliquota. Snap-congelare le aliquote e conservare a -80 ° C fino a quando la misura. Per un esperimento è necessario 20 - 50 mg di standard di Super-SILAC. Fare attenzione a preparare la standard in eccesso come cambiare la norma all'interno di una serie di esperimenti deve essere evitato.
4. Misura di proteine Concentrazione e Spike-in
- Tumori liquidi.
- Utilizzando proteine test quantificazione determinare le concentrazioni di proteine dei rispettivi lisati cellulari e lo standard Super-SILAC.
- Mescolare pari quantità di lisato cellulare eliminato e lo standard Super-SILAC e successivamente aggiungere litio dodecil solfato (LDS) tampone (25% del volume del campione) e agente riducente (10% del volume del campione).
- Riscaldare la soluzione risultante in un blocco di riscaldamento a 72 ° C per 10 min. Facoltativamente, memorizzare le proteine denaturate risultanti a -80 ° C.
- Tumori solidi.
- Per la misurazione della concentrazione di proteine su un lettore di piastre, mescolare uno standard di albumina sierica bovina (BSA) Serie soluzione di diluizione, il lisato e lo standard Super-SILAC con una proteina disponibile in commercio unssay in una piastra a 96 pozzetti e agitare per 1 min, incubare per tempo indicato e misurare l'assorbanza come indicato dal produttore.
NOTA: L'elevata concentrazione di SDS e ditiotreitolo (DTT) nel lisato è un problema per la maggior parte dei saggi per la determinazione della concentrazione proteica. - Mescolare pari quantità di lisato chiarificato e lo standard Super-SILAC con 200 microlitri di urea nell'unità filtro e centrifugare a 14.000 xg per 30 min a 20 ° C. Non utilizzare più di 50 ml di lisato chiarificato e lo standard Super-SILAC. Evitare temperature inferiori ai 15 ° C, in modo che l'urea non cristallizza out.
- Per la misurazione della concentrazione di proteine su un lettore di piastre, mescolare uno standard di albumina sierica bovina (BSA) Serie soluzione di diluizione, il lisato e lo standard Super-SILAC con una proteina disponibile in commercio unssay in una piastra a 96 pozzetti e agitare per 1 min, incubare per tempo indicato e misurare l'assorbanza come indicato dal produttore.
5. La separazione del campione e proteine Digest
- Tumori liquidi.
- Indipendente 30-100 mg di proteine totali per corsia su un 4 - gradiente gel SDS-PAGE 12%. Proteine Macchia con Coomassie Blu O / N. Rimuovere l'eccesso macchia Coomassie da due successivi lavaggi con acqua.
- Tagliare ogni corsia dal gel e dividerloin 23 fette di dimensioni uguali a prescindere dal modello di gel di colorazione. Elaborare le fette di gel separatamente, ciascuno in un in 0,6 ml polipropilene flaconcino.
- Lavare le fette di gel con acqua e metanolo / acqua (50:50, v / v), ridurre con DTT 10 mM incubando per 30 minuti a 56 ° C. Alchilata le fette di gel con 55 mm iodoacetamide (IAA) incubando a 60 min a RT in scuro.
- Tra passaggi-campione manipolazione, lavare le fette con acetonitrile per 15 minuti e secco in un SpeedVac per rimuovere l'eccesso di solvente e di migliorare l'assorbimento di soluzione reagente.
- Eseguire proteasi clivaggio da reidratazione fette gel essiccato con la minima quantità di soluzione di tripsina porcina (12,5 ng / ml in 0,025 M di bicarbonato di ammonio acquoso) per 16 ore a 37 ° C.
- Aggiungere acqua 10 ml di fetta gel e incubare per 15 minuti a 37 ° C. Aggiungere 80 microlitri acetonitrile e incubare per 15 minuti a 37 ° C. Centrifugare a 15.800 xg, per 1 min. Raccogliere il surnatante e conservare in separata 0,6 mltube.
- Aggiungere 65 ml di 5% soluzione di acido formico, vortex e incubare per 15 min a 37 ° C. Aggiungere 65 microlitri acetonitrile e incubare per 15 minuti a 37 ° C. Centrifugare a 15.800 g per 1 min. Raccogliere il surnatante e aggiungere al supernatante dal passaggio precedente. Evaporare il surnatante combinato a secchezza in un concentratore a vuoto.
- Tumori solidi.
- Per rimuovere SDS dal lisato tissutale utilizzare il seguente protocollo FASP, anche descritto come riferimento 15.
- Dopo la prima centrifugazione descritto al punto 5.2.3 aggiungere altri 200 ml di 8 M urea sul filtro, e in seguito centrifugare a 14.000 xg per 20 min a 20 ° C. Gettare il filtrato a flusso continuo.
- Aggiungere 100 microlitri IAA e mescolare in un Thermomixer a 600 rpm per 1 min. Incubare il filtro per 20 min a 20 ° C al buio. Centrifugare il filtro a 14000 xg per 10 min a 20 ° C.
- Aggiungere 100 ml di urea al filtro e centrifuge a 14.000 xg per 15 min a 20 ° C. Ripetere questo passaggio ancora una volta.
- Aggiungere 100 ml di NH 4 HCO 3 al filtro e centrifugare a 14.000 xg per 10 min a 20 ° C. Ripetere questa operazione due volte.
- Aggiungere 40 ml di NH 4 HCO 3 + 1 ml (= 0,4 mg) tripsina e mescolare in thermomixer a 20 ° C per 600 rpm, 1 min. Incubare il filtro O / N in una camera umida a 37 ° C. Trasferire il filtro di nuovi tubi di raccolta.
- Centrifugare il filtro a 14000 xg per 10 min a 20 ° C. Aggiungere 50 ml di NH 4 HCO 3 e centrifugare il filtro a 14.000 g per 10 min a 20 ° C. Conservare i peptidi risultanti a -20 ° C fino a quando la misura spettrometria di massa.
- Per la misurazione della concentrazione del peptide, erogare 50 microlitri della risultante flusso continuo e una serie di diluizioni appropriate di triptofano ordinata in una piastra a 96 pozzetti. Misurare la fluorescenza del triptofano. Convertire la concentrazione risultantetriptofano a concentrazione peptide, come 0,1 mg di triptofano corrisponde a 9 mg di proteine.
Analisi 6. cromatografia liquida e la Messa spettrometrica
- Riprendere peptidi in tampone di caricamento 30 microlitri per 5 minuti in un bagno di sonicazione. Spin giù in una centrifuga a 15.800 xg per 1 min e pipetta la soluzione limpida in un flaconcino di MS campionatore automatico.
- Iniettare 5 ml di campione per l'analisi utilizzando il campionatore automatico del sistema nanoLC-MS / MS. Concentrato e peptidi desalt online su un fase inversa C18 pre-colonna (0,15 millimetri ID x 20 mm con 5 micron dimensione dei pori materiale C18) montato sia in una configurazione di colonna ventilato o una configurazione di commutazione precolonna.
- Peptidi separati su una fase inversa C18 microcolonna (0,075 millimetri ID x 200 mm auto-compresso con 3 micron o più piccolo dei pori del materiale formato C18 in una colonna nanospray auto-pack come PicoFrit, con un 90 min pendenza 5> 35% acetonitrile vs . 0,1% soluzione di acido formico a 300 nl /min. Trasferire eluente in un quadrupolo ibrido / spettrometro di massa Orbitrap tramite una fonte nanospray ioni.
- Analizzare peptidi con un Top15 dati dipendente metodo di acquisizione (MS m / gamma z 350 -. 1600, risoluzione bersaglio 70.000 FWHM, AGC obiettivo 1 x 10 6, tempo di riempimento max 60 msec MS / MS partenza di massa 100, risoluzione bersaglio 17.500 FWHM, AGC Target 2 x 10 5, tempo max di riempimento 60 msec MS / MS soglia 3 x 10 4, includere stati di carica 2 -. 5, normalizzato Collision Energia NCE 25%, dinamico esclusione 15 sec).
Analisi 7. I dati
- Per l'analisi dei dati utilizzare il software MaxQuant 16 liberamente disponibile. Un protocollo dettagliato per l'analisi dei dati bioinformatica è descritta in 16,17. Per l'analisi, combinare file di dati grezzi provenienti da tutte le sezioni di una corsia di SDS-PAGE in un singolo esperimento 17.
Risultati
Profiling proteomica di tumori liquidi e solidi da pazienti è un approccio promettente per la scoperta di nuovi biomarcatori diagnostici e predittivi. Tuttavia, la procedura di preparazione del campione e la spettrometria di massa sono difficili, a causa della complessità dei campioni e la necessità di quantificazione accurata proteine in grandi serie di campioni. La procedura sperimentale descritta sopra inizia con l'isolamento di cellule di interesse, sia per fluorescenza-attivato cell sorting o laser cattura microdissezione.
A questo scopo, le cellule da aspirati di midollo osseo-derivate pazienti o campioni di sangue sono macchiati con anticorpi fluorescenza marcati contro marcatori di superficie definiti di interesse prima di ordinamento basato su FACS e lisi cellulare.
Per arricchire sottoinsiemi cellulari da sezioni di tessuto, questi primi devono essere montati su slitte appropriate per consentire laser cattura microdissezione. A questo scopo montare tha sezioni di tessuto su adeguati scivoli membrana e utilizzare un laser-capture microdissector secondo le istruzioni del produttore. Selezione delle aree e le cellule di interesse richiede conoscenze adeguate circa la istomorfologia di tessuto sano e neoplastico. Le cellule estratte vengono poi trasferiti in una provetta di raccolta appropriato. Esperimenti iniziali devono essere eseguite in modo da definire la quantità di tessuto necessaria per l'estrazione di quantità sufficienti di proteina (minimo proteina totale 10 mg) per ogni nuovo tessuto di interesse. Ogni campione microdissezione viene trattato con 60 ml di tampone di lisi tissutale. Per lisi efficace, cellule da campioni di tessuto devono essere sonicato per 3 min e dopo l'aggiunta di 15 microlitri di campione SDS vengono incubati in un thermomixer a 99 ° C per 1 ora per rimuovere reticolazioni proteine formalina-indotta. Centrifugazione finalmente facilitare la raccolta del lisato cellulare cancellato.
Per ogni malattia di interest (liquidi e tumori solidi) un adeguato standard di quantificazione Super-SILAC deve stabilire 14. Uno standard appropriati dovrebbe rappresentare più del 90% delle proteine espresse nei campioni di interesse. Per la preparazione di uno standard di quantificazione, linee cellulari relative al tipo di cancro di interesse dovrebbero prima essere analizzati mediante spettrometria di massa per determinare i loro profili di espressione proteica e successivamente analisi delle componenti principali devono essere eseguite. 4 - 6 differenti linee cellulari con modelli di espressione proteica differenziale dovrebbero essere scelti e SILAC-etichettati con arginina "pesante" e lisina. Efficienza etichettatura deve essere controllato da nanoLC-MS / MS e dovrebbe essere superiore al 98%. Successivamente, linee cellulari devono essere lisate nello stesso modo come il rispettivo campione derivato paziente e, successivamente, proteine equimolari quantità di ciascun campione e lo standard picco-in SILAC devono essere miscelati. Un passo fondamentale per precisa proteina quantificazione dei campioni clinici ointeresse f è la miscela accurata di proteine equimolari quantità di campioni derivati da pazienti e il picco-in standard quantificazione SILAC. Per determinare la concentrazione di proteina di lisati derivati da cellule filtrate, possono essere utilizzati vari metodi di quantificazione della proteina. Per lisati cellulari da cellule dei tessuti di derivazione è necessario un test in grado di affrontare con alte concentrazioni di SDS e DTT nel lisato. Concentrazione proteica dei campioni derivati da pazienti e lo standard picco-in SILAC devono essere misurate ogni momento prima combinazione con i rispettivi campioni clinici per garantire la corretta miscela, che è cruciale nel fare anche centinaia di campioni comparabili.
Dopo che la miscela di quantità uguali di picco-a campioni standard e lisato, ottenuti da tumori liquidi sono mescolati con LDS, riscaldate in un thermomixer a 72 ° C per 10 minuti e successivamente sottoposti a 1D-PAGE in gel digestione del separato proteine con tripsina. I campioni ottenuti daltessuti sono liberati da SDS e digerito con tripsina utilizzando l'approccio FASP come stabilito dalla Wisniewski et al. 15
I peptidi triptici ottenuti possono essere quantificato misurando la fluorescenza del triptofano, che è di particolare interesse per i campioni di tessuto come la quantità di peptidi rilasciati dal filtro differisce tra il 15% e il 75% rispetto ai valori proteici inizialmente caricati sul filtro. A questo scopo si misura la fluorescenza di triptofano. Il confronto con un appropriato diluizioni triptofano consente la quantificazione dei peptidi, come 1,1 mg di triptofano corrisponde a circa 100 mg di peptide. Se necessario, i campioni provenienti soprattutto dalla lavorazione FASP possono essere pre-purificati su entrambi i suggerimenti palco commerciali o artigianali confezionati con la fase-C18 (RP-C18) materiale rovesciato prima di caricare sul sistema nanoLC / MS / MS 18.
Analisi Mass-spettrometria viene eseguito su un alto-Risoluzione, alta sensibilità del sistema spettrometria di massa. In breve, i campioni sono peptidi dissalato preconcentrato su una precolonna RP-C18, e separati su una colonna analitica RP-C18 direttamente accoppiato allo spettrometro di massa. Per raggiungere una sufficiente profondità di analisi, ci avvaliamo sia una combinazione di SDS-PAGE proteina prefrazionamento con 40 min gradienti RP-C18 (per i tumori liquidi) o iniezioni singole con lunghi 2 - 3 hr RP-C18 gradienti (per i tumori solidi trattati da FASP ). MS vengono acquisite ad una risoluzione di 70.000 FWHM o meglio, per consentire la quantificazione accurata di coppie SILAC mediante integrazione del picco profili cromatografici. Per l'identificazione di proteine, un metodo di acquisizione dati dipendente Top15 viene utilizzato per generare un gran numero di peptidi MS / MS per peptidi e identificazione delle proteine.
Dai dati grezzi risultanti, l'identificazione delle proteine e la quantificazione si ottengono con database di ricerca con software MaxQuant contro una UniProt Knowledgebase umano Proteome completo database di sequenze 17. Nel software MaxQuant (versione attuale 1.5.0.25) peptidi sono identificati dalla acquisito MS / MS per peptide frammento-matching contro spettri derivata in silico dal database sequenza della proteina. Allo stesso tempo, precursore ione profili isotopiche sono estratti intorno ai tempi di ritenzione cromatografiche, e le loro aree dei picchi integrati vengono utilizzati per la quantificazione relativa della luce: coppie peptidici pesanti generati mediante l'etichettatura SILAC. Identità peptidi e intensità relative vengono poi assegnati alle proprietà delle proteine corrispondenti. Software Perseus (versione attuale 1.5.0.15) viene quindi utilizzato per eseguire un'ulteriore valutazione statistica valle dei risultati di elaborazione MaxQuant, compresi campione a campione confronti, PCA e clustering gerarchico.
Utilizzando il setup sperimentale descritto abbiamo identificato e quantificato fino a 8.000 proteine da un minimo di 30 mg di proteine totaliderivate da tumori liquidi.
Fino a 2.500 proteine da campioni solidi-tumorali possono essere identificati e quantificati in un approccio di proteomica fucile con un gradiente LC di soli 2 ore, permettendo l'analisi di centinaia di campioni clinici in un tempo relativamente breve.

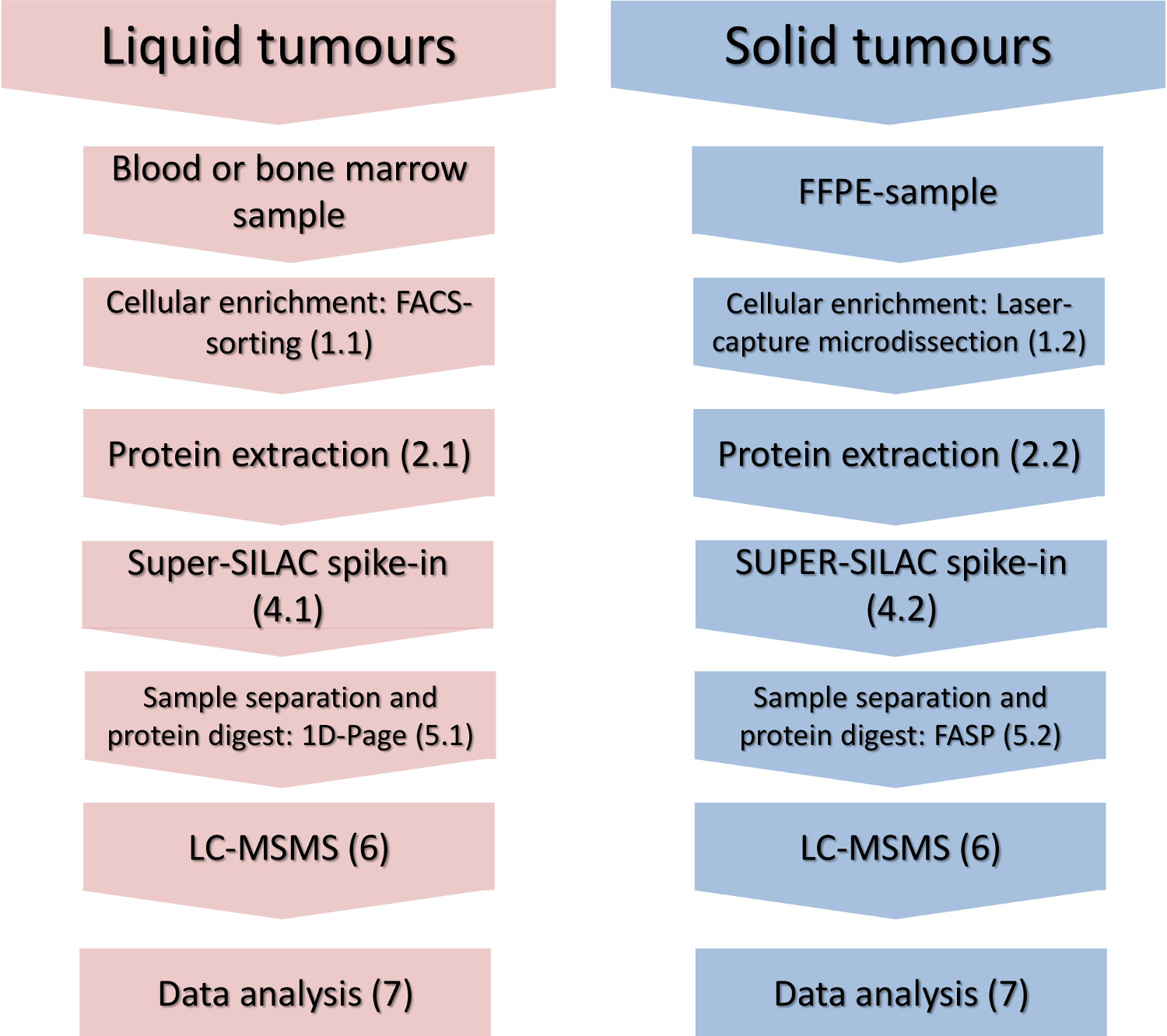
Figura 1. Flusso di lavoro sperimentale. I passi principali di arricchimento cellulari-sottoinsieme, isolamento delle proteine, picco-in dello standard di quantificazione e l'analisi di massa-spettrometria sono indicati per i tumori solidi e liquidi. Cliccate qui per vedere una versione più grande di questa figura.
{kind=link}

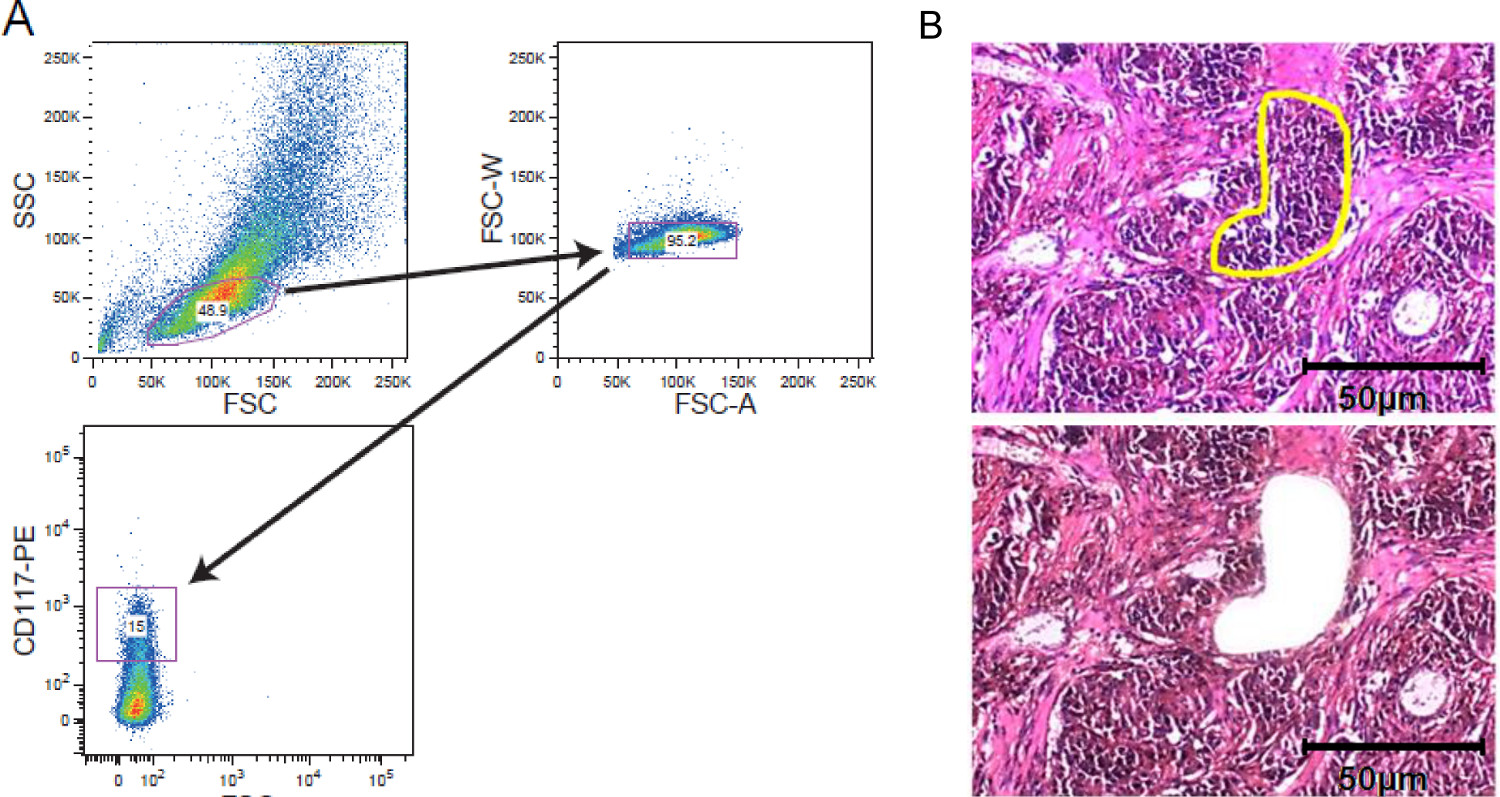
Figura 2. Cellular arricchimento strategies. FACS ordinamento e LCM. (A) strategia di gating esemplare raffigurante gating su una popolazione di cellule leucemiche CD117 macchiato acquisita da un cell sorter. (B) microdissezione laser di tessuto tumorale solido. Le sezioni di tessuto di 5-10 micron di spessore sono stati montati su film coperti diapositive membrana prima della colorazione. Regione di interessi è stata selezionata manualmente per microdissezione laser. Vengono mostrati sezioni prima microdissezione con la regione di interesse segnato giallo e dopo microdissezione. Una barra di scala di 50 micron è incluso nella figura. Clicca qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 3. Analisi Cluster di proteomi cancro delle cellule umane. Cluster analysis non supervisionata di prprofili di espressione otein di tumori solidi e liquidi è stata eseguita utilizzando la piattaforma computazionale Perseo. AC - Adenocarcinoma, SCC - carcinoma a cellule squamose, SCC-Met - squamose metastasi da carcinoma della testa e del collo, R - replica tecnica.
Discussione
Occorre Mass spettrometria profiling di proteomi cancro cellule derivati da pazienti per la scoperta di nuovi biomarcatori diagnostici / predittivi e per dare una migliore comprensione della biologia del cancro delle cellule, che potrebbe a sua volta portare alla identificazione di nuovi potenziali bersagli farmacologici. Tuttavia, tali analisi di spettrometria di massa sono molto impegnativo, in particolare a causa vari problemi pre-analitiche devono essere risolti, se si vuole ottenere risultati solidi e biologicamente rilevanti.
Il flusso di lavoro sperimentale qui descritto consente per la caratterizzazione proteomica quantitativa dei proteomi derivati da sottopopolazioni cellulari di tumori sia liquidi che solidi. È necessaria l'arricchimento iniziale di cellule tumorali o FACS-cell-based sortingor microdissezione per evitare la contaminazione da parte delle cellule del microambiente tumorale. Inoltre, queste tecniche permettono di isolare sottopopolazioni cellulari di interesse. Recenti studi sulle cellule biologiche hanno demonestrata che certe sottopopolazioni cellulari hanno proprietà-tumorali iniziare e sono quindi di grande rilevanza per il cancro patogenesi 19,20. Come spettrometria di massa è diventata più sensibile negli ultimi anni, le analisi quantitative proteomica sono possibili per le piccole quantità di proteine che possono essere derivati da poche migliaia di cellule, rendendo possibile concentrarsi su popolazioni cellulari funzionalmente rilevanti.
Il set-up qui presentata può essere utilizzato per identificare e validare nuovi biomarcatori diagnostici in campioni FFPE. Quindi, promette di bea utile strumento per il miglioramento della diagnostica clinica, in quanto ad oggi non vi è ancora una mancanza di biomarcatori molecolari in numero e qualità sufficiente per molti tipi di cancro. Importanti esempi di diagnosi differenziale difficili, per i quali biomarcatori sono carenti, sono la discriminazione tra cancro polmonare primario da metastasi del polmone, carcinoma cholangiocellular intrapancreatica e adenocarcinoma pancreatico, così come sono diverseziazione di neurofibroma benigni da tumori del nervo-guaina periferiche altamente maligne. Inoltre, noi e altri hanno dimostrato che delucidazione quantitativa delle firme di proteomica può essere utile nello studio della biologia delle cellule del cancro in generale, e per rivelare biomarcatori predittivi della risposta terapeutica in pazienti affetti da cancro 21.
Due attuali svantaggi del metodo presentato qui sono i requisiti per svariati processi di lavorazione manuale del campione e la domanda sul nanoLC-MS / MS tempo di acquisizione. Mentre i primi possono essere affrontati spostando preparazione del campione per esempio, i formati da 96 pozzetti e con trattamento robotico, quest'ultimo richiederà un cambiamento nella strategia di acquisizione di massa spettrometria. Una volta sottoinsiemi di proteine bersaglio sono stati identificati che possono essere connessi ad esempio, la classificazione del tumore, prevediamo la progettazione di metodi di spettrometria di massa mirati che forniscono letture quantitativi per questi sottoinsiemi con uno sforzo di separazione notevolmente ridotto, equindi con un tempo di acquisizione corrispondentemente ridotta. Se il tempo di acquisizione richiesto richiesto potrebbe essere ridotto 24-36 ore (tumori liquidi) o 3 ore (tumori solidi) per esempio, 1 ora utilizzando mirati spettrometria di massa e una semplice separazione unidimensionale di peptidi, quindi il guadagno conseguente rendimento potrebbero essere utilizzati per aumentare in modo significativo il numero di repliche biologiche e tecniche esaminate, con corrispondente miglioramento della significatività dei risultati di quantificazione. Approcci di spettrometria di massa mirati sono già dimostrato di essere uno strumento adeguato per la verifica di cancro associato proteine biomarcatori-candidati 22, e sono stati sviluppati per un punto in cui essi mostrano promessa per la validazione o addirittura come un potenziale strumento per l'uso clinico di routine 23 , 24.
Divulgazioni
Gli autori non hanno alcun conflitto di interessi o altri problemi di rivelare.
Riconoscimenti
The authors thank Uwe Plessmann, Monika Raabe und Silvia Münch for technical support.
Materiali
| Name | Company | Catalog Number | Comments |
| 660 nm Kit | Thermo scientific | 22662 | |
| Cell culture medium depleted of arginine and lysine | Thermo Scientific | 88421 | |
| Coomassie Brilliant Blue R-250 staining solution | Bio Rad | 161-0436 | |
| Dialyzed fetal calf serum (FCS) | PAA | A15-107 | |
| Diffuser caps for microdissection | MMI | 50202 | |
| FACS-sorter | BD | FACSAria III | |
| Ionic Detergent Compatibility Reagent | Thermo scientific | 22663 | |
| Laser-capture microdissector | MMI | cell cut plus | |
| LDS buffer | Life Technologies | NP0009 | |
| Membrane slides for microdissection | MMI | 50103 | |
| Microcon YM-30 | Millipore | MRCF0R030 | |
| NuPAGE 4-12% Bis-Tris Mini Gels | Life Technologies | NP0335PK2 | |
| Picofrit Self-Pack Columns | New Objective | PF360-75-15-N-5 | Mass Spectrometry Column/Emitter |
| Reducing agent | Life Technologies | NP0007 | |
| Reprosil-Pur LC/MS/MS Column stationary phase | Dr. Maisch | 120 C18-AQ, 3 µm | |
| Reprosil-Pur LC/MS/MS Precolumn stationary phase | Dr. Maisch | 120 C18-AQ, 5 µm | |
| SILAC-labeled arginine | Eurisotop | CLM-2265-H-0.1 | |
| SILAC-labeled lysine | Eurisotop | DLM-2640-0.25 | |
| Trypsin, NB Sequencing Grade | Serva | 3728301 | for in-gel digests |
| Trypsin, Sequencing Grade | Promega | V5111 | for in-solution digests |
| Buffer and solutions | |||
| Cell lysis buffer: 150 mM NaCl, 50 mM Tris/HCl pH 7.8, 5 mM NaF, 0.5% NP40, 0.1% laurylmaltoside, Roche complete protease inhibitor, 1 mM Na3VO4 | |||
| Tissue lysis buffer: 100 mM Tris/HCl pH 7.8, 0.1 M DTT | |||
| Urea: 8 M urea in 0.1 M Tris-HCl, pH 8.5 | for FASP-protocoll | ||
| IAA: 0.05 M iodoacetamide, 8 M urea, 0.1 M Tris-HCl, pH 8.5 | for FASP-protocoll | ||
| 0.05 M NH4HCO3 | |||
| 10 mM dithiothreitol (DTT) in 0.1 M ammonium bicarbonate | for in-gel digest | ||
| 55 mM iodoacetamide (IAA) in 0.1 mM ammonium bicarbonate | for in-gel digest | ||
| 5% aqueous formic acid. |
Riferimenti
- Walther, T. C., Mann, M. Mass spectrometry-based proteomics in cell biology. J Cell Biol. 190 (4), 491-500 (2010).
- Lenz, C., Urlaub, H. Separation methodology to improve proteome coverage depth. Expert Rev Proteomics. 11 (4), 409-414 (2014).
- Olsen, J. V., Mann, M. Status of large-scale analysis of post-translational modifications by mass spectrometry. Mol Cell Proteomics. 12 (12), 3444-3452 (2013).
- Ong, S. E., et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 1 (5), 376-386 (2002).
- Jimenez, C. R., Verheul, H. M. Mass spectrometry-based proteomics: from cancer biology to protein biomarkers, drug targets, and clinical applications. Am Soc Clin Oncol Educ Book. , e504-e510 (2014).
- Tang, D. G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 22 (3), 457-472 (2012).
- Evans, C., et al. An insight into iTRAQ: where do we stand now. Anal Bioanal Chem. 404 (4), 1011-1027 (2012).
- Ostasiewicz, P., Zielinska, D. F., Mann, M., Wisniewski, J. R. Proteome, phosphoproteome, and N-glycoproteome are quantitatively preserved in formalin-fixed paraffin-embedded tissue and analyzable by high-resolution mass spectrometry. J Proteome Res. 9 (7), 3688-3700 (2010).
- Malmström, J., Picotti, P., Aebersold, R. Perspectives of targeted mass spectrometry for protein biomarker verification. Curr Opin Chem Biol. 13 (5-6), 518-525 (2009).
- Gillet, L. C., et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 11 (6), (2012).
- Basu, S., Campbell, H. M., Dittel, B. N., Ray, A. Purification of specific cell population by fluorescence activated cell sorting (FACS). J Vis Exp. 41, (2010).
- Edwards, R. A. Laser capture microdissection of mammalian tissue. J Vis Exp. 8, 309 (2007).
- Liu, N. Q., et al. Proteomics pipeline for biomarker discovery of laser capture microdissected breast cancer tissue. J Mammary Gland Biol Neoplasia. 17 (2), 155-164 (2012).
- Geiger, T., et al. Use of stable isotope labeling by amino acids in cell culture as a spike-in standard in quantitative proteomics. Nat Protoc. 6 (2), 147-157 (2011).
- Wisniewski, J. R. Proteomic sample preparation from formalin fixed and paraffin embedded tissue. J Vis Exp. (79), (2013).
- Cox, J., Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 26 (12), 1367-1372 (2008).
- Cox, J., et al. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat Protoc. 4 (5), 698-705 (2009).
- Rappsilber, J., Mann, M., Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2 (8), 1896-1906 (2007).
- Sarvi, S., et al. CD133+ cancer stem-like cells in small cell lung cancer are highly tumorigenic and chemoresistant but sensitive to a novel neuropeptide antagonist. Cancer Res. 74 (5), 1554-1565 (2014).
- Shlush, L. I., et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 506 (7488), 328-333 (2014).
- Schaab, C., et al. Global phosphoproteome analysis of human bone marrow reveals predictive phosphorylation markers for the treatment of acute myeloid leukemia with quizartinib. Leukemia. 28 (3), 716-719 (2014).
- Hüttenhain, R., et al. Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Sci Transl Med. 4 (142), 142ra94 (2012).
- Burgess, M. W., et al. Simplified and efficient quantification of low-abundance proteins at very high multiplex via targeted mass spectrometry. Mol Cell Proteomics. 13 (4), 1137-1149 (2014).
- Boja, E. S., et al. Analytical Validation Considerations of Multiplex Mass Spectrometry-based Proteomic Platforms for Measuring Protein Biomarkers. J Proteome Res. , (2014).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati