
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Microscopía de localización de varios colores solo de proteínas de membrana en organelos de células mamíferas vivas
En este artículo
Resumen
Aquí, presentamos un protocolo para la localización de varios color de proteínas de membrana único en organelos de células vivas. Para colocar fluoróforos, se usan proteínas auto etiquetadas. Proteínas, situadas en compartimientos diferentes membranas de la mismo orgánulo, se pueden localizar con una precisión de ~ 18 nm.
Resumen
Conocimiento sobre la localización de proteínas en subcompartimientos celulares es crucial para entender su función específica. Aquí, presentamos una técnica de súper-resolución que permite la determinación de la microcompartments que son accesibles para las proteínas generando localización y seguimiento de los mapas de estas proteínas. Por otra parte, por microscopia de la localización de varios colores, la localización y seguimiento de perfiles de proteínas en subcompartimientos diferentes se obtienen simultáneamente. La técnica es específica para las células vivas y se basa en la proyección de imagen repetitiva de proteínas de membrana móvil único. Proteínas de interés son genéticamente fusionadas con etiquetas auto etiquetados específicos, supuestos. Estas etiquetas son enzimas que reaccionan con el sustrato de manera covalente. Conjugado a estos sustratos son tintes fluorescentes. Reacción de las proteínas de la enzima etiquetada con la fluorescencia etiquetada resultados de sustratos en proteínas etiquetadas. Aquí, Tetramethylrhodamine (TMR) y silicio rodamina (SiR) se utilizan como tintes fluorescentes a los sustratos de las enzimas. Usando las concentraciones de sustrato en la pM a rango nM, etiquetado sub-estequiométrica se consigue que se traduce en señales distintas. Estas señales se localizan con ~ 15 – 27 precisión nm. La técnica permite la proyección de imagen de varios color de moléculas individuales, por el que el número de colores es limitado por los tintes disponibles permeable a la membrana y el repertorio de auto etiquetado de las enzimas. Mostramos la viabilidad de la técnica en la determinación de la localización de la enzima de control de calidad (Pten)-inducida quinasa 1 (PINK1) en diferentes compartimentos mitocondriales durante su proceso en relación con otras proteínas de membrana. La prueba verdadera interacciones físicas entre proteínas diferentemente marcadas solo por sola molécula traste o seguimiento Co es restringida, sin embargo, debido a los bajos grados de etiquetado disminuyen la probabilidad de tener dos proteínas adyacentes con la etiqueta al mismo tiempo. Mientras que la técnica es fuerte para las proteínas en los compartimientos de la membrana de la proyección de imagen, en la mayoría de los casos no es apropiado determinar la localización de proteínas solubles altamente móviles.
Introducción
El objetivo de este protocolo es proporcionar un método de imagen para localizar y rastrear las proteínas de membrana solo dentro de células vivas. Llamamos a este método de seguimiento y localización de la microscopia (TALM)1,2. Como microscopia de reconstrucción óptica estocástica (tormenta)3 y microscopía de fluorescencia fotoactivación localización ((F) PALM)4,5, TALM es una técnica de localización de fluorescencia basada en la molécula sola. Sin embargo, es distinta de la manera que la movilidad de las proteínas de la membrana en combinación con imágenes repetitivas de la misma etiqueta molécula en diversas posiciones revela el microcompartment que es accesible para la proteína móvil. En otras palabras, las localizaciones posibles de la proteína se encuentran en la arquitectura de las organelas y por la movilidad de la proteína1. El método es complementario a varios otros súper resolución técnicas6,7,8 porque revela la trayectoria y localización mapas por proyección de imagen móvil proteínas. El etiquetado se basa en el uso de proteínas de fusión genéticamente que son fluorescentes por sí no. Estas proteínas de fusión son auto etiquetado enzimas que reaccionan covalentemente con un substrato conjugado a un tinte. Este procedimiento tiene la ventaja de que el grado del etiquetado puede ser controlado por la cantidad de sustrato añadido. Además, permite variar el color de la fluorescencia, dependiendo del tinte conjugado solicitada. Varios auto etiquetados enzima-tags están disponibles9. Otra ventaja de usar auto etiquetado enzima etiquetas es, que los tintes conjugados suelen ser más estables y más brillante de proteínas fluorescentes1 y proteínas individuales por lo tanto se pueden grabar más y más precisamente hasta que se blanquean. Esto permite la grabación de las trayectorias de proteínas móviles y la extracción de coeficientes de difusión10,11.
Aquí, demostramos la factibilidad de TALM con proteínas de la membrana mitocondrial, pero también puede ser aplicado para otras proteínas de membrana cellular intra y extraurbano, incluyendo células diferentes tipos12,13. Mostramos que TALM multicolor más permite la distinción simultánea de proteínas en diferentes subcompartimientos en complementación a los súper resolución fluorescencia microscopía técnicas14,15, 16. TALM es compatible con celular directo imágenes17. La física de la foto del elegido rhodamines Tetramethylrhodamine (TMR) y silicio-Rhodamien (SiR), en particular su brillo y su estabilidad, permite a las proteínas de membrana único registro varios fotogramas proporcionar mapas de trayectoria (y localización). Sin embargo, TALM está limitada para la localización de proteínas solubles con coeficientes de difusión alta puesto que el desenfoque de movimiento es demasiado alto y los fotones recogidos por frame son demasiado bajos para la localización correcta. Además, TALM requiere menos energía de la excitación que por ejemplo tormenta o agotamiento estimulado de emisión (STED) microscopía6,7, reduciendo efectos phototoxic. Esto es importante, puesto que estrés fototóxico a menudo afecta la morfología organellar18 y así movilidad análisis19. En suma, presentamos varios color TALM en células vivas como una técnica que llena un vacío entre los métodos de microscopía de localización tormenta/STED/Palma (F) y las técnicas que analizan la movilidad de la proteína tales como recuperación de fluorescencia Tras Fotoblanqueo (FRAP)20 ,21, correlación de fluorescencia (FCS) la espectroscopia22y fluorescencia cross correlación espectroscopia (FCCS)11,23.
Access restricted. Please log in or start a trial to view this content.
Protocolo
El siguiente protocolo sigue las directrices de la Comisión de ética de investigación institución local.
1. métodos
-
Cultivo de células
- Cultivar células, por ejemplo las células HeLa (carcinoma de cérvix humano) en un frasco de cultura de célula T25 que contenga 5 mL de medio de crecimiento a 37 ° C y 5% CO2.
Nota: Para la proyección de imagen, dividir las células sobre cubreobjetos preparado (véanse los pasos 1.3 y 1.4) y mantener en medio de la proyección de imagen.
- Cultivar células, por ejemplo las células HeLa (carcinoma de cérvix humano) en un frasco de cultura de célula T25 que contenga 5 mL de medio de crecimiento a 37 ° C y 5% CO2.
-
Transfección de la célula
Nota: Utilice las líneas celulares que expresan de forma estable las proteínas etiquetadas siempre que sea posible24 para evitar fuerte sobreexpresión. Para transitorios de la transfección, adaptar la cantidad de plásmido utilizada para la transfección de la DNA. Por ejemplo, cuando se usa la transfección de fosfato de Ca2 + 25 , transfectar las células (confluencia de 80-90%) en una placa de cultivo celular de 3,5 cm con 2,5 a 5 μg de ADN plásmido. Cuando se realiza doble transfección, utilizar 2,5 μg por cada construcción de plásmidos.- Para los experimentos de doble color, utilice una línea de células con expresión estable de una proteína por etiquetado y transitoriamente transfectar con el plásmido codificación el otro auto etiquetado proteína17.
Nota: Aquí, para experimentos de dos colores, las células HeLa fueron utilizadas que estable las proteínas auto etiquetadas PINK1-Halo-etiqueta y etiqueta de fSNAP Tom20.
- Para los experimentos de doble color, utilice una línea de células con expresión estable de una proteína por etiquetado y transitoriamente transfectar con el plásmido codificación el otro auto etiquetado proteína17.
-
Limpieza de cubreobjetos
- Coloque el cubreobjetos en un vaso de precipitados. Añadir 30 mL de H2O en el vaso que contiene el cubreobjetos y agítelo suavemente para eliminar el polvo de su superficie.
- Se reúnen el cubreobjetos con la pinza y séquelos con una corriente de nitrógeno.
- Eliminar cualquier contaminación orgánica en la superficie del cubreobjetos, por ejemplo, limpieza de plasma.
Nota: Para evitar una mayor contaminación del material de vidrio, use guantes durante la manipulación de los cubreobjetos.
PRECAUCIÓN: Cuando cubreobjetos se limpian por plasma de limpieza, se limpia sólo la parte superior de los cubreobjetos; Utilice esta parte para recubrimiento con poli-L-lisina-polietileno glicol-arginina-glicina-aspartato (PLL-PEG-RGD) (sección 1.4) y siembra (sección 1.5) de la célula.
-
Capa del cubreobjetos con PLL-PEG-RGD
Nota: PLL-PEG-RGD es un derivado de (PLL) de poli-l-lisina Unido con un polietilenglicol (Da 3.000) y un péptido (CGRGDS) cysteine-glycine-arginine-glycine-aspartate-serine. PLL se une a la superficie de vidrio cargada negativamente y forma un cepillo de PEG. Esto reduce drásticamente la Unión inespecífica de tintes fluorescentes cargados. Además, el motivo RGD imita el péptido señal de los receptores de integrina y promueve así la adhesión mediada por integrinas de las células que de lo contrario no se adhieren fácilmente.- Preparar PLL-PEG-RGD como se describió anteriormente26. En Resumen, disolver 0,8 mg de PLL-PEG-RGD en 1 mL de PBS. Añadir 10 μl de la solución de PLL-PEG-RGD en la parte superior de un cubreobjetos limpio.
- Tomar un segundo cubreobjetos y colocarlo con su superficie limpia boca abajo sobre el cubreobjetos primero (que tiene la gota de PLL-PEG-RGD en la parte superior); se consigue colocando la solución de PLL-PEG-RGD entre dos cubreobjetos.
- Con cuidado coloque el cubreobjetos intercalado en un vaso de precipitados e incubar durante 1 h a temperatura ambiente en un ambiente libre de polvo seco.
- Después de 1 h, Añadir 30 mL de H2O en el vaso para poder cubrir totalmente el cubreobjetos con agua.
- Sacuda suavemente el vaso hasta que el cubreobjetos separan unos de otros.
- Utilice unas pinzas para recoger el cubreobjetos fuera del agua y secarlos con un chorro de gas nitrógeno.
Nota: El cubreobjetos recubierto pueden almacenarse en un vaso seco y estéril con tapa de Petri durante un par de días.
-
Preparación de la muestra para la proyección de imagen
- Transferir el cubreobjetos recubierto solo en una placa de cultivo celular de 35 mm, con la superficie recubierta de PLL-PEG-RGD hacia arriba y añadir 2 mL de medio en la parte superior de la imagen.
- Añadir ~ 500.000 tripsinizaron células (200 – 500 μl) que expresan las uno mismo-etiquetadas etiquetas en las proteínas de membrana respectivos a 2 mL de medio en la placa de cultivo celular con el cubreobjetos recubierto. Agitar suavemente a mano para asegurar una distribución homogénea de las células para obtener una capa uniforme de la célula.
- Incube las células a 37 ° C y 5% de CO2 hasta que se alcanza el 80% de confluencia.
Nota: Muestras de células deben ser sembradas 3 días antes de la proyección de imagen y 1 día antes de la transfección. Las células, que estable expresan la proteína de interés, pueden ser sembradas 2 días antes de la proyección de imagen. Más tarde, sólo las células cultivadas en el cubreobjetos están reflejadas.
-
Etiquetado de proteínas etiquetadas
Nota: Sustratos más fluorescentes deban ser disuelto en DMSO sin agua. Recomendamos utilizar las soluciones madre de sustrato fluorescente de 1 μm cuando la concentración final de etiquetado es 0.2-30 nM17. Para proteínas de la membrana interior de las células de proyección de imagen, utilice sustratos fluorescentes permeable la membrana.- El medio de imágenes a 37 ° C en un baño de agua caliente.
- Pipetear 1 mL del medio de proyección de imagen previamente calentado en un tubo de 2 mL con tapa. Añadir 0,2 – 30 μl de sustratos fluorescentes de las soluciones madre para preparar la solución de etiquetado final 1 μm (concentración final: 0.2-30 nM).
- Vórtice de la solución de etiquetado para 10 s.
- Sustituir el medio de la placa de cultivo de 35 mm con las células sobre cubreobjetos (ver paso 1.5) 1 ml de preparado de solución de etiquetado.
- Incubar las células en la solución de etiquetado a 37 ° C y 5% CO2 para 20-30 min.
- Lavar las células con 2 mL de PBS una vez, luego con 2 mL de medio de la proyección de imagen dos veces. Finalmente, Pipetear 1 mL de medio fresco de imágenes al plato celular y vuelva a colocar la muestra en la incubadora a 37 ° C y 5% CO2 durante al menos 1 h. Antes de la proyección de imagen, cambiar el medio de imágenes una vez más.
Nota: Cuando se ejecuta el experimento por primera vez, confirmar la orientación correcta de auto-etiquetadas proteínas a membranas organellar manchando los organelos con organelas disponibles en el mercado tintes específicos27,28. En este caso, también utilizar 100-200 nM de sustrato para las enzimas auto etiquetadas para producir señales fuertes.
-
Preparación de una muestra de grano fluorescente
Nota: Para determinar la dispersión óptica y alinear imágenes de los canales, se utilizan varios colores fluorescentes granos (0.1 μm). Con las imágenes grabadas, se genera una matriz de transformación afín para los canales de dos emisión.- Diluir la solución de los abalorios al 1% con puro H2O.
- Lugar 5 gotas de la solución preparada con los granos de la fluorescencia en cinco posiciones diferentes en un cubreobjetos limpio (vea el paso 1.3).
- Que la muestra fluorescente grano seco en una mesa de trabajo limpia.
Nota: La muestra se puede volver a utilizar; por lo tanto, cubrir la muestra con papel de aluminio para evitar la contaminación y conservar a 4 ° c.
2. microscopía
-
Disposición experimental
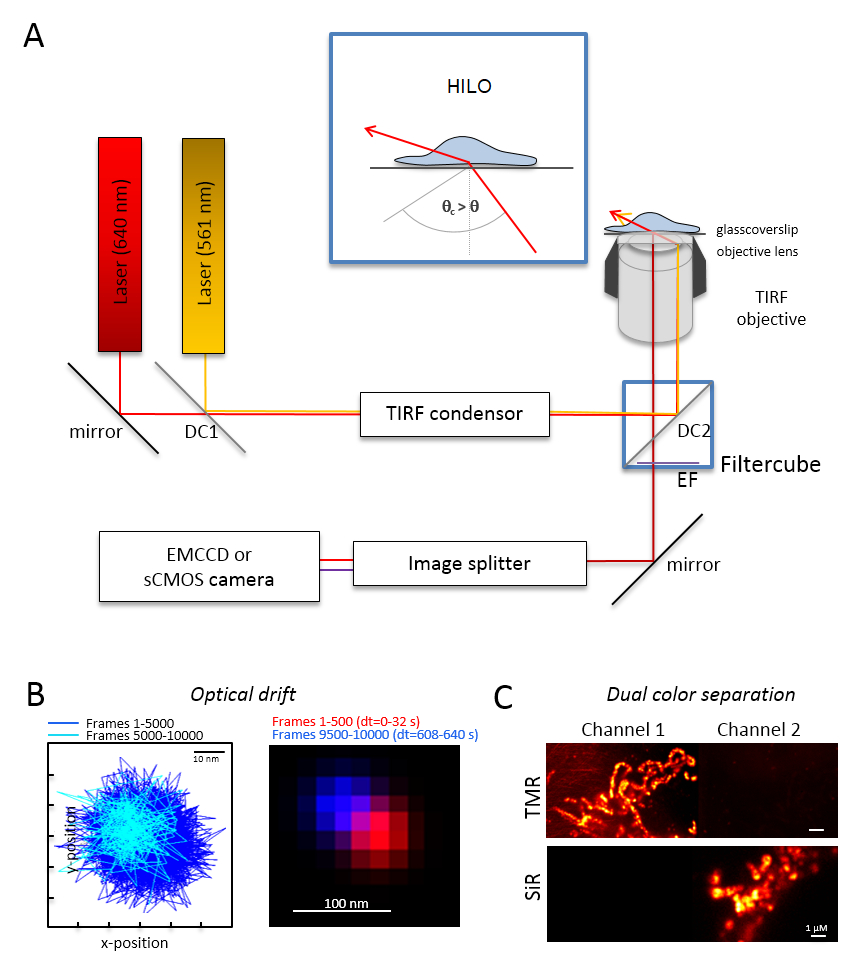
Nota: Un sistema de microscopía básica para la proyección de imagen de color dual sola molécula se basa en un microscopio invertido: está equipado con dos rayos láser acoplado por medio de una polarización modos-óptica mantiene monomodo fibra en una sola reflexión interna total (TIR) condensador, un objetivo de inmersión de aceite diseñado para TIRF, un filtros de emisión polyband, un divisor de imagen y una cámara de alta sensibilidad (figura 1). Un condensador TIR es necesario que permite la adaptación continua del ángulo incidente para cambiar entre el epi-, muy inclinado y hoja laminada de óptica (iluminación fina altamente inclinado (HILO)29) y el modo de la excitación de TIRF con optimizado profundidad de la penetración. Se adquieren imágenes con un sistema altamente sensible detector enfriado, por ejemplo, una parte trasera iluminada multiplicando electrones carga cámara dispositivo acoplado (EMCCD) (eficiencia cuántica QE > 90%) o una cámara sCMOS (FC > 80-90%).- Determinar la dispersión óptica, perlas fluorescentes (véase el paso 2.2) bajo las mismas condiciones que las que se utilizará más adelante para el experimento, por ejemplo, cuando se registran 10.000 marcos en el experimento, también de 10.000 marcos con la muestra de talón de registro de imagen. Para la determinación de la dispersión óptica, comparar la posición de los granos en el primer fotograma y el último fotograma adquirido (figura 1B). Si es necesario, posteriormente corregir la serie de imagen óptica deriva30 o utilice entornos estables deriva.
- Equipar el cubo de filtro con el divisor de viga dicroico apropiado, por ejemplo, para fluorescencia naranja y roja además de los filtros de la adecuada emisión de fluorescencia naranja y fluorescencia roja. Equipar el divisor de imagen con el filtro adecuado. Compruebe la posible fuga de señales de un canal en el otro canal mediante el registro de las muestras de color individuales en ambos canales (figura 1).

Figura 1 : Diseño óptico para multicolor seguimiento y localización de microscopía (TALM) con emisores de naranja y rojos. (A) instalación de microscopio invertido con por lo menos dos láseres de excitación, un condensador TIRF, un objetivo conveniente TIRF, un divisor de imagen y una cámara sensible. Recuadro: para excitar a los organelos dentro de células, el ángulo del haz incidente debe establecerse más pequeño que el ángulo crítico para TIRF alcanzar altamente inclinado y había laminado iluminación (HILO) de la hoja óptica. DC1: Espejo dicroico 1; DC2: Espejo dicroico 2. EF: filtro de emisión. (B) prueba óptica deriva por posiciones de una capa fluorescente para 10.000 marcos con la misma velocidad de fotogramas a partir de los siguientes experimentos de proyección de imagen (aquí: 15 Hz). Posiciones conectados de los primeros 500 marcos y los últimos 500 marcos demuestran la deriva. También, una imagen combinada con la posición del primero y el último fotograma en rojo y azul muestran una dispersión mínima. La deriva es que la distancia entre el centro de las señales dividido por el tiempo de grabación total, aquí 125 pm/s. (C) comprobación de la separación clara de las señales, TMR y SiR. Para ambos canales, se obtuvieron imágenes de suma acumulada de 3.000 marcos (TMR en canal 1) y SiR en canal 2. SiRHTL fue unido al HaloTag Tom20 y TMRHTL a OxPhos complejo V-HaloTag. Los colores son colores falsos. Barras de escala = 100 nm (B) y 1 μm (C). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
-
Alineación física de divisor de imagen imágenes generadas
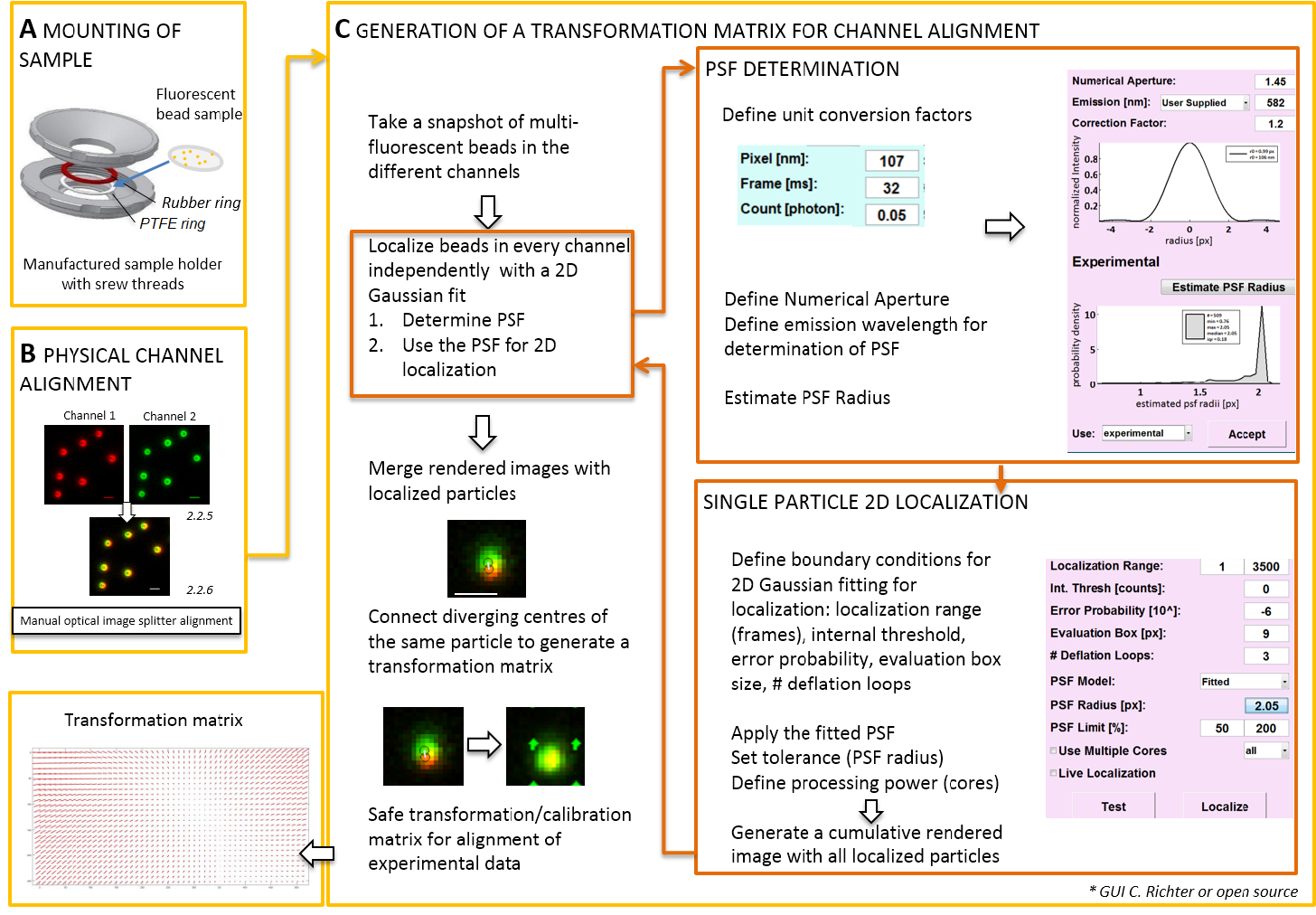
Nota: Para el montaje de la muestra preparada en un cubreobjetos, un portamuestras hecho a sí mismo pueden ser utilizado (figura 2A). Para evitar el polvo, etc. cayendo en la muestra, coloque la tapa de la placa de cultivo libremente encima de la cámara, montado. El titular de la misma muestra puede utilizarse para montar el cubreobjetos con perlas fluorescentes o células; Cuando las células son imágenes, Añadir 0,5 – 0,8 mL de medio de la proyección de imagen. El separador de la imagen divide la imagen en dos o más espectral separaron los canales y los side-by-side proyectan en la misma cámara. Este proceso potencialmente introduce distorsiones sistemáticas entre los canales por distintos caminos ópticos atravesados y obstruye análisis de colocalización directa. Por lo tanto, en primer lugar realizar alineación física y segundo, corrección de la alineación con una matriz de transformación. Para ambos procesos de alineación, perlas fluorescentes deberían distribuirse homogéneamente a lo largo del campo de visión.- Montaje de la muestra preparada con los granos fluorescentes en el sostenedor de la muestra entre el politetrafluoroetileno (PTFE)-anillo y el anillo de goma roja (figura 2A).
- Inicio el microscopio, todos los componentes de hardware y todo el software necesario para microscopía.
- Limpie el objetivo y la parte inferior del cubreobjetos con una toallita de pelusa humedecida con isopropanol. Luego secar ambos elementos con un tejido fresco de pelusa. Coloque una gota de aceite de inmersión sobre la pupila de la lente del objetivo.
- Coloque el portamuestras con la muestra de grano en la platina del microscopio para que el aceite en contacto con la parte inferior del cubreobjetos. Se centran en los granos mediante el uso de una línea de láser o luz de transmisión.
- Ajustar la potencia de los dos láseres de excitación para lograr la intensidad de señal similar en los dos canales de fluorescencia. Busque un área con muchas distintas señales fluorescentes.
- Generar una vista combinada de los canales fluorescentes mediante el software de control de cámara. Luego utilice los tornillos en el divisor de imagen para inclinar manualmente los espejos internos del divisor de imagen para lograr el mejor recubrimiento de las señales de los dos canales fluorescentes (figura 2B).
Nota: atención! No exceda el rango dinámico de la cámara.
-
Alineación de canales separados espectralmente por software realizar transformación espacial
Nota: La parte siguiente muestra la corrección de la alineación y el procedimiento de localización con nuestro plugin de software (disponible a petición).- Iniciar el microscopio TIRF software de control y elija Mostrar canales individuales en el modo de transmisión en vivo. Tomar una instantánea imagen emocionante de la fluorescencia en todos los canales (figura 2).
- Utilizar esta imagen instantánea para producir la matriz de transformación (ver figura 2).
Nota: Se utiliza la matriz de transformación para una transformación espacial, normalmente un uno afín, que corrige para la traducción (divergencia de las señales de un solo punto de origen entre dos canales). - Iniciar el complemento de análisis de software (puede obtenerse a petición de nuestro laboratorio, ver figura 2).
- Cargar las imágenes previamente grabadas bicolor (vea el paso 2.2) de perlas fluorescentes en el software. Especificar la orientación de uso de los canales de fluorescentes. A continuación, haga clic en 'sí' cuando se le preguntó por 'calibrar imágenes' y seleccione la instantánea tomada previamente.
- Abra el "Administrador de unidad" para definir factores de conversión (tamaño de píxel, velocidad de fotogramas, factor de conversión de fotones).
- Abra el "Gestor de localización". Determinar que el punto de extensión función (PSF) primero. Presione el botón: "Radio PSF". En la ventana de "Estimador de PSF" que se abre, definir la apertura numérica y la emisión máxima. Inicio «estimación PSF radio "haciendo clic en. Aceptar la obtenida experimental PSF. definir el cuadro de evaluación, el número de bucles de deflación, y cómo muchos corazones de la computadora se utilizan para el cálculo. Pulse "localizar" para iniciar el montaje de la distribución de la intensidad de las partículas individuales de una función Gaussiana simétrica 2D (figura 2).
- "Aceptar" la obtenida experimental PSF. definir el cuadro de evaluación, el número de bucles de deflación, y cómo muchos corazones de la computadora se utilizan para el cálculo. Pulse "localizar" para iniciar el montaje de la distribución de la intensidad de las partículas individuales de una función Gaussiana simétrica 2D (figura 2).
- Abra el "Administrador de calibración". En la imagen combinada de los dos canales, se muestran las señales originales y los centros localizados. Seleccione el modo "afín". Conecte manualmente los correspondientes pares de centros localizados en los dos canales que se han originado desde el mismo grano fluorescente dibujando una línea de conexión.
- Conecte las señales correspondientes distribuidas por todo el campo de visión. Después de esto, pulse "Aceptar". Guardar la calibración.
Nota: La transformación espacial es muestreada en cada grano fluorescente e interpolada en el medio. La función de transformación extraído representa un Δr(x,y) de campo de desplazamiento que se utiliza para corregir posteriormente las localizaciones experimental dos colores sola molécula que recubrimiento dentro de la precisión de localización. La matriz de transformación espacial es típicamente afín que corrige para la traducción, escalado y rotación entre canales con precisión de nanómetros, y se puede deducir de esta asignación uno a uno manual (figura 2).

Figura 2 : Flujo de trabajo para la alineación de doble color. (A) el cubreobjetos con las perlas fluorescentes se monta en un soporte de muestra entre un PTFE y un anillo de goma. Luego la parte superior e inferior de la cámara están atornillados entre sí. (B) alineación física de las vistas del canal que se generan por el separador de la imagen. Registran las señales fluorescentes de granos (0,1 μm) en dos canales (verde y rojo, colores) se fusionan. Los tornillos correspondientes en el divisor óptico de la imagen están activados manualmente hasta logra la mejor plantilla de las diferentes señales (color amarillo, panel inferior). (C) generación de una matriz de transformación para la alineación del canal post-processive. Para la localización exacta de una partícula, es necesario determinar que el punto de función (PSF) separó en dependencia de la longitud de onda de emisión y la apertura numérica del objetivo. El centro de una PSF puede determinarse por su perfil de intensidad por una gaussiana bidimensional simétrico ajuste. La localización resultante del pico de la señal entonces se proyecta en las señales originales, borrosas. En una imagen combinada, los centros localizados de las señales de los dos canales se conectan para generar una matriz de transformación que más tarde se utiliza para la alineación del post-processive de los datos experimentales. Barras de escala = 1 μm (B, C). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
-
Proyección de imagen de una molécula de proteínas de la membrana mitocondrial
Nota: Todos los experimentos se llevan a cabo a temperatura ambiente. Las células T o células no adherente deben inmovilizadas en agarosa antes de la proyección de imagen31.- Montaje de la muestra con células adherentes sobre el cubreobjetos entre el caucho y los anillos de PTFE (Figura 3A). Llene la cámara con 0,5 – 0,8 mL de medio.
- Repita los pasos 2.2.2–2.2.5.
- Ajustar el microscopio TIRF de ángulo de iluminación control de software para crear un ángulo de incidencia es menor que el ángulo crítico para el modo de TIR excitar la región específica de interés mediante un HILO hoja32 (modo de HILO, figura 1A).
PRECAUCIÓN: Evite el contacto directo del ojo con el rayo láser! - Establece la ganancia de EM y un tiempo de exposición adecuado para el experimento que recoge suficientes fotones por marco.
- Ajustar la potencia del láser para lograr una alta señal a ruido (S) relación (figura 3B), puesto que la precisión de la localización directamente corresponde a S/N33 (figura 3).
- Encontrar una zona en la periferia de la célula con mitocondrias no superpuestos, alargadas y señales de molécula única (figura 3D; Video complementario 1). Si ninguna señal sola molécula es visible, espere hasta blanqueo resulta en la aparición de señales de molécula única (figura 3E).
- Registro hasta que el número de señales es demasiado baja para continuación razonable (generalmente 1.000-10.000 marcos dependiendo el comportamiento de decoloración del tinte fluorescente, figura 3F).
- Iniciar la proyección de imagen de software de procesamiento y busque estructuras mitocondriales generando una imagen suma representada acumulado de al menos 1.000 fotogramas grabados (figura 3).
Nota: La velocidad de fotogramas posible más rápida es dictada por el área de lectura. El campo de visión para un canal es reducido por un divisor de imagen de doble color (512 x 512 pixeles) a 256 x 512 píxeles y para un Quad color a 256 x 256 píxeles. Así, para el uso de un divisor de imagen para dos colores, esto es 30 Hz. Fije el modo de transferencia de marco para lograr el menor tiempo de lectura posible. - Inicio software análisis plugin y cargar crudos datos. Seleccione las imágenes de orientación y de la carga del canal. Utilizar la matriz de transformación de paso 2.3.9 cuando pidió "Calibrar imágenes". Canales se mostrará por separado.
- Abra el "Administrador de unidad" como antes para definir factores de conversión para cada canal. Abra el "Gestor de localización" para cada canal. Definir el cuadro de evaluación, número de bucles de deflación, añadir a continuación el PSF teórica para las condiciones de utiliza y establecer cuántos núcleos de la computadora se utilizan para el cálculo. Por último, pulse "localizar" para obtener partículas individuales localizadas (figura 3 H; Video complementario 2).
- Nota que el programa generará finalmente una imagen acumulativa superresolution todos localizados las partículas (figura 3I).
- Realizar el análisis, por ejemplo, software de código abierto o nuestro software disponible bajo petición.
- Seguimiento de las moléculas individuales en ambos canales localizados, por ejemplo, con el indicador de objetivo múltiple10
Nota: Paso 2.4.13 tiene conocimiento (experimental) preliminar sobre la difusibilidad de las proteínas de interés para establecer correctamente las condiciones de límite. Por lo general, encontrar las correctas condiciones de límite es un proceso iterativo.

Figura 3 : Pasos durante sola molécula de microscopía de localización. (A) A cubreobjetos con el espécimen está montado entre la parte superior e inferior (gris) del porta muestra casera (diseñado por J. Bereiter-Hahn). Un anillo de goma (rojo) y un anillo PTFE (blanco) sellan el sistema de arriba y abajo el cubreobjetos, cuando el sostenedor de la muestra se perno juntos. (B) relación señal a ruido de la señal de la TMR. (C) calcula el histograma de precisión de localización de partículas todo localizadas. (D) elección de una región razonable para la proyección de imagen, aquí, la periferia de la célula con mitocondrias claramente separadas. (E) grabación y procesamiento de imágenes: un solo cuadro con señales diferentes a la sola molécula se muestra (aquí, las moléculas individuales de la CV-HaloTag/TMRHTL se registraron). (F) intensidad de TMR en el tiempo de grabación. (G) imagen de la suma acumulada de 3.000 cuadros, sin procesar. (H) las partículas de CV-HaloTag/TMRHTL localizada con una función Gaussiana 2D de un solo cuadro. () Acumulado, prestados imagen suma todas las partículas localizadas de la CV-HaloTag/TMRHTL de 3.000 marcos. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Access restricted. Please log in or start a trial to view this content.
Resultados
Análisis de la proyección de imagen y colocalización multicolor puede ayudar a determinar la localización sub-organellar de proteínas. Hemos demostrado esto antes con el citosol fosfatasa y tensina homólogo, PINK1, que tiene diferentes lugares sub-mitocondrial debido a su procesamiento por las proteasas mitocondrial17. PINK1 es un factor importante que garantiza la funcionalidad mitocondrial34,35. Par...
Access restricted. Please log in or start a trial to view this content.
Discusión
Aquí, se presentó una técnica para la localización de la sola molécula bicolor de proteínas de membrana móvil. Tras el protocolo, proteínas de membrana están fusionadas a auto etiquetado de proteínas que reaccionan con los tintes del rhodamine TMR y SiR conjugado con sus respectivos sustratos. Rodamina tintes son brillante y Fotoestables y así permitir repetitiva imagen1. Para un rendimiento exitoso, varias condiciones y temas críticos tienen que ser tenidas en cuenta.
Access restricted. Please log in or start a trial to view this content.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Los autores desean agradecer al grupo de Biofísica y Jacob Piehler en la Universidad de Osnabrück para apoyo continuo, Wladislaw Kohl para asistencia técnica y preparación del material y la Junta de CellNanOs para proporcionar microscopios para uso. El proyecto fue financiado por el SFB 944.
Access restricted. Please log in or start a trial to view this content.
Materiales
| Name | Company | Catalog Number | Comments |
| (2-(4-(2-hydroxyethyl)-1-piperazinyl)-ethanesulfonic acid, 1 M) (HEPES) | Biochrom | #1104E | |
| DC1: Dichroid beam splitter | Chroma | 640 dcxr | NC506031 |
| DC2: Polychroic Mirror, beamsplitter | Chroma | zt405/488/561/640rpc | discontinued |
| Dulbecco´s Phosphate-Buffered Saline (PBS) 1x (w/o Ca & Mg) | Sigma-Aldrich & Co. | #RNBF8311 | |
| Earle´s MEM without phenol red, without L-Glutamine and without NaHCO3 containing 1% FBS, 0.1% HEPES, 0.1% NEAA, 0.1% Alanyl-L-Glutamine and 34.78% sodium hydrogen carbonate (NaHCO3 0.75g/l) | Imaging medium | ||
| Earle´s minimum essential medium (MEM) with phenol red, containing 1% Fetal Bovine Serum Superior (FBS), 0.1% HEPES (2-(4-(2-hydroxyethyl)-1-piperazinyl)-ethanesulfonic acid, 1 M), and 0.1% non-essential amino acids (NEAA) | Growth medium | ||
| EF: Emission filter quadbandpass | AHF analysentechnik | F72-866 | Brightline HC 446 nm/523 nm/600 nm/677 nm |
| EMCCD camera | Andor | Andor iXON 897 | EMCCD camera |
| Emission filter QuadView filter cubes, orange | AHF analysentechnik | F39-637 | bandpass 582 - 619 nm |
| Emission filter QuadView filter cubes, red | Chroma | bandpass 655 - 725 nm (HQ 690/70) | |
| FBS (Fetal bovine serum) superior | Biochrom | S0615 | |
| Fluorescent beads: TetraSpeck™ Microspheres, 0.1 µm, fluorescent blue/green/orange/dark red | Thermo Fisher Scientific | T7279 | fluorescent microspheres |
| Glutamine | Biochrom | #0951C | |
| HeLa cells | DSMZ | ACC-57 | Cervical carcinoma cells from patient Henrietta Lacks |
| Hela cells CI::paGFP, stable | Muster et al., PLOSOne 2010 | ||
| Hela cells CV g::Halo7-Tag, stable | Appelhans et al., NanoLett 2012 | ||
| Hela cells Tom20::Halo7-Tag, stable | Appelhans et al., NanoLett 2012 | ||
| Image splitter | Photometrics | Dual-View QV2 | image splitter emission |
| Imaging processing software | ImageJ2 / Fiji | freeware | |
| Immersion Oil - ImmersolTM 518 F (ne = 1.518, ve = 45) | Carl Zeiss Jena GmbH | 444960-0000-000 | |
| Inverted epifluorescence microscope | Olympus IX-71/73/83 | ||
| Laser 561 nm, 200 mW | CrystaLaser | CL-561-200 | 561 nm emission |
| Laser 642 nm, 140 mW | Omicron | Luxx-642-140 | 642 nm emission |
| MATLAB | MathWorks | version R2013a | |
| MEM with Earle's Balanced Salt Solution 2.2 g/L NaHCO3, stable glutamine w/o PR | Biochrom | FG-0385 | |
| MEM with Earle's Balanced Salt Solution with 2.2 g/L NaHCO3, stable glutamine, Phenolred | Biochrom | FG-0325 | |
| MitoTracker® Deep Red FM | Thermo Fisher Scientific | M22426 | dye |
| MitoTracker® Green FM | Thermo Fisher Scientific | M7514 | dye |
| Multi-mode-optical polarization maintaining monomode fiber | Pointsource/Qioptiq | KineFLEX | |
| NHS-PEG-MAL, Rapp Polymer | Rapp Polymere GmbH Tübingen | coverslip coating | |
| non-essential amino acids (NEAA) | Biochrom | #0802E | |
| PEG 800 (Polyethylene glycol) 10 % | Carl Roth GmbH | Art No. 0263.1 | coverslip coating |
| Penicillin/Streptomycin | Biochrom | #0122E | |
| Plasmid for PINK1-Halo7-Tag expression | Beinlich et al., ACS Chemical Biology 2015 | ||
| Poly-L-lysine (1.2 mg/ml) | Sigma-Aldrich & Co. | Cat. No.P9155 | coverslip coating |
| RGD Peptide (Ac-CGRGDS-COOH) | Coring System Diagnostix GmbH, Gernsheim | coverslip coating / Intergrin receptor motif | |
| Silicon Rhodamine linked to HaloTag®-Ligand (SiRHTL) | personal gift from Kai Johnson | dye | |
| Software analysis plugin | self-written C. P. Richter, Biophysik Osnabrück | SLIMFAST 16g | |
| Tetramethylrhodamine / SNAP-Cell® TMR-Star linked to SNAP-Ligand (TMRstar) | New England Biolab® | S9105S | dye |
| Tetramethylrhodamine linked to HaloTag®-Ligand (TMRHTL) | Promega | G8251 | dye |
| TIRF condensor | Olympus | Cell^TIRF MITICO System | TIRF condensor |
| TIRF microscope controlling software | Olympus cellSens 1.12 | ||
| TIRF objective | Olympus | 150x oil objective (N.A. 1.45; Olympus UAPO) | |
| Trypsin/EDTA 10x | Biochrom | #0266 | |
| Water H2O 99,5 % Rotipuran® Low organic | Carl Roth GmbH | Art. No. HN57.1 |
Referencias
- Appelhans, T., Richter, C., Wilkens, V., Hess, S., Piehler, J., Busch, K. Nanoscale organization of mitochondrial microcompartments revealed by combining tracking and localization microscopy. Nano Letters. 12 (2), 610-616 (2012).
- Appelhans, T., Busch, K. Single Molecule Tracking and Localization of Mitochondrial Protein Complexes in Live Cells. Methods Mol Biol. 1567, 273-291 (2017).
- Rust, M. J., Bates, M., Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods. 3 (10), 793-795 (2006).
- Gould, T. J., Verkhusha, V. V., Hess, S. T. Imaging biological structures with fluorescence photoactivation localization microscopy. Nat Protoc. 4 (3), 291-308 (2009).
- Pennacchietti, F., Gould, T. J., Hess, S. T. The Role of Probe Photophysics in Localization-Based Superresolution Microscopy. Biophys J. 113 (9), 2037-2054 (2017).
- Wegel, E., Göhler, A., Lagerholm, B. C., Wainman, A. Imaging cellular structures in super-resolution with SIM, STED and Localisation Microscopy: A practical comparison. Scientific reports. , Available from: https://www.nature.com/articles/srep27290?WT.feed_name=subjects_physical-sciences (2016).
- Pellett, P., et al. Two-color STED microscopy in living cells. Biomedical Optics Express. 2 (8), (2011).
- Ishigaki, M., et al. STED super-resolution imaging of mitochondria labeled with TMRM in living cells. Mitochondrion. 28, 79(2016).
- Liss, V., Barlag, B., Nietschke, M., Hensel, M. Self-labelling enzymes as universal tags for fluorescence microscopy, super-resolution microscopy and electron microscopy. Scientific Reports. 5, 17740(2015).
- Sergé, A., Bertaux, N., Rigneault, H., Marguet, D. Dynamic multiple-target tracing to probe spatiotemporal cartography of cell membranes. Nat Methods. 5 (8), (2008).
- Appelhans, T., Busch, K. B. Dynamic imaging of mitochondrial membrane proteins in specific sub-organelle membrane locations. Biophysical reviews. 9 (4), 345-352 (2017).
- Wilmes, S., et al. Triple-color super-resolution imaging of live cells: resolving submicroscopic receptor organization in the plasma membrane. Angewandte Chemie. 51 (20), 4868-4871 (2012).
- Niewidok, B., et al. Single-molecule imaging reveals dynamic biphasic partition of RNA-binding proteins in stress granules. J Cell Biol. , (2018).
- Wurm, C. A., Jakobs, S. Differential protein distributions define two sub-compartments of the mitochondrial inner membrane in yeast. FEBS Lett. 580 (24), 5628-5634 (2006).
- Schmidt, R., Wurm, C. A., Punge, A., Egner, A., Jakobs, S., Hell, S. W. Mitochondrial cristae revealed with focused light. Nano Lett. 9 (6), 2508-2510 (2009).
- Kukat, C., Wurm, C. A., Spahr, H., Falkenberg, M., Larsson, N. G., Jakobs, S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proceedings of the National Academy of Sciences of the United States of America. 108 (33), 13534-13539 (2011).
- Beinlich, F., Drees, C., Piehler, J., Busch, K. Shuttling of PINK1 between Mitochondrial Microcompartments Resolved by Triple-Color Superresolution Microscopy. ACS chemical biology. 10 (9), 1970-1976 (2015).
- Shim, S., et al. Super-resolution fluorescence imaging of organelles in live cells with photoswitchable membrane probes. Proceedings of the National Academy of Sciences. 109 (35), 13978-13983 (2012).
- Sbalzarini, I., Mezzacasa, A., Helenius, A., Koumoutsakos, P. Effects of Organelle Shape on Fluorescence Recovery after Photobleaching. Biophysical Journal. 89 (3), 1482-1492 (2005).
- Reits, E., Neefjes, J. From fixed to FRAP: measuring protein mobility and activity in living cells. Nature Cell Biology. 3 (6), E145-E147 (2001).
- Goehring, N., Chowdhury, D., Hyman, A., Grill, S. FRAP Analysis of Membrane-Associated Proteins: Lateral Diffusion and Membrane-Cytoplasmic Exchange. Biophysical Journal. 99 (8), 2443-2452 (2010).
- Bacia, K., Haustein, E., Schwille, P. Fluorescence correlation spectroscopy: principles and applications. Cold Spring Harbor protocols. 2014 (7), 709-725 (2014).
- Sukhorukov, V., Dikov, D., Busch, K., Strecker, V., Wittig, I., Bereiter-Hahn, J. Determination of protein mobility in mitochondrial membranes of living cells. Biochimica et biophysica acta. 1798 (11), 2022-2032 (2010).
- Kim, T. K., Eberwine, J. H. Mammalian cell transfection: the present and the future. Anal Bioanal Chem. 397 (8), 3173-3178 (2010).
- Graham, F. L., van der Eb, A. J. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 52 (2), 456-467 (1973).
- Wedeking, T., et al. Spatiotemporally Controlled Reorganization of Signaling Complexes in the Plasma Membrane of Living Cells. Small. 11 (44), 5912-5918 (2015).
- Mironov, S. L., Ivannikov, M. V., Johansson, M. [Ca2+]i signaling between mitochondria and endoplasmic reticulum in neurons is regulated by microtubules. From mitochondrial permeability transition pore to Ca2+-induced Ca2+ release. J Biol Chem. 280 (1), 715-721 (2005).
- Poot, M., et al. Analysis of mitochondrial morphology and function with novel fixable fluorescent stains. J Histochem Cytochem. 44 (12), 1363-1372 (1996).
- Tokunaga, M., Imamoto, N., Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells (vol 5, pg 159). Nature Methods. 5 (5), 455(2008).
- Elmokadem, A., Yu, J. Optimal Drift Correction for Superresolution Localization Microscopy with Bayesian Inference. Biophys J. 109 (9), 1772-1780 (2015).
- Barlag, B., et al. Single molecule super-resolution imaging of proteins in living Salmonella enterica using self-labelling enzymes. Sci Rep. 6, 31601(2016).
- Tokunaga, M., Imamoto, N., Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nature Methods. 5 (2), 159-161 (2008).
- Mortensen, K. I., Churchman, L. S., Spudich, J. A., Flyvbjerg, H. Optimized localization analysis for single-molecule tracking and super-resolution microscopy. Nat Methods. 7 (5), 377-381 (2010).
- Jin, S., Youle, R. J. PINK1- and Parkin-Mediated Mitophagy at a Glance. Journal of Cell Science. 125, 795-799 (2013).
- Yamano, K., Youle, R. J. PINK1 is degraded through the N-end rule pathway. Autophagy. 9 (11), 1758-1758 (2013).
- Wiedemann, N., et al. Machinery for protein sorting and assembly in the mitochondrial outer membrane. Nature. 424 (6948), 565-571 (2003).
- Thompson, R., Larson, D., Webb, W. Precise Nanometer Localization Analysis for Individual Fluorescent Probes. Biophysical Journal. 82 (5), 2775-2783 (2002).
Access restricted. Please log in or start a trial to view this content.
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados