
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Multi-Color-Lokalisierung Mikroskopie des einzigen Membranproteine in Organellen der lebenden Säugetier-Zellen
In diesem Artikel
Zusammenfassung
Hier präsentieren wir ein Protokoll für die multi-Color Lokalisierung der einzelnen Membranproteine in Organellen der lebenden Zellen. Um Fluorophore zu befestigen, sind selbst Kennzeichnung Proteine verwendet. Proteine, befindet sich in verschiedenen Membranen Kompartimenten des die gleichen Organellen lokalisiert werden können, mit einer Genauigkeit von ~ 18 nm.
Zusammenfassung
Wissen über die Lokalisierung von Proteinen in zellulären Subcompartments ist entscheidend für ihre spezifische Funktion zu verstehen. Hier präsentieren wir Ihnen eine super-Resolution-Technik, die für die Bestimmung der Microcompartments, die zugänglich für Proteine sind ermöglicht durch Erzeugung von Lokalisierung und tracking-Karten dieser Proteine. Darüber hinaus werden durch multi-Color-Lokalisierung-Mikroskopie, die Lokalisierung und Überwachungsprofile von Proteinen in unterschiedlichen Subcompartments gleichzeitig erhalten. Die Technik ist speziell für lebende Zellen und basiert auf die sich wiederholenden Darstellung der einzigen mobilen Membranproteine. Interessierenden Proteine sind genetisch mit bestimmten, sogenannten Self Labelling Tags fusioniert. Diese Tags sind Enzyme, die mit einem Substrat kovalent zu reagieren. Auf diese Substrate konjugiert sind fluoreszierende Farbstoffe. Reaktion der Enzym-markierten Proteine mit der Fluoreszenz beschriftet Substrate Ergebnisse in markierte Proteine. Hier werden Tetramethylrhodamine (TMR) und Silizium Rhodamine (SiR) als Fluoreszenz-Farbstoffen beigefügt, die Substrate der Enzyme eingesetzt. Substrat-Konzentrationen in der pM, nM-Bereich verwenden, wird Sub stöchiometrischen Kennzeichnung erreicht, der deutliche Signale führt. Diese Signale werden lokalisiert, mit ~ 15 – 27 nm Präzision. Die Technik ermöglicht multi-Color Imaging von einzelne Moleküle, wobei die Anzahl der Farben durch die verfügbaren Membran durchlässig Farbstoffe und das Repertoire der selbstbeschriftung Enzyme begrenzt wird. Wir zeigen die Machbarkeit der Technik durch die Bestimmung der Lokalisation des Enzyms Qualitätskontrolle (Pten)-induzierte Kinase 1 (PINK1) in verschiedenen mitochondrialen Kompartimenten während seiner Verarbeitung in Bezug auf andere Membranproteine. Der Test für echte physikalische Wechselwirkungen zwischen unterschiedlich markierte einzelne Proteine durch Einzelmolekül Bund oder Co-Tracking ist jedoch beschränkt, weil die niedrigen Kennzeichnung Grad die Wahrscheinlichkeit verringern dafür, dass zwei benachbarte Proteine gleichzeitig beschriftet. Während die Technik für imaging-Proteine in Membran Fächer stark ist, ist in den meisten Fällen es nicht angebracht, die Lokalisierung der hochmobilen lösliche Proteine bestimmen.
Einleitung
Das Ziel dieses Protokolls ist, bieten ein bildgebendes Verfahren zu lokalisieren und einzelne Membranproteine in lebenden Zellen zu verfolgen. Wir nennen diese Methode Tracking und Localization Microscopy (Talmor)1,2. Talmor ist wie stochastische optische Rekonstruktion Mikroskopie (Sturm)3 und Photoaktivierungen Lokalisierung Fluoreszenzmikroskopie ((F) PALM)4,5ein einzelnes Molekül-basierte Fluoreszenz Lokalisierung Technik. Es unterscheidet sich jedoch in der Weise, dass die Mobilität von Membranproteinen in Kombination mit sich wiederholenden Darstellung desselben Moleküls an verschiedenen Positionen zeigt die Microcompartment beschriftet, die zugänglich für das mobile Protein ist. Das heißt, werden die möglichen Lokalisierungen des Proteins von der Architektur der Organellen und durch die Mobilität der Protein-1festgelegt. Die Methode ist komplementär zu verschiedenen anderen Höchstauflösung Techniken6,7,8 , weil es zeigt, Lokalisierung und die Flugbahn Karten durch bildgebende mobile Proteine. Die Kennzeichnung basiert auf der Verwendung gentechnisch veränderter Fusionsproteine, die per se nicht fluoreszierenden sind. Diese Schmelzverfahren Proteine sind Enzyme selbstbeschriftung, die kovalent mit einem Substrat zu einem Farbstoff konjugiert zu reagieren. Dieses Verfahren hat den Vorteil, dass die Kennzeichnung Grad kann durch die Menge an zusätzlichen Substrat gesteuert. Darüber hinaus ermöglicht es die Farbe der Fluoreszenz, abhängig von der gewählten konjugierten Farbstoff variieren. Einige selbst Kennzeichnung Enzym-Tags sind verfügbar9. Ein weiterer Vorteil der Verwendung selbstbeschriftung Enzym-Tags ist, dass die konjugierte Farbstoffe sind in der Regel stabiler und heller als fluoreszierende Proteine1 und einzelne Proteine daher mehr aufgezeichnet werden können und vieles mehr, eben bis sie gebleicht werden. Dies ermöglicht die Aufnahme von Trajektorien von mobilen Proteinen und der Gewinnung von Diffusion Koeffizienten10,11.
Hier zeigen wir die Machbarkeit der Talmor mit mitochondrialen Membranproteine, aber es kann auch für andere Intra und extra zellulare Membranproteine, einschließlich andere Zelle Arten12,13angewendet werden. Wir zeigen, dass multi-Color Talmor weiter die gleichzeitige Unterscheidung von Proteinen in verschiedenen Subcompartments in Ergänzung zur bestehenden Höchstauflösung Fluoreszenz-Mikroskopie-Techniken14,15, ermöglicht 16. Talmor ist kompatibel mit live Cell imaging-17. Die Foto-Physik von der gewählten Rhodamine Tetramethylrhodamine (TMR) und Silikon-Rhodamien (SiR), insbesondere ihre Helligkeit und Stabilität, ermöglicht Rekord einzelne Membranproteine über mehrere Frames mit Lokalisierung (und die Flugbahn) Karten. Talmor ist jedoch für die Lokalisierung von löslichen Proteinen mit hohen Diffusionskoeffizienten beschränkt, da die Bewegungsunschärfe zu hoch ist und die gesammelten Photonen pro Frame zu niedrig für die korrekte Lokalisierung sind. Außerdem erfordert Talmor weniger erregerleistung als z.B. Sturm oder stimulierte Emission Depletion (STED) Mikroskopie6,7, phototoxische Effekte reduzieren. Dies ist wichtig, da phototoxische Stress oft Organellen Morphologie18 und damit Mobilität Analyse19beeinflusst. Zusammenfassend lässt sich sagen, wir präsentieren mehrfarbige Talmor in lebenden Zellen als eine Technik, die eine Lücke zwischen der Lokalisierung Mikroskopieverfahren Sturm/STED füllt / (F) PALM und Techniken, die Protein Mobilität wie Fluoreszenz Erholung nach Immunofluoreszenz analysieren (FRAP)20 ,21, Fluoreszenz Korrelation Spektroskopie (FCS)22und Fluoreszenz überqueren Korrelation Spektroskopie (FCC)11,23.
Access restricted. Please log in or start a trial to view this content.
Protokoll
Das folgende Protokoll folgt den Richtlinien der lokalen Institution Forschung Ethik-Kommission.
1. Methoden
-
Zellkultur
- Zellen, z. B. HeLa-Zellen (menschliche Zervix-Karzinom), in ein T25 Zelle Kultur Fläschchen mit 5 mL Nährmedium bei 37 ° C und 5 % CO2zu kultivieren.
Hinweis: Für die Bildgebung, teilen Sie die Zellen auf vorbereiteten Deckgläsern (siehe Schritte 1.3 und 1.4) und Imaging-Medium halten.
- Zellen, z. B. HeLa-Zellen (menschliche Zervix-Karzinom), in ein T25 Zelle Kultur Fläschchen mit 5 mL Nährmedium bei 37 ° C und 5 % CO2zu kultivieren.
-
Zelle Transfektion
Hinweis: Verwenden Sie Zell-Linien, die die tagged Proteine möglichst stabil Ausdrücken24 , starke Überexpression zu vermeiden. Passen Sie für Transiente Transfektion die Höhe der Plasmid-DNA für die Transfektion verwendet. Beispielsweise wenn Ca2 + Phosphat Transfektion25 verwendet wird, transfizieren Sie (80 – 90 % Konfluenz) Zellen in der Kulturschale 3,5 cm Zelle mit 2,5 – 5 µg von Plasmid DNA. Wenn Sie doppelte Transfektion durchführen, verwenden Sie 2,5 µg pro jedes Plasmid-Konstrukt.- Verwenden Sie für dual Farbe Experimente eine Zell-Linie mit stabilen Expression von einem selbst Kennzeichnung Protein und vorübergehend mit dem Plasmid Codierung der anderen selbst Kennzeichnung Protein17transfizieren.
Hinweis: Hier wurden für dual Farbe Experimente, HeLa-Zellen verwendet, die stabil selbst Kennzeichnung Proteine PINK1-Halo-Tag und Tom20-fSNAP-Tag zum Ausdruck.
- Verwenden Sie für dual Farbe Experimente eine Zell-Linie mit stabilen Expression von einem selbst Kennzeichnung Protein und vorübergehend mit dem Plasmid Codierung der anderen selbst Kennzeichnung Protein17transfizieren.
-
Reinigung von Deckgläsern
- Legen Sie die Deckgläsern in ein Becherglas. 30 mL H2O in den Becher mit den Deckgläsern und schütteln Sie sanft, um Staub von der Oberfläche zu entfernen.
- Sammeln Sie die Deckgläsern mit einer Pinzette zu und trocknen Sie sie mit einem Strom von Stickstoff.
- Entfernen Sie organische Verunreinigungen auf der Oberfläche von Deckgläsern, z. B.durch Plasmareinigung.
Hinweis: Um weitere Kontamination des Werkstoffes Glas zu vermeiden, tragen Sie Handschuhe beim Umgang mit den Deckgläsern.
Achtung: Deckgläsern von Plasma Reinigung gereinigt werden, wird nur die obere Seite der Deckgläsern gereinigt; Verwenden Sie diese Seite für Beschichtung mit Poly-L-Lysin-Polyethylen Glycol-Arginin-Glycin-Aspartat-(PLL-PEG-RGD) (Abschnitt 1.4) und Cell seeding (Abschnitt 1.5).
-
Deckglas Beschichtung mit PLL-PEG-RGD
Hinweis: PLL-PEG-RGD ist ein Poly-L-Lysin (PLL) Derivat mit einem Polyethylenglykol (3.000 Da) und ein cysteine-glycine-arginine-glycine-aspartate-serine Peptid (CGRGDS) angebracht. PLL bindet an die negativ geladenen Glasoberfläche und bildet einen PEG-Pinsel. Dies reduziert drastisch die unspezifische Bindung des geladenen Fluoreszenzfarbstoffen. Darüber hinaus die RGD-Motiv imitiert das Signalpeptid des Rezeptors Integrin und fördert dadurch Integrin-vermittelte Einhaltung von Zellen, die sonst nicht gut haften.- PLL-PEG-RGD als zuvor beschriebenen26vorbereiten. Kurzum, lösen Sie PLL-PEG-RGD 0,8 mg in 1 mL PBS auf. Fügen Sie 10 µL der PLL-PEG-RGD-Lösung auf der Oberseite von einem sauberen deckgläschen.
- Nehmen Sie eine zweite Deckglas und legen Sie es mit einer sauberen Oberfläche kopfüber auf die erste deckgläschen (das PLL-PEG-RGD Tropfen an der Spitze); Dadurch wird die PLL-PEG-RGD-Lösung zwischen zwei Deckgläsern sandwiching.
- Setzen Sie die Sandwich Deckgläsern in ein Becherglas vorsichtig und Inkubation für 1 h bei Raumtemperatur in einer staubfreien trockenen Umgebung.
- Fügen Sie nach 1 h 30 mL von H2O das Becherglas auf Deckgläsern mit Wasser vollständig bedecken hinzu.
- Schütteln Sie das Becherglas bis die Deckgläsern voneinander lösen.
- Benutzen Sie eine Pinzette zu sammeln Deckgläsern aus dem Wasser und trocknen Sie sie unter einem Strom von Stickstoffgas.
Hinweis: Die beschichteten Deckgläsern können für ein paar Tage in einem Glas trockenen, sterile Petrischale mit Deckel gespeichert werden.
-
Vorbereitung der Probe für die Bildgebung
- Übertragen der einzelnen beschichteten Deckgläsern in einer 35 mm-Zelle-Kulturschale mit der PLL-PEG-RGD beschichtete Oberfläche nach oben und 2 mL von imaging-Medium an der Spitze.
- Fügen Sie hinzu ~ 500.000 trypsiniert Zellen (200 – 500 µL), die den selbst Kennzeichnung Tags an die jeweiligen Membranproteine, die 2 mL Medium in die Zelle Kulturschale mit beschichteten Deckglas imaging Ausdrücken. Schütteln sanft mit der Hand um eine homogene Verteilung der Zellen zu gewährleisten eine gleichmäßig Zellschicht.
- Inkubieren Sie Zellen bei 37 ° C und 5 % CO2 , bis 80 % Konfluenz erreicht ist.
Hinweis: Zellproben sollte 3 Tage vor Bildgebung und 1 Tag vor Transfektion ausgesät werden. Zellen, die das Protein des Interesses stabil zum Ausdruck bringen, können 2 Tage vor Bildgebung ausgesät werden. Später werden nur Zellen gewachsen auf dem Deckglas abgebildet.
-
Kennzeichnung von tagged Proteine
Hinweis: Die meisten fluoreszierenden Substrate müssen in wasserfreiem DMSO aufgelöst werden. Wir empfehlen Ihnen, Stammlösungen von 1 µM fluoreszierende Substrat verwenden, wenn die Kennzeichnung Endkonzentration 0,2 – 30 nM17ist. Verwenden Sie für imaging-Membranproteine innerhalb der Zellen, Membran durchlässig fluoreszierende Substrate.- Das bildgebendes Medium auf 37 ° C im Wasserbad erwärmen.
- Pipette 1 mL vorgewärmten bildgebendes Medium in ein 2 mL-Gefäß mit Deckel. Fügen Sie 0,2 – 30 µL fluoreszierende Substrate von 1 µM Stammlösungen, die endgültige Kennzeichnung Lösung vorzubereiten (Endkonzentration: 0,2 – 30 nM).
- Wirbel die Kennzeichnung Lösung für 10 s.
- Das Medium in der Kulturschale 35 mm mit den Zellen auf einem deckgläschen zu ersetzen (siehe Punkt 1.5) von 1 mL der vorbereiteten Kennzeichnung Lösung.
- Inkubieren Sie die Zellen in der Kennzeichnung Lösung bei 37 ° C und 5 % CO2 für 20-30 min.
- Waschen Sie die Zellen mit 2 mL PBS einmal, dann mit 2 mL Medium zweimal imaging. Zu guter Letzt pipette 1 mL frisches bildgebendes Medium in die Zelle-Schale und setzen Sie die Probe wieder in den Brutschrank bei 37 ° C und 5 % CO2 für mindestens 1 h. Tauschen Sie vor Bildgebung bildgebende Medium noch einmal.
Hinweis: Wenn das Experiment zum ersten Mal ausgeführt wird, bestätigen Sie korrekte Ausrichtung der selbst markierte Proteine, Membranen der Organellen durch Färbung der Organellen mit handelsüblichen Organellen bestimmte Farbstoffe27,28. In diesem Fall auch 100 – 200 nM von Substrat für die selbst-Kennzeichnung Enzyme produzieren starke Signale.
-
Vorbereitung einer fluoreszierenden Bead Probe
Hinweis: Um die optische Drift zu bestimmen und Bilder der verschiedenen Kanäle ausrichten, werden Multi-Color fluoreszierende Perlen (0,1 µm) verwendet. Mit den aufgenommenen Bildern wird ein affinen Transformationsmatrix für die zwei Emission Kanäle generiert werden.- Verdünnen Sie die Lösung von Perlen auf 1 % mit reinen H2O.
- Platz 5 Tropfen die fertige Lösung mit der Fluoreszenz-Perlen an fünf verschiedenen Positionen auf einem gereinigten deckgläschen (siehe Punkt 1.3).
- Lassen Sie fluoreszierende Bead Probe auf einem sauberen Werkbank trocknen.
Hinweis: Die Probe kann wiederverwendet werden; Daher decken Sie die Probe mit Alufolie zu Kontamination zu vermeiden und halten es bei einer 4 ° C.
2. Mikroskopie
-
Versuchsaufbau
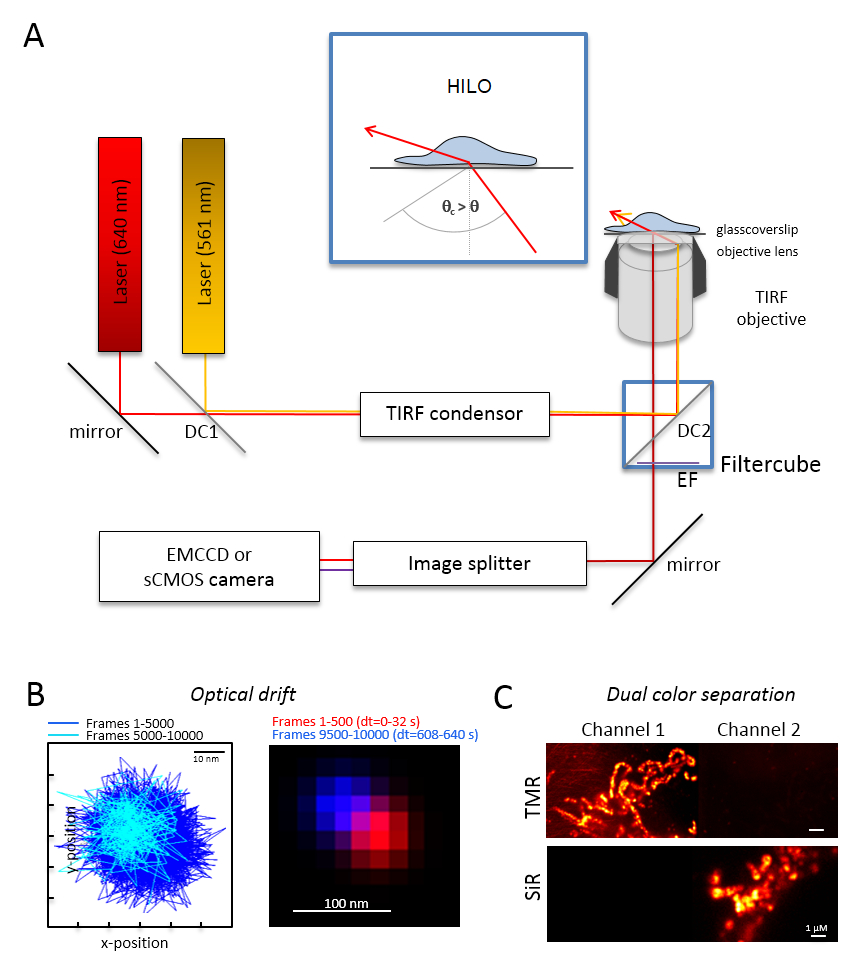
Hinweis: Eine grundlegende Mikroskopie-System für die zweifarbige Einzelmolekül Bildgebung basiert auf einem inversen Mikroskop: Es ist ausgestattet mit zwei Lasern über eine multi-Modus-optische Polarisation Aufrechterhaltung Monomode-Faser in einem einzigen Totalreflexion (TIR) gekoppelt Kondensator, objektives Öl Immersion entworfen für TIRF, ein Polyband Emission Filter, eine Bild-Splitter und eine hochempfindliche Kamera (Abbildung 1). Ein TIR-Kondensator ist notwendig, das Wechseln zwischen den Epi-, stark geneigt für die kontinuierliche Optimierung von der Einfallswinkel ermöglicht und laminierte optische Blatt (stark geneigten dünne Beleuchtung (HILO)29) und der TIRF Erregung Modus mit optimiert Eindringtiefe. Bilder werden mit einem hochempfindlichen gekühlten Detektorsystem erworben, z. B. eine Rückseite beleuchteten Elektron Multiplikation Kamera gekoppelten Geräts (EMCCD) in Rechnung gestellt (Quantenausbeute QE > 90 %) oder eine Kamera sCMOS (QE > 80 – 90 %).- Bestimmen Sie die optische Drift durch bildgebende fluoreszierende Perlen (siehe Punkt 2.2) unter den gleichen Bedingungen wie die, die später verwendet werden für das Experiment, z.B., wenn 10.000 Bilder im Experiment erfasst werden, aufzeichnen auch 10.000 Frames mit der Wulst-Probe. Vergleichen Sie für die Bestimmung der optischen Drift die Position der Perlen in den ersten Frame und dem letzten erfassten Frame (Abbildung 1 b). Falls erforderlich, später korrigieren Sie die Bildserien für optische Drift30 und/oder verwenden Sie Drift stabile Umgebungen.
- Statten Sie den Filter-Würfel mit der entsprechenden dichroitischer Strahlteiler, z.B.für orange und rote Fluoreszenz plus die entsprechende Emission Filter für orange Fluoreszenz und rote Fluoreszenz. Die Bild-Splitter mit geeigneten Filtern auszustatten. Überprüfen Sie die mögliche Leckage von Signalen von einem Kanal in den anderen Kanal durch die Aufnahme von einzelnen Farbmuster in beiden Kanälen (Abbildung 1).

Abbildung 1 : Optische Layout für Multi-Color Tracking und Lokalisierung Mikroskopie (Talmor) mit orange und Rot Strahler. (A) Inverted Mikroskop Setup mit mindestens zwei Erregung Laser, TIRF Kondensator ein TIRF geeignet Ziel, ein Bild-Splitter und eine sensible Kamera. Einschub: Organellen innerhalb der Zellen zu begeistern, muss der Winkel des einfallenden Strahls festgelegt werden kleiner als der Grenzwinkel für TIRF zu sehr geneigt und optische Blatt Beleuchtung (HILO kaschiert). DC1: Dichroitischen Spiegel 1; DC2: Dichroitischen Spiegel 2. EF: Emission Filter. (B) Prüfung auf optische Drift durch bildgebende Positionen einer fluoreszierenden Raupe für 10.000 Frames mit den gleichen Frame-Rate ab den folgenden Experimenten (hier: 15 Hz). Verbundenen Positionen der ersten 500 Rahmen und die letzten 500 Bilder zeigen die Drift. Auch eine zusammengefügte Bild mit der Position des ersten und dem letzten Frame in rot und blau zeigen einen minimalen Drift. Die Drift ist, dass der Abstand zwischen dem Zentrum der Signale durch die Gesamtaufnahmezeit hier 125 pm/s. (C) Check auf die klare Trennung von Signalen, hier TMR und SiR dividiert. Für beide Kanäle wurden kumulative Summe Bilder von 3.000 Frames (TMR in Kanal 1) und SiR in Kanal 2 erzeugt. SiRHTL hing an Tom20-HaloTag und TMRHTL , OxPhos Komplex V-HaloTag. Farben sind falsche Farben. Skalieren von Balken = 100 nm (B) und 1 µm (C). Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}
-
Physische Ausrichtung des Bildes Splitter computergenerierte Bilder
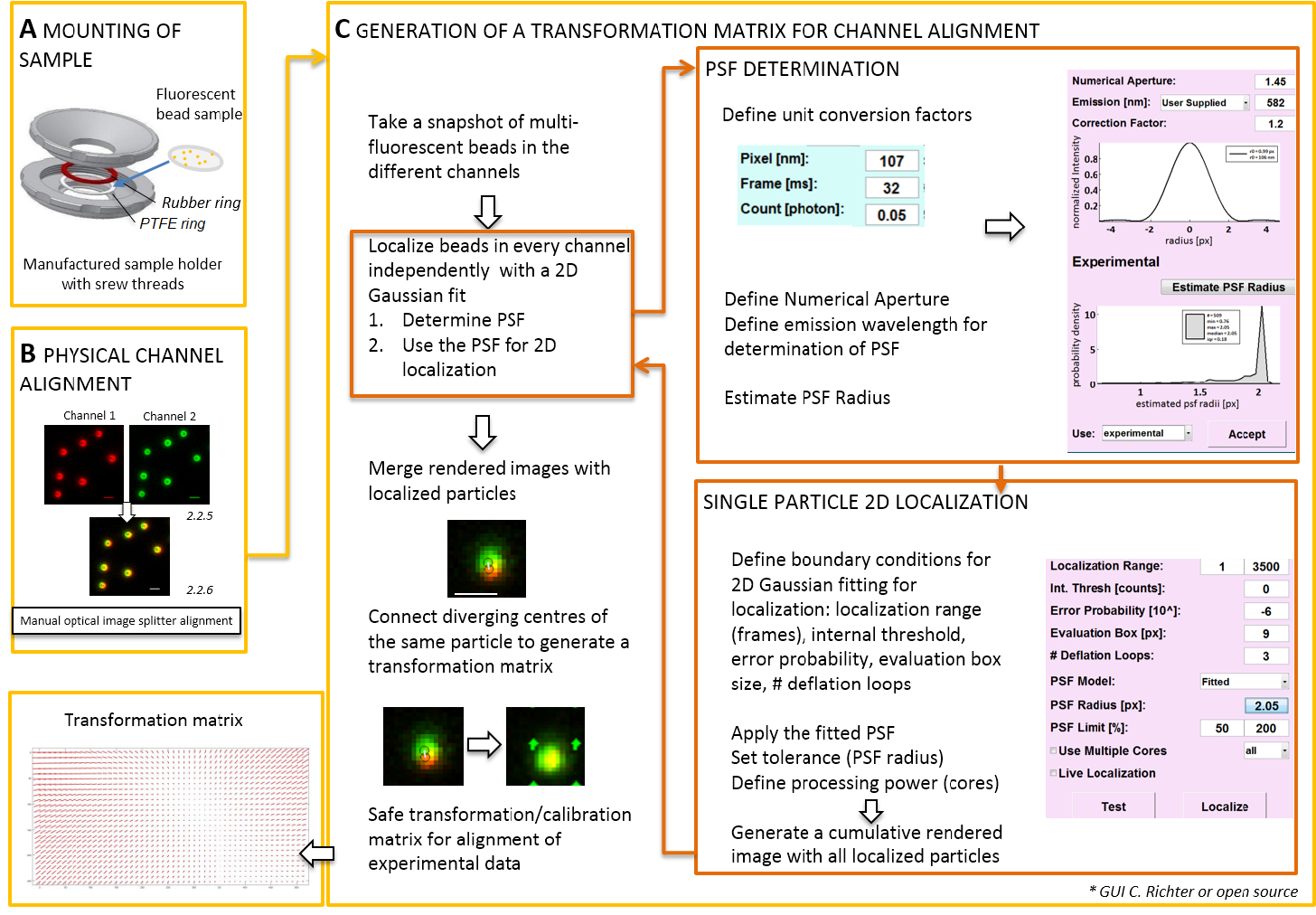
Bemerkung: Für die Montage der Probe auf einem deckgläschen vorbereitet, ein self-made Probenhalter kann verwendet (Abbildung 2A). Zur Vermeidung von Staub setzen etc. fallen in die Probe Sie den Deckel von der Kulturschale lose auf die Kammer, wenn montiert. Der gleichen Probenhalter kann verwendet werden, um das Deckglas mit fluoreszierenden Perlen oder Zellen zu montieren; Wenn Zellen abgebildet werden, fügen Sie 0,5-0,8 mL von imaging-Medium. Bild-Splitter teilt das Bild in zwei oder mehrere Spektral Kanäle getrennt und projiziert sie Side-by-Side auf die gleiche Kamera. Dieser Prozess möglicherweise stellt systematische Verzerrungen zwischen den Kanälen durch unterschiedliche optische Wege durchzogen und behindert direkte ns1 Analyse. Daher zuerst durchführen, körperliche Ausrichtung und zweitens nach Korrektur Ausrichtung mit einer Transformationsmatrix. Für beide Prozesse Ausrichtung sollten fluoreszierende Perlen über das Sichtfeld homogen verteilt werden.- Montieren Sie die vorbereitete Probe mit fluoreszierenden Perlen in der Probenhalter zwischen Polytetrafluorethylen (PTFE)-Ring und den roten Gummiring (Abbildung 2A).
- Starten Sie das Mikroskop, alle Hardware-Komponenten und alle Software für Mikroskopie.
- Reinigen Sie das Ziel und dem unteren Rand des Deckglases mit einem fusselfreien Gewebe Abwischen mit Isopropanol benetzt. Trocknen Sie anschließend beide Elemente mit einem frischen, fusselfreien Tuch. Legen Sie einen Tropfen Immersionsöl auf die Pupille der Objektivlinse.
- Der Probenhalter mit der Wulst Probe auf den Mikroskoptisch zu platzieren, so dass die Unterseite der das Deckglas das Öl berührt. Perlen mit Übertragung Licht oder eine Laserlinie im Mittelpunkt.
- Passen Sie die Kraft der beiden Erregung Laser ähnlich Signalintensität in den beiden Fluoreszenz-Kanälen zu erreichen. Suche für ein Gebiet mit vielen verschiedenen fluoreszierenden Signale.
- Generieren Sie eine zusammengeführte Ansicht der fluoreszierenden Kanäle mithilfe der Kamera-Steuerungssoftware. Dann verwenden Sie die Schrauben an den Bild-Splitter manuell kippen, die internen Spiegel der Bild-Splitter, die beste Überlagerung der Signale von fluoreszierenden zweikanalig (Abb. 2 b) zu erreichen.
Hinweis: Achtung! Überschreiten Sie den Dynamikbereich der Kamera nicht.
-
Ausrichtung der Spektral getrennte Kanäle durch Software räumliche Transformation durchführen
Hinweis: Der folgende Abschnitt zeigt die Post-Korrektur Ausrichtung und Lokalisierung Verfahren mit unserem Software-Plugin (auf Anfrage erhältlich).- Beginnen Sie die TIRF Mikroskop Steuerungssoftware und anzeigen Sie einzelne Kanäle in den live-Stream-Modus. Nehmen Sie eine Snapshot-Bild spannende Fluoreszenz in allen Kanälen (Abbildung 2).
- Dieser Snapshotbild verwenden, um die Transformationsmatrix zu produzieren (siehe Abbildung 2).
Hinweis: Die Transformationsmatrix eine räumliche Transformation, in der Regel eine affine, dient, die für Übersetzung (Divergenz von Signalen aus einer Hand Punkt zwischen zwei Kanälen) korrigiert. - Starten Sie das Software-Analyse-Plugin (erhalten Sie auf Anfrage von unserem Labor, siehe Abbildung 2).
- Laden Sie die zuvor aufgenommene dual Farbbilder (siehe Punkt 2.2) von fluoreszierenden Perlen in die Software. Wählen Sie die verwendete Ausrichtung der fluoreszierenden Kanäle. Dann klicken Sie auf "Ja", wenn gefordert, "Kalibrieren Bilder" und wählen Sie die zuvor aufgenommenen Schnappschuss.
- Öffnen Sie den "UNIT MANAGER" um Einheit Umrechnungsfaktoren (Pixelgröße, Frame-Rate, Photon Umrechnungsfaktor) zu definieren.
- Öffnen Sie den "LOCALIZATION MANAGER". Bestimmen Sie der Punkt zuerst Funktion (PSF) verteilt. Drücken Sie die Taste: "PSF Radius". Definieren Sie im "PSF Estimator" Fenster, das geöffnet wird die numerische Apertur und die maximale Emission. Starten Sie "Schätzung PSF Radius" klickst. Akzeptieren Sie die erhaltenen experimentellen PSF definieren Feld Bewertung, Anzahl der Deflation Schleifen und wie viele Kerne des Computers für die Berechnung verwendet werden. Drücken Sie die Taste "lokalisieren", um die Intensitätsverteilung der einzelnen Partikel durch eine 2D symmetrischen Gaußsche Funktion (Abbildung 2) passend zu starten.
- "Accept" erhaltenen experimentellen PSF definieren Feld Bewertung, Anzahl der Deflation Schleifen und wie viele Kerne des Computers für die Berechnung verwendet werden. Drücken Sie die Taste "lokalisieren", um die Intensitätsverteilung der einzelnen Partikel durch eine 2D symmetrischen Gaußsche Funktion (Abbildung 2) passend zu starten.
- Öffnen Sie die "KALIBRIERUNGSMANAGER". Im zusammengeführten gerenderten Bild der beiden Kanäle werden die ursprünglichen Signale und die lokalisierten Centers angezeigt. Wählen Sie die "affinen" Modus. Manuell verbinden Sie die entsprechenden Paare von lokalisierten Zentren in den beiden Kanälen, die entstanden sind aus der gleichen fluoreszierende Wulst durch eine Verbindungslinie ziehen.
- Verbinden Sie die entsprechenden Signale verteilt das Sichtfeld. Danach drücken Sie auf "akzeptieren". Speichern Sie die Kalibrierung.
Hinweis: Die räumliche Transformation auf jedes fluoreszierende Kügelchen abgetastet und dazwischen interpoliert. Die extrahierten Transformationsfunktion stellt eine Verschiebung-Feld-Δr(x,y), die verwendet wird, um anschließend die experimentelle zweifarbige Einzelmolekül-Lokalisierungen korrigieren, sodass sie innerhalb ihrer Lokalisierung Präzision überlagern. Die räumliche Transformationsmatrix ist in der Regel eine affine, die für Translation, Skalierung und Rotation zwischen den Kanälen mit Nanometer-Genauigkeit korrigiert, und es kann diese manuelle 1: 1-Zuordnung (Abbildung 2) entnommen werden.

Abbildung 2 : Workflow für dual Farbausrichtung. (A) das Deckglas mit fluoreszierenden Perlen in einen Probenhalter zwischen einem PTFE und einem Gummiring angebracht ist. Dann die obere und untere Teil der Kammer sind miteinander verschraubt. (B) physische Ausrichtung des Kanal-Ansichten, die von der Bild-Splitter erzeugt werden. Fluoreszierende Signale aus Perlen (0,1 µm) in zwei Kanälen aufgezeichnet (grün und rot, Falschfarben) werden zusammengeführt. Die entsprechenden Schrauben an das optische Bild-Splitter sind manuell gedreht, bis die beste Überlagerung der verschiedenen Signale erreicht ist (gelbe Farbe, untere Leiste). (C) Generation einer Transformationsmatrix für post-processive Kanal Ausrichtung. Für die präzise Lokalisierung eines Teilchens ist es notwendig, um festzustellen, dass der Punkt Funktion (PSF) in Abhängigkeit der Emissionswellenlänge und die numerische Apertur des Objektivs zu verbreiten. Das Zentrum von einem PSF kann durch seine Intensität Profil analysiert durch einen symmetrischen zweidimensionalen Gaußschen passen bestimmt werden. Die daraus resultierende Lokalisierung des Peak Signal wird dann auf die ursprünglichen, verschwommene Signale projiziert. In einem zusammengeführten Bild sind die lokalisierten Zentren der Signale von den beiden Kanälen verbunden, um eine Transformationsmatrix zu erzeugen, die später für die post-processive Ausrichtung der experimentellen Daten verwendet wird. Skalieren von Balken = 1 µm (B, C). Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}
-
Einzelmolekül-Bildgebung der Mitochondrien-Membranproteine
Hinweis: Alle Experimente sind bei Raumtemperatur durchgeführt. T-Zellen oder nicht-anhaftende Zellen müssen in Agarose vor imaging31immobilisiert werden.- Montieren Sie die Probe mit adhärenten Zellen auf das Deckglas zwischen Gummi- und PTFE-Ringe (Abb. 3A). Füllen Sie die Kammer mit 0,5 – 0,8 mL Medium imaging.
- Wiederholen Sie die Schritte 2.2.2–2.2.5.
- Passen Sie die Beleuchtung Winkel TIRF Mikroskop controlling Software ein Einfallswinkel zu erstellen, die kleiner ist als der Grenzwinkel für TIR-Modus die jeweiligen Region von Interesse über eine HILO Blatt32 (HILO-Modus, Abb. 1A) zu begeistern.
Achtung: Vermeiden Sie direkten Blickkontakt mit dem Laserstrahl! - Die EM-Verstärkung und wählen Sie eine Belichtungszeit geeignet für das Experiment, das genügende Photonen pro Frame sammelt.
- Legen Sie die Laserleistung erreichen einen hohen Signal-Rausch Verhältnis (S/N) (Abb. 3 b), da die Lokalisierung Präzision direkt S/N33 (Abbildung 3) entspricht.
- Suchen Sie einen Bereich in der Zelle Peripherie mit nicht überlappenden, längliche Mitochondrien und Einzelmolekül-Signale (Abbildung 3D; Zusätzliche Video 1). Wenn kein einziges Molekül Signale sichtbar sind, warten Sie, bis die bleichen Ergebnisse in der Erscheinung der Einzelmolekül-Signale (Abbildung 3E).
- Zeichnen Sie auf, bis die Anzahl der Signale für angemessene Fortsetzung (in der Regel 1.000 – 10.000 Bilder je nach dem Bleichen Verhalten der Fluoreszenzfarbstoff, Abbildung 3F) zu niedrig ist.
- Starten Sie die Imaging Verarbeitung Software und suchen Sie nach mitochondriale Strukturen durch eine kumulative Summe gerenderten Bild von mindestens 1.000 aufgezeichneten Frames (Abbildung 3).
Hinweis: Die schnellste Bildrate wird durch die Anzeige-Bereich vorgegeben. Das Sichtfeld für einen Kanal ist durch eine zweifarbige Bild-Splitter (512 x 512 Pixel), 256 x 512 Pixel und für ein Quad-Color, 256 x 256 Pixel reduziert. So ist dies für die Verwendung eines Bild-Splitters für zwei Farben, 30 Hz. Set Frame-Transfer-Modus auf die niedrigste mögliche Anzeige Zeit zu erreichen. - Software-Analyse-Plugin und Last Rohdaten zu starten. Wählen Sie den Kanal Orientierung und laden Bilder. Verwenden Sie die Transformationsmatrix aus Schritt 2.3.9 gefragt, für "Calibrate Bilder". Kanäle werden separat angezeigt.
- Öffnen Sie "UNIT MANAGER" wie zuvor um Einheit Umrechnungsfaktoren für jeden Kanal zu definieren. Öffnen Sie die "LOCALIZATION MANAGER" für jeden Kanal. Dann fügen Sie Feld Bewertung, Anzahl der Deflation Schleifen definieren die theoretische PSF für die Bedingungen verwendet und festlegen, wie viele Kerne des Computers für die Berechnung verwendet werden. Schließlich drücken Sie die Taste "lokalisieren", um lokalisierte einzelne Teilchen (Abbildung 3 H; Zusätzliche Video 2).
- Beachten Sie, dass das Programm eine kumulative hochauflösendes Bild zeigt schließlich generieren wird lokalisiert alle Partikel (Abbildung 3I).
- Führen Sie Analysen, z. B.durch open-Source-Software oder unsere Software auf Anfrage erhältlich.
- Verfolgen Sie die einzelnen Moleküle in beiden lokalisierten Kanälen, z. B.mit dem Multi-Ziel Tracer-10
Hinweis: Schritt 2.4.13 braucht (experimentell) Vorkenntnisse über die Diffusionsfähigkeit der Proteine von Interesse für die Randbedingungen korrekt eingestellt. In der Regel ist die Suche nach der richtigen Randbedingungen ein iterativer Prozess.

Abbildung 3 : Schritte während Einzelmolekül Lokalisierung Mikroskopie. (A) A Deckglas mit der Probe wird zwischen der oberen und unteren Teil (grau) der hausgemachte Probenhalter (entworfen durch J. Bereiter-Hahn) montiert. Ein Gummiring (rot) und einem PTFE-Ring (weiß) versiegeln das System von oben und unten das Deckglas, wenn die Probenhalter Teile Schraube zusammen sind. (B) Signal-Rausch-Verhältnis des TMR-Signals. (C) Lokalisation Präzision Histogramm für alle lokalisierten Teilchen berechnet. (D) Wahl einer angemessenen Region für imaging, hier der Zelle Peripherie mit klar getrennten Mitochondrien. (E) Aufnahme und Bildverarbeitung: ein Einzelbild mit unterschiedlichen Einzelmolekül-Signale angezeigt (hier einzelne Moleküle des CV-HaloTag/TMRHTL aufgenommen wurden). (F) Intensität der TMR über die Aufnahmezeit. (G) kumulative Summe von 3.000 Frames, unverarbeitete Bild. (H) Partikel des CV-HaloTag/TMRHTL mit einer 2D Gaußsche Funktion von einem einzelnen Frame lokalisiert. (ich) kumuliert, gerendert Summe Bild zeigt alle lokalisierten CV-HaloTag/TMRHTL -Partikel aus 3.000 Frames. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}
Access restricted. Please log in or start a trial to view this content.
Ergebnisse
Multi-Color Imaging und ns1 Analyse kann helfen, die Sub-Organellen Lokalisierung der Proteine festzustellen. Wir haben das früher mit der cytosolischen Phosphatase und tensin Homolog, PINK1, das Sub-mitochondriale Standorten aufgrund der Verarbeitung von mitochondrialen Proteasen17hat bewiesen. PINK1 ist ein wichtiger Faktor, die Gewährleistung der mitochondrialen Funktion34,35. Um festzustellen, dass di...
Access restricted. Please log in or start a trial to view this content.
Diskussion
Hier wurde eine Technik für dual Farbe Einzelmolekül Lokalisierung von mobilen Membranproteine vorgestellt. Nach dem Protokoll Membranproteine sind verschmolzen, zur selbstbeschriftung Proteine, die mit Rhodamin Farbstoffe TMR und SiR konjugiert zu ihrer jeweiligen Substrate reagieren. Rhodamin Farbstoffe sind hell und Photostabil und damit für sich wiederholende bildgebenden1. Für eine erfolgreiche Leistung müssen mehrere Bedingungen und kritische Themen im Auge behalten werden.
Access restricted. Please log in or start a trial to view this content.
Offenlegungen
Die Autoren haben nichts preisgeben.
Danksagungen
Die Autoren möchten die Biophysik Gruppe und Jacob Piehler an der Universität Osnabrück für kontinuierliche Unterstützung, Wladislaw Kohl für technische Hilfe und Vorbereitung des Materials und der CellNanOs-Vorstand für die Bereitstellung von Mikroskopen für den Einsatz danken. Das Projekt wurde von der SFB 944 finanziert.
Access restricted. Please log in or start a trial to view this content.
Materialien
| Name | Company | Catalog Number | Comments |
| (2-(4-(2-hydroxyethyl)-1-piperazinyl)-ethanesulfonic acid, 1 M) (HEPES) | Biochrom | #1104E | |
| DC1: Dichroid beam splitter | Chroma | 640 dcxr | NC506031 |
| DC2: Polychroic Mirror, beamsplitter | Chroma | zt405/488/561/640rpc | discontinued |
| Dulbecco´s Phosphate-Buffered Saline (PBS) 1x (w/o Ca & Mg) | Sigma-Aldrich & Co. | #RNBF8311 | |
| Earle´s MEM without phenol red, without L-Glutamine and without NaHCO3 containing 1% FBS, 0.1% HEPES, 0.1% NEAA, 0.1% Alanyl-L-Glutamine and 34.78% sodium hydrogen carbonate (NaHCO3 0.75g/l) | Imaging medium | ||
| Earle´s minimum essential medium (MEM) with phenol red, containing 1% Fetal Bovine Serum Superior (FBS), 0.1% HEPES (2-(4-(2-hydroxyethyl)-1-piperazinyl)-ethanesulfonic acid, 1 M), and 0.1% non-essential amino acids (NEAA) | Growth medium | ||
| EF: Emission filter quadbandpass | AHF analysentechnik | F72-866 | Brightline HC 446 nm/523 nm/600 nm/677 nm |
| EMCCD camera | Andor | Andor iXON 897 | EMCCD camera |
| Emission filter QuadView filter cubes, orange | AHF analysentechnik | F39-637 | bandpass 582 - 619 nm |
| Emission filter QuadView filter cubes, red | Chroma | bandpass 655 - 725 nm (HQ 690/70) | |
| FBS (Fetal bovine serum) superior | Biochrom | S0615 | |
| Fluorescent beads: TetraSpeck™ Microspheres, 0.1 µm, fluorescent blue/green/orange/dark red | Thermo Fisher Scientific | T7279 | fluorescent microspheres |
| Glutamine | Biochrom | #0951C | |
| HeLa cells | DSMZ | ACC-57 | Cervical carcinoma cells from patient Henrietta Lacks |
| Hela cells CI::paGFP, stable | Muster et al., PLOSOne 2010 | ||
| Hela cells CV g::Halo7-Tag, stable | Appelhans et al., NanoLett 2012 | ||
| Hela cells Tom20::Halo7-Tag, stable | Appelhans et al., NanoLett 2012 | ||
| Image splitter | Photometrics | Dual-View QV2 | image splitter emission |
| Imaging processing software | ImageJ2 / Fiji | freeware | |
| Immersion Oil - ImmersolTM 518 F (ne = 1.518, ve = 45) | Carl Zeiss Jena GmbH | 444960-0000-000 | |
| Inverted epifluorescence microscope | Olympus IX-71/73/83 | ||
| Laser 561 nm, 200 mW | CrystaLaser | CL-561-200 | 561 nm emission |
| Laser 642 nm, 140 mW | Omicron | Luxx-642-140 | 642 nm emission |
| MATLAB | MathWorks | version R2013a | |
| MEM with Earle's Balanced Salt Solution 2.2 g/L NaHCO3, stable glutamine w/o PR | Biochrom | FG-0385 | |
| MEM with Earle's Balanced Salt Solution with 2.2 g/L NaHCO3, stable glutamine, Phenolred | Biochrom | FG-0325 | |
| MitoTracker® Deep Red FM | Thermo Fisher Scientific | M22426 | dye |
| MitoTracker® Green FM | Thermo Fisher Scientific | M7514 | dye |
| Multi-mode-optical polarization maintaining monomode fiber | Pointsource/Qioptiq | KineFLEX | |
| NHS-PEG-MAL, Rapp Polymer | Rapp Polymere GmbH Tübingen | coverslip coating | |
| non-essential amino acids (NEAA) | Biochrom | #0802E | |
| PEG 800 (Polyethylene glycol) 10 % | Carl Roth GmbH | Art No. 0263.1 | coverslip coating |
| Penicillin/Streptomycin | Biochrom | #0122E | |
| Plasmid for PINK1-Halo7-Tag expression | Beinlich et al., ACS Chemical Biology 2015 | ||
| Poly-L-lysine (1.2 mg/ml) | Sigma-Aldrich & Co. | Cat. No.P9155 | coverslip coating |
| RGD Peptide (Ac-CGRGDS-COOH) | Coring System Diagnostix GmbH, Gernsheim | coverslip coating / Intergrin receptor motif | |
| Silicon Rhodamine linked to HaloTag®-Ligand (SiRHTL) | personal gift from Kai Johnson | dye | |
| Software analysis plugin | self-written C. P. Richter, Biophysik Osnabrück | SLIMFAST 16g | |
| Tetramethylrhodamine / SNAP-Cell® TMR-Star linked to SNAP-Ligand (TMRstar) | New England Biolab® | S9105S | dye |
| Tetramethylrhodamine linked to HaloTag®-Ligand (TMRHTL) | Promega | G8251 | dye |
| TIRF condensor | Olympus | Cell^TIRF MITICO System | TIRF condensor |
| TIRF microscope controlling software | Olympus cellSens 1.12 | ||
| TIRF objective | Olympus | 150x oil objective (N.A. 1.45; Olympus UAPO) | |
| Trypsin/EDTA 10x | Biochrom | #0266 | |
| Water H2O 99,5 % Rotipuran® Low organic | Carl Roth GmbH | Art. No. HN57.1 |
Referenzen
- Appelhans, T., Richter, C., Wilkens, V., Hess, S., Piehler, J., Busch, K. Nanoscale organization of mitochondrial microcompartments revealed by combining tracking and localization microscopy. Nano Letters. 12 (2), 610-616 (2012).
- Appelhans, T., Busch, K. Single Molecule Tracking and Localization of Mitochondrial Protein Complexes in Live Cells. Methods Mol Biol. 1567, 273-291 (2017).
- Rust, M. J., Bates, M., Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods. 3 (10), 793-795 (2006).
- Gould, T. J., Verkhusha, V. V., Hess, S. T. Imaging biological structures with fluorescence photoactivation localization microscopy. Nat Protoc. 4 (3), 291-308 (2009).
- Pennacchietti, F., Gould, T. J., Hess, S. T. The Role of Probe Photophysics in Localization-Based Superresolution Microscopy. Biophys J. 113 (9), 2037-2054 (2017).
- Wegel, E., Göhler, A., Lagerholm, B. C., Wainman, A. Imaging cellular structures in super-resolution with SIM, STED and Localisation Microscopy: A practical comparison. Scientific reports. , Available from: https://www.nature.com/articles/srep27290?WT.feed_name=subjects_physical-sciences (2016).
- Pellett, P., et al. Two-color STED microscopy in living cells. Biomedical Optics Express. 2 (8), (2011).
- Ishigaki, M., et al. STED super-resolution imaging of mitochondria labeled with TMRM in living cells. Mitochondrion. 28, 79(2016).
- Liss, V., Barlag, B., Nietschke, M., Hensel, M. Self-labelling enzymes as universal tags for fluorescence microscopy, super-resolution microscopy and electron microscopy. Scientific Reports. 5, 17740(2015).
- Sergé, A., Bertaux, N., Rigneault, H., Marguet, D. Dynamic multiple-target tracing to probe spatiotemporal cartography of cell membranes. Nat Methods. 5 (8), (2008).
- Appelhans, T., Busch, K. B. Dynamic imaging of mitochondrial membrane proteins in specific sub-organelle membrane locations. Biophysical reviews. 9 (4), 345-352 (2017).
- Wilmes, S., et al. Triple-color super-resolution imaging of live cells: resolving submicroscopic receptor organization in the plasma membrane. Angewandte Chemie. 51 (20), 4868-4871 (2012).
- Niewidok, B., et al. Single-molecule imaging reveals dynamic biphasic partition of RNA-binding proteins in stress granules. J Cell Biol. , (2018).
- Wurm, C. A., Jakobs, S. Differential protein distributions define two sub-compartments of the mitochondrial inner membrane in yeast. FEBS Lett. 580 (24), 5628-5634 (2006).
- Schmidt, R., Wurm, C. A., Punge, A., Egner, A., Jakobs, S., Hell, S. W. Mitochondrial cristae revealed with focused light. Nano Lett. 9 (6), 2508-2510 (2009).
- Kukat, C., Wurm, C. A., Spahr, H., Falkenberg, M., Larsson, N. G., Jakobs, S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proceedings of the National Academy of Sciences of the United States of America. 108 (33), 13534-13539 (2011).
- Beinlich, F., Drees, C., Piehler, J., Busch, K. Shuttling of PINK1 between Mitochondrial Microcompartments Resolved by Triple-Color Superresolution Microscopy. ACS chemical biology. 10 (9), 1970-1976 (2015).
- Shim, S., et al. Super-resolution fluorescence imaging of organelles in live cells with photoswitchable membrane probes. Proceedings of the National Academy of Sciences. 109 (35), 13978-13983 (2012).
- Sbalzarini, I., Mezzacasa, A., Helenius, A., Koumoutsakos, P. Effects of Organelle Shape on Fluorescence Recovery after Photobleaching. Biophysical Journal. 89 (3), 1482-1492 (2005).
- Reits, E., Neefjes, J. From fixed to FRAP: measuring protein mobility and activity in living cells. Nature Cell Biology. 3 (6), E145-E147 (2001).
- Goehring, N., Chowdhury, D., Hyman, A., Grill, S. FRAP Analysis of Membrane-Associated Proteins: Lateral Diffusion and Membrane-Cytoplasmic Exchange. Biophysical Journal. 99 (8), 2443-2452 (2010).
- Bacia, K., Haustein, E., Schwille, P. Fluorescence correlation spectroscopy: principles and applications. Cold Spring Harbor protocols. 2014 (7), 709-725 (2014).
- Sukhorukov, V., Dikov, D., Busch, K., Strecker, V., Wittig, I., Bereiter-Hahn, J. Determination of protein mobility in mitochondrial membranes of living cells. Biochimica et biophysica acta. 1798 (11), 2022-2032 (2010).
- Kim, T. K., Eberwine, J. H. Mammalian cell transfection: the present and the future. Anal Bioanal Chem. 397 (8), 3173-3178 (2010).
- Graham, F. L., van der Eb, A. J. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 52 (2), 456-467 (1973).
- Wedeking, T., et al. Spatiotemporally Controlled Reorganization of Signaling Complexes in the Plasma Membrane of Living Cells. Small. 11 (44), 5912-5918 (2015).
- Mironov, S. L., Ivannikov, M. V., Johansson, M. [Ca2+]i signaling between mitochondria and endoplasmic reticulum in neurons is regulated by microtubules. From mitochondrial permeability transition pore to Ca2+-induced Ca2+ release. J Biol Chem. 280 (1), 715-721 (2005).
- Poot, M., et al. Analysis of mitochondrial morphology and function with novel fixable fluorescent stains. J Histochem Cytochem. 44 (12), 1363-1372 (1996).
- Tokunaga, M., Imamoto, N., Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells (vol 5, pg 159). Nature Methods. 5 (5), 455(2008).
- Elmokadem, A., Yu, J. Optimal Drift Correction for Superresolution Localization Microscopy with Bayesian Inference. Biophys J. 109 (9), 1772-1780 (2015).
- Barlag, B., et al. Single molecule super-resolution imaging of proteins in living Salmonella enterica using self-labelling enzymes. Sci Rep. 6, 31601(2016).
- Tokunaga, M., Imamoto, N., Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nature Methods. 5 (2), 159-161 (2008).
- Mortensen, K. I., Churchman, L. S., Spudich, J. A., Flyvbjerg, H. Optimized localization analysis for single-molecule tracking and super-resolution microscopy. Nat Methods. 7 (5), 377-381 (2010).
- Jin, S., Youle, R. J. PINK1- and Parkin-Mediated Mitophagy at a Glance. Journal of Cell Science. 125, 795-799 (2013).
- Yamano, K., Youle, R. J. PINK1 is degraded through the N-end rule pathway. Autophagy. 9 (11), 1758-1758 (2013).
- Wiedemann, N., et al. Machinery for protein sorting and assembly in the mitochondrial outer membrane. Nature. 424 (6948), 565-571 (2003).
- Thompson, R., Larson, D., Webb, W. Precise Nanometer Localization Analysis for Individual Fluorescent Probes. Biophysical Journal. 82 (5), 2775-2783 (2002).
Access restricted. Please log in or start a trial to view this content.
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten