Method Article
Caracterización de las glicoproteínas con el doblez de la inmunoglobulina por cristalografía de rayos x y técnicas biofísicas
En este artículo
Resumen
Presentamos los enfoques para la caracterización Biofísica y estructural de las glicoproteínas con el doblez de la inmunoglobulina por interferometría de biocapa, calorimetría isotérmica de titulación y Cristalografía de rayos x.
Resumen
Glicoproteínas en la superficie de las células desempeñan un papel crítico en la función celular, incluyendo señalización, transporte y adherencia. En leucocitos, varias de estas glicoproteínas poseen pliegues de inmunoglobulina (Ig) y son esenciales para la regulación y reconocimiento inmune. Aquí, presentamos una plataforma para el diseño, expresión y caracterización biofísica del dominio extracelular del receptor de células B humanas CD22. Proponemos que estos enfoques son ampliamente aplicables a la caracterización de ectodomains de mamíferos de la glicoproteína que contiene dominios Ig. Dos suspensión riñón embrionario humano (HEK) líneas celulares, HEK293F y HEK293S, se utilizan para expresar glicoproteínas dan cobijo glycans complejo y de alta manosa, respectivamente. Estas glicoproteínas recombinantes con diferentes glicoformas permiten investigar el efecto de tamaño de glicanos y composición en el atascamiento del ligand. Discutimos protocolos para el estudio de la cinética y la termodinámica de unión de la glicoproteína a ligandos biológicamente relevantes y anticuerpo terapéutico candidatos. Glicoproteínas recombinantes que se producen en las células HEK293S son susceptibles de cristalización debido a la homogeneidad de glicanos, flexibilidad reducida y susceptibilidad a endoglycosidase H. Presentan métodos de remojo cristales glicoproteína con átomos pesados y moléculas pequeñas para determinación de fase y análisis de ligando, respectivamente. Los protocolos experimentales discutidos aquí son prometedoras para la caracterización de las glicoproteínas mamíferos para dar información sobre su función e investigar el mecanismo de acción de la terapéutica.
Introducción
Proteínas de superficie juegan papeles críticos en la función celular. A través de sus dominios extracelulares, estas proteínas de membrana pueden modular las interacciones célula-célula, adherencia, transporte y señalización1,2. La localización extracelular de estas proteínas es objetivos atractivos para el desarrollo de la terapéutica para el tratamiento de una amplia gama de enfermedades, incluyendo cáncer y enfermedades autoinmunes3,4,5 , 6 , 7. uno de los dobleces más comunes de ectodomains de proteína de la membrana humana es el pliegue de inmunoglobulina-como (Ig), que está formado por siete o más β-filamentos dispuestos en dos β-hojas8,9. Glicoproteínas que contienen Ig son típicamente, estructuras multidominio con dominios Ig secuencialmente dispuestas en la porción extracelular de la proteína de la membrana10. Modificaciones postraduccionales de estas proteínas de superficie celular, particularmente N - y O-ligado glycosylation, han demostrado que desempeñan un papel fundamental en su regulación, plegable, secreción y función11. Para mejorar nuestra comprensión de su función y a la mejor terapéutica de diseño que les puede orientar, se requieren técnicas que permiten su caracterización molecular detallada. Aquí, presentamos una combinación de técnicas que permiten el biofísico (biocapa interferometría (BLI) y calorimetría isoterma de titulación (ITC)) y caracterización estructural (cristalografía de rayos x) el dominio extracelular de Ig glicoproteínas de membrana, solas y en conjunto con sus ligandos biológicamente relevantes y moléculas terapéuticas (figura 1).
N-ligado glycosylation es una de las modificaciones post traducción más común en las proteínas mamíferas y se produce durante la maduración de la proteína en el retículo endoplasmático y Golgi12,13. Líneas celulares, como células de riñón embrionario humano (HEK) 293, se han desarrollado para la expresión recombinante de grandes cantidades de glycosylated proteínas mamíferas14,15. Esta línea celular se ha desarrollado en un formato de suspensión, que permite la facilidad de escalado de producción de proteínas en grandes cantidades en comparación con líneas de células adherentes. Aquí, utilizamos dos líneas de células HEK293: HEK293F y HEK293 Gnt I- / - (HEK293S), que se diferencian por la ausencia de N-acetylglucosaminyl transferasa I (Gnt yo) en el último. A su vez, producción de glycans complejo (como visto en HEK293F) no es posible y en su lugar alta manosa-tipo glicanos (predominante hombre5GlcNAc2) residen en N-ligados glycan sitios18,19,20 . Estas dos variedades de células en paralelo permite estudiar el efecto de glicanos tamaño y la complejidad sobre la función biológica y orientación terapéutica. En efecto, glicoproteínas producidas en células de HEK293F tendrá glicanos más grande, más compleja en comparación con la misma glicoproteína producida en las células HEK293S. Glicoproteínas que se producen en las células HEK293S son más susceptibles de cristalización, debido a la menor heterogeneidad química y conformacional de los glycans N-ligados. Para mejorar aún más el crystallizability, glicoproteínas producidas en células HEK293S (pero no HEK293F) pueden ser tratados con la enzima endoglycosidase H (Endo H), que se traduce en la ruptura de glicanos de alta manosa tal que solamente una sola N-acetilglucosamina (GlcNAc) parte se mantiene en cada sitio de glicosilación N-ligados21,22. También pueden utilizar otros métodos para limitar el proceso de N-glicanos dentro de las células, tales como la adición de inhibidores de glicosidas durante la expresión de la glicoproteína, incluyendo kifunensine23. Alternativas implican la expresión de glicoproteínas nativos (en las células HEK293F) seguida por deglycosylation enzimática utilizando péptidos N-glucosidasa F (PNGaseF). Sin embargo, deglycosylation con PNGaseF ha demostrado ser menos eficaz bajo condiciones nativas y aumenta la agregación de algunas proteínas; en casos cuando la proteína permanece soluble después del tratamiento, adquiere cargas negativas en su superficie debido a la deamidation del residuo Asparagina Ácido aspártico24, que podría ser perjudicial para su cristalización. Sitios de N-glicosilación predijo también pueden ser transformados, más a menudo a los residuos de alanina o glutamina, para prevenir N-ligado glycosylation en estos sitios y para generar muestras de glicoproteína de alta homogeneidad. Alternativamente, se pueden producir glicoproteínas en otras culturas de células eucariotas, incluyendo levaduras, insectos y sistemas de planta u otras líneas de células de mamífero como el hámster chino ovario (CHO) células16,17.
Muchos vectores de expresión mamíferos, incluyendo pHLsec, permiten la secreción de ectodomains de la glicoproteína recombinante en la célula media25. Secreción de glicoproteínas de las células HEK293 permite la purificación rápida y fácil sin necesidad de lisis celular. Adición de etiquetas de purificación (por ejemplo, su etiqueta, Strep-tag, bandera, Myc-etiqueta, etiqueta de HA) a la N o C terminal del destino glicoproteína permite la purificación mediante una cromatografía de afinidad solo paso. Posteriormente, cromatografía por exclusión de tamaño puede utilizarse para obtener una muestra de monodispersa de caracterización Biofísica y estructural.
Una muestra de glicoproteína altamente puro y homogéneo en condiciones adecuadas puede dar lugar a cristales bien diffracting. Una vez que se cuente con un conjunto de datos de difracción de rayos x completo de tales cristales, fases iniciales deben determinarse para el cálculo de la densidad del electrón de la glicoproteína. Gracias a un número cada vez mayor de estructuras en el Banco de datos de la proteína (PDB), el método más comúnmente utilizado para la eliminación en gran medida ha convertido en reemplazo molecular (MR), que utiliza una estructura de la proteína relacionada para obtener fases iniciales26. Sin embargo, cuando Señor es incapaz de resolver el problema de fase, como en ocasiones ha sido el caso de multi-Ig dominio glicoproteínas27,28,29, métodos alternativos son necesarios. En este artículo detallamos un método para disfrutar de cristales con átomos pesados (HA) para la eliminación, que se requería para resolver la estructura del ectodomain CD2228. Identificación de la derecha HA para la eliminación es un proceso iterativo que depende de la reactividad de la HA, átomos disponibles en la glicoproteína en un enrejado cristalino determinado y la cristalización solución30,31. Como alternativa, átomos de azufre natural en residuos de cisteína y metionina pueden utilizarse para la eliminación si está presente en una proporción bastante alta a otros átomos en la glicoproteína, y si se recogerán datos de difracción de rayos x con suficiente redundancia32, 33.
La función biológica de glicoproteínas de membrana está a menudo mediada por interacciones proteína-proteína o las interacciones proteína-ligando, como con hidratos de carbono. Cuando el ligando es lo suficientemente pequeño como para la difusión de la solución para el sitio de unión de la glicoproteína en el enrejado cristalino, experimentos de remojo puede ser exitoso para obtener una estructura de cristal Co glicoproteína ligando para entender mejor el reconocimiento de ligando.
Los protocolos aquí presentados también son relevantes para la comprensión de las interacciones de glicoproteínas de superficie con ligandos sintéticos de terapéutica34,35 y anticuerpo terapéutica36,37. Cuando se combina con información estructural, Unión cinética y la termodinámica puede ser poderosa para entender y mejorar sus mecanismos de acción. Una técnica que permite el análisis cinético de anticuerpos terapéuticos a una glicoproteína es BLI38,39. BLI utiliza biosensores con un ligando inmovilizado para medir la cinética de asociación y disociación con un socio de la Unión, en última instancia, determinar una constante de disociación de equilibrio (KD). BLI es un enfoque atractivo ya que se requieren pequeñas cantidades de glicoproteínas (< 100 μg), tiempo del experimento es rápido (~ 10-15 min por ejecutar), y puede ser automatizado. ITC es también útil para el estudio de las afinidades entre glicoproteínas y Unión socios40,41,42,43. Mientras que ITC es más tiempo y reactivo intensivo, puede obtenerse información valiosa sobre la termodinámica de la interacción (ΔG, ΔH, ΔS y estequiometría). ITC es también muy útil para el estudio de las interacciones débiles que a menudo están asociadas con la unión transitoria de glicoproteínas de superficie a ligandos. Además, estas técnicas se pueden utilizar en conjunto para evaluar la Unión de varias construcciones y evaluar el efecto de diferentes glicoformas N-ligados de expresión de la glicoproteína en diferentes líneas celulares. Realizar BLI y ITC con glicoproteínas producidas en HEK293F, HEK293S y tratados con Endo H puede proporcionar una vista detallada del papel de glycans en actividad biológica y el compromiso terapéutico.
Hemos aplicado con éxito estos protocolos para caracterizar el dominio extracelular (ECD) del humano CD2228, miembro de la glicoproteína de la familia de lectinas (Siglecs) de Ig-como Unión a ácido siálico que es esencial para mantener la homeostasis de la célula de B44 . Realizamos diseño de fondo de construcción para facilitar la cristalización y gradualmente el conjunto de datos de rayos x por HA con Hg. También empapado CD22 cristales con su ligando el ácido siálico (α2-6 sialyllactose) para obtener una estructura del complejo ligando-receptor inmune y así proporcionan los planos para el diseño de estructura guiada de glicanos miméticos45,46. Además, genera el atascamiento del antígeno del fragmento (Fab) del epratuzumab anticuerpo terapéutico de anti-CD22 - un candidato terapéutico actualmente en ensayos clínicos de fase III para de linfoma de los non-Hodgkin47- para determinar su afinidad de Unión por BLI y CCI a diferencialmente glucosilada CD22 ECD construye. Estos estudios revelaron un papel crítico para el N-ligado glycosylation en compromiso epratuzumab, con posibles consecuencias para el reconocimiento de CD22 en células B disfuncionales.
Protocolo
1. construir el diseño para la ECD de la glicoproteína
- Evaluar la secuencia de aminoácidos de humano CD22 (Uniprot) utilizando los servidores de InterPro y Phyre2 para identificar elementos de dominio previstas y los límites situado dentro de la proteína48,49.
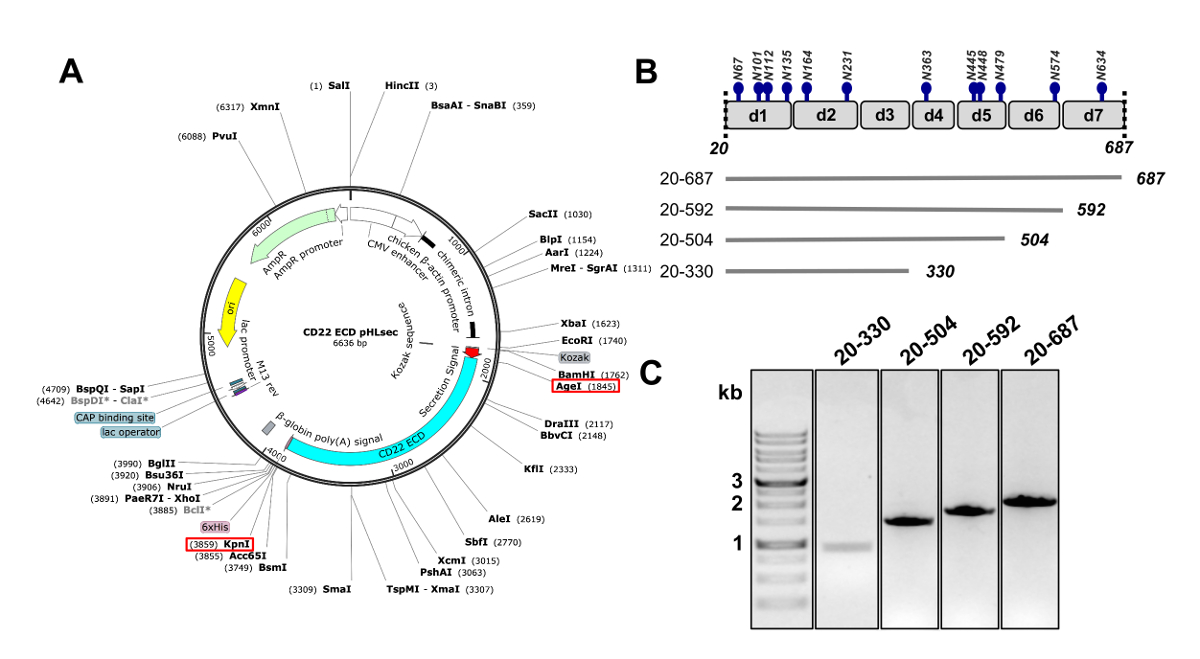
- Copia la secuencia de humano CD22, careciendo el péptido señal, dominios transmembranales y citosólicas (residuos 20-687, de ahora en adelante dominio extracelular CD22 CD22 ECD) en el pHLsec expresión mamífera vector25 usando enzimas de restricción africanas y KpnI ( Figura 2 A) 50.
Nota: El pHLsec vector es optimizado para la sobreexpresión de proteínas solubles, secretadas en las células mamíferas25. Este vector contiene una señal de secreción para permitir la secreción extracelular de glicoproteínas solubles. pHLsec contiene una etiqueta dex 6 C-terminal (su) para facilitar la purificación de la afinidad de sobrenadantes de células utilizando métodos de cromatografía de afinidad metálica inmovilizados. - Clon truncadas construcciones de ECD CD22 con canceladuras secuenciales de los dominios c-terminal Ig: dominios de 1 a 6 (residuos 20-687), los dominios 1-5 (residuos 20-592), dominios 1 a 4 (residuos 20-504) y dominios 1-3 (residuos 20-330) (figuras 2B y 2C)50 .
- Evaluar la secuencia primaria de CD22 DIT utilizando el servidor de NetNGlyc para identificar sitios de glicosilación de N-ligados prevista en la construcción de51.
- Mediante mutagénesis sitio-dirigida, mediante protocolos estándar52 o traslapo PCR53, mutar cada sitio previsto N-ligado glycosylation (Asn a Gln o Asn a Ala) para crear construcciones de CD22 ECD que contienen uno o varios Mutaciones de N-ligado glycosylation.
- Después de la verificación de la secuencia de construcciones clonadas, transformar en competentes de e. coli DH5α células54 y maxi-preparación del ADN (según las instrucciones del fabricante) para prepararse para la transfección.
2. HEK293F y HEK293S celular establecimiento
Nota: Toda manipulación de las células HEK293F o HEK293S con el equipo y reactivos necesarios debe realizarse en un centro de nivel 2 de bioseguridad en un gabinete de bioseguridad apropiado. La superficie externa de todos los elementos debe ser esterilizada con una solución de etanol 70% o un reactivo equivalente.
- Obtención de células suspensión HEK293F y HEK293S (véase Tabla de materiales) y almacenar a-80 ° C hasta que esté listo para su uso.
- Medios calientes (véase Tabla de materiales) por 1 h a 37 ° C baño. Transferencia 24 mL de medio caliente a un matraz de cultura celular desconcertado de 125 mL con una tapa ventilada.
- Obtener celular 1 mL alícuota de-80 ° C y la transferencia de hielo.
- Incubar las células en un baño de agua de 37 ° C durante aproximadamente 1 minuto, para descongelar parcialmente las células. Transferir 1 mL de las células del frasco al matraz 125 mL desconcertado de la célula cultura con los medios de comunicación.
- Cierre el frasco de cultivo celular con la tapa de ventilación y matraz de lugar en una coctelera a 37 ° C, 130 rpm, 70% de humedad y 8% CO2.
3. HEK293 Mantenimiento de células
Nota: La densidad celular y la viabilidad de las células deben ser revisado aproximadamente 24 h después de la descongelación. Este paso asegura que las células se están recuperando después de la inoculación; viabilidad inicial debe ser > 80%.
- Con cuidado Extraiga 10 μl de las células del frasco de 125 mL que contiene las células de suspensión fresca y transferirlo en un microtubo de 1.5 mL estéril. Cerrar el frasco y volver a la incubadora.
- Pipetear 10 μl de solución de azul tripán en el microtubo de 1.5 mL que contiene las células, homogeneizar y transferir 10 μl de la cámara del portaobjetos de conteo.
- Coloque el portaobjetos de conteo en un contador celular automático y obtener los valores de densidad celular (en unidades de células mL-1) y viabilidad de las células (en porcentaje).
- Calcular el volumen de las células que serían necesarios para sembrar una cultura fresca 200 mL a una densidad final de ~0.8 x 106 células mL-1 usando las siguientes ecuaciones:
 (1)
(1) (2)
(2)
Nota: Puede tomar ~ 5 d para obtener una densidad celular adecuado para inoculación en una cultura de 200 mL. - Una vez que la densidad celular es suficiente para la inoculación de una cultura de 200 mL, medios de calentamiento de 1 h a 37 ° C baño de agua y transferir los medios caliente en el gabinete de seguridad de la biotecnología.
- Con una pipeta serológica, transferir cuidadosamente el volumen requerido de los medios de comunicación (según lo calculado en la ecuación 2) en un matraz de 500 mL desconcertado de la célula cultura con una tapa ventilada.
- Con una pipeta serológica, transferir el volumen requerido de suspensión de células (calculado en la ecuación 1) en el matraz de 500 mL desconcertado de la célula cultura con los medios de comunicación.
- Tapa de la nueva acción de mantenimiento de 200 mL y volver a la incubadora. Crecen las células a una densidad de aproximadamente 3 x 106 células mL-1. Las células a una densidad de 0.8 x 106 células mL-1 de paso cada 2-3 d para mantener un cultivo estable de células (como se describe en la sección 3.4-3.7). No permita que las células exceder una densidad de ~ 4 x 106 células mL-1.
4. transfección de células HEK293 para la expresión de la glicoproteína
- Calcular el volumen de las células y los medios de comunicación que se requiere para una cultura de 200 mL para transfección en 0.8 x 106 células mL-1 (usando las ecuaciones 1 y 2 de la sección 3.4).
Nota: El número de transfecciones 200 mL que se pueden realizar depende de la densidad celular de las acciones de mantenimiento. - Transferir el volumen requerido de los medios de comunicación y células para la transfección en un nuevo frasco de cultura celular 500 mL con una tapa ventilada y retomar la acción celular de la incubadora.
- Incube las células durante 1 h antes de transfección para permitir la aclimatación después de partir de las células.
- Transferencia de 50 μg de ADN en un tubo cónico estéril de 50 mL y diluir con 5 mL de medio. Filtro de vacío del ADN diluido usando un sistema de filtración de 0,22 μm en otro tubo estéril.
- Mezcla diluida, había filtrado de ADN con una relación masa: volumen 1:1 con el reactivo de transfección. Agite suavemente la solución de reactivo de ADN: transfección para mezclar e incubar la solución a temperatura ambiente durante 10 minutos.
- Añadir solución de reactivo de transfección: DNA directamente a las células. Incube las células transfected a 37 ° C, 130 rpm, 70% de humedad y 8% CO2 en un agitador durante 5-7 días.
5. optimización de condiciones de la transfección de la célula
Nota: Para optimizar las condiciones de transfección celular para el rendimiento máximo de la glicoproteína, transfectar las células de una variedad de densidades de células iniciales y evaluar rendimiento de proteína en el tiempo (figura 3A). Transfectar las células como se describe en la sección 4, en la densidad celular inicial de 0.5 x 106 2 x 106 células mL-1 55. Transfecciones ensayo pueden ser reducidos a 25 mL de volumen total (en frasco de cultivo celular desconcertado 125 mL) con 6 μg de ADN para ahorrar espacio y reactivos. La cantidad de ADN también puede ser optimizado55.
- La transfección de post de cada día (días 1-7), transferir una alícuota de 500 μl de cultivo celular en un microtubo de 1.5 mL estériles (en el gabinete de seguridad de la biotecnología).
- Girar las células alícuotas a 12.000 x g durante 5 minutos en una microcentrífuga inmediatamente después de su obtención. Transferir el sobrenadante a un nuevo microtubo de 1.5 mL y conservar a 4 ° C hasta que todas las muestras se obtienen.
- Cuantificar la glicoproteína secretada por densitometría
- Una vez obtenidas todas las muestras, alícuotas de 20 μl de cada muestra en un microtubo de 1.5 mL nueva y mezclar con 6 μl de tampón de muestra x Laemmli no reductores 4.
- Hervir las muestras por 5 min a 95 ° C en un termo-block. Girar las muestras durante 1 min a 12.000 g de x en una microcentrífuga.
- 20 μl de cada muestra por pocillo en un gel de SDS-PAGE del gradiente bien 10 4-15% de la carga. Incluyen un carril para los marcadores de tamaño de la proteína. Correr el gel a 250 V por 20 min en un tampón de Tris/glicina/SDS.
- Después ejecutar, transferir el gel a Coomassie mancha (véase Tabla de materiales) para 20 minutos la mancha gel en ddH2O 20 min gel de imagen.
- Realizar densitometría con ImageJ, siguiendo protocolos estándar57,58.
- Compilar y trama de datos con 'días después de la transfección' en el eje x y ' densitometría ósea ' en el eje y (figura 3A).
Nota: Alternativamente, si la expresión de la proteína es insuficiente para la visualización por SDS-PAGE, técnicas como el Western Blot pueden ser utilizado56.
- Cuantificar la glicoproteína secretada por BLI
- Uso de biosensores de Ni-NTA, cuantificar la cantidad de la glicoproteína secretada con BLI59.
- Compilar y trama de datos con 'días después de la transfección' en el eje x y la 'concentración de proteína (μg/mL)' en el eje y (figura 3A).
6. purificación de la glicoproteína Soluble de HEK293 sobrenadante
- La cosecha de las células por centrifugación a 6.371 x g por 20 min a 4 ° C. Sobrenadante que contiene secretada CD22 ECD de retener y filtrar con un filtro de 0,22 μm.
- Sobrenadante en 4 mL min-1 en el equilibrado de carga columna de Ni-NTA (20 mM Tris, pH 9.0, 150 mM NaCl, imidazol 5 mM) (5 mL de volumen) con un sistema de cromatografía de sobremesa.
Nota: Otras técnicas de purificación basadas en la afinidad pueden usarse, basados en las etiquetas de afinidad incluidas en el diseño de la construcción en la sección 1. - Después de carga flotante, lavar la columna de afinidad con volúmenes de columna de 3-4 (CV) de tampón de lavado (20 mM Tris, pH 9.0, 150 mM NaCl, imidazol 5 mM).
- Eluir la glicoproteína purificada de la columna con un gradiente de 4-100% (4 CV) de tampón de elución (20 mM Tris, pH 9.0, 150 mM NaCl, imidazol de 500 mM) mientras que recogiendo fracciones (figura 3B).
- Fracciones de piscina que contiene el pico eluído de un dispositivo de filtración centrífuga con 10 kDa peso molecular nominal límite (NMWL) y concentrado por centrifugación a 4.000 x g a 4 ° C por 15 minutos o hasta que la muestra alcanza un volumen de 500 μl.
- Inyectar la glicoproteína concentrada en una 500 μl muestra lazo y carga en 0,5 mL min-1 previamente equilibrado (20 mm Tris, pH 9.0, 150 mM NaCl) columna de exclusión de tamaño de alto rendimiento (aproximadamente 24 mL de volumen) de una cromatografía líquida de la proteína rápida (FPLC) sistema a 4 ° C mientras que la recogida de fracciones (figura 3C).
- Correr el gel SDS-PAGE de las fracciones eluídas a identificar fracciones que contiene la glicoproteína y piscina fracciones correspondientes. El gel de SDS-PAGE se puede ejecutar como se describe en la sección 560.
7. Deglycosylation de la glicoproteína purificada
- Medir la concentración de la proteína purificada después de cromatografía por exclusión de tamaño mediante el uso de la absorbancia a 280 nm se divide por el coeficiente de extinción (por ejemplo, 1,418 M-1 cm-1 para CD22 ECD).
Nota: El coeficiente de extinción teórica de proteínas de interés se puede calcular utilizando los servidores como ExPASy ProtParam61. - Incubar la proteína purificada con Endo H por 1 h a 37 ° C, a razón de 1 mg de proteína purificada a 10 μl de enzima comercial en 1 buffer X Endo H (según las instrucciones del fabricante).
Nota: Endo H hiende glicanos de alta manosa producido en HEK293S dejando una sola molécula GlcNAc en cada sitio de glicosilación21. Endo H desdoblan glycans en proteínas producidas en las células de HEK293F22, sin embargo, otras enzimas pueden ser utilizados para este propósito (por ejemplo,PNGaseF24). - Concentrado deglycosylated ECD a 500 μl y ejecutar cromatografía de gel filtración en una columna de exclusión de tamaño de alto rendimiento (aproximadamente 24 mL de volumen) en 0.5 mL min-1 en un FPLC Endo H de eliminar y separar cualquier agregados resultantes.
- Almacenar la proteína deglycosylated a 4 ° C hasta su utilización en posteriores experimentos.
8. cristalización de glicoproteínas
Nota: Realizar ensayos de cristalización mediante pantallas disponibles en el mercado y establecer sesión caída de experimentos usando un robot de cristalización.
- Concentrado puro, deglycosylated ECD a 10 mg mL-1 , con un dispositivo de filtración centrífuga 10 kDa NMWL a 4.000 x g (4 ° C) hasta obtener la concentración deseada.
- Determinar la concentración de proteína usando la absorbancia a 280 nm y dividiendo por el coeficiente de extinción.
- Muestra de centrifugar a 12.000 x g durante 5 min a 4 ° C antes de ensayos de cristalización para quitar polvo no deseado u otros contaminantes de la muestra.
- Llenar el depósito de pozos de 96 pocillos sesión gota placas de cristalización con 80 μl de solución de cristalización desde una pantalla comercial de la cristalización.
Nota: Utilizamos escasa matriz comerciales pantallas que han sido diseñados basados en las condiciones de cristalización más exitosas con respecto a las estructuras en el PDB. - Utilizando un robot de cristalización, dispensar gotas en el pocillo de la placa de cristalización con un volumen total de gota de 200 nL en una proporción de la solución purificada: cristalización de proteínas de 1:1.
- Una vez que ha salido toda la placa, sello de la placa con cinta y colóquelos en un toner de la placa para su inspección por luz visible y ultravioleta.
- Inspeccione la cristalización las placas inmediatamente después de la instalación y en las semanas siguientes, utilizando luz visible y ULTRAVIOLETA para identificar las condiciones que dan glicoproteína inicial cristal hits.
- Optimizar aún más los cristales obtenidos de cristalización inicial de golpes usando pantallas finas basados en la condición del éxito de cristal o matriz al azar siembra de micro métodos62,63,64,65.
- Cryo-proteger los cristales carecer suficiente crio-protector dentro de las condiciones de cristalización empapando el cristal madre licor solución enriquecida con solución de glicerol al 20% (v/v) (o equivalente crio-protector, tales como glicol de etileno o polietilenglicol 400).
- Montaje de cristales en cryoloops y flash les congelan en nitrógeno líquido antes de la recogida de datos en un difractómetro fuente casera o usando radiación de sincrotrón.
9. eliminación mediante derivatización de átomo pesado
Nota: Antes de cualquier manipulación de compuestos HA, deben considerarse aspectos de seguridad. HA compuestos usados en Cristalografía de la proteína son seleccionados para su fuerte afinidad a moléculas biológicas y riesgos para la salud humana de la exposición prolongada. Tomar medidas de seguridad apropiadas para compuestos HA como se ha mencionado en sus hojas de datos de seguridad Material.
- Para probar diferentes HA compuestos, las concentraciones y tiempos de incubación, reproducen bien diffracting cristales obtenidos en la sección 8 en una placa de 24 pocillos cristalización mediante la difusión de vapor de gota colgante método66.
- Decidir que se HA utilizado para la derivatización de cristal. Servidores (por ejemplo, el átomo pesado sistema de base de datos,67) pueden ayudar a con HA compuesto selección, asegurando que son apropiados para la condición de la proteína y la cristalización.
Nota: HA pantallas también están disponibles comercialmente para la fácil detección de compuestos HA más eficaces para la eliminación. Un conjunto de "magia siete" HA compuestos se han descrito previamente para tener alta probabilidad de éxito para HA derivatización68. - Configurar estaciones de trabajo HA remojo (figura 4A). Usando un cryoloop, transfiera rápidamente cristales a un 0.2 μl gota en un cubreobjetos de 22 mm que contenga solución HA diluido en la condición de la cristalización, tal que la concentración final de HA oscila entre 1-20 mM. La caída del sello e incubar durante diferentes periodos de tiempo (figura 4B). Un buen punto de partida es de 5, 10, 60 y 90 minutos y durante la noche.
- Inspeccione visualmente los cristales con un microscopio de luz para identificar posibles fisuras o cambios en el color, que pueden indicar efectos adversos a la glicoproteína cristal o derivatización de cristal.
- Montar cristales en cryoloops y parte posterior remojo cristales para 30 s en tres consecutivos de 0,2 μl gotas que contengan solución de licor madre suplementado con 20% (v/v) de glicerol (o suplente crio-protector)69. Espalda-remojo los cristales quita HA compuesto que debía no específica y reduce la ocupación parcial causada por la débil Unión de HA. Flash cristales de congelación en nitrógeno líquido (figura 4B).
- Para la recolección de datos, procesamiento, solución de la estructura y refinamiento, utilizan protocolos previamente descritos26,70,71,72.
10. la inmersión de la glicoproteína cristales con su ligando
- Se reproducen bien diffracting cristales obtenidos en la sección 8 en una placa de 24 pocillos cristalización utilizando el método de difusión de vapor de la gota colgante.
- Prepare una solución del ligando de 50 mM en 20 mM Tris, pH 9.0, 150 mM NaCl.
Nota: La concentración del ligando debe elaborarse según la afinidad por su glicoproteína. Si la afinidad es desconocida, podría ser necesario utilizar un método como ITC (sección 12.2) para determinar la afinidad antes Inicio remojo experimentos. Asegúrese de que el ligando es soluble a la concentración deseada en el buffer requerido. - Añadir diversas concentraciones de ligando a los gota que contiene cristales ECD y sello de la gota para la incubación en longitudes de tiempo que oscilan entre los 5 min d 5.
- Cristales la pista visualmente con un microscopio de luz para identificar posibles cambios en la morfología.
- Montar cristales en cryoloops y cryo-protegerlos en la solución de licor madre suplementado con 20% (v/v) de glicerol (u otro crio-protector como etilenglicol o glicol de polietileno de bajo peso molecular 400)69.
- Para la recolección de datos, procesamiento, solución de la estructura y refinamiento, utilizar protocolos previamente descritos73,74,75.
11. producción de fragmento de antígeno (Fab) de enlace
- Subclone genes que corresponden a la cadena pesada Fab (HC) y secuencias de la cadena ligera (LC) de los anticuerpos anti-ECD, por ejemplo,epratuzumab.
Nota: Como alternativa, IgG puede ser dividido por la papaína enzima para generar fragmentos Fab76. - Transfectar las células como se describe en la sección 4, con las siguientes modificaciones:
- Utilice una masa total de DNA para la transfección de fragmentos Fab de 90 μg por cada 200 mL de la cultura.
- Transfectar plásmidos HC y cl en una proporción de 2:1 para reducir la cantidad de formación de dímeros de LC.
- Después de 7 d de incubación, cosechar células, conservar el sobrenadante y filtrar con un dispositivo de filtración de vacío impulsados de 0,22 μm.
- Equilibrar columnas de afinidad de anti-LC (kappa o lambda) en tampón PBS con un sistema de cromatografía de sobremesa.
Nota: Si la formación de dímeros de LC es un problema durante la purificación, cromatografía de afinidad de la proteína G puede utilizarse como alternativa a la purificación de afinidad kappa/lambda LC. - Carga de sobrenadante en columna de afinidad en 4 mL min-1. Tras la carga de la muestra, lavar la columna con 3-4 CV de PBS.
- Eluir la proteína de la columna usando una elución isocrática con glicina 100 mM, pH 2.2, inmediatamente neutralizando las fracciones eluídas con 10% (v/v) 1 M Tris, pH 9.0 en cada fracción.
Nota: La Fab eluída puede ser más purificada por cromatografía de intercambio iónico o cromatografía por exclusión de tamaño utilizando un FPLC a 4 ° C.
12. Caracterización de la Fab y pequeña molécula vinculante a la glicoproteína
- Interferometría de biocapa
- Preparar 50 mL de 1 x de buffer de cinética (1 x PBS, 0.002% Tween-20, 0.01% (p/v) BSA (v/v)).
- Hidratar seis biosensores de Ni-NTA en 200 μL de tampón de cinética de 1 x durante 10 minutos en una placa de humectación previa.
- Diluir su etiquetado ECD en 1 mL de tampón de cinética de 1 x a una concentración final de 25 ng μl-1. Pipeta de diluciones seriadas de Fab purificado en 200 μL de 1 x de buffer de cinética, con una alta concentración de 500 nM y subsiguientes diluciones seriadas de 250 nM, 125 nM y 62.5 nM.
- Reactivos alícuotas en polipropileno microplacas de 96 pocillos fondo plano negro como se muestra en la figura 5A, donde cada uno bien contiene 200 μL de la solución indicada.
- Recopilar datos mediante los ensayos de cinética en el software de adquisición de datos, como se describió anteriormente38,39,77 (figura 5A).
- Brevemente, transferir los biosensores en pocillos que contengan 1 x buffer de cinética a línea de fondo por 60 s antes de cargar 25 ng μl-1 de la glicoproteína para 240 s (o hasta un límite de 1.0 nm se alcanza) a 1.000 rpm.
- Después de una segunda base de los 60 s en 1 x buffer de cinética, transferir los biosensores en pozos que contiene la dilución seriada de la Fab. La fase de Asociación de s 180 posteriormente es seguido por un paso de disociación s 180 en 1 x buffer de cinética.
Nota: Biosensores pueden ser reutilizados si el protocolo anterior es seguido por un paso de la regeneración, que consiste en tres ciclos de lavado de los biosensores en stripping buffer (PBS con imidazol de 500 mM) 5 s seguido de 5 s en el 1 x de buffer de la cinética de neutralización. Biosensores pueden reutilizarse hasta ~ 10-20 veces durante el mismo día, o hasta datos de mala calidad se observa.
- Analizar los datos utilizando el software de análisis (figura 5A):
- En la ficha 1, importación y seleccionarlos datos.
- Sección 2, paso 1: Selección de datos, seleccione 'Sensor selección' y destaca referencia pozos (filas E y F, figura 5A), haga clic derecho y hacer referencia bien a. Bajo paso 2: Resta, seleccione 'pozos de referencia'. En paso 3: Alinear el eje, seleccione 'Instantánea' de rango de tiempo de 0.1 a 59,8 s. En el paso 4: Paso entre corrección, seleccione 'Alinear a disociación'. Bajo paso 5: El proceso, seleccione 'Filtro de Savitzky-Golay' y pulse datos de proceso.
- Bajo la ficha 3, seleccione 'Asociación y disociación' en paso a analizar con un modelo 1:1. Seleccione 'Conexión Global' y 'Grupo de Color'. Haga clic con el botón derecho curvas, seleccione 'Cambiar color', establece todas las curvas en el color de su elección. Seleccione 'Ajuste curvas'. Si bien se cuentan los datos, se puede exportar un informe seleccionando 'Guardar informe'.
- Repite el experimento con la glicoproteína producida en las células HEK293F y HEK293S (sección 5) y después del tratamiento Endo H (sección 7) para evaluar el efecto, si los hubiere, de diferentes glicoformas reconocimiento Fab. Además, repetir el experimento con truncamientos de ECD para proporcionar la penetración en los dominios vinculados por la Fab.
- Calorimetría isotérmica de la titulación de la interacción de la glicoproteína de la Fab
Nota: Experimentos ITC descritos aquí se realizan utilizando un instrumento automatizado de ITC. Los experimentos se llevan a cabo en un 1 mL redondo inferior bloque de 96 pocillos.- Dializan los ECD y Fab en un solo vaso de 4 L de 20 mM Tris, pH 8.0, 150 mM NaCl a 4 ° C durante la noche con una barra de agitación.
- Concentrado de dializado ECD y Fab 5 μm y 50 μm, respectivamente, con un filtro centrífugo NMWL de 10 kDa, asegúrese de lavar las membranas concentrador tres veces con 5 mL de diálisis tampón a 4.000 x g durante 5 minutos a 4 ° C antes de usar.
Nota: Cualquier desajuste en almacenador intermediario entre las muestras en la célula y la jeringa puede causar calor no deseado ser lanzado durante el experimento ITC y el resultado en datos de mala calidad. - Para el experimento 1: Añadir 400 μL de ECD a A1 que se cargue en la celda y 120 μl de la Fab a A2 bien a ser cargado en la jeringa. Bien A3 se deja vacío para volver la siguiente muestra mixta experimenta realización. Cada experimento posterior se puede Agregar al plato en el mismo orden (es decir, el experimento 2: pozo del vacío de la célula - A4, jeringa - A5, - A6; Figura 5 B).
Nota: Incluye buffer en controles de buffer (para confirmar que el instrumento se comporta bien) al principio y al final de cada gestión, así como ligando (en jeringa) en controles de buffer (en celda) para calcular el calor de dilución para la muestra en la jeringa. Esto calcula el calor de dilución entonces debe restarse de datos experimentales durante el análisis de los datos (figura 5B). - Ejecutar un total de 16 inyecciones con un volumen de 2.5 μL para cada inyección. La duración de la inyección es de 5 s, con 180 separación s entre las inyecciones. Ajustar la temperatura de la célula a 25 ° C, con una velocidad de 750 rpm de agitación y un período de filtro de 5 s.
Nota: Basado en la afinidad y la termodinámica de la interacción ECD:Fab, puede ser necesario cambiar la concentración de la muestra, número de inyecciones o temperatura de la célula. - Analizar los datos con el software de análisis, como se describió anteriormente40,41,43 (figura 5B).
- Repetir el experimento por lo menos por duplicado, cálculo de medias KD valores y errores estándar. Repite el experimento con ECD de diferentes glicoformas (secciones 5 y 6) para evaluar el efecto, si los hubiere, de glicoformas en la termodinámica de la interacción de la Fab: glicoproteína.
- Para la calorimetría isotérmica de la titulación de las interacciones ligando-glicoproteína, configurar el experimento ITC como se describe en la sección 12.2, con los siguientes cambios:
- Dializan ECD en 4 L de buffer de diálisis durante la noche. Disolver el ligando utilizando buffer de diálisis tras la finalización de la diálisis.
- Realizar experimentos ITC en concentraciones significativamente más altas para poder detectar las interacciones de baja afinidad. Para la interacción ECD y ligando, realizar experimentos ITC en concentraciones de 100 μm de ECD en la célula y 1 mM de ligando en la jeringa.
Resultados
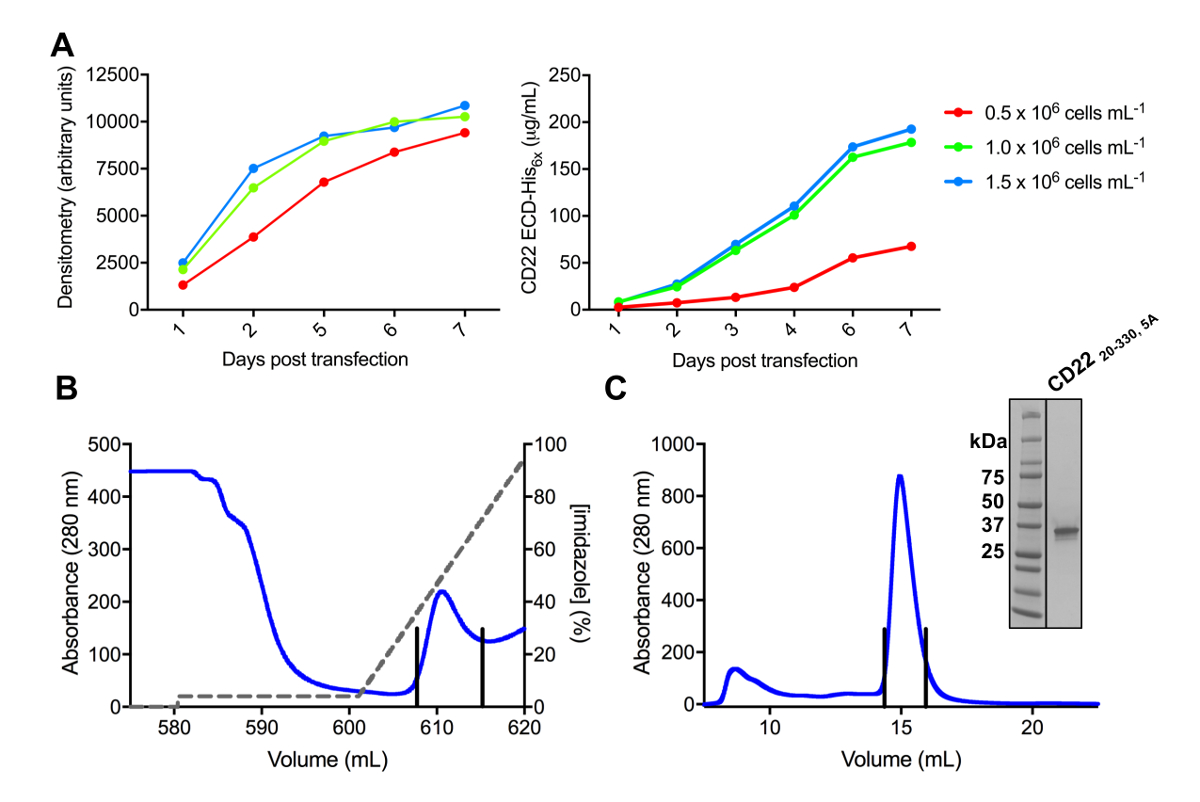
Varias construcciones de ECD CD22 con éxito fueron clonadas en el vector de expresión pHLsec y overexpressed en mamíferos HEK293F y HEK293S líneas celulares (figura 2 y 3A). Todas las construcciones eran purificadas a homogeneidad de tamaño por cromatografía por exclusión de tamaño y rindió una muestra muy pura para los estudios de cristalización (figura 3B y 3C). La construcción de CD22 que llevó a cristales bien diffracting era el truncamiento de d1-d3 (residuos 20-330), con cinco de los seis sitios previstos N-ligado glycosylation mutados de ADN a Ala (N67A, N112A, N135A, N164A y N231A), producido en las células HEK293S, tales que solamente el sitio de glicosilación en la posición se mantuvo el N101 (esta construcción se denomina CD2220-330, 5A). Cristales obtenidos en diversas condiciones de la pantalla de matriz sparse MCSG-1, pero los mejores cristales eran de una condición que contiene 30% (p/v) de polietilenglicol 4000, cloruro de litio de 0,2 M y 0,1 M de Tris, pH 8.5. Estos cristales nativos difractados a 2.1 resolución Å; utilizando estructuras conocidas de dominios Ig de proteínas relacionadas Siglec no dió ninguna solución en busca del Señor.
Para adquirir información puesta en fase, hemos empapado cristales nativos con un grupo de compuestos HA que incluye Hg, Pt, Os, Ta y Br en concentraciones que van desde 1-20 mM de HA compuesto para un tiempo de incubación de 5 min a 1 d (figura 4). Seguimiento cristales para cambios en la morfología y encontró que cristales empapados con HA compuesto en 20 mM resultaron en el rápido resquebrajamiento y la disolución del cristal. Nos congeló un total de 63 cristales que conservan su forma después de tiempos de incubación establecido que fueron empapados con racimo de bromuro de tantalio, platino cloruro, acetato de mercurio y cloruro de mercurio. Cristales empapados con 7 mM de cloruro de mercurio 30 minutos demostrada señal anómala en una exploración de fluorescencia en la fuente de luz canadiense (CLS) 08-BM beamline (Saskatoon, Canadá) y permitieron la dispersión anómala onda recogida de datos de rayos x en un solo cristal. Estos conjuntos de datos nos ha permitido resolver la subestructura de mercurio de CD225A 20-330, que reveló un átomo de mercurio solo limitado a una cisteína libre en posición C308 y en última instancia nos permitió construir la estructura de CD225A 20-330, en las fases mapa de densidad de electrones utilizando autocompilación78.
Una vez que se resolvió la estructura unliganded, estábamos interesados en la solución de la estructura de CD22 a su ligando, α2-6 siallylactose. Primero calculamos la afinidad de CD22 hacia sialyllactose α2-6 uso de ITC para caracterizar la termodinámica de unión de la interacción. Observamos una afinidad de ~ 280 μm y utiliza esta información para identificar una concentración inicial (~ 100 x KD) de ligando para remojo de nuestros nativos CD2220-330 5A cristales. Empapado el CD2220-330, 5A cristales siallylactose de 25 mM para 5 min, 2 h, 14 h, 40 h y 5 d y controlar cambios en la morfología cristalina. Un total de ~ 75 cristales fueron congelados desde varios puntos de tiempo y enviado a Sincrotrón CLS línea 08-ID (Saskatoon, Canadá) para la recolección remota de datos. Se recolectaron un total de seis conjuntos de datos de rayos x de cristales bien diffracting. La estructura de cada conjunto de datos de rayos x fue resuelto por el Señor usando unliganded CD2220-330, 5A estructura como un modelo de búsqueda inicial. La densidad del electrón que resulta para todos los datasets entonces fue inspeccionada para densidad positiva en el mapa de Fo-Fc que correspondería para destino α2-6 sialyllactose en el sitio de unión de CD22. Notablemente, los conjuntos de datos recogido, incluso los cristales empapados después de sólo 5 minutos de tiempo de incubación, contenido positivo densidad correspondiente a ligand en el sitio de Unión. Estructuras generales del unliganded y CD22 liganded fueron muy similares con mínimos cambios conformacionales, que podrían explicar el éxito de remojo experimentos con α2-6 sialyllactose.
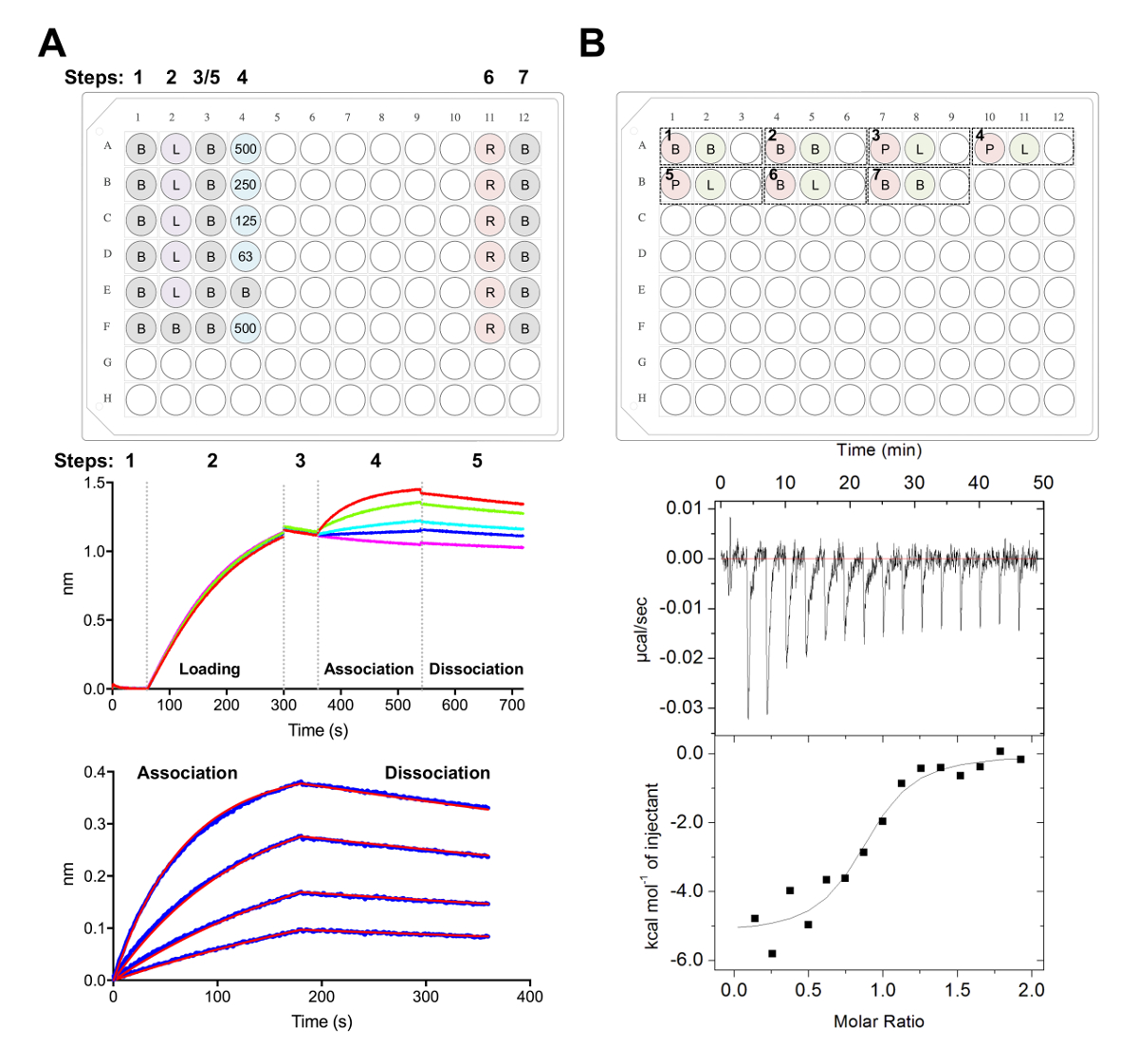
A continuación caracterizamos la superficie antigénica de CD22 reconocido por epratuzumab anticuerpo terapéutico en experimentos BLI y CCI (figura 5). Cinética y termodinámica perfiles de Unión Fab epratuzumab a CD22 construcciones con diferentes glicoformas revelaron una afinidad creciente para CD22 con tamaño reducido glycan N-ligados, con hasta una 14-fold mejora en afinidad por glicanos más pequeña (327 nM vs 24 nM en BLI; 188 nM vs 58 nM en ITC). Lo glicanos CD22 N-ligados restrinjan el acceso del anticuerpo a su epítopo fue identificado por BLI utilizando a mutantes de un solo punto de CD22 y resolviendo el epratuzumab Fab-CD22 d1-d3 cristal Co estructura28.

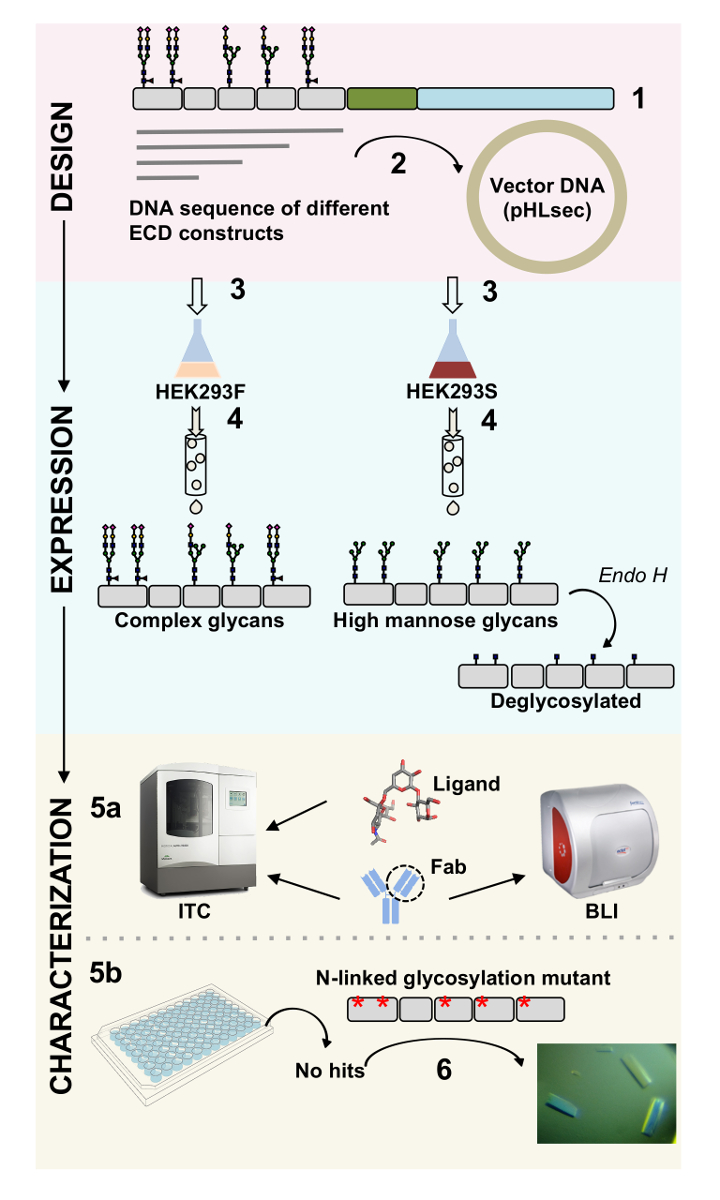
Figura 1 . Resumen de caracterización de glicoproteína de construcción diseño de caracterización Biofísica y estructural. Primaria (1) análisis de la glicoproteína representativa de la secuencia. En gris, el dominio extracelular (ECD); en verde, segmento transmembrana (TM); y en azul, el dominio citosólico de la glicoproteína. Predijo los glycans N-ligados están etiquetados. (2) reproducción de construcciones de la primera infancia. (3) expresión de la primera infancia construye, en células de mamíferos. (4) purificación de la glicoproteína. Mientras que las proteínas expresadas en HEK293F contendrá glycans complejo, las proteínas expresadas en HEK293S tendrá glicanos de alta manosa. Tratamiento enzimático de glicoproteínas producida en células HEK293S con Endo H resultados en glicoproteínas con sólo una molécula GlcNAc en sitios de glicosilación N-ligados. Glicoproteínas (5a) se analizan para su unión los anticuerpos a por interferometría de la biocapa (BLI) y calorimetría isoterma de titulación (ITC). Afinidad a ligandos puede medirse también por el CCI. (5b) ensayos de cristalización de las glicoproteínas con homogénea glycans N-ligados, como las expresadas en HEK293S y deglycosylated con Endo H. (6) en algunos casos, la mutación de sitios de glicosilación N-ligado es necesario para obtener cristales. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2 . Diseño del ectodominio de CD22 ADN construye para su expresión en células de mamífero. A) representación del plásmido pHLsec utilizado por transfección transitoria de construcciones CD22 ECD. Africanas y KpnI sitios utilizados para la clonación se indican con cajas rojas. B) el DIT CD22 contiene siete dominios de Ig (d1-d7) y 12 sitios de glicosilación predicha de N-ligados (en azul). Se diseñaron cuatro constructos de la ECD CD22. C) 1% gel de agarosa mostrando los amplicones de PCR de CD22 ECD construye para el clonado en el vector de expresión mamífero pHLsec. Primer carril contiene marcadores de DNA de 1 kb. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3 . Expresión y purificación de glicoproteínas. A) efecto de la densidad celular en los rendimientos de la expresión. Expresión de la glicoproteína en la cultura de pequeña escala 25 mL de HEK293F suspensión células transfectadas con tres diferentes densidades a partir de las células (0,5 x 106 células mL-11.0 x 106 células mL-1y 1.5 x 106 células mL -1). Cuantificación realizada por densitometría de SDS-PAGE en el panel de la izquierda y cuantitativa BLI en el panel de la derecha. Valores son representativos de una preparación de glicoproteína. B) cromatograma del primer paso de purificación para construir CD2220-330, 5A de 600 mL de sobrenadante usando una columna de afinidad de Ni-NTA. La glicoproteína eluyó utilizando un gradiente de imidazol (línea gris), donde 100% corresponde al buffer de elución, que contiene imidazol de 500 mM. Combinado fracciones se representan con líneas verticales. C) cromatograma de exclusión de tamaño para construir CD2220-330, 5A con una potente columna de filtración de gel. Fracciones agrupados desde el pico de elución son representados con líneas verticales. Recuadro: Gel tinción de Coomassie SDS-PAGE que muestra la pureza de la glicoproteína. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4 . Cristal con átomos pesados. A) estación de trabajo muestra de remojo cristales nativos HA compuestos. Todo requiere herramientas que están etiquetadas. B) pasos que se siguieron para remojo cristales de construcción CD2220-330, 5A con compuestos HA. Paso 1, abrir bien con cristales y cristales de transferencia utilizando un bucle a una caída de 0,2 μL en un cubreobjetos con solución HA diluido en la condición de la cristalización, tal que la concentración final de HA oscila entre 1-10 mM. Paso 2, sello de la gota en la placa de cristalización e incubar cristales con el HA compuesto para diferentes periodos de tiempo. Paso 3, montar el cristal empapado en el lazo y espalda-remoje durante 30 s en tres consecutivos 0,2 μl las gotas que contiene la solución del licor madre suplementada con glicerol 20% (v/v) dispensado en un cubreobjetos. Paso 4, flash congele el cristal montado en un circuito con nitrógeno líquido y colocar en un disco para su envío a la línea de sincrotrón. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5 . Biocapa interferometría e isotérmico titulación calorimetría mediciones. A) experimento representante BLI. Panel superior: ejemplo de configuración de la placa para un experimento de cinética, donde están marcados los siguientes: 1 cinética x buffer (B), su6 x-glicoproteína etiquetada carga (L), representante de concentraciones Fab (500, 250, 125, 62.5 nM), PBS + regeneración de 500 mM buffer (R) y 1 x la neutralización de la cinética del almacenador intermediario (B). Bien, cada una contiene 200 μL de solución. Número de paso para el experimento de cinética se indica en la parte superior de la placa. Panel de centro: Datos crudos representativos de BLI experimento realizado usando biosensores de Ni-NTA y la placa descrito en el panel superior. Paso números corresponden a la línea de base (1) su glicoproteínax 6 carga (2), base (3), Asociación de dilución seriada del Fab (4) y disociación (5). Medidas de regeneración no son representados (pasos 6-7). Panel inferior: Datos representativos mostrando cruda asociación y disociación (línea azul) con el correspondiente de 1:1 ajuste (línea roja). B) panel superior: configuración de placa representativa de un ITC solo ejecutar en un instrumento automatizado de ITC con siete experimentos en un 96-pozo redondo del bloque inferior. Cada experimento está compuesto por tres pozos. El primer pozo (rojo) corresponde a la muestra de la celda (400 μL), el segundo pozo (verde) corresponde a la muestra de la jeringa (120 μL). La tercera también queda vacía, y las muestras mixtas se devolverán a este experimento bien finalizado. Experimentos 1, 2 y 7 son buffer en controles de búfer. Experimentos 3-5 representan los experimentos por triplicado con glicoproteína (P) en la celda y Fab o ligando (L) en la jeringa. Experimento 6 representa un calor ligando de control de dilución y debe restarse de experimentos 3-5 durante el análisis de datos. Panel inferior: Representante crudo (arriba) y procesados (abajo) ITC datos de Fab (epratuzumab) atar a ECD CD22 se producen en células de HEK293F. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Discusión
Glicoproteínas ancladas a membrana son críticos para la función de la célula y atractivo dianas terapéuticas. Aquí, presentamos un protocolo para la caracterización estructural y Biofísica de la ECD de glicoproteínas de membrana, tanto solos como en complejos con ligandos de molécula pequeña y los fragmentos Fab. Con éxito hemos utilizado este protocolo para determinar la estructura cristalina de los tres dominios de Ig N-terminal-la mayor parte de la porción extracelular del humano CD2228, un correceptor crítico en las células B involucradas en mantener inmunidad humoral check79. También hemos caracterizado el sitio de unión de CD22 con su ligando natural α2-6 sialyllactose y define el modo de reconocimiento de un anticuerpo terapéutico hacia humano CD22. Estos resultados proporcionan ideas sobre la relación estructura-función de miembro de la familia de Siglecs que ha restringido la expresión en las células de B y una hoja de ruta molecular hacia el desarrollo de la nueva molécula pequeña dirigida CD22 y basados en anticuerpos terapéutica. Mientras que este protocolo fue utilizado con éxito para un receptor de la célula de B que contienen Ig, proponemos que nuestro enfoque puede ser aplicado para la caracterización estructural y Biofísica de cualquier glicoproteína de membrana con una organización de dominio distintos. En tales casos, la construcción diseño y combinatoria glycan N-ligado pueden evaluarse las mutaciones (ya sea a Gln o Ala) para encontrar una construcción conveniente para el crecimiento cristalino y difracción de alta resolución.
Obtención de una muestra homogénea y pura de la glicoproteína es de vital importancia para crecimiento cristalino y difracción de rayos x, así como para la caracterización biofísica aguas abajo. Glycans N-ligados en glicoproteínas son inherentemente heterogéneos y pueden causar heterogeneidad conformacional y química dentro de la glicoproteína que puede impedir la formación de cristales. Para reducir esta heterogeneidad micro, estrategias que introducen mutaciones puntuales para eliminar los residuos de Asn predichos para abrigar glycans N-ligados, o usando líneas celulares mutantes (como HEK293S) seguido de tratamiento con endoglycosidases (como EndoH) pueden considerablemente mejorar la cristalización éxito15,21,22. En este protocolo, se discute la purificación de glicoproteínas solubles y fábricas que son secretadas en el sobrenadante de la célula. La secreción de glicoproteínas proporciona una ruta relativamente sencilla hacia la pureza, sin la necesidad de lisis celular o la adición de productos químicos fuertes o detergentes. La célula sobrenadante, obtenido siguientes célula cosecha luego corre directamente sobre una columna que tiene afinidad por la proteína de interés (por ejemplo, Ni-NTA para su etiquetado glicoproteínas o afinidad de LC con fragmentos Fab). Sin embargo, dependiendo de la columna de uso y las condiciones de la célula sobrenadante (e.g., pH), puede verse afectada la capacidad de unión de la proteína de interés a la columna. Si este es el caso, puede ser necesario concentrar y cambio el sobrenadante celular para mejorar el enlace a la columna de búfer. Además, se recomienda emplear medidas de control de calidad durante la purificación para ayudar a evaluar la pureza de la proteína. Correr un gel de SDS-PAGE o Western blot de todas las muestras (antes, durante y después de pasos de purificación) puede producir información sobre si el esquema de purificación propuesto es adecuado para la proteína de interés. Si las bandas contaminantes están visibles en SDS-PAGE, o si varias especies se obtienen durante la purificación (por ejemplo, varios picos sobre la exclusión de tamaño), pasos de purificación adicional se debe considerar, por ejemplo, cromatografía de intercambio iónico, para obtener en pureza y aumento de las posibilidades de cristalización aguas abajo80.
De cristalización macromolecular, a menudo es crítica para obtener altos rendimientos de la proteína de interés para permitir la proyección de un gran número de posibles condiciones de cristalización en las concentraciones de proteína alta para encontrar hits de cristal adecuado. Generalmente, las HEK293 líneas celulares aquí (HEK293F y HEK293S) son sistemas de expresión sólida y pueden fácilmente ampliarse para producir más muestra según sea necesario. Sin embargo, es posible que la proteína de interés no puede expresar suficientemente en estas líneas celulares. En estos casos, otras líneas celulares, como células de Expi293 de81,82, se han encontrado para demostrar niveles superiores de expresión de la proteína y deben ser considerados como una alternativa.
Si bien ordenados, diffracting los cristales no se después de pruebas de varias construcciones de la proteína de interés a pesar de alta pureza, puede ser necesario ampliar técnicas de cristalización para promover la formación de cristales. Se ha demostrado que fragmentos Fab de anticuerpos y nanobodies pueden ser potenciadores de excelente cristalización y promover cristal ordenado embalaje83,84,85. Estos fragmentos pueden expresados y purificados a homogeneidad y utilizados en un complejo con la proteína de interés para promover la cristalización. Lo importante, fragmentos Fab producidas como se describe en la sección 10 pueden tener una tendencia a formar no funcionales LC dímeros86. Estos dímeros son contaminantes y deben eliminarse durante la purificación. En nuestra experiencia, dímeros de LC a menudo tengan un volumen de retención diferentes de exclusión de tamaño, o responsables como un pico distinto en la cromatografía de intercambio iónico y así pueden extraerse la purificación Fab - sin embargo esto no es siempre el caso. Si estas técnicas no son suficientes para eliminar dímeros de LC de la purificación de la Fab, métodos de purificación adicional, como la purificación de la afinidad de la proteína G, pueden ser empleados para mejorar la pureza.
Alternativa a la co-complejación con fragmentos Fab, técnicas bien documentadas como microseeding de la matriz al azar pueden mejorar las posibilidades de obtención de cristales bien ordenado63,70. Este método implica la adición de pequeñas cantidades de cristales aplastados, subóptimos en la condición de la cristalización, proporcionando un cristal nuclea a promover el crecimiento del cristal. Esto puede realizarse usando cristales de la proteína de interés, o con estructura similar dominio de arquitectura y terciario. Además, microseeding de la matriz al azar se puede realizar en un intento de cristalizar la proteína sola o en conjunto con un fragmento Fab o pequeñas moléculas de interés. Los recientes avances en microscopia del cryo-electrón también hacen esta técnica una alternativa atractiva a la cristalografía de rayos x para obtener información estructural de alta resolución para moléculas con características apropiadas87,88, 89,90,91.
Cuando eliminación de conjuntos de datos de difracción de rayos x por el Señor, HA remojo puede ser necesaria para resolver el problema de la fase dispersión anómala o sustitución isomórfica. Inspección de la secuencia de aminoácidos de la proteína puede proporcionar pistas sobre la estrategia para la derivatización de HA, incluyendo el pH óptimo para el enlace. En particular, cisteínas impares dentro de la proteína pueden atar específicamente HA compuestos que contienen mercurio. Remojo cristales nativos HA compuestos es un proceso iterativo para determinar la identidad del compuesto HA óptima, su concentración y el tiempo de incubación requerido. Si los intentos de remojo iniciales no den bien diffracting cristales que contienen una HA adecuado para la eliminación, puede ser necesario introducir sustituciones de aminoácidos para mejorar la probabilidad de la Unión HA y señal anómala. Los ejemplos incluyen mutaciones para incluir un residuo de cisteína libre para enlazar eficientemente Au, Pt, Hg y Pb. expresión de proteínas para eliminación anómala en un medio de seleno-metionina suplido en e. coli se utiliza ampliamente para la eliminación anómala, sin embargo un sistema equivalente que incorpora confiablemente seleno-metionina no está fácilmente disponible para células de mamífero en suspensión92,93y es una zona de futuro desarrollo.
Una vez que se obtiene la estructura unliganded de glicoproteínas de interés, tomando los cristales con ligandos de molécula pequeña puede realizarse para obtener una estructura del complejo ligando-receptor inmune. Estos datos proporcionan un modelo para el diseño racional de ligandos más específicos y de alta afinidad que puede utilizarse como terapéutica de molécula pequeña, como también ofrece ideas de alta resolución sobre la función biológica de la glicoproteína. Al intentar remojo cristales glicoproteína con ligandos de pequeña molécula de interés, inspección de la estructura de cristal unliganded puede indicar si el remojo debe ser posible. Si cierre cristal-embalaje contactos se encuentran alrededor del sitio de unión a ligando o en regiones que sufren cambios conformacionales al ligando vinculante, tomando probablemente ser problemático. En este caso, se deben realizar otros métodos como la cristalización del complejo proteína-ligando.
Divulgaciones
Los autores declaran a no hay intereses contrapuestos.
Agradecimientos
Experimentos de difracción de rayos x se describe en este documento fueron realizados utilizando líneas ID-08 y 08-BM en la fuente de luz canadiense, que es apoyado por la Fundación de Canadá para la innovación, ciencias naturales y Consejo de investigación de ingeniería de Canadá, la Universidad de Saskatchewan, el gobierno de Saskatchewan, diversificación económica occidental Canadá, el Consejo de investigación nacional Canadá y los institutos canadienses de investigación en salud. Nos gustaría reconocer la estructurales y biofísicas instalaciones centrales, el Hospital para niños enfermos, para el acceso a los instrumentos de ITC y BLI. J.E.O. fue apoyado por Banting Postdoctoral Fellowship BPF-144483 de los institutos canadienses de investigación en salud. T.S. es un receptor de Canadá postgrado becas Master Award y un Vanier Canadá becas postgrado de los institutos canadienses de investigación en salud. Este trabajo fue apoyado por funcionamiento PJT-148811 (J.-P.J.) la subvención de los institutos canadienses de investigación en salud. Esta investigación fue realizada, en parte, gracias a la financiación del programa de Cátedras de investigación de Canadá (J.-P.J.).
Materiales
| Name | Company | Catalog Number | Comments |
| 0.22 μm Steritop filter | EMD Millipore | SCGPS02RE | |
| 10 well 4-15% gradient SDS-PAGE gel | Bio-Rad | 4561084 | |
| 10x glycobuffer 3 | New England Biolabs | P0702S | Comes with Endo H reagent |
| 10x Kinetics Buffer | PALL FortéBio | 18-1092 | |
| 10x Tris/Glycine/SDS Buffer | Bio-Rad | 1610732 | |
| 1 mL round bottom 96 well block | ThermoFisher | 260251 | |
| 22 mm cover slip | Hampton research | HR3-231 | |
| 4x Laemmli Sample Buffer | Bio-Rad | 1610747 | |
| 96-3 well INTELLIPLATE low volume reservior | Art Robbins Instruments | 102-0001-03 | |
| AgeI | New England Biolabs | R0552S | |
| ÄKTA Pure | GE Healthcare | ||
| ÄKTA Start | GE Healthcare | ||
| Amicon Ultra 15 centrifugal filtration device 10KDa MWCO | Millipore | UFC901008 | |
| Amicon Ultra 4 centrifugal filtration device 10KDa MWCO | Millipore | UFC801008 | |
| Auto-iTC200 | Malvern | ||
| Axygen MaxyClear Snaplock 1.5 mL microtubes | Fisher Scientific | MCT150C | |
| Countess Cell Counting Chamber Slides | Thermo Fisher Scientific | C10228 | |
| CryoLoop 18 x 0.05-0.1 mm | Hampton research | HR4-945 | |
| CryoLoop 18 x 0.1-0.2 mm | Hampton research | HR4-947 | |
| CryoLoop 18 x 0.2-0.3 mm | Hampton research | HR4-970 | |
| Digital Dry Bath | Bio-Rad | 1660562EDU | |
| E. coli DH5α | Invitrogen | 18258012 | |
| Endo H | New England Biolabs | P0702S | |
| Erlenmeyer flask (baffled base), polycarbonate, sterile, 500 mL, DuoCAP | TriForest Labware | FBC05000S | |
| Erlenmeyer flask 125 mL (baffled base), polycarbonate, sterile, 125 mL with vented cap | VWR | 89095-258 | |
| Falcon Disposable sterile serological pipet, non-pyrogenic, 10 mL | Greiner Bio-One | 607180 | |
| Falcon Disposable sterile serological pipet, non-pyrogenic, 25 mL | Greiner Bio-One | 760180 | |
| Falcon Disposable sterile serological pipet, non-pyrogenic, 5 mL | Greiner Bio-One | 606180 | |
| Falcon Disposable sterile serological pipet, non-pyrogenic, 50 mL | Greiner Bio-One | 768180 | |
| FectoPRO DNA Transfection Reagent, Polyplus | VWR | 10118-842 | |
| Freestyle 293F cells | Thermo Fisher Scientific | R79007 | |
| Freestyle Expression medium | Thermo Fisher Scientific | 12338001 | |
| Heavy Atom Screens Au | Hampton research | HR2-444 | |
| Heavy Atom Screens Hg | Hampton research | HR2-446 | |
| Heavy Atom Screens M1 | Hampton research | HR2-448 | |
| Heavy Atom Screens M2 | Hampton research | HR2-450 | |
| Heavy Atom Screens Pt | Hampton research | HR2-442 | |
| HEK 293S | ATCC | ATCC CRL-3022 | |
| HisTrap Affinity Column | GE Healthcare | 17525501 | |
| HiTrap KappaSelect Affinity Columns | GE Healthcare | 17545811 | |
| HiTrap LambdaSelect Affinity Columns | GE Healthcare | 17548211 | |
| KpnI | New England Biolabs | R0142S | |
| MCSG-1 Crystal Screen 1.7 mL block | Anatrace | MCSG-1 | |
| MCSG-2 Crystal Screen 1.7 mL block | Anatrace | MCSG-2 | |
| MCSG-3 Crystal Screen 1.7 mL block | Anatrace | MCSG-3 | |
| MCSG-4 Crystal Screen 1.7 mL block | Anatrace | MCSG-4 | |
| Mercuric chloride | Sigma | 1044170100 | |
| Microplate, 96 well, polypropelene, flat bottom, black | Greiner Bio-One | 655209 | |
| Minstrel DT UV | Formulatrix | ||
| Multitron Pro shaker | Infors HT | MP25-TA-CO2HB | |
| Nanodrop 2000/2000c Spectrophotometer | Thermo Fisher Scientific | ND-2000 | |
| Nanosep 3K Omega centrifugal device | PALL Life Science | OD003C33 | |
| Ni-NTA biosensors | PALL FortéBio | 18-5102 | |
| Octet RED96 | PALL ForteBio | ||
| Oryx 4 crystallizaiton robot | Douglas Instrument | ORY-4/1 | |
| Platinum chloride | Sigma | 520632-1g | |
| Precision Plus Protein Standard | Bio-Rad | 161-0374 | |
| PureLink HiPure Plasmid Maxiprep Kit | Invitrogen | K210006 | |
| Quick Coomassie Stain | Protein Ark | GEN-QC-STAIN-1L | |
| Steriflip Sterile 50 mL Disposable Vacuum Filtration System 0.22 µm Millipore Express | EMD Millipore | SCGP00525 | |
| Superdex 200 Increase 10/300 GL | GE Healthcare | 28990944 | |
| Superose 6 10/300 GL | GE Healthcare | 17517201 | |
| Tantalum bromide cluster | Jena bioscience | PK-103 | |
| Top96 Crystallization Screen | Rigaku Reagents | 1009846 | |
| Tryphan Blue | Thermo Fisher Scientific | T10282 | |
| VDX 24-well with sealant | Hampton research | HR3-172 | |
| α2-6 sialyllactose | Sigma Aldrich | A8556-1mg |
Referencias
- Sachs, J. N., Engelman, D. M. Introduction to the membrane protein reviews: The interplay of structure, dynamics, and environment in membrane protein function. Annu Rev Biochem. 75 (1), 707-712 (2006).
- Cournia, Z., et al. Membrane protein structure, function, and dynamics: A perspective from experiments and theory. J Membr Biol. 248 (4), 611-640 (2015).
- Macauley, M. S., et al. Antigenic liposomes displaying CD22 ligands induce antigen-specific B cell apoptosis. J Clin Invest. 123 (7), 3074-3083 (2013).
- Hyde, C. A. C., et al. Targeting extracellular domains D4 and D7 of vascular endothelial growth factor receptor 2 reveals allosteric receptor regulatory sites. Mol Cell Biol. 32 (19), 3802-3813 (2012).
- Tai, W., Mahato, R., Cheng, K. The role of HER2 in cancer therapy and targeted drug delivery. J Control Release. 146 (3), 264-275 (2010).
- Zarei, O., Benvenuti, S., Ustun-Alkan, F., Hamzeh-Mivehroud, M., Dastmalchi, S. Strategies of targeting the extracellular domain of RON tyrosine kinase receptor for cancer therapy and drug delivery. J Cancer Res Clin Oncol. 142 (12), 2429-2446 (2016).
- Rosman, Z., Shoenfeld, Y., Zandman-Goddard, G. Biologic therapy for autoimmune diseases: an update. BMC Med. 11 (1), 88 (2013).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Barclay, A. N. Membrane proteins with immunoglobulin-like domains - A master superfamily of interaction molecules. Semin Immunol. 15 (4), 215-223 (2003).
- Barclay, A. N. Ig-like domains: evolution from simple interaction molecules to sophisticated antigen recognition. Proc Natl Acad Sci. 96 (26), 14672-14674 (1999).
- Aebi, M. N-linked protein glycosylation in the ER. Biochim Biophys Acta - Mol Cell Res. 1833 (11), 2430-2437 (2013).
- Ohtsubo, K., Marth, J. D. Glycosylation in cellular mechanisms of health and disease. Cell. 126 (5), 855-867 (2006).
- Lodish, H., Berk, A., Zipursky, S., Al, E. Glycosylation in the ER and Golgi complex. Mol Cell Biol. (4), (2000).
- Thomas, P., Smart, T. G. HEK293 cell line: A vehicle for the expression of recombinant proteins. J Pharmacol Toxicol Methods. 51 (3), 187-200 (2005).
- Lee, J. E., Fusco, M. L., Ollmann Saphire, E. An efficient platform for screening expression and crystallization of glycoproteins produced in human cells. Nat Protoc. 4 (4), 592-604 (2009).
- Betenbaugh, M. J., Tomiya, N., Narang, S., Hsu, J. T. A., Lee, Y. C. Biosynthesis of human-type N-glycans in heterologous systems. Curr Opin Struct Biol. 14 (5), 601-606 (2004).
- Yang, Z., et al. Engineered CHO cells for production of diverse, homogeneous glycoproteins. Nat Biotechnol. 33 (8), 842-844 (2015).
- Bláha, J., Kalousková, B., Skořepa, O., Pažický, S., Novák, P., Vaněk, O. High-level expression and purification of soluble form of human natural killer cell receptor NKR-P1 in HEK293S GnTI-cells. Protein Expr Purif. 140, 36-43 (2017).
- Bláha, J., Pachl, P., Novák, P., Vaněk, O. Expression and purification of soluble and stable ectodomain of natural killer cell receptor LLT1 through high-density transfection of suspension adapted HEK293S GnTI- cells. Protein Expr Purif. 109, 7-13 (2015).
- Chaudhary, S., Pak, J. E., Gruswitz, F., Sharma, V., Stroud, R. M. Overexpressing human membrane proteins in stably transfected and clonal human embryonic kidney 293S cells. Nat Protoc. 7 (3), 453-466 (2012).
- Chang, V. T., et al. Glycoprotein structural genomics: Solving the glycosylation problem. Structure. 15 (3), 267-273 (2007).
- Davis, S. J., Crispin, M. Solutions to the glycosylation problem for low- and high-throughput structural glycoproteomics. Funct Struct Proteomics Glycoproteins. , 127-158 (2011).
- Elbein, A. D., Tropea, J. E., Mitchell, M., Kaushal, G. P. Kifunensine, a potent inhibitor of the glycoprotein processing mannosidase I. J Biol Chem. 265 (26), 15599-15605 (1990).
- Zheng, K., Bantog, C., Bayer, R. The impact of glycosylation on monoclonal antibody conformation and stability. MAbs. 3 (6), 568-576 (2011).
- Aricescu, A. R., Lu, W., Jones, E. Y. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr Sect D Biol Crystallogr. 62 (10), 1243-1250 (2006).
- Adams, P. D., et al. The Phenix software for automated determination of macromolecular structures. Methods. 55 (1), 94-106 (2011).
- May, A. P., Robinson, R. C., Vinson, M., Crocker, P. R., Jones, E. Y. Crystal structure of the N-terminal domain of sialoadhesin in complex with 3' sialyllactose at 1.85 Å resolution. Mol Cell. 1 (5), 719-728 (1998).
- Ereño-Orbea, J., et al. Molecular basis of human CD22 function and therapeutic targeting. Nat Commun. 8 (1), 764 (2017).
- Yu, X. -. L., et al. Crystal structure of HAb18G/CD147: implications for immunoglobulin superfamily homophilic adhesion. J Biol Chem. 283 (26), 18056-18065 (2008).
- Garman, E., Murray, J. W. Heavy-atom derivatization. Acta Crystallogr - Sect D Biol Crystallogr. 59 (11), 1903-1913 (2003).
- Agniswamy, J., Joyce, M. G., Hammer, C. H., Sun, P. D. Towards a rational approach for heavy-atom derivative screening in protein crystallography. Acta Crystallogr Sect D Biol Crystallogr. 64 (4), 354-367 (2008).
- Rose, J. P., Wang, B. C., Weiss, M. S. Native SAD is maturing. IUCrJ. 2 (20), 431-440 (2015).
- Olieric, V., et al. Data-collection strategy for challenging native SAD phasing. Acta Crystallogr Sect D Struct Biol. 72 (3), 421-429 (2016).
- Rillahan, C. D., et al. Disubstituted sialic acid ligands targeting Siglecs CD33 and CD22 associated with myeloid leukaemias and B cell lymphomas. Chem Sci. 5 (6), 2398-2406 (2014).
- Mesch, S., et al. From a library of MAG antagonists to nanomolar CD22 ligands. ChemMedChem. 7 (1), 134-143 (2012).
- Chiu, M. L., Gilliland, G. L. Engineering antibody therapeutics. Curr Opin Struct Biol. 38, 163-173 (2016).
- Elgundi, Z., Reslan, M., Cruz, E., Sifniotis, V., Kayser, V. The state-of-play and future of antibody therapeutics. Adv Drug Deliv Rev. 122 (2016), 2-19 (2017).
- Yang, D., Singh, A., Wu, H., Kroe-Barrett, R. Determination of high-affinity antibody-antigen binding kinetics using four biosensor platforms. J Vis Exp. (122), e55659 (2017).
- Kamat, V., Rafique, A. Designing binding kinetic assay on the bio-layer interferometry (BLI) biosensor to characterize antibody-antigen interactions. Anal Biochem. 536, 16-31 (2017).
- Brautigam, C. A., Zhao, H., Vargas, C., Keller, S., Schuck, P. Integration and global analysis of isothermal titration calorimetry data for studying macromolecular interactions. Nat Protoc. 11 (5), 882-894 (2016).
- Duff, M. R., Grubbs, J., Howell, E. E. Isothermal titration calorimetry for measuring macromolecule-ligand affinity. J Vis Exp. (55), e2796 (2011).
- Livingstone, J. R. Antibody characterization by isothermal titration calorimetry. Nature. 384 (6608), 491-492 (1996).
- Freyer, M. W., Lewis, E. A. Isothermal titration calorimetry: Experimental design, data analysis, and probing macromolecule/ligand binding and kinetic interactions. Methods Cell Biol. 84, 79-113 (2008).
- Macauley, M. S., Crocker, P. R., Paulson, J. C. Siglec-mediated regulation of immune cell function in disease. Nat Rev Immunol. 14 (10), 653-666 (2014).
- Zaccai, N. R., et al. Structure-guided design of sialic acid-based Siglec inhibitors and crystallographic analysis in complex with sialoadhesin. Structure. 11 (5), 557-567 (2003).
- Pantophlet, R., et al. Bacterially derived synthetic mimetics of mammalian oligomannose prime antibody responses that neutralize HIV infectivity. Nat Commun. 8 (1), 1601 (2017).
- Leonard, J. P., et al. Epratuzumab, a humanized anti-CD22 antibody, in aggressive non-Hodgkin's lymphoma: phase I/II clinical trial results. Clin Cancer Res. 10 (16), 5327-5334 (2004).
- Finn, R. D., et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 45, 190-199 (2017).
- Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., Sternberg, M. J. E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 10 (6), 845-858 (2015).
- Lessard, J. C. Molecular cloning. Methods Enzymol. 529, 85-98 (2013).
- Gupta, R., Jung, E., Brunak, S. . NetNGlyc: Prediction of N-glycosylation sites in human proteins. , (2004).
- Liu, H., Naismith, J. H. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 8, 91 (2008).
- Heckman, K. L., Pease, L. R. Gene splicing and mutagenesis by PCR-driven overlap extension. Nat Protoc. 2 (4), 924-932 (2007).
- Froger, A., Hall, J. E. Transformation of plasmid DNA into E. coli using the heat shock method. J Vis Exp. (6), e253 (2007).
- . Optimization of glycoprotein expression by transient transfection in HEK293 F/S suspension cells Available from: https://www.polyplus-transfection.com/wp-content/uploads/2015/09/FectoPRO-Technical-Note-031716.pdf (2015)
- Taylor, S. C., Berkelman, T., Yadav, G., Hammond, M. A defined methodology for reliable quantification of western blot data. Mol Biotechnol. 55 (3), 217-226 (2013).
- Tan, H. Y., Ng, T. W. Accurate step wedge calibration for densitometry of electrophoresis gels. Opt Commun. 281 (10), 3013-3017 (2008).
- Gassmann, M., Grenacher, B., Rohde, B., Vogel, J. Quantifying Western blots: pitfalls of densitometry. Electrophoresis. 30 (11), 1845-1855 (2009).
- . Rapid, reliable quantitation of Fc-fusion protein in cell culture supernatants Available from: https://www.fortebio.com/documents/ForteBio_App_Note_13.pdf (2018)
- JoVE Science Education Database. Basic methods in cellular and molecular biology: Separating protein with SDS-PAGE. J Vis Exp. , (2018).
- Wilkins, M. R., et al. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol. 112, 531-552 (1999).
- Till, M., et al. Improving the success rate of protein crystallization by random microseed matrix screening. J Vis Exp. (78), e50548 (2013).
- Obmolova, G., Malia, T. J., Teplyakov, A., Sweet, R., Gilliland, G. L. Promoting crystallization of antibody-antigen complexes via microseed matrix screening. Acta Crystallogr Sect D Biol Crystallogr. 66 (8), 927-933 (2010).
- D'Arcy, A., Bergfors, T., Cowan-Jacob, S. W., Marsh, M. Microseed matrix screening for optimization in protein crystallization: What have we learned. Acta Crystallogr Sect F, Struct Biol Commun. 70 (9), 1117-1126 (2014).
- Luft, J. R., et al. Efficient optimization of crystallization conditions by manipulation of drop volume ratio and temperature. Protein Sci. 16 (4), 715-722 (2007).
- Dessau, M. A., Modis, Y. Protein crystallization for X-ray crystallography. J Vis Exp. (47), e2285 (2011).
- Sugahara, M., Asada, Y., Ayama, H., Ukawa, H., Taka, H., Kunishima, N. Heavy-atom Database System: A tool for the preparation of heavy-atom derivatives of protein crystals based on amino-acid sequence and crystallization conditions. Acta Crystallogr D Biol Crystallogr. 61 (9), 1302-1305 (2005).
- Boggon, T. J., Shapiro, L. Screening for phasing atoms in protein crystallography. Structure. 8 (7), 143-149 (2000).
- Vera, L., Stura, E. A. Strategies for protein cryocrystallography. Cryst Growth Des. 14 (2), 427-435 (2014).
- Pichlo, C., Montada, A. A., Schacherl, M., Baumann, U. Production, crystallization and structure determination of C. difficile PPEP-1 via microseeding and Zinc-SAD. J Vis Exp. (118), e55022 (2016).
- Leslie, A. G. W., et al. Automation of the collection and processing of X-ray diffraction data - a generic approach. Acta Crystallogr Sect D Biol Crystallogr. 58 (11), 1924-1928 (2002).
- Pike, A. C. W., Garman, E. F., Krojer, T., Von Delft, F., Carpenter, E. P. An overview of heavy-atom derivatization of protein crystals. Acta Crystallogr Sect D Struct Biol. 72 (3), 303-318 (2016).
- Cooper, D. R., Porebski, P. J., Chruszcz, M., Minor, W. X-ray crystallography: Assessment and validation of protein-small molecule complexes for drug discovery. Expert Opin Drug Discov. 6 (8), 771-782 (2011).
- Hassell, A. M., et al. Crystallization of protein-ligand complexes. Acta Crystallogr Sect D Biol Crystallogr. 63 (1), 72-79 (2006).
- Muller, I., et al. Guidelines for the successful generation of protein-ligand complex crystals. Acta Crystallogr Sect D Struct Biol. 73 (2), 79-92 (2017).
- Zhao, Y., et al. Two routes for production and purification of Fab fragments in biopharmaceutical discovery research: Papain digestion of mAb and transient expression in mammalian cells. Protein Expr Purif. 67 (2), 182-189 (2009).
- Shah, N. B., Duncan, T. M. Bio-layer interferometry for measuring kinetics of protein-protein interactions and allosteric ligand effects. J Vis Exp. (84), e51383 (2014).
- Terwilliger, T. C., et al. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr Sect D Biol Crystallogr. 64 (1), 61-69 (2008).
- Walker, J. A., Smith, K. G. C. CD22: An inhibitory enigma. Immunology. 123 (3), 314-325 (2008).
- Gräslund, S., et al. Protein production and purification. Nat Methods. 5 (2), 135-146 (2008).
- Jain, N. K., et al. A high density CHO-S transient transfection system: Comparison of ExpiCHO and Expi293. Protein Expr Purif. 134, 38-46 (2017).
- Fang, X. T., Sehlin, D., Lannfelt, L., Syvänen, S., Hultqvist, G. Efficient and inexpensive transient expression of multispecific multivalent antibodies in Expi293 cells. Biol Proced Online. 19 (1), 11 (2017).
- Löw, C., et al. Nanobody mediated crystallization of an archeal mechanosensitive channel. PLoS One. 8 (10), 77984 (2013).
- Hunte, C., Michel, H. Crystallization of membrane proteins mediated by antibody fragments. Curr Opin Struct Biol. 12 (4), 503-508 (2002).
- Ereño-Orbea, J., Sicard, T., Cui, H., Carson, J., Hermans, P., Julien, J. -. P. Structural basis of enhanced crystallizability induced by a molecular chaperone for antibody antigen-binding fragments. J Mol Biol. 430 (3), 322-336 (2018).
- Spooner, J., et al. Evaluation of strategies to control Fab light chain dimer during mammalian expression and purification: A universal one-step process for purification of correctly assembled Fab. Biotechnol Bioeng. 112 (7), 1472-1477 (2015).
- Elmlund, D., Le, S. N., Elmlund, H. High-resolution cryo-EM: The nuts and bolts. Curr Opin Struct Biol. 46, 1-6 (2017).
- Merk, A., et al. Breaking cryo-EM resolution barriers to facilitate drug discovery. Cell. 165 (7), 1698-1707 (2016).
- Bai, X. C., McMullan, G., Scheres, S. H. W. How cryo-EM is revolutionizing structural biology. Trends Biochem Sci. 40 (1), 49-57 (2015).
- Wlodawer, A., Li, M., Dauter, Z. High-resolution cryo-EM maps and models: A crystallographer's perspective. Structure. 25 (10), 1589-1597 (2017).
- Bartesaghi, A., et al. 2.2 Å resolution cryo-EM structure of β-galactosidase in complex with a cell-permeant inhibitor. Science. 348 (6239), 1147-1151 (2015).
- Hendrickson, W. a., Horton, J. R., LeMaster, D. M. Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): A vehicle for direct determination of three-dimensional structure. EMBO J. 9 (5), 1665-1672 (1990).
- Walden, H. Selenium incorporation using recombinant techniques. Acta Crystallogr Sect D Biol Crystallogr. 66 (4), 352-357 (2010).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados