
Method Article
Caractérisation des glycoprotéines dont le pli d’immunoglobuline par cristallographie aux rayons x et Techniques biophysiques
Dans cet article
Résumé
Nous présentons des approches pour la caractérisation biophysique et structurelle des glycoprotéines avec le pli de l’immunoglobuline par interférométrie biolayer, calorimétrie isotherme de titration et cristallographie aux rayons x.
Résumé
Glycoprotéines sur la surface des cellules jouent un rôle crucial dans le fonctionnement cellulaire, y compris la signalisation, l’adhérence et le transport. Sur les leucocytes, plusieurs de ces glycoprotéines possèdent des plis de l’immunoglobuline (Ig) et sont au cœur de la réglementation et de reconnaissance immunitaire. Nous présentons ici, une plate-forme pour la conception, l’expression et la caractérisation biophysique du domaine extracellulaire du récepteur des cellules B humaines CD22. Nous proposons que ces approches sont largement applicables à la caractérisation des mammifères glycoprotéine glycoprotéines contenant les domaines de l’Ig. Deux suspension rein embryonnaire humain (HEK) lignées cellulaires, HEK293F et HEK293S, servent à exprimer des glycoprotéines abriter des glycans complexes et riches en mannose, respectivement. Ces glycoprotéines recombinantes avec différentes glycoformes permettent sur les effets de glycane taille et composition sur la liaison du ligand. Nous discutons des protocoles pour l’étude de la cinétique et thermodynamique de la liaison de la glycoprotéine à ligands biologiquement pertinentes et candidats d’anticorps thérapeutiques. Glycoprotéines recombinantes produites dans les cellules HEK293S sont prêtent à la cristallisation en raison de la homogénéité de glycan, flexibilité réduite et la susceptibilité au traitement endoglycosidase H. Nous présentons des méthodes pour le trempage des cristaux de glycoprotéine avec des atomes lourds et petites molécules pour la détermination de la phase et l’analyse de liaison du ligand, respectivement. Les protocoles expérimentaux discutées ici sont prometteurs pour la caractérisation des glycoprotéines mammifères à donner un aperçu de leur fonction et d’étudier le mécanisme d’action de la thérapeutique.
Introduction
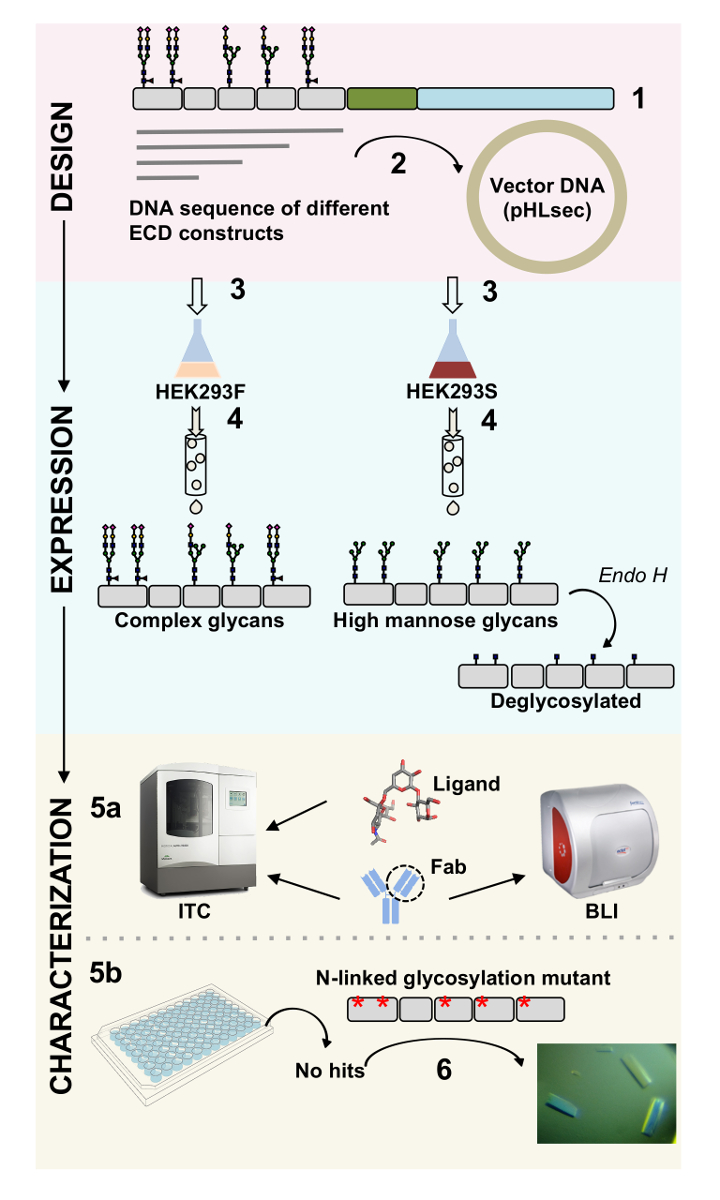
Protéines de surface jouent un rôle crucial dans le fonctionnement cellulaire. Par le biais de leurs domaines extracellulaires, ces protéines membranaires peuvent moduler les interactions cellule-cellule, adhésion, transport et signalisation1,2. La localisation extracellulaire de ces protéines rend les cibles intéressantes pour le développement d’agents thérapeutiques pour le traitement d’un large éventail de maladies, notamment le cancer et les maladies auto-immunes3,4,5 , 6 , 7. les plis plus courantes de glycoprotéines de protéine de membrane humaine est le pli immunoglobuline-like (Ig), qui est formé de sept ou plusieurs β-brins disposés en deux β-feuilles8,9. En général, glycoprotéines contenant Ig sont des structures de multi-domaine avec domaines Ig disposés séquentiellement sur la portion extracellulaire de la protéine de membrane10. Les modifications post-traductionnelles de ces protéines de surface cellulaire, en particulier N - et O-liés glycosylation, ont démontré que jouent un rôle essentiel dans leur règlement, pliage, sécrétion et fonction11. Afin d’améliorer notre compréhension de leur fonction et à la meilleure thérapeutique de conception qui peut cibler leur, ne sont obligatoires qui permettent leur caractérisation moléculaire détaillée. Nous présentons ici une combinaison de techniques qui permettent la biophysique (biolayer interférométrie (BLI) et la calorimétrie isotherme titration (ITC)) et la caractérisation structurale (diffraction) du domaine extracellulaire Ig contenant du glycoprotéines membranaires, seuls et en complexe avec leurs ligands biologiquement pertinentes et les molécules thérapeutiques (Figure 1).
N-lié glycosylation est une des modifications après traduction plus courantes sur les protéines de mammifères et se produit au cours de la maturation de la protéine dans le réticulum endoplasmique et le Golgi12,13. Des lignées de cellules, telles que les 293 cellules rein embryonnaire humain (HEK), ont été développées pour l’expression recombinante de grandes quantités de glycosylée protéines de mammifères14,15. Cette lignée cellulaire a été développée sous forme de suspension qui permet pour la facilité d’intensification de la production de protéines de plus grandes quantités par rapport aux lignées de cellules adhérentes. Ici, nous utilisons deux lignées de cellules HEK293 : HEK293F et HEK293 Gnt j’ai- / - (HEK293S), qui se distinguent par l’absence de N-acétylglucosaminyl transférase I (Gnt je) dans le second. À son tour, production de glycanes complexes (comme on le voit dans HEK293F) n’est pas possible et mannose-type plutôt élevé glycanes (principalement Man5GlcNAc2) résident à glycan N-linked sites18,19,20 . À l’aide de ces deux lignées cellulaires en parallèle permet d’étudier l’effet de glycane de taille et de complexité sur la fonction biologique et ciblage thérapeutique. En effet, glycoprotéines produites dans les cellules de HEK293F aura des glycanes plus volumineux et plus complexes par rapport à la même glycoprotéine produite dans les cellules HEK293S. Glycoprotéines produites dans les cellules HEK293S sont plus propices à la cristallisation, en raison de l’hétérogénéité chimique et conformationnelle réduite de leur N-glycanes. Afin d’améliorer crystallizability, glycoprotéines produites dans les cellules HEK293S (mais pas HEK293F) peuvent être traitées avec l’enzyme endoglycosidase H (Endo H), qui se traduit par le clivage des glycanes riche en mannose tel que seul un unique N-acétylglucosamine (GlcNAc) portion reste à chaque site de glycosylation N-liés21,22. Autres méthodes permettent également de limiter le traitement N-glycanes dans les cellules, telles que l’ajout des inhibiteurs de la glycosyltransférase au cours de l’expression de glycoprotéine, y compris le caddie23. Approches alternatives impliquent l’expression des glycoprotéines natifs (dans les cellules HEK293F) suivi de déglycosylation enzymatique à l’aide de peptide N-glycosidase F (PNGaseF). Cependant, déglycosylation avec PNGaseF s’est avérée moins efficace dans des conditions natives et augmente l’agrégation dans certaines protéines ; dans le cas lorsque la protéine reste soluble après le traitement, il acquiert des charges négatives sur sa surface, en raison de la désamidation du résidu asparagine, acide aspartique24, qui pourrait être préjudiciable pour sa cristallisation. Sites de N-glycosylation prédites peuvent également être mutés, plus souvent à des résidus alanine ou la glutamine, pour empêcher la glycosylation N-liée à ces sites et de générer des échantillons de la glycoprotéine de grande homogénéité. Alternativement, les glycoprotéines peuvent être produites dans d’autres cultures de cellules eucaryotes, y compris les levures, insecte et systèmes de plantes ou autres lignées cellulaires de mammifères tels que les hamsters chinois ovarienne (CHO) cellules16,17.
De nombreux vecteurs d’expression chez les mammifères, y compris pHLsec, permettent la sécrétion des glycoprotéines recombinantes glycoprotéine dans la cellule moyenne25. Sécrétion de glycoprotéines de cellules HEK293 permet une purification rapide et facile sans besoin de lyse cellulaire. Ajout de balises de purification (par exemple, His-tag, Strep-tag, drapeau-tag, tag-Myc, HA-tag) jusqu’au terminus de N ou C de la cible glycoprotéine permet la purification par une chromatographie d’affinité de la seule étape. Par la suite, chromatographie d’exclusion peut être utilisée pour obtenir un échantillon de monodispersés pour la caractérisation biophysique et structurel.
Un échantillon de glycoprotéine très pure et homogène dans des conditions appropriées peut entraîner des cristaux bien diffractants. Une fois un ensemble de données complet de diffraction des rayons x a été obtenue de ces cristaux, phases initiales doivent être déterminé pour calculer la densité d’électrons de la glycoprotéine. Grâce à un nombre toujours croissant des structures dans la Banque de données de protéine (PDB), la méthode couramment utilisée pour l’élimination est devenue de loin de remplacement moléculaire (MR), qui utilise une structure protéique connexes pour obtenir des phases initiales de26. Toutefois, lorsque M. ne parvient pas à résoudre le problème de phase, comme ce fut parfois le cas pour multi-Ig domaine glycoprotéines27,28,29, méthodes alternatives sont nécessaires. Dans cet article, nous détaillons une méthode à tremper des cristaux avec des atomes lourds (HA) pour l’élimination, qui était nécessaire pour résoudre la structure du CD22 ectodomaine28. Identifier le droit HA pour l’élimination est un processus itératif qui dépend HA réactivité, atomes disponibles dans la glycoprotéine dans un réseau cristallin donné et la cristallisation solution30,31. Alternativement, les atomes de soufre naturel dans les résidus de cystéine et méthionine peuvent être utilisés pour progressive s’il est présent à un taux assez élevé d’autres atomes dans la glycoprotéine, et si les données de la diffraction des rayons x peuvent être collectées avec redondance assez élevé32, 33.
La fonction biologique des glycoprotéines membranaires est souvent véhiculée par les interactions protéine-protéine ou des interactions de protéine-ligand, comme contenant des glucides. Quand le ligand est assez petit pour diffuser de la solution vers le site de liaison de glycoprotéine dans le réseau cristallin, trempage des expériences peut réussir à obtenir une structure en cristal co glycoprotéine-ligand pour mieux comprendre la reconnaissance ligand.
Les protocoles présentés ici sont également pertinentes pour comprendre les interactions des glycoprotéines de surface avec des ligands thérapeutiques synthétiques34,35 et anticorps thérapeutique36,37. Lorsqu’il est combiné avec l’information structurale, cinétique et thermodynamique contraignants peuvent être puissants pour comprendre et améliorer leurs mécanismes d’action. Une technique qui permet l’analyse cinétique des anticorps thérapeutiques liant à une glycoprotéine est BLI38,,39. BLI utilise des biocapteurs avec un ligand immobilisé pour mesurer la cinétique association et dissociation avec un partenaire de liaison, déterminant en fin de compte une constante de dissociation de l’équilibre (KD). BLI est une approche intéressante car de petites quantités de glycoprotéines sont requises (< 100µg), temps d’expérience est rapide (~ 10-15 min par course), et il peut être automatisé. ITC est également utile pour l’étude des affinités entre les glycoprotéines et liaison partenaires40,41,42,43. ITC est plus de temps et de réactif intensive, des renseignements précieux peuvent être obtenus au sujet de la thermodynamique de l’interaction (ΔG, ΔH, ΔS et stoechiométrie). ITC est également très utile pour l’étude des interactions faibles qui sont souvent associées à la liaison passagère des glycoprotéines de surface aux ligands. En outre, ces techniques peuvent servir en même temps à la liaison de diverses constructions et d’évaluer l’effet de différents N-liés glycoformes obtenues d’exprimer la glycoprotéine dans différentes lignées cellulaires. Produisant de BLI et ITC avec glycoprotéines produites dans HEK293F, HEK293S et traités avec Endo H peut fournir une vision approfondie du rôle des glycanes dans l’activité biologique et engagement thérapeutique.
Nous avons appliqué avec succès ces protocoles pour caractériser le domaine extracellulaire (DPE) humaine CD22 contre28, un membre de la glycoprotéine de la famille des lectines (Siglecs) acide sialique liant Ig-like qui est essentielle au maintien de l’homéostasie de B-cellule44 . Nous avons effectué conception construction approfondie pour faciliter la cristallisation et progressivement le dataset des rayons x par HA trempage avec Hg. Nous avons aussi trempé CD22 cristaux avec son ligand l’acide sialique (α2-6 sialyllactose) pour obtenir une structure du complexe récepteur-ligand immunitaire et donc fourni les plans pour la conception de structure guidée de glycane mimétiques45,46. En outre, nous avons généré la liaison de l’antigène de fragment (Fab) de l’epratuzumab d’anticorps thérapeutiques anti-CD22 - un candidat actuellement en essais cliniques de phase III de lymphome non hodgkinien47- afin de déterminer son affinité par BLI et ITC à différentiellement glycosylée CD22 DPE construit. Ces études ont révélé un rôle essentiel pour la glycosylation N-liés dans epratuzumab engagement, avec des implications potentielles pour CD22 reconnaissance sur les cellules B dysfonctionnels.
Protocole
1. construire Design glycoprotéine DPE
- Évaluer la séquence d’acides aminés CD22 humaine (Uniprot) en utilisant les serveurs InterPro et Phyre2 pour identifier les éléments de domaine prédites et limites agrave la protéine48,49.
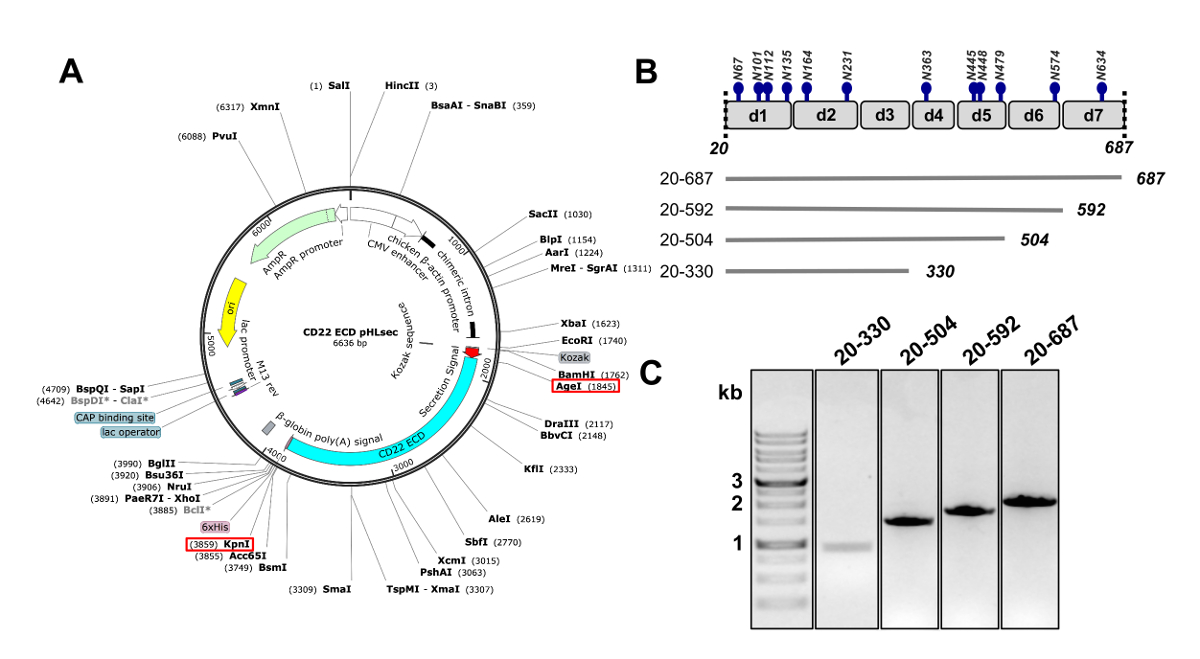
- Clone de la séquence des humains CD22, manque le peptide signal, domaines transmembranaires et cytosoliques (résidus 20-687, ci-après dénommé le domaine extracellulaire de CD22, CD22 DPE) dans le pHLsec expression mammifère vector25 à l’aide d’enzymes de restriction AgeI et KpnI) Figure 2 A) 50.
Remarque : Le vecteur pHLsec est optimisé pour la surexpression de protéines solubles sécrétés dans les cellules de mammifères,25. Ce vecteur contient un signal de sécrétion pour permettre la sécrétion extracellulaire de glycoprotéines solubles. pHLsec contient une balise de6 x C-terminal (sa) pour faciliter la purification d’affinité de surnageants de cellule à l’aide de méthodes chromatographie d’affinité immobilisée de métal. - Cloner des constructions tronquées du CD22 DJE avec destructions séquentielles des domaines Ig C-terminal : domaines 1 à 6 (résidus 20-687), domaines 1 à 5 (résidus 20-592), domaines 1-4 (résidus 20-504) et domaines 1-3 (résidus 20-330) (Figures 2 b et 2C)50 .
- Évaluer la séquence primaire de DPE CD22 utilisant le serveur NetNGlyc pour identifier les sites de glycosylation de N-liés prédit présents dans la construction de51.
- À l’aide de mutagenèse dirigée, par des protocoles standard52 ou en superposant des PCR53, muter chaque site de glycosylation de N-liés prédites (Asn à Gln et/ou Asn à Ala) pour créer des constructions du CD22 DJE qui contiennent un ou plusieurs Mutations de glycosylation N-liés.
- Après vérification de séquence des constructions clonées, transformer en compétent e. coli DH5α cellules54 et maxi-prep l’ADN (selon les instructions du fabricant) afin de préparer pour la transfection.
2. HEK293F et création de cellule de HEK293S
Remarque : Toutes les manipulations de cellules HEK293F ou HEK293S avec les réactifs nécessaires et l’équipement doivent être effectuées dans un établissement de niveau 2 de biosafety dans une armoire de biosécurité approprié. La surface externe de tous les éléments doit être stérilisée avec une solution d’éthanol à 70 % ou équivalent réactif.
- Obtenir les cellules HEK293F et HEK293S de suspension (voir Table des matières) et stocker à-80 ° C jusqu’au moment de l’utilisation.
- Médias chauds (voir Table des matières) dans un bain-marie à 37 ° C pendant 1 heure. Transférer 24 mL de médias chauffé dans un flacon de culture cellulaire dérouté de 125 mL avec un bouchon ventilé.
- Obtenir 1 mL cellule aliquote à-80 ° C et transfert à la glace.
- Incuber les cellules dans un bain-marie à 37 ° C pendant environ 1 min, pour décongeler partiellement les cellules. Transférer 1 mL de cellules à partir du flacon dans le flacon de culture cellulaire dérouté 125 mL contenant les médias.
- Fermer le flacon de culture cellulaire avec le capuchon ventilé et lieu fiole dans un shaker égale à 37 ° C, 130 tr/min, 70 % d’humidité et 8 % CO2.
3. HEK293 Entretien de cellules
Remarque : La densité cellulaire et la viabilité des cellules doivent être vérifiée environ 24h après décongélation. Cette étape garantit que les cellules sont rétablissent après l’inoculation ; viabilité initiale devrait être > 80 %.
- Soigneusement retirer 10 µL de cellules du flacon de 125 mL contenant les cellules fraîches de suspension et le transférer dans un microtube stérile de 1,5 mL. Boucher le flacon et renvoyez-le à l’incubateur.
- Dans les microtubes de 1,5 mL contenant les cellules déposer 10µl de solution de bleu de Trypan, bien mélanger et transférer 10 µL dans la chambre de la diapositive de comptage.
- Mettez la diapositive comptage dans un compteur de cellule automatique et obtenir les valeurs de densité cellulaire (par incréments de cellules mL-1) et la viabilité des cellules (en pourcentage).
- Calculer le volume des cellules qui seraient nécessaires pour ensemencer une culture fraîche 200 mL à une densité finale de ~0.8 x 106 cellules mL-1 à l’aide des équations suivantes :
 (1)
(1) (2)
(2)
Remarque : Il peut prendre ~ 5D pour obtenir une densité cellulaire appropriée pour l’inoculation dans une culture de 200 mL. - Lorsque la densité cellulaire est suffisante pour l’inoculation d’une culture de 200 mL, médias de warm-up dans un 37 ° C pendant 1 heure bain d’eau et transférer les médias réchauffés dans la biosécurité du cabinet.
- À l’aide d’une pipette sérologique, transvaser avec soin le volume requis de médias (tel que calculé dans l’équation 2) dans un flacon de culture cellulaire dérouté 500 mL avec un bouchon ventilé.
- À l’aide d’une pipette sérologique, transférer le volume requis de cellules en suspension (calculé dans l’équation 1) dans le flacon de culture cellulaire dérouté 500 mL contenant les médias.
- Boucher le nouveau stock de maintenance 200 mL et renvoyez-le à l’incubateur. La croissance de cellules à une densité d’environ 3 x 106 cellules mL-1. Le passage des cellules à une densité de 0. 8 x 106 cellules mL-1 chaque 2-3 jours pour maintenir une culture stable de cellules (comme décrit à la section 3.4-3.7). Ne laissez pas les cellules à dépasser une densité d’environ 4 x 106 cellules mL-1.
4. la transfection des cellules HEK293 pour l’Expression de la glycoprotéine
- Calculer le volume des cellules et des médias qui est nécessaires pour une culture de 200 mL pour la transfection à 0,8 x 106 cellules mL-1 (avec les équations 1 et 2 de l’article 3.4).
Remarque : Le nombre de transfections 200 mL qui peut être effectuée dépend de la densité cellulaire du stock maintenance. - Transférer le volume requis des médias et pour la transfection des cellules dans un nouveau flacon de culture cellulaire 500 mL avec un bouchon ventilé et remettez le stock de cellules dans l’incubateur.
- Incuber les cellules pendant 1 h avant la transfection pour permettre aux cellules de s’y acclimater après fractionnement.
- Transférer 50 µg d’ADN dans un tube conique stérile de 50 mL et diluer avec 5 mL de médias. Aspirateur filtre l’ADN diluée à l’aide d’un système de filtration de 0,22 µm dans un autre tube stérile.
- Mélange dilué, filtré d’ADN dans un ratio de masse : volume 1:1 avec le réactif de transfection. Agiter doucement de la solution de réactif d’ADN : transfection pour mélanger et incuber la solution à température ambiante pendant 10 min.
- Ajouter la solution de réactif ADN : transfection directement aux cellules. Incuber à 37 ° C, 130 tr/min, 70 % d’humidité et 8 % CO2 dans un shaker pendant 5-7 jours, les cellules transfectées.
5. optimisation des Conditions de Transfection de cellules
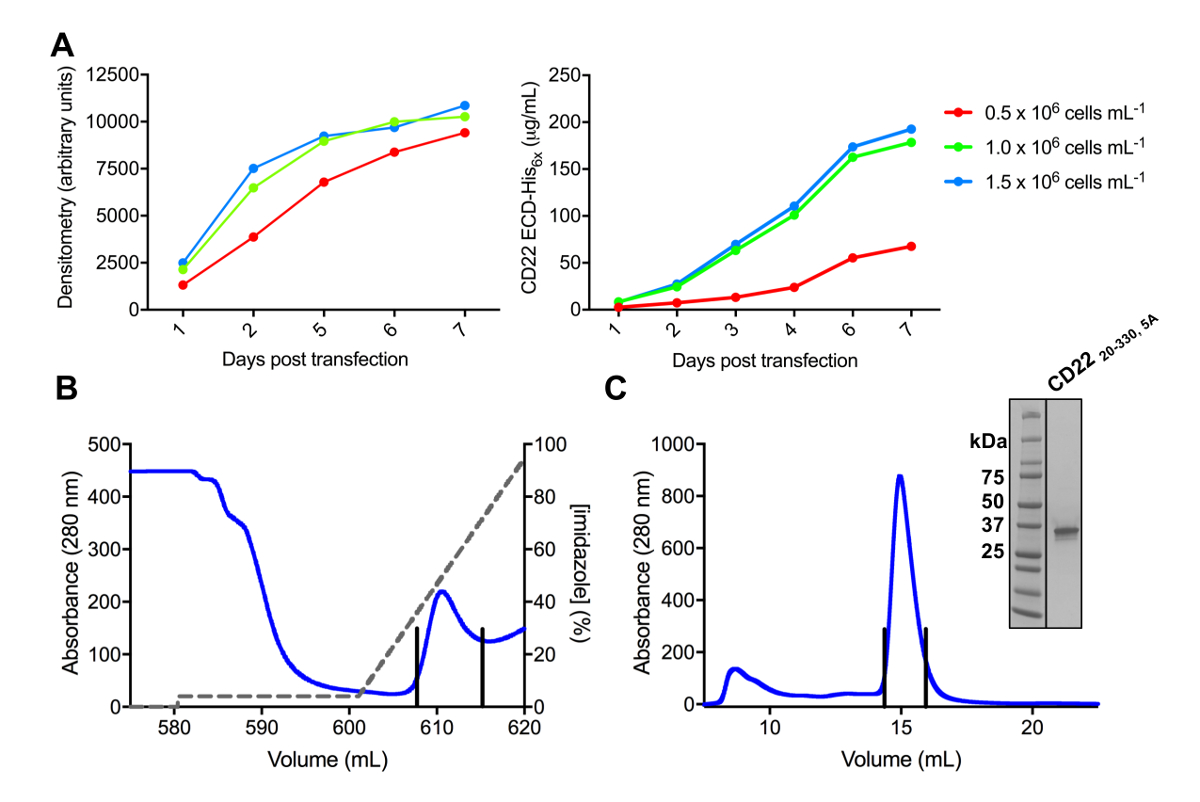
Remarque : Pour optimiser les conditions de transfection de cellules pour un rendement maximal de glycoprotéine, transfecter les cellules à une variété de densités cellulaires initiales et évaluer le rendement de protéine au fil du temps (Figure 3A). Transfecter cellules tel que décrit à l’article 4, à la densité des cellules initiales comprises entre 0,5 x 106 à 2 x 106 cellules mL-1 55. Transfections du procès peuvent être réduites au volume total de 25 mL (en flacon de 125 mL dérouté cell culture) avec 6 µg d’ADN pour économiser espace et réactifs. La quantité d’ADN peut également être optimisé55.
- La transfection après chaque jour (jours 1 à 7), transférer une quantité de 500 µL de la culture de cellules dans un microtube stérile de 1,5 mL (dans le cabinet de prévention des risques biotechnologiques).
- Spin aliquotés cellules à 12 000 x g pendant 5 min dans une micro-centrifugeuse immédiatement après le prélèvement. Transférer surnageant dans un nouveau microtubes de 1,5 mL et de la conserver à 4 ° C jusqu'à ce que tous les échantillons sont obtenus.
- Quantifier la glycoprotéine sécrétée par densitométrie
- Une fois que tous les échantillons sont prélevés, aliquote 20 µL de chaque échantillon dans un microtube 1,5 mL nouveau et mélanger avec 6 µL de non réducteur 4 x Laemmli solution tampon.
- Faire bouillir les échantillons pendant 5 min à 95 ° C dans un thermo-bloc. Tissent des échantillons pendant 1 min à 12 000 x g dans une micro-centrifugeuse.
- Charger 20 µL de chaque échantillon / puits dans un gel SDS-PAGE gradient de 10 puits 4 à 15 %. Inclure une voie pour les marqueurs de taille de protéine. Exécutez le gel à 250 V pendant 20 min dans un tampon Tris/Glycine/SDS.
- Après exécution, transférer le gel au bleu de Coomassie tache (voir Table des matières) pendant 20 min. tache hors gel dans ddH2O pendant 20 min. de gel d’Image.
- Effectuer la densitométrie avec ImageJ, suite de protocoles standard57,58.
- Compiler et tracer des données avec « transfection après jours » sur l’axe des x et des « valeurs de densitométrie » sur l’axe y (Figure 3A).
Remarque : Par ailleurs, si l’expression de la protéine est insuffisante pour la visualisation par SDS-PAGE, techniques telles que le Western Blot peuvent être utilisé56.
- Quantifier la glycoprotéine sécrétée par BLI
- À l’aide de biocapteurs Ni-NTA, doser la quantité de glycoprotéine sécrétée en utilisant BLI59.
- Compiler et tracer des données avec « transfection après jours » sur l’axe des abscisses et « concentration protéique (µg/mL) », sur l’axe y (Figure 3A).
6. purification de glycoprotéine Soluble HEK293 surnageant
- Récolter les cellules par centrifugation à 6 371 x g pendant 20 min à 4 ° C. Conserver le surnageant contenant sécrétées CD22 DPE et filtrer à l’aide d’un filtre de 0,22 µm.
- Charger le surnageant à 4 mL min-1 sur un pré équilibré colonne de Ni-NTA (20 mM Tris pH 9.0, 150 mM NaCl, imidazole de 5 mM) (volume de 5 mL) à l’aide d’un système de chromatographie de benchtop.
Remarque : Autres techniques de purification d’affinité-basé peuvent servir, basée sur les balises d’affinité inclus dans la conception de la construction dans la Section 1. - Après chargement surnageant, laver la colonne d’affinité avec 3-4 volumes de colonne (CV) du tampon de lavage (20 mM Tris pH 9.0, 150 mM NaCl, imidazole de 5 mM).
- Éluer la glycoprotéine purifiée de la colonne à l’aide d’un dégradé de 4 à 100 % (4 CV) du tampon d’élution (20 mM Tris pH 9.0, NaCl, imidazole de 500 mM à 150 mM) tout en recueillant des fractions (Figure 3B).
- Fractions de piscine contenant le pic éluée dans un dispositif de filtration centrifuge avec une limite de poids moléculaire nominal kDa (sera) et concentré 10 par centrifugation à 4 000 x g à 4 ° C pendant 15 min ou jusqu'à ce que l’échantillon atteint un volume de 500 µL.
- Glycoprotéine concentré d’injecter dans une boucle d’échantillonnage µL 500 et la charge à 0,5 mL min-1 sur un pré équilibré (20 mM Tris, pH 9.0, NaCl 150 mM) colonne d’exclusion de taille haute performance (environ le volume 24 mL) sur une chromatographie en phase liquide protéine rapide (FPLC) système à 4 ° C, tout en recueillant des fractions (Figure 3C).
- Exécutez le gel SDS-PAGE des fractions éluées à identifier les fractions contenant la glycoprotéine et poule fractions correspondantes. Le gel SDS-PAGE peut être exécuté comme décrit dans la Section 560.
7. déglycosylation de glycoprotéine purifiée
- Mesurer la concentration de protéine purifiée après chromatographie d’exclusion à l’aide d’absorbance à 280 nm, divisé par le coefficient d’extinction (p. ex., 1,418 M-1 cm-1 de CD22 DPE).
Remarque : Le coefficient d’extinction théorique des protéines d’intérêt peut être calculé à l’aide de serveurs comme ExPASy ProtParam61. - Incuber une protéine purifiée avec Endo H pendant 1 h à 37 ° C, à raison de 1 mg de protéine purifiée à 10 µL d’enzyme commerciale dans un 1 tampon X Endo H (selon les instructions du fabricant).
NOTE : Endo H fend riche en mannose glycanes produites dans HEK293S laissant une portion GlcNAc unique à chaque site de glycosylation21. Endo H ne pas Cliver glycanes sur protéines produites dans les cellules de HEK293F22, cependant autres enzymes peuvent être utilisées à cette fin (p. ex.,PNGaseF,24). - Concentrer déglycosylée DPE à 500 µL et exécuter la chromatographie par filtration sur gel sur une colonne d’exclusion de taille haute performance (environ le volume 24 mL) à 0,5 mL min-1 sur un FPLC d’enlever Endo H et séparer les agrégats qui en résulte.
- Conserver la protéine déglycosylée à 4 ° C jusqu'à l’utilisation dans des expériences en aval.
8. la cristallisation des glycoprotéines
Remarque : Effectuer des essais de cristallisation à l’aide d’écrans disponibles dans le commerce et mis en place assise expériences goutte à l’aide d’un robot de cristallisation.
- Concentré pur, déglycosylée DPE à mg 10 mL-1 à l’aide d’un dispositif de filtration centrifuge avec 10 kDa sera à 4 000 x g (4 ° C) jusqu'à ce que la concentration désirée est obtenue.
- Déterminer la concentration de la protéine à l’aide d’absorbance à 280 nm et en divisant par le coefficient d’extinction.
- Échantillon de centrifugation à 12 000 x g pendant 5 min à 4 ° C avant les essais de cristallisation pour enlever la poussière indésirables ou autres contaminants de l’échantillon.
- Remplir le réservoir de puits de 96 puits séance drop plaques de cristallisation avec 80 µL de solution de cristallisation d’un écran de cristallisation commerciale.
Remarque : Nous utilisons les matrices creuses écrans commerciaux qui ont été conçus selon les conditions de cristallisation plus de succès en ce qui concerne les structures déposées dans l’APB. - À l’aide d’un robot de cristallisation, déposer des gouttes dans le puits de la plaque de la cristallisation d’un volume total baisse de 200 nL à un ratio de solution : cristallisation de protéine purifiée de 1:1.
- Une fois la plaque entière a été prélevée, sceller la plaque avec ruban et placez dans un imageur de plaque pour l’inspection par la lumière visible et ultraviolette.
- Inspecter les plaques de cristallisation immédiatement après l’installation et dans les semaines suivantes, en utilisant la lumière visible et ultraviolette pour identifier les conditions qui donnent la glycoprotéine initiale crystal hits.
- Optimisez les cristaux obtenus à partir des hits de cristallisation initial à l’aide d’écrans fines basées sur l’état du hit de cristal ou matrice aléatoire micro-ensemencement méthodes62,63,64,65.
- Cryo-protéger les cristaux manquant suffisamment cryo-protecteur dans la condition de cristallisation en trempant le cristal dans la mère d’alcool solution additionnée de solution de glycérol à 20 % (v/v) (ou équivalent cryo-protecteur, telles que l’éthylène glycol ou polyéthylèneglycol 400).
- Cristaux de Mont en cryoloops et flash congelez-les dans l’azote liquide avant la collecte de données sur un diffractomètre source maison ou à l’aide de rayonnement synchrotron.
9. progressive à l’aide de dérivation aux atomes lourds
NOTE : Avant toute manipulation de HA composés, des aspects de sécurité doivent être considérées. HA composés utilisés en cristallographie des protéines sont sélectionnés pour leur forte affinité pour les molécules biologiques et présentant des risques pour la santé humaine d’une exposition prolongée. Prendre des mesures de sécurité appropriées pour HA composés comme mentionné dans leurs fiches signalétiques.
- Pour tester différents HA composés, les concentrations et les temps d’incubation, se reproduisent bien diffractants cristaux obtenus au chapitre 8 dans une plaque de 24 puits cristallisation à l’aide de la pendaison-goutte vapeur diffusion méthode66.
- Décidez qui HA sera utilisé pour la dérivation de cristal. Serveurs (par exemple, les atomes lourds Database System67) peuvent aider à HA composée sélection, assurant qu’ils ne conviennent pas à la condition de protéine et de cristallisation.
NOTE : HA écrans sont également disponibles dans le commerce pour le dépistage facile de composés HA plus efficaces pour l’élimination. Un ensemble de « magie sept » HA composés ont été décrits précédemment pour la forte probabilité de succès pour HA dérivatisation68. - Mis en place le poste de travail pour HA trempage (Figure 4A). À l’aide d’un cryoloop, transférer rapidement des cristaux à un 0.2 µL drop sur une lamelle de 22 mm contenant HA solution diluée dans la condition de la cristallisation, telle que la concentration finale de HA varie de 1 à 20 mM. Sceller la goutte et incuber pendant différentes périodes de temps (Figure 4B). Un bon point de départ est de 5, 10, 60 et 90 min et Pendant la nuit.
- Inspecter visuellement les cristaux avec un microscope optique pour identifier de possibles fissures ou des changements de couleur, qui peut indiquer des effets indésirables à la glycoprotéine cristal ou de la dérivation de cristal.
- Monter les cristaux dans cryoloops et cristaux de dos-trempage pendant 30 s dans trois consécutives µL 0,2 gouttes de solution de liqueur mère contenant additionnée de 20 % (v/v) de glycérol (ou remplaçant cryo-protecteur)69. Dos-tremper les cristaux supprime HA composé qui était non spécifiquement liée et réduit l’occupation partielle provoquée par la faible liaison HA. Flash gel des cristaux dans l’azote liquide (Figure 4B).
- Collecte, traitement, solution de structure, et raffinement, utilisez les protocoles décrits antérieurement26,70,71,72.
10. trempage glycoprotéine cristaux avec son Ligand
- Reproduire des cristaux bien diffraction obtenues au chapitre 8 dans une plaque de 24 puits cristallisation à l’aide de la méthode de diffusion de vapeur de pendaison-goutte.
- Préparer une solution de ligand de 50 mM à 20 mM Tris, pH 9.0, NaCl 150 mM.
Remarque : La concentration de ligand doit être préparée selon l’affinité pour la glycoprotéine. Si on ne connaît pas l’affinité, il pourrait être obligatoire d’utiliser une méthode telle que l’ITC (Section 12.2) afin de déterminer l’affinité avant le début des expériences de trempage. Veiller à ce que le ligand est soluble à la concentration voulue dans la mémoire tampon requise. - Ajouter différentes concentrations du ligand pour les goutte contenant des cristaux de DPE et sceller la goutte pour incubation à longueurs de temps variant entre 5 min pour 5D.
- Suivre visuellement les cristaux avec un microscope optique pour identifier d’éventuels changements dans la morphologie.
- Monter des cristaux cryoloops et cryo-Protégez-les en solution de liqueur mère additionnée de 20 % (v/v) de glycérol (ou autre protectant-cryo telles que l’éthylène glycol ou de faible poids moléculaire polyéthylèneglycol 400)69.
- Collecte, traitement, solution de structure et raffinement, utiliser des protocoles décrits antérieurement73,74,75.
11. production du Fragment Antigen Binding (Fab)
- Subclone gènes qui correspondent à la chaîne lourde de Fab (HC) et des séquences de chaîne légère (LC) d’anticorps anti-DCE, par exemple,epratuzumab.
Remarque : Vous pouvez également IgG peut être clivée par la papaïne enzyme pour générer des fragments Fab76. - Transfecter cellules tel que décrit à la Section 4, avec les modifications suivantes :
- Utiliser une masse totale de l’ADN pour la transfection des fragments Fab de 90 µg / 200 mL de culture.
- Transfecter HC et LC plasmides dans un rapport de 2:1 pour réduire le montant de la formation de dimères de LC.
- Après 7 jours d’incubation, récolter les cellules, conserver le liquide surnageant et filtrer avec un dispositif de filtration axée sur le vide de 0,22 µm.
- Equilibrer les colonnes d’affinité anti-LC (kappa ou lambda) dans le tampon PBS à l’aide d’un système de chromatographie de benchtop.
Remarque : Si la formation de dimères LC est un problème durant la purification, chromatographie d’affinité de protéines G peut servir comme alternative à la purification d’affinité LC kappa/lambda. - Charger le surnageant sur colonne d’affinité à 4 mL min-1. Après le chargement de l’échantillon, laver la colonne avec 3-4 CV de PBS.
- Éluer les protéines de la colonne à l’aide d’une élution isocratique avec glycine 100 mM, pH 2.2, neutraliser immédiatement les fractions éluées avec 10 % (v/v) 1 M Tris, pH 9.0 dans chacune des fractions.
Remarque : Le Fab éluée peut être encore purifié par chromatographie échangeuse d’ions ou chromatographie d’exclusion à l’aide d’un FPLC à 4 ° C.
12. caractérisation des Fab et petite molécule contraignant à la glycoprotéine
- Interférométrie de Biolayer
- Préparer 50 mL de 1 x tampon de cinétique (1 x PBS, 0,002 % (v/v) de Tween-20, 0,01 % (p/v) BSA).
- Hydrater les six biocapteurs Ni-NTA dans 200 µL de tampon de cinétique x 1 pendant 10 min dans une plaque de prémouillage.
- Diluer son étiquette DPE dans 1 mL de tampon de cinétique de 1 x à une concentration finale de 25 ng µL-1. Pipetter dilutions successives du Fab purifié dans 200 µL de 1 x tampon de cinétique, avec une forte concentration de 500 nM et les dilutions en série de 250 nM, 125 nM et 62,5 nM.
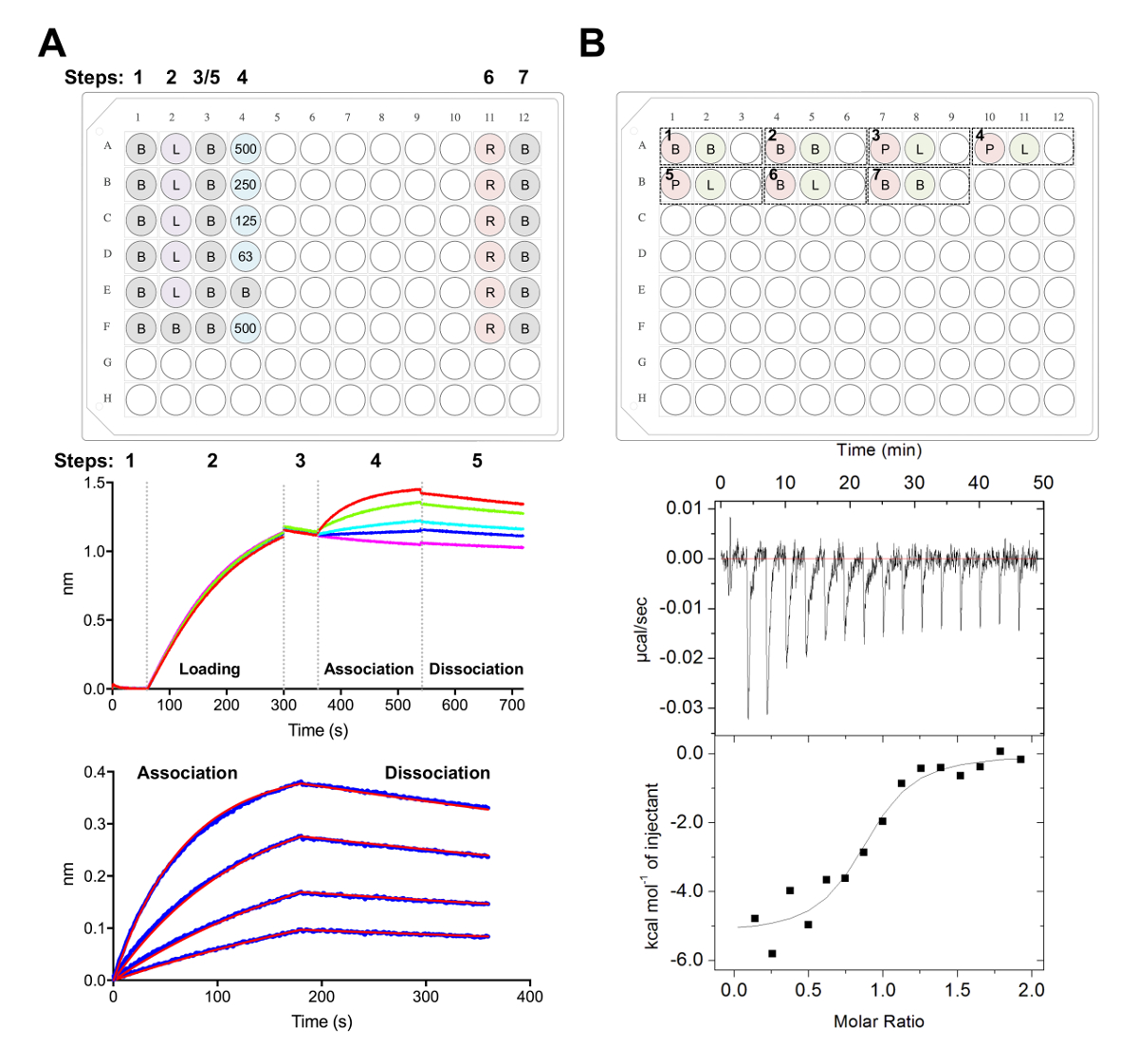
- Aliquotes réactifs en microplaque de 96 puits en polypropylène noir fond plat tel qu’illustré à la Figure 5A, dont chacun contient bien 200 µL de la solution indiquée.
- Collecter des données à l’aide de la cinétique des essais dans le logiciel d’Acquisition de données, comme décrit plus haut38,,du3977 (Figure 5A).
- Brièvement, transférer les biocapteurs dans puits contenant 1 x tampon de cinétique à ligne de base pour 60 s avant le chargement de 25 ng µL-1 glycoprotéine pour 240 s (ou jusqu'à ce qu’un seuil de 1,0 nm est atteint) à 1 000 tr/min.
- Après une deuxième ligne de base de 60 s à 1 x tampon de cinétique, transférer les biocapteurs dans puits contenant les dilutions de Fab. La phase de liaison s 180 est ensuite suivie d’une étape de dissociation 180 s dans 1 x tampon de cinétique.
Remarque : Biocapteurs peuvent être réutilisées si le protocole ci-dessus est suivi d’une étape de régénération, qui se compose de trois cycles de lavage les biocapteurs dans le tampon (PBS avec imidazole 500 mM) se déshabillant pour 5 s suivie de 5 s à 1 x tampon de cinétique pour la neutralisation. Biocapteurs peuvent être réutilisés jusqu'à environ 10-20 fois pendant le même jour, ou jusqu'à ce que de mauvaises données qualité est observée.
- Analyser les données en utilisant le logiciel d’analyse (Figure 5A) :
- Sous l’onglet 1, l’importation et sélectionner des données.
- Sous l’onglet 2, étape 1 : Sélection de données, sélectionnez « capteur de sélection » et point culminant référence puits (lignes E et F, Figure 5A), faites un clic droit et de la valeur de référence bien. Sous étape 2 : Soustraction, sélectionnez « puits de référence ». Sous étape 3 : Aligner l’axe des ordonnées, sélectionnez « Base » dans la plage horaire de 0,1 à 59,8 s. Sous étape 4 : inter-Étape Correction, sélectionnez « Aligner à la dissociation. » Sous étape 5 : Processus, sélectionnez « Savitzky-Golay filtrage » et appuyez sur traiter les données.
- Sous l’onglet 3, sélectionnez « Association et Dissociation » sous étape à analyser avec un modèle 1:1. Sélectionnez « Ajustement Global » et « Groupe de couleur ». Faites un clic droit de courbes, sélectionnez « Changer de couleur », définissez toutes les courbes à la couleur de votre choix. Sélectionnez « Ajuster les courbes ». Si les données sont bien montées, un rapport peut être exporté en sélectionnant « Enregistrer le rapport ».
- Réitérer l’expérience avec la glycoprotéine produite dans les cellules HEK293F et HEK293S (Section 5) et après traitement Endo H (Section 7) pour évaluer les répercussions, le cas échéant, des différentes glycoformes sur reconnaissance Fab. En outre, répétez l’expérience avec troncatures de DPE à fournir la perspicacité dans les domaines liés par la Fab.
- Calorimétrie isotherme de titration de l’interaction de Fab-glycoprotéine
Remarque : ITC expériences décrites ici réalisées à l’aide d’un instrument automatisé de la CCI. Des expériences sont effectuées dans un 1 mL rond moufle de 96 puits.- Dialyser le DPE et la Fab dans un bécher 4 L unique de 20 mM Tris, pH 8,0, NaCl 150 mM à 4 ° C durant la nuit avec une barre de remuer.
- Concentré dialysée DPE et Fab à 5 µM et 50 µM, respectivement, à l’aide d’un filtre centrifuge avec 10 kDa sera, assurant pour laver les membranes concentrateur trois fois avec 5 mL de dialyse tampon à 4 000 x g pendant 5 min à 4 ° C avant de l’utiliser.
Remarque : Toute incompatibilité dans un tampon entre les échantillons dans la cellule et la seringue peut causer chaleur indésirable qui sortira au cours de l’expérience de l’ITC et le résultat dans les données de mauvaise qualité. - Pour l’expérience 1 : ajouter 400 µL de DPE a1 à charger dans la cellule et 120 µL de la Fab à bien A2 doit être chargé dans la seringue. Bien A3 est laissé vide pour retourner qui suit l’échantillon mélangé expérimenter achèvement. Chaque expérience ultérieure peut être ajouté à la plaque dans le même ordre (p. ex., expérience 2 : puits vide de cellule - A4, seringue - A5, - A6 ; Figure 5 ( B).
Note : Inclus tampon contrôles de tampon (pour confirmer l’instrument est bien se comporter) en début et en fin de chaque course, ainsi que le ligand (en seringues) contrôles de tampon (dans la cellule) pour calculer la chaleur de dilution de l’échantillon dans la seringue. Cette calculée chaleur de dilution doit ensuite être soustraites des données expérimentales brutes au cours de l’analyse de données (Figure 5B). - Exécuter un total de 16 injections avec un volume de 2,5 μL pour chaque injection. La durée d’injection est de 5 s, avec un espacement s 180 entre les injections. Régler la température de cellule de 25 ° C, avec une vitesse d’agitation de 750 tr/min et une période de filtre de 5 s.
Remarque : Selon l’affinité et la thermodynamique de l’interaction de ECD:Fab, il peut être nécessaire de modifier la concentration de l’échantillon, nombre d’injections ou de la température de la cellule. - Analyser les données avec le logiciel d’analyse, comme décrit plus haut40,41,43 (Figure 5B).
- Répéter l’expérience au moins de doublons, calculer les valeurs de KD moyennes et les écarts-types. Répétez l’expérience avec DPE de différentes glycoformes (Articles 5 et 6) pour évaluer les répercussions, le cas échéant, de glycoformes sur la thermodynamique de l’interaction de Fab : glycoprotéine.
- Pour la calorimétrie isotherme titration des interactions ligand-glycoprotéine, mis en place l’expérience de la CCI comme décrit dans l’article 12.2, avec les modifications suivantes :
- Dialyser DPE dans la 4 L de tampon de dialyse pendant la nuit. Dissoudre le ligand à l’aide de tampons de dialyse après l’achèvement de la dialyse.
- Réaliser des expériences de la CCI à des concentrations considérablement plus élevées pour pouvoir détecter des interactions de faible affinité. Pour l’interaction de la DPE et de ligand, réaliser des expériences de la CCI à des concentrations de 100 µM de DPE dans la cellule et 1 mM de ligand dans la seringue.
Résultats
Plusieurs constructions de CD22 DPE ont satisfait clonées dans le vecteur d’expression pHLsec et surexprimés dans mammifères HEK293F et HEK293S lignées de cellules (Figure 2 et 3 a). Toutes les constructions ont été purifiées jusqu'à homogénéité de taille par chromatographie d’exclusion et a donné un échantillon très pur pour les études de cristallisation (Figure 3 b et 3C). La construction de CD22 qui a mené à bien diffractants cristaux était la troncature de d1-d3 (résidus 20-330), avec cinq des six sites prédit N lié glycosylation mutés de Asn à Ala (N67A, N112A, N135A, N164A et N231A), produite dans les cellules HEK293S, tels que seul le site de glycosylation postées sur N101 a été retenu (cette construction se nomme CD2220-330, 5 a). Cristaux ont été obtenus dans plusieurs conditions de l’écran de matrices creuses MCSG-1, mais les meilleurs cristaux proviennent d’un état contenant 30 % (p/v) de polyéthylène glycol 4000, chlorure de lithium de 0,2 M et 0,1 M Tris, pH 8,5. Ces cristaux native diffractés à 2.1 résolution Å ; à l’aide de structures connues de domaines connexes Siglec protéines Ig n’a donné aucune solution dans les recherches de Monsieur.
Pour obtenir des informations "phasage", nous imbibé natives cristaux avec un panel de HA composés comprenant Hg, Pt, Os, Ta et Br à des concentrations allant de 1 à 20 mM de HA composé pour une durée d’incubation de 5 min à 1 d (Figure 4). Nous avons surveillé les cristaux pour les changements dans la morphologie et constaté que les cristaux imbibé HA composé de 20 mM a entraîné la fissuration rapide et la dissolution du cristal. Nous avons gelé un total de 63 cristaux qui ont conservé leur forme après des temps de jeu d’incubation qui ont été imbibés de cluster de tantale bromure, le chlorure de platine, acétate mercurique et chlorure mercurique. Cristaux imbibé de 7 mM de chlorure mercurique pour 30 min signal anormal ne présentaient un scan de fluorescence à la Canadian Light Source (CLS) 08-BM beamline (Saskatoon, Canada) et a permis à dispersion anomale multi-longueur d’onde collecte de données de rayons x sur un seul Crystal. Ces ensembles de données nous a permis de résoudre la sous-structure de mercure de CD2220-330, 5 a, qui a révélé un atome de mercure unique lié à une cystéine libre en position C308 et finalement nous a permis de construire la structure du CD225 a 20-330, dans la progressive carte de densité des électrons à l’aide de AutoBuild78.
Une fois qu’on a résolu la structure d’aboutissent, nous étions intéressés à résoudre la structure du CD22 lié à son ligand, α2-6 siallylactose. Tout d’abord, nous avons calculé l’affinité de CD22 vers sialyllactose α2-6 à l’aide de ITC pour caractériser la thermodynamique de liaison de l’interaction. Nous avons observé une affinité de ~ 280 µM et utilisé ces informations pour identifier une concentration initiale (~ 100 x KD) du ligand à utiliser pour le trempage de nos cristaux de20-330, 5 a CD22 native. Nous tremper les cristaux de CD2220-330, 5 a avec siallylactose de 25 mM pour 5 min, 2 h, 14 h, 40 h et 5D et suivi des changements dans la morphologie des cristaux. Un total de 75 ~ cristaux ont été gelé de divers points dans le temps et envoyés à la CLS synchrotron beamline 08-ID (Saskatoon, Canada) pour la collecte de données à distance. Un total de six ensembles de données de rayons x ont été prélevés de cristaux bien diffractants. La structure de chaque jeu de données de rayons x a été résolue par M. utilisant l’aboutissent CD2220-330, 5 a la structure comme un modèle de recherche initiale. La densité d’électrons qui en résulte pour tous les ensembles de données a été ensuite inspectée pour densité positive dans la carte Fo-Fc qui correspondrait pour consolidé sialyllactose α2-6 dans le site de liaison du CD22. Fait remarquable, tous les ensembles de données recueillies, même ceux de cristaux trempés après seulement 5 min de temps d’incubation, contenue densité positive correspondant au ligand dans le site de liaison. Les structures globales de l’aboutissent et CD22 liés ont été très similaires avec des changements conformationnels minimales, ce qui pourraient expliquer le succès de trempage expérimente le sialyllactose α2-6.
Ensuite, nous avons caractérisé la surface antigénique du CD22 reconnus par des anticorps thérapeutiques epratuzumab dans les expériences de BLI et le CCI (Figure 5). Thermodynamique et cinétique des profils de liaison Fab epratuzumab pour des constructions de CD22 avec différent glycoformes a révélé une affinité croissante à CD22 avec taille réduite glycane N-liés, avec jusqu'à 14 fois en affinité pour les plus petits glycanes (327 nM vs 24 nM dans BLI ; 188 nM nM de vs 58 au CCI). Le CD22 N-liés glycane limitant l’accès de l’anticorps à l’épitope a été identifié par BLI utilisant monopoint mutants de CD22 et en résolvant l’epratuzumab Fab-CD22 d1-d3 co crystal structure28.

Figure 1 . Vue d’ensemble de caractérisation de la glycoprotéine de la conception de la construction de caractérisation biophysique et structurelle. (1) analyse de la séquence primaire de glycoprotéine représentatif. En gris, le domaine extracellulaire (DCE) ; en vert, le segment transmembranaire de (TM) ; et en bleu, le domaine cytosolique de la glycoprotéine. Prédites N-glycanes sont étiquetés. (2) de clonage de constructions de DPE. (3) expression de DPE construit en cellules mammifères. (4) purification de glycoprotéine. Tandis que les protéines exprimées en HEK293F contiendra glycans complexes, protéines exprimées en HEK293S aura riche en mannose glycanes. Le traitement enzymatique des glycoprotéines produites dans les cellules HEK293S Endo H résultats chez glycoprotéines avec seulement une portion GlcNAc aux sites de glycosylation N-liés. glycoprotéines (5 a) sont testés pour leur liaison aux anticorps par interférométrie biolayer (BLI) et calorimétrie isotherme titration (ITC). Affinité pour les ligands petits peut également être mesurée par CCI. (5 b) les essais de cristallisation des glycoprotéines avec homogène N-glycanes, tels que ceux exprimés en HEK293S et déglycosylée avec Endo H. (6) dans certains cas, mutation des sites de glycosylation N-liés est nécessaire pour obtenir des cristaux. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 2 . Conception de CD22 ectodomaine ADN construit pour l’expression en cellules mammifères. A) représentation du plasmide pHLsec utilisé pour la transfection transitoire de CD22 DPE constructions. AgeI et KpnI sites utilisés pour le clonage sont indiquées par des cases rouges. B) The CD22 DPE contient sept domaines Ig (d1-d7) et 12 sites de glycosylation de N-liés prédites (en bleu). Quatre concepts ont été conçus dès le DPE CD22. C) gel d’agarose à 1 % montrant amplimères PCR du CD22 DJE construit pour le clonage dans le vecteur d’expression chez les mammifères pHLsec. Première voie contient le marqueur d’ADN 1 Ko. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 3 . Expression et purification des glycoprotéines. A) effet de la densité des cellules sur les rendements de l’expression. Expression de la glycoprotéine dans la culture à petite échelle 25 mL de HEK293F suspension de cellules transfectées à l’aide de trois densités différentes de départ des cellules (0,5 x 106 cellules mL-1, 1,0 x 106 cellules mL-1et 1. 5 x 106 cellules mL -1). Quantification réalisée par densitométrie de SDS-PAGE dans le panneau de gauche et par BLI quantitative dans le panneau de droite. Valeurs sont représentatives d’une préparation de glycoprotéine. B) chromatogramme de la première étape de purification pour construire CD2220-330, 5 a de 600 mL du surnageant à l’aide d’une colonne d’affinité Ni-NTA. La glycoprotéine a été éluée utilisant un gradient de l’imidazole (ligne grise), où 100 % correspond à la mémoire tampon d’élution, qui contient l’imidazole de 500 mM. Mise en commun des fractions sont dépeints avec des lignes verticales. C) chromatogramme d’exclusion stérique pour construire CD2220-330, 5 a , à l’aide d’une haute performance gel colonne de filtration. Mise en commun des fractions après avoir culminé à élution sont dépeints avec des lignes verticales. En médaillon : Gel de coloration au bleu de Coomassie SDS-PAGE montrant la pureté de la glycoprotéine. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 4 . Cristal de trempage en atomes lourds. A) station de travail échantillon pour le trempage des cristaux natives avec HA composés. Tous les requis outils sont étiquetés. B) les étapes suivies pour tremper les cristaux de construire CD2220-330, 5 a avec HA composés. Étape 1, ouvrez cupule contenant des cristaux et des cristaux de transfert en utilisant une boucle à une baisse de 0.2 µL sur une lamelle couvre-objet contenant HA solution diluée dans la condition de la cristallisation, telle que la concentration finale de HA varie de 1 à 10 mM. Étape 2, scelle la chute de la plaque de cristallisation et incuber les cristaux avec le HA composé pour différentes périodes de temps. Étape 3, monter le cristal trempé dans la boucle et le dos-laisser tremper pendant 30 s dans trois baisses consécutives de µl 0,2 contenant la solution de liqueur mère additionnée de 20 % (v/v) de glycérol distribué sur une lamelle couvre-objet. Étape 4, flash geler le cristal monté sur une boucle à l’azote liquide et placez-le dans une rondelle pour envoi à la source de rayonnement synchrotron. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 5 . Biolayer, mesures de calorimétrie titration interférométrie et isotherme. A) expérience représentant BLI. Panneau supérieur : exemple de plaque de montage pour une expérience de cinétique, où ce qui suit est étiqueté de : 1 cinétique x buffer (B), sonx 6-glycoprotéine tagged chargement (L), les concentrations de Fab représentatives (62,5, 125, 250, 500 nM), PBS + régénération de 500 mM tampon (R) et 1 x neutralisation cinétique tampon (B). Eh bien, chacun contient 200 µL de solution. Numéro de l’étape pour l’expérience cinétique est indiqué en haut de la plaque. Panneau milieu : Données brutes représentante d’expérience BLI effectuée à l’aide de biocapteurs Ni-NTA et la plaque dans le panneau supérieur. Étape numéros correspondent à la base (1), ses6 x glycoprotéine chargement (2), référence (3), association en dilution en série de Fab (4) et la dissociation (5). Étapes de la régénération ne sont pas représentés (étapes 6 et 7). Panneau inférieur : Représentatifs ont analysé les données liste brute association et dissociation (ligne bleue) avec le correspondant 1:1 monter (ligne rouge). B) panneau supérieur : installation de plaque représentant une seule CCI se faire sur un instrument automatisé de ITC avec sept expériences dans un 96 puits rond moufle. Chaque expérience est composé de trois puits. Le premier puits (rouge) correspond à l’échantillon pour la cellule (400 µL), le deuxième puits (vert) d’échantillon pour la seringue (120 µL). Le troisième bien est laissé vide, et les échantillons mixtes seront retournés à ceci bien après l’achèvement de l’expérience. Expériences de 1, 2 et 7 sont tampon dans des contrôles de la mémoire tampon. Expériences de 3-5 représentent en triple expérimente la glycoprotéine (P) dans la cellule et la Fab ou le ligand (L) dans la seringue. Expérience 6 représente une chaleur de ligand du contrôle de la dilution et devrait être soustrait du 3-5 des expériences au cours de l’analyse des données. Panneau inférieur : Représentant brut (en haut) et transformés (en bas) CCI données montrant Fab (epratuzumab) liant à CD22 DPE produites dans les cellules HEK293F. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
Discussion
Glycoprotéines membranaires ancrés sont critiques pour la fonction cellulaire et cibles thérapeutiques intéressantes. Ici, nous présentons un protocole pour la caractérisation structurale et biophysique de la JEC de glycoprotéines membranaires, les deux seuls ou en complexe avec des ligands de petites molécules et de fragments Fab. Nous avons utilisé avec succès ce protocole pour déterminer la structure cristalline des trois domaines Ig N-terminal-la plupart de la partie extracellulaire du humain CD2228, un corécepteur critique sur les cellules B impliqués dans le maintien de l’immunité humorale en cocher79. Nous avons également caractérisé l’accepteur de CD22 avec son ligand naturel α2-6 sialyllactose et défini le mode de reconnaissance d’un anticorps thérapeutique vers CD22 humaine. Ces résultats fournissent des aperçus de la relation structure-fonction de membre clé de la famille Siglecs qui a limité l’expression sur les lymphocytes B et une feuille de route moléculaire pour le développement de nouvelles petites molécules ciblées de CD22 et d’anticorps méthodes thérapeutiques. Alors que ce protocole a été utilisé avec succès pour un récepteur des cellules B contenant de l’Ig, nous proposons que notre approche peut être appliquée pour la caractérisation structurale et biophysique d’une glycoprotéine membranaire avec une organisation de domaine distincts. Dans ce cas, construire design et combinatoire glycane N-liés des mutations (que ce soit à Gln ou Ala) peuvent être évaluées pour trouver une construction approprié pour la croissance de cristaux et de diffraction à haute résolution.
Obtenir un échantillon homogène et pur glycoprotéine est d’une importance cruciale pour la croissance de cristaux et de diffraction des rayons x, ainsi que pour la caractérisation biophysique en aval. N-glycanes présents sur les glycoprotéines sont par nature hétérogènes et peuvent causer une hétérogénéité conformationnelle et chimique au sein de la glycoprotéine qui peut décourager la formation de cristaux. Pour réduire cette micro-hétérogénéité, stratégies qui introduisent des mutations ponctuelles pour éliminer les résidus Asn prévues pour héberger des N-glycanes, ou en utilisant des lignées de cellules mutantes (comme HEK293S) suivi du traitement par endoglycosidases (par exemple EndoH) peuvent considérablement améliorer la cristallisation succès15,21,22. Dans ce protocole, nous discutons de la purification de glycoprotéines solubles et Fabs qui sont sécrétées dans la cellule surnageante. Sécrétion de glycoprotéine fournit un itinéraire relativement simple vers la pureté, sans la nécessité de la lyse cellulaire ou l’ajout de produits chimiques ou détergents. La cellule surnageant, obtenu suivant cellule récolte est ensuite exécutée directement sur une colonne qui a une affinité pour la protéine d’intérêt (par exemple, Ni-NTA de glycoprotéines His-tag, ou de l’affinité de la LC pour les fragments Fab). Toutefois, selon la colonne d’utilisation et les conditions de la cellule surnageante (p. ex., pH), la fonctionnalité de liaison de la protéine d’intérêt pour la colonne peut être affectée. Si c’est le cas, il peut être nécessaire de se concentrer et de mettre en mémoire tampon la cellule surnageante d’échange afin d’améliorer la liaison à la colonne. En outre, il est fortement recommandé que des mesures de contrôle de la qualité durant la purification être employées pour évaluer la pureté de la protéine. Exécute un gel SDS-PAGE ou Western blot de tous les échantillons (avant, pendant et après les étapes de purification) peut donner un aperçu si le schéma de purification proposée convient à la protéine d’intérêt. Si contaminantes bandes sont visibles sur SDS-PAGE, ou si plusieurs espèces sont obtenus au cours de la purification (par exemple, plusieurs sommets sur l’exclusion de taille), étapes de purification supplémentaires devraient être envisagées, par exemple, chromatographie échangeuse d’ions, à gagner dans la pureté et l’augmentation des chances de cristallisation en aval80.
De cristallisation macromoléculaire, il est souvent essentiel d’obtenir des rendements élevés de la protéine d’intérêt pour permettre la projection d’un grand nombre des conditions de cristallisation potentiels à des concentrations de protéines pour trouver approprié crystal hits. Généralement, les lignées de cellules HEK293 discutées ici (HEK293F et HEK293S) sont des systèmes d’expression robuste et peuvent être facilement transposées de produire un échantillon plus que nécessaire. Cependant, il est possible que la protéine d’intérêt ne peut pas exprimer suffisamment au sein de ces lignées cellulaires. Dans ces cas, autres lignées de cellules, telles que les cellules de Expi29381,82, ont été trouvées pour montrer des niveaux supérieurs d’expression de la protéine et devraient être considérées comme une alternative.
Si bien ordonnées, diffraction de cristaux n’est pas obtenus après essais de plusieurs constructions de la protéine d’intérêt malgré le haut degré de pureté, il peut être nécessaire de développer des techniques de cristallisation pour promouvoir la formation de cristaux. Il a été démontré que des fragments Fab d’anticorps et nanocorps peuvent être exhausteurs de cristallisation excellent et promouvoir le cristal bien ordonnée d’emballage83,84,85. Ces fragments peuvent être exprimées et purifiés jusqu'à homogénéité et utilisés dans un complexe avec la protéine d’intérêt pour favoriser la cristallisation. Ce qui est important, des fragments Fab produites tel que décrit à l’article 10 peuvent ont tendance à former non-fonctionnel LC dimères86. Ces dimères sont des contaminants et devraient être supprimés au cours de la purification. Dans notre expérience, LC dimères souvent ont un volume de rétention différents sur l’exclusion de taille, ou éluer comme un pic distinct sur chromatographie échangeuse d’ions et donc peuvent être supprimés de la purification Fab - mais ce n’est pas toujours le cas. Si ces techniques ne suffisent pas à enlever les dimères de LC de la purification Fab, méthodes de purification supplémentaires, tels que de la purification d’affinité G protéines, peuvent être utilisées pour améliorer la pureté.
Alternative aux co-complexation avec les fragments Fab, techniques bien documentés comme matrice aléatoire microseeding peuvent améliorer ses chances d’obtenir des cristaux bien ordonnée63,70. Cette méthode implique l’addition de petites quantités de cristaux écrasé, sous-optimale dans la condition de la cristallisation, fournissant un cristal nuclée pour promouvoir la croissance des cristaux. Ceci peut être effectué à l’aide de cristaux de la protéine d’intérêt, ou ceux avec une structure similaire de domaine architecture et tertiaire. En outre, microseeding matrice aléatoire peut être effectuée pour tenter de cristalliser la protéine seule, ou en complexe avec un fragment Fab ou petite molécule d’intérêt. Les progrès récents en cryo-microscopie électronique aussi faire cette technique une alternative intéressante à la cristallographie par rayons x pour obtenir des informations structurales à haute résolution pour les molécules avec des caractéristiques appropriées87,88, 89,90,91.
Lorsque progressive des ensembles de données de diffraction des rayons x n’est pas par M., HA trempage peut être nécessaire pour résoudre le problème de la phase de dispersion anomale ou remplacement isomorphe. Inspection de la séquence d’acides aminés de la protéine peut fournir des indices sur la stratégie pour dérivatisation HA, y compris le pH optimum pour la liaison. En particulier, des cystéines non appariés au sein de la protéine peuvent lier spécifiquement HA composés contenant du mercure. Le trempage de cristaux natives avec HA composés est un processus itératif pour déterminer l’identité du composé HA optimale, sa concentration et le temps d’incubation nécessaire. Si les premières tentatives de trempage ne donnent pas de cristaux bien diffractants contenant un HA adapté pour l’élimination, il peut être nécessaire d’introduire des substitutions d’acides aminés pour améliorer la probabilité de liaison HA et signal anormal. Les exemples incluent des mutations pour inclure un résidu cystéine libre pour lier efficacement Hg, Au, Pt ou Pb. Expression de protéines pour élimination anormale dans un média séléno-méthionine complétée par e. coli est intensivement employé pour l’élimination anormale, cependant une un système équivalent qui intègre avec fiabilité séléno-méthionine n’est pas facilement disponible pour les cellules de mammifères en suspension92,93et est une zone de développement futur.
Une fois obtenue la structure aboutissent de la glycoprotéine d’intérêt, tremper les cristaux avec des ligands de petites molécules peut être effectuée pour obtenir une structure du complexe récepteur-ligand immunitaire. Ces données fournissent un modèle pour la conception rationnelle de plus spécifiques et de haute affinité des ligands qui peuvent servir de petit-molécule thérapeutique mais aussi permet de mieux comprendre la fonction biologique de la glycoprotéine haute résolution. Lorsque vous tentez de faire tremper les cristaux de glycoprotéine avec des ligands de petites molécules d’intérêt, inspection de la structure cristalline d’aboutissent peut indiquer si le trempage doit être possible. Si proches-l’entassement du cristal contacts se trouvent autour du site de liaison du ligand ou autour de régions devrait subir des modifications conformationnelles lors de la liaison ligands, trempage sera probablement être problématique. Dans ce cas, les autres méthodes telles que cocristallisation du complexe protéine-ligand doivent être effectuées.
Déclarations de divulgation
Les auteurs ne déclarent aucun intérêts opposés.
Remerciements
Des expériences de diffraction des rayons x décrits dans cet article ont été effectués avec ces faisceaux ID 08 et 08-BM au canadien de rayonnement synchrotron, qui est soutenu par la Fondation canadienne pour l’Innovation, Sciences naturelles et en génie conseil de recherches du Canada, le Université de la Saskatchewan, le gouvernement de la Saskatchewan, Diversification de l’économie de l’Ouest Canada, le Conseil National de recherches du Canada et les Instituts canadiens de recherche en santé. Nous tenons à remercier la construction & la biophysique Core Facility, l’hôpital pour enfants malades, accès aux instruments de CCI et BLI. Sergent a été pris en charge par Banting Postdoctoral Fellowship BPF-144483 de l’instituts de recherche en santé du Canada. T.S. est récipiendaire du Canada Graduate Scholarship maîtrise Award et une bourse Canada de Vanier de l’instituts de recherche en santé du Canada. Ce travail a été soutenu par Subv PJT-148811 (J.-P.J.) de l’instituts de recherche en santé du Canada. Cette recherche a été entreprise, en partie, grâce au financement du programme des chaires de recherche du Canada (J.-P.J.).
matériels
| Name | Company | Catalog Number | Comments |
| 0.22 μm Steritop filter | EMD Millipore | SCGPS02RE | |
| 10 well 4-15% gradient SDS-PAGE gel | Bio-Rad | 4561084 | |
| 10x glycobuffer 3 | New England Biolabs | P0702S | Comes with Endo H reagent |
| 10x Kinetics Buffer | PALL FortéBio | 18-1092 | |
| 10x Tris/Glycine/SDS Buffer | Bio-Rad | 1610732 | |
| 1 mL round bottom 96 well block | ThermoFisher | 260251 | |
| 22 mm cover slip | Hampton research | HR3-231 | |
| 4x Laemmli Sample Buffer | Bio-Rad | 1610747 | |
| 96-3 well INTELLIPLATE low volume reservior | Art Robbins Instruments | 102-0001-03 | |
| AgeI | New England Biolabs | R0552S | |
| ÄKTA Pure | GE Healthcare | ||
| ÄKTA Start | GE Healthcare | ||
| Amicon Ultra 15 centrifugal filtration device 10KDa MWCO | Millipore | UFC901008 | |
| Amicon Ultra 4 centrifugal filtration device 10KDa MWCO | Millipore | UFC801008 | |
| Auto-iTC200 | Malvern | ||
| Axygen MaxyClear Snaplock 1.5 mL microtubes | Fisher Scientific | MCT150C | |
| Countess Cell Counting Chamber Slides | Thermo Fisher Scientific | C10228 | |
| CryoLoop 18 x 0.05-0.1 mm | Hampton research | HR4-945 | |
| CryoLoop 18 x 0.1-0.2 mm | Hampton research | HR4-947 | |
| CryoLoop 18 x 0.2-0.3 mm | Hampton research | HR4-970 | |
| Digital Dry Bath | Bio-Rad | 1660562EDU | |
| E. coli DH5α | Invitrogen | 18258012 | |
| Endo H | New England Biolabs | P0702S | |
| Erlenmeyer flask (baffled base), polycarbonate, sterile, 500 mL, DuoCAP | TriForest Labware | FBC05000S | |
| Erlenmeyer flask 125 mL (baffled base), polycarbonate, sterile, 125 mL with vented cap | VWR | 89095-258 | |
| Falcon Disposable sterile serological pipet, non-pyrogenic, 10 mL | Greiner Bio-One | 607180 | |
| Falcon Disposable sterile serological pipet, non-pyrogenic, 25 mL | Greiner Bio-One | 760180 | |
| Falcon Disposable sterile serological pipet, non-pyrogenic, 5 mL | Greiner Bio-One | 606180 | |
| Falcon Disposable sterile serological pipet, non-pyrogenic, 50 mL | Greiner Bio-One | 768180 | |
| FectoPRO DNA Transfection Reagent, Polyplus | VWR | 10118-842 | |
| Freestyle 293F cells | Thermo Fisher Scientific | R79007 | |
| Freestyle Expression medium | Thermo Fisher Scientific | 12338001 | |
| Heavy Atom Screens Au | Hampton research | HR2-444 | |
| Heavy Atom Screens Hg | Hampton research | HR2-446 | |
| Heavy Atom Screens M1 | Hampton research | HR2-448 | |
| Heavy Atom Screens M2 | Hampton research | HR2-450 | |
| Heavy Atom Screens Pt | Hampton research | HR2-442 | |
| HEK 293S | ATCC | ATCC CRL-3022 | |
| HisTrap Affinity Column | GE Healthcare | 17525501 | |
| HiTrap KappaSelect Affinity Columns | GE Healthcare | 17545811 | |
| HiTrap LambdaSelect Affinity Columns | GE Healthcare | 17548211 | |
| KpnI | New England Biolabs | R0142S | |
| MCSG-1 Crystal Screen 1.7 mL block | Anatrace | MCSG-1 | |
| MCSG-2 Crystal Screen 1.7 mL block | Anatrace | MCSG-2 | |
| MCSG-3 Crystal Screen 1.7 mL block | Anatrace | MCSG-3 | |
| MCSG-4 Crystal Screen 1.7 mL block | Anatrace | MCSG-4 | |
| Mercuric chloride | Sigma | 1044170100 | |
| Microplate, 96 well, polypropelene, flat bottom, black | Greiner Bio-One | 655209 | |
| Minstrel DT UV | Formulatrix | ||
| Multitron Pro shaker | Infors HT | MP25-TA-CO2HB | |
| Nanodrop 2000/2000c Spectrophotometer | Thermo Fisher Scientific | ND-2000 | |
| Nanosep 3K Omega centrifugal device | PALL Life Science | OD003C33 | |
| Ni-NTA biosensors | PALL FortéBio | 18-5102 | |
| Octet RED96 | PALL ForteBio | ||
| Oryx 4 crystallizaiton robot | Douglas Instrument | ORY-4/1 | |
| Platinum chloride | Sigma | 520632-1g | |
| Precision Plus Protein Standard | Bio-Rad | 161-0374 | |
| PureLink HiPure Plasmid Maxiprep Kit | Invitrogen | K210006 | |
| Quick Coomassie Stain | Protein Ark | GEN-QC-STAIN-1L | |
| Steriflip Sterile 50 mL Disposable Vacuum Filtration System 0.22 µm Millipore Express | EMD Millipore | SCGP00525 | |
| Superdex 200 Increase 10/300 GL | GE Healthcare | 28990944 | |
| Superose 6 10/300 GL | GE Healthcare | 17517201 | |
| Tantalum bromide cluster | Jena bioscience | PK-103 | |
| Top96 Crystallization Screen | Rigaku Reagents | 1009846 | |
| Tryphan Blue | Thermo Fisher Scientific | T10282 | |
| VDX 24-well with sealant | Hampton research | HR3-172 | |
| α2-6 sialyllactose | Sigma Aldrich | A8556-1mg |
Références
- Sachs, J. N., Engelman, D. M. Introduction to the membrane protein reviews: The interplay of structure, dynamics, and environment in membrane protein function. Annu Rev Biochem. 75 (1), 707-712 (2006).
- Cournia, Z., et al. Membrane protein structure, function, and dynamics: A perspective from experiments and theory. J Membr Biol. 248 (4), 611-640 (2015).
- Macauley, M. S., et al. Antigenic liposomes displaying CD22 ligands induce antigen-specific B cell apoptosis. J Clin Invest. 123 (7), 3074-3083 (2013).
- Hyde, C. A. C., et al. Targeting extracellular domains D4 and D7 of vascular endothelial growth factor receptor 2 reveals allosteric receptor regulatory sites. Mol Cell Biol. 32 (19), 3802-3813 (2012).
- Tai, W., Mahato, R., Cheng, K. The role of HER2 in cancer therapy and targeted drug delivery. J Control Release. 146 (3), 264-275 (2010).
- Zarei, O., Benvenuti, S., Ustun-Alkan, F., Hamzeh-Mivehroud, M., Dastmalchi, S. Strategies of targeting the extracellular domain of RON tyrosine kinase receptor for cancer therapy and drug delivery. J Cancer Res Clin Oncol. 142 (12), 2429-2446 (2016).
- Rosman, Z., Shoenfeld, Y., Zandman-Goddard, G. Biologic therapy for autoimmune diseases: an update. BMC Med. 11 (1), 88(2013).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Barclay, A. N. Membrane proteins with immunoglobulin-like domains - A master superfamily of interaction molecules. Semin Immunol. 15 (4), 215-223 (2003).
- Barclay, A. N. Ig-like domains: evolution from simple interaction molecules to sophisticated antigen recognition. Proc Natl Acad Sci. 96 (26), 14672-14674 (1999).
- Aebi, M. N-linked protein glycosylation in the ER. Biochim Biophys Acta - Mol Cell Res. 1833 (11), 2430-2437 (2013).
- Ohtsubo, K., Marth, J. D. Glycosylation in cellular mechanisms of health and disease. Cell. 126 (5), 855-867 (2006).
- Lodish, H., Berk, A., Zipursky, S., Al, E. Glycosylation in the ER and Golgi complex. Mol Cell Biol. (4), Section 17.7 (2000).
- Thomas, P., Smart, T. G. HEK293 cell line: A vehicle for the expression of recombinant proteins. J Pharmacol Toxicol Methods. 51 (3), 187-200 (2005).
- Lee, J. E., Fusco, M. L., Ollmann Saphire, E. An efficient platform for screening expression and crystallization of glycoproteins produced in human cells. Nat Protoc. 4 (4), 592-604 (2009).
- Betenbaugh, M. J., Tomiya, N., Narang, S., Hsu, J. T. A., Lee, Y. C. Biosynthesis of human-type N-glycans in heterologous systems. Curr Opin Struct Biol. 14 (5), 601-606 (2004).
- Yang, Z., et al. Engineered CHO cells for production of diverse, homogeneous glycoproteins. Nat Biotechnol. 33 (8), 842-844 (2015).
- Bláha, J., Kalousková, B., Skořepa, O., Pažický, S., Novák, P., Vaněk, O. High-level expression and purification of soluble form of human natural killer cell receptor NKR-P1 in HEK293S GnTI-cells. Protein Expr Purif. 140, 36-43 (2017).
- Bláha, J., Pachl, P., Novák, P., Vaněk, O. Expression and purification of soluble and stable ectodomain of natural killer cell receptor LLT1 through high-density transfection of suspension adapted HEK293S GnTI- cells. Protein Expr Purif. 109, 7-13 (2015).
- Chaudhary, S., Pak, J. E., Gruswitz, F., Sharma, V., Stroud, R. M. Overexpressing human membrane proteins in stably transfected and clonal human embryonic kidney 293S cells. Nat Protoc. 7 (3), 453-466 (2012).
- Chang, V. T., et al. Glycoprotein structural genomics: Solving the glycosylation problem. Structure. 15 (3), 267-273 (2007).
- Davis, S. J., Crispin, M. Solutions to the glycosylation problem for low- and high-throughput structural glycoproteomics. Funct Struct Proteomics Glycoproteins. , 127-158 (2011).
- Elbein, A. D., Tropea, J. E., Mitchell, M., Kaushal, G. P. Kifunensine, a potent inhibitor of the glycoprotein processing mannosidase I. J Biol Chem. 265 (26), 15599-15605 (1990).
- Zheng, K., Bantog, C., Bayer, R. The impact of glycosylation on monoclonal antibody conformation and stability. MAbs. 3 (6), 568-576 (2011).
- Aricescu, A. R., Lu, W., Jones, E. Y. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr Sect D Biol Crystallogr. 62 (10), 1243-1250 (2006).
- Adams, P. D., et al. The Phenix software for automated determination of macromolecular structures. Methods. 55 (1), 94-106 (2011).
- May, A. P., Robinson, R. C., Vinson, M., Crocker, P. R., Jones, E. Y. Crystal structure of the N-terminal domain of sialoadhesin in complex with 3' sialyllactose at 1.85 Å resolution. Mol Cell. 1 (5), 719-728 (1998).
- Ereño-Orbea, J., et al. Molecular basis of human CD22 function and therapeutic targeting. Nat Commun. 8 (1), 764(2017).
- Yu, X. -L., et al. Crystal structure of HAb18G/CD147: implications for immunoglobulin superfamily homophilic adhesion. J Biol Chem. 283 (26), 18056-18065 (2008).
- Garman, E., Murray, J. W. Heavy-atom derivatization. Acta Crystallogr - Sect D Biol Crystallogr. 59 (11), 1903-1913 (2003).
- Agniswamy, J., Joyce, M. G., Hammer, C. H., Sun, P. D. Towards a rational approach for heavy-atom derivative screening in protein crystallography. Acta Crystallogr Sect D Biol Crystallogr. 64 (4), 354-367 (2008).
- Rose, J. P., Wang, B. C., Weiss, M. S. Native SAD is maturing. IUCrJ. 2 (20), 431-440 (2015).
- Olieric, V., et al. Data-collection strategy for challenging native SAD phasing. Acta Crystallogr Sect D Struct Biol. 72 (3), 421-429 (2016).
- Rillahan, C. D., et al. Disubstituted sialic acid ligands targeting Siglecs CD33 and CD22 associated with myeloid leukaemias and B cell lymphomas. Chem Sci. 5 (6), 2398-2406 (2014).
- Mesch, S., et al. From a library of MAG antagonists to nanomolar CD22 ligands. ChemMedChem. 7 (1), 134-143 (2012).
- Chiu, M. L., Gilliland, G. L. Engineering antibody therapeutics. Curr Opin Struct Biol. 38, 163-173 (2016).
- Elgundi, Z., Reslan, M., Cruz, E., Sifniotis, V., Kayser, V. The state-of-play and future of antibody therapeutics. Adv Drug Deliv Rev. 122 (2016), 2-19 (2017).
- Yang, D., Singh, A., Wu, H., Kroe-Barrett, R. Determination of high-affinity antibody-antigen binding kinetics using four biosensor platforms. J Vis Exp. (122), e55659(2017).
- Kamat, V., Rafique, A. Designing binding kinetic assay on the bio-layer interferometry (BLI) biosensor to characterize antibody-antigen interactions. Anal Biochem. 536, 16-31 (2017).
- Brautigam, C. A., Zhao, H., Vargas, C., Keller, S., Schuck, P. Integration and global analysis of isothermal titration calorimetry data for studying macromolecular interactions. Nat Protoc. 11 (5), 882-894 (2016).
- Duff, M. R., Grubbs, J., Howell, E. E. Isothermal titration calorimetry for measuring macromolecule-ligand affinity. J Vis Exp. (55), e2796(2011).
- Livingstone, J. R. Antibody characterization by isothermal titration calorimetry. Nature. 384 (6608), 491-492 (1996).
- Freyer, M. W., Lewis, E. A. Isothermal titration calorimetry: Experimental design, data analysis, and probing macromolecule/ligand binding and kinetic interactions. Methods Cell Biol. 84, 79-113 (2008).
- Macauley, M. S., Crocker, P. R., Paulson, J. C. Siglec-mediated regulation of immune cell function in disease. Nat Rev Immunol. 14 (10), 653-666 (2014).
- Zaccai, N. R., et al. Structure-guided design of sialic acid-based Siglec inhibitors and crystallographic analysis in complex with sialoadhesin. Structure. 11 (5), 557-567 (2003).
- Pantophlet, R., et al. Bacterially derived synthetic mimetics of mammalian oligomannose prime antibody responses that neutralize HIV infectivity. Nat Commun. 8 (1), 1601(2017).
- Leonard, J. P., et al. Epratuzumab, a humanized anti-CD22 antibody, in aggressive non-Hodgkin's lymphoma: phase I/II clinical trial results. Clin Cancer Res. 10 (16), 5327-5334 (2004).
- Finn, R. D., et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 45, 190-199 (2017).
- Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., Sternberg, M. J. E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 10 (6), 845-858 (2015).
- Lessard, J. C. Molecular cloning. Methods Enzymol. 529, 85-98 (2013).
- Gupta, R., Jung, E., Brunak, S. NetNGlyc: Prediction of N-glycosylation sites in human proteins. , (2004).
- Liu, H., Naismith, J. H. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 8, 91(2008).
- Heckman, K. L., Pease, L. R. Gene splicing and mutagenesis by PCR-driven overlap extension. Nat Protoc. 2 (4), 924-932 (2007).
- Froger, A., Hall, J. E. Transformation of plasmid DNA into E. coli using the heat shock method. J Vis Exp. (6), e253(2007).
- Akula, I., Julien, J. -P. Optimization of glycoprotein expression by transient transfection in HEK293 F/S suspension cells. , Available from: https://www.polyplus-transfection.com/wp-content/uploads/2015/09/FectoPRO-Technical-Note-031716.pdf (2015).
- Taylor, S. C., Berkelman, T., Yadav, G., Hammond, M. A defined methodology for reliable quantification of western blot data. Mol Biotechnol. 55 (3), 217-226 (2013).
- Tan, H. Y., Ng, T. W. Accurate step wedge calibration for densitometry of electrophoresis gels. Opt Commun. 281 (10), 3013-3017 (2008).
- Gassmann, M., Grenacher, B., Rohde, B., Vogel, J. Quantifying Western blots: pitfalls of densitometry. Electrophoresis. 30 (11), 1845-1855 (2009).
- Jonnalgadda, K., Markley, L., Estes, S., Prajapati, S., Takkar, R., Kumaraswamy, S. Rapid, reliable quantitation of Fc-fusion protein in cell culture supernatants. , Available from: https://www.fortebio.com/documents/ForteBio_App_Note_13.pdf (2018).
- JoVE Science Education Database. Basic methods in cellular and molecular biology: Separating protein with SDS-PAGE. J Vis Exp. , (2018).
- Wilkins, M. R., et al. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol. 112, 531-552 (1999).
- Till, M., et al. Improving the success rate of protein crystallization by random microseed matrix screening. J Vis Exp. (78), e50548(2013).
- Obmolova, G., Malia, T. J., Teplyakov, A., Sweet, R., Gilliland, G. L. Promoting crystallization of antibody-antigen complexes via microseed matrix screening. Acta Crystallogr Sect D Biol Crystallogr. 66 (8), 927-933 (2010).
- D'Arcy, A., Bergfors, T., Cowan-Jacob, S. W., Marsh, M. Microseed matrix screening for optimization in protein crystallization: What have we learned. Acta Crystallogr Sect F, Struct Biol Commun. 70 (9), 1117-1126 (2014).
- Luft, J. R., et al. Efficient optimization of crystallization conditions by manipulation of drop volume ratio and temperature. Protein Sci. 16 (4), 715-722 (2007).
- Dessau, M. A., Modis, Y. Protein crystallization for X-ray crystallography. J Vis Exp. (47), e2285(2011).
- Sugahara, M., Asada, Y., Ayama, H., Ukawa, H., Taka, H., Kunishima, N. Heavy-atom Database System: A tool for the preparation of heavy-atom derivatives of protein crystals based on amino-acid sequence and crystallization conditions. Acta Crystallogr D Biol Crystallogr. 61 (9), 1302-1305 (2005).
- Boggon, T. J., Shapiro, L. Screening for phasing atoms in protein crystallography. Structure. 8 (7), 143-149 (2000).
- Vera, L., Stura, E. A. Strategies for protein cryocrystallography. Cryst Growth Des. 14 (2), 427-435 (2014).
- Pichlo, C., Montada, A. A., Schacherl, M., Baumann, U. Production, crystallization and structure determination of C. difficile PPEP-1 via microseeding and Zinc-SAD. J Vis Exp. (118), e55022(2016).
- Leslie, A. G. W., et al. Automation of the collection and processing of X-ray diffraction data - a generic approach. Acta Crystallogr Sect D Biol Crystallogr. 58 (11), 1924-1928 (2002).
- Pike, A. C. W., Garman, E. F., Krojer, T., Von Delft, F., Carpenter, E. P. An overview of heavy-atom derivatization of protein crystals. Acta Crystallogr Sect D Struct Biol. 72 (3), 303-318 (2016).
- Cooper, D. R., Porebski, P. J., Chruszcz, M., Minor, W. X-ray crystallography: Assessment and validation of protein-small molecule complexes for drug discovery. Expert Opin Drug Discov. 6 (8), 771-782 (2011).
- Hassell, A. M., et al. Crystallization of protein-ligand complexes. Acta Crystallogr Sect D Biol Crystallogr. 63 (1), 72-79 (2006).
- Muller, I., et al. Guidelines for the successful generation of protein-ligand complex crystals. Acta Crystallogr Sect D Struct Biol. 73 (2), 79-92 (2017).
- Zhao, Y., et al. Two routes for production and purification of Fab fragments in biopharmaceutical discovery research: Papain digestion of mAb and transient expression in mammalian cells. Protein Expr Purif. 67 (2), 182-189 (2009).
- Shah, N. B., Duncan, T. M. Bio-layer interferometry for measuring kinetics of protein-protein interactions and allosteric ligand effects. J Vis Exp. (84), e51383(2014).
- Terwilliger, T. C., et al. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr Sect D Biol Crystallogr. 64 (1), 61-69 (2008).
- Walker, J. A., Smith, K. G. C. CD22: An inhibitory enigma. Immunology. 123 (3), 314-325 (2008).
- Gräslund, S., et al. Protein production and purification. Nat Methods. 5 (2), 135-146 (2008).
- Jain, N. K., et al. A high density CHO-S transient transfection system: Comparison of ExpiCHO and Expi293. Protein Expr Purif. 134, 38-46 (2017).
- Fang, X. T., Sehlin, D., Lannfelt, L., Syvänen, S., Hultqvist, G. Efficient and inexpensive transient expression of multispecific multivalent antibodies in Expi293 cells. Biol Proced Online. 19 (1), 11(2017).
- Löw, C., et al. Nanobody mediated crystallization of an archeal mechanosensitive channel. PLoS One. 8 (10), 77984(2013).
- Hunte, C., Michel, H. Crystallization of membrane proteins mediated by antibody fragments. Curr Opin Struct Biol. 12 (4), 503-508 (2002).
- Ereño-Orbea, J., Sicard, T., Cui, H., Carson, J., Hermans, P., Julien, J. -P. Structural basis of enhanced crystallizability induced by a molecular chaperone for antibody antigen-binding fragments. J Mol Biol. 430 (3), 322-336 (2018).
- Spooner, J., et al. Evaluation of strategies to control Fab light chain dimer during mammalian expression and purification: A universal one-step process for purification of correctly assembled Fab. Biotechnol Bioeng. 112 (7), 1472-1477 (2015).
- Elmlund, D., Le, S. N., Elmlund, H. High-resolution cryo-EM: The nuts and bolts. Curr Opin Struct Biol. 46, 1-6 (2017).
- Merk, A., et al. Breaking cryo-EM resolution barriers to facilitate drug discovery. Cell. 165 (7), 1698-1707 (2016).
- Bai, X. C., McMullan, G., Scheres, S. H. W. How cryo-EM is revolutionizing structural biology. Trends Biochem Sci. 40 (1), 49-57 (2015).
- Wlodawer, A., Li, M., Dauter, Z. High-resolution cryo-EM maps and models: A crystallographer's perspective. Structure. 25 (10), 1589-1597 (2017).
- Bartesaghi, A., et al. 2.2 Å resolution cryo-EM structure of β-galactosidase in complex with a cell-permeant inhibitor. Science. 348 (6239), 1147-1151 (2015).
- Hendrickson, W. a, Horton, J. R., LeMaster, D. M. Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): A vehicle for direct determination of three-dimensional structure. EMBO J. 9 (5), 1665-1672 (1990).
- Walden, H. Selenium incorporation using recombinant techniques. Acta Crystallogr Sect D Biol Crystallogr. 66 (4), 352-357 (2010).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.