
Method Article
Charakterisierung von Glykoproteinen mit dem Immunglobulin-Falz durch Röntgen-Kristallographie und biophysikalischen Techniken
* Diese Autoren haben gleichermaßen beigetragen
In diesem Artikel
Zusammenfassung
Ansätze für die biophysikalischen und strukturelle Charakterisierung von Glykoproteinen präsentieren wir mit der Immunglobulin-Falte Biolayer Interferometrie, Isothermen Titration Kalorimetrie und Röntgen-Kristallographie.
Zusammenfassung
Glykoproteine auf der Oberfläche der Zellen spielen wichtige Rollen in zelluläre Funktion, einschließlich der Signalisierung, Haftung und Transport. Auf Leukozyten einige der diese Glykoproteine besitzen Immunglobulin (Ig) Falten und zentralen immun Anerkennung und Regulierung. Hier präsentieren wir Ihnen eine Plattform für Design, Ausdruck und biophysikalischen Charakterisierung der extrazellulären Domäne des menschlichen B-Zell-Rezeptors CD22. Wir schlagen vor, dass diese Ansätze bis hin zur Charakterisierung von Säugetieren Glykoprotein Ectodomains Ig Domänen mit breit anwendbar sind. Zwei menschliche embryonale Nieren (HEK) Zelle Fangleinen, HEK293F und HEK293S, werden verwendet, um Glykoproteine mit komplexen und hoch-Mannose Glykoproteinen bzw. zum Ausdruck bringen. Diese rekombinanten Glykoproteine mit verschiedenen Glycoforms ermöglichen Untersuchung der Wirkung von Glycan Größe und Zusammensetzung für Ligand-Bindung. Wir besprechen Sie Protokolle für die Untersuchung der Kinetik und Thermodynamik Glykoprotein Bindung an biologisch relevante Liganden und therapeutische Antikörper Kandidaten. Rekombinanten Glykoproteine produziert in HEK293S Zellen sind Kristallisation durch Glycan Homogenität, geringere Flexibilität und Anfälligkeit für Endoglycosidase H Behandlung zugänglich. Wir präsentieren Ihnen Methoden zum Einweichen Glykoprotein Kristalle mit schweren Atome und kleine Moleküle für Phase-Ermittlung und Analyse der Ligand-Bindung, beziehungsweise. Die experimentelle Protokolle diskutiert versprechen hier zur Charakterisierung von Säugetieren Glykoproteine geben Einblick in ihre Funktion und den Wirkmechanismus von Therapeutika zu untersuchen.
Einleitung
Oberflächenproteine spielen wichtige Rollen in zelluläre Funktion. Durch ihren extrazellulären Domänen können diese Membranproteine Zell-Zell-Interaktionen, Adhäsion, Transport und1,2Signalisierung modulieren. Die extrazelluläre Lokalisation dieser Proteine macht sie attraktive Ziele für die Entwicklung von Therapeutika zur Behandlung einer Vielzahl von Krankheiten, einschließlich Krebs und Autoimmunerkrankungen3,4,5 , 6 , 7. einer der häufigsten Falten der menschlichen Membrane Protein Ectodomains ist die Immunglobulin-ähnlichen (Ig) Falte, die von sieben oder mehr β-Stränge in zwei β-Blätter8,9angeordnet gebildet wird. In der Regel sind Ig-haltigen Glykoproteine Multidomain-Strukturen mit Ig Domains nacheinander auf den extrazellulären Teil der Membrane Protein10angeordnet. Post-translationalen Modifikationen dieser Zelle Oberfläche Proteine, besonders N und O verbunden Glykosylierung, wurden wesentliche Rollen in ihrer Regulation, Falten, Sekretion und Funktion11spielen gezeigt. Um zu einem besseren Verständnis ihrer Funktion und bessere Design-Therapeutika, die ihnen zugeordnet werden kann, Techniken erforderlich sind, mit denen für ihre genaue molekulare Charakterisierung. Hier präsentieren wir eine Kombination von Techniken, mit denen für die biophysikalischen (Biolayer Interferometrie (BLI) und Isotherme Titration Kalorimetrie (ITC)) und strukturelle (Röntgen-Kristallographie) Charakterisierung der extrazellulären Domäne des Ig-haltigen Membran Glykoproteine, allein und in Anlage mit ihrer biologisch relevante Liganden und therapeutische Moleküle (Abbildung 1).
N-linked Glykosylierung ist eines der am häufigsten verwendeten Post-Übersetzungen Änderungen auf Säugetier-Proteine und tritt während Protein Reifung im endoplasmatischen Retikulum und Golgi12,13. Zell-Linien, wie menschliche embryonale (HEK) 293 Nierenzellen, wurden für die rekombinante Expression von großen Mengen des glykosylierten Säugetier-Proteine14,15entwickelt. Diese Zelllinie wurde in einem Fahrwerk-Format entwickelt, die für die Leichtigkeit des Protein-Produktion in größeren Mengen im Vergleich zu adhärenten Zelllinien Skalierung ermöglicht. Hier nutzen wir zwei HEK293-Zell-Linien: HEK293F und HEK293 Gnt ich- / - (HEK293S), die durch das Fehlen von N-Acetylglucosaminyl-Transferase unterscheide ich mich (Gnt ich) in der letzteren. Wiederum, Herstellung von komplexen Glykoproteinen (wie gesehen in HEK293F) ist nicht möglich und stattdessen hohe Mannose-Typ Glykoproteinen (überwiegend Mann5GlcNAc2) befinden sich in N-linked Glycan Seiten18,19,20 . Mit diesen beiden Zelllinien parallel ermöglicht die Untersuchung der Wirkung von Glycan Größe und Komplexität auf biologische Funktion und therapeutischen Zielen. Glykoproteine in HEK293F Zellen produziert haben sogar größere, komplexere Glykane im Vergleich zu den gleichen Glykoprotein in HEK293S Zellen produziert. Glykoproteine in HEK293S Zellen produziert sind zugänglicher, Kristallisation, wegen der reduzierten chemischen und Konformationsänderungen Heterogenität der ihre N-Verbindung handelt. Zur weiteren Verbesserung der Crystallizability in HEK293S (aber nicht HEK293F) Zellen produzierten Glykoproteine mit Enzym Endoglycosidase H (Endo H), die solche in der Spaltung der hohen Mannose Glykane Ergebnisse behandelt werden können, dass nur eine einzelne N-Acetylglucosamin (GlcNAc) Abstimmungsunterlagen bleibt bei jeder N-linked Glykosylierung Seite21,22. Andere Methoden können auch verwendet werden, um N-Glycan Verarbeitung innerhalb der Zellen, wie die Zugabe von Glycosyltransferase Inhibitoren während Glykoprotein Ausdruck, einschließlich Kifunensine23zu begrenzen. Alternative Ansätze beinhalten den Ausdruck von native Glykoproteinen (in HEK293F Zellen) gefolgt von enzymatischen Deglycosylation Peptid N-Glucosidase F (PNGaseF) verwenden. Deglycosylation mit PNGaseF ist jedoch nachweislich weniger wirksam unter heimischen Bedingungen und erhöht Aggregation in einigen Proteinen; in Fällen wenn das Protein lösliche nach der Behandlung bleibt erwirbt es negative Ladungen an der Oberfläche aufgrund der deamidierung des Rückstands Asparagin, Asparaginsäure24, die möglicherweise nachteilig für die Kristallisation. Vorhergesagten N-Glykosylierung Websites können auch mutiert, in den meisten Fällen zu Alanin und Glutamin Rückstände, N-linked Glycosylation an diesen Standorten zu verhindern und Glykoprotein Proben von hohe Homogenität zu generieren. Alternativ können Glykoproteine in anderen eukaryontischen Zellkulturen, einschließlich Hefe, Insekt, und Anlagensysteme oder anderen Säugetieren Zelllinien wie chinesische Hamster Eierstock (CHO) Zellen16,17hergestellt werden.
Viele Säugetiere Expressionsvektoren, einschließlich pHLsec, ermöglichen die Sekretion von rekombinantes Glykoprotein Ectodomains in die Zelle mittlere25. Sekretion von Glykoproteinen aus HEK293 Zellen ermöglicht eine schnelle und einfache Reinigung ohne die Notwendigkeit einer Zelle Lysis. Neben der Reinigung Tags (z.B. His-Tag, Strep-Tag, Flag-Tag, Myc-Tag, HA-Tag) an den N oder C-Terminus des Ziels Glykoprotein ermöglicht Reinigung durch einen einstufigen Affinitätschromatographie. Anschließend kann Größe Ausgrenzung Chromatographie verwendet werden, eine Monodisperse Probe für biophysikalische und strukturelle Charakterisierung liefern.
Eine sehr reine und homogene Glykoprotein-Probe unter geeigneten Bedingungen kann gut diffracting Kristalle führen. Sobald eine vollständige Röntgenbeugung Dataset aus solche Kristalle gewonnen worden sind, müssen Anfangsphase bestimmt werden, um die Elektronendichte an das Glykoprotein zu berechnen. Dank einer ständig wachsenden Anzahl von Strukturen in der Protein Data Bank (PDB) ist die am häufigsten verwendete Methode zur schrittweisen bei weitem Molekulare Ersatz (MR), geworden, die eine verwandte Protein-Struktur verwendet, um Anfangsphase26zu erhalten. Wenn Herr nicht zur Lösung des Problems Phase wie gelegentlich bei Multi-Ig Domäne Glykoproteine27,28,29der Fall gewesen ist, sind alternative Methoden jedoch erforderlich. In diesem Artikel zeigen wir eine Methode, um Kristalle mit schweren Atomen (HA) Einweichen auslaufen, die für die Struktur der CD22 ektodomäne28Lösung erforderlich war. Identifiziert das Recht HA für den schrittweisen ist ein iterativer Prozess, der HA Reaktivität zur Verfügung Atome in das Glykoprotein in einem gegebenen Kristallgitter und die Kristallisation Lösung30,31abhängt. Alternativ können natürlichen Schwefel Atome in Cystein und Methionin Rückstände verwendet werden, für den schrittweisen wenn anwesend in einem hoch genug Verhältnis zu anderen Atomen in das Glykoprotein und x-ray Diffraction Daten mit ausreichender Redundanz32gesammelt werden können, 33.
Die biologische Funktion der Membran Glykoproteine wird oft von Protein-Protein-Wechselwirkungen oder Protein-Ligand-Interaktionen, wie z. B. mit Kohlenhydraten vermittelt. Wenn die Liganden klein genug ist, um aus der Lösung an die Glykoprotein-Bindungsstelle im Kristallgitter zu verbreiten, kann das Einweichen Experimente erfolgreich, eine Glykoprotein-Liganden Co Kristallstruktur um besser zu verstehen, Liganden Anerkennung zu erhalten sein.
Die hier vorgestellten Protokolle sind auch relevant für das Verständnis der Wechselwirkungen der Oberfläche Glykoproteine mit synthetischen therapeutische Liganden34,35 und Antikörper Therapeutika36,37. In Kombination mit Strukturinformationen kann verbindliche Kinetik und Thermodynamik werden stark, um zu verstehen und zu verbessern ihre Wirkmechanismen. Eine Technik, die für die kinetische Analyse der therapeutische Antikörper binden an ein Glykoprotein ermöglicht ist BLI38,39. BLI verwendet Biosensoren mit einem immobilisierten Liganden, um die Assoziation und Dissoziation Kinetik mit einen Bindungspartner, bestimmen letztlich ein Gleichgewicht Dissoziationskonstante (KD) zu messen. BLI ist ein attraktiver Ansatz, da geringe Mengen von Glykoproteinen erforderlich sind (< 100 µg), Experiment ist schnell (~ 10-15 min pro Durchlauf), und es kann automatisiert werden. ITC eignet sich auch für das Studium Affinitäten zwischen Glykoproteine und verbindliche Partner40,41,42,43. ITC mehr Zeit und intensive Reagenz ist, erhalten Sie wertvolle Informationen über die Thermodynamik der Interaktion (ΔG, ΔH, ΔS und Stöchiometrie). ITC ist auch sehr nützlich für das Studium der schwachen Wechselwirkung, die oft mit der vorübergehenden Bindung Oberfläche Glykoproteine, Liganden verbunden sind. Darüber hinaus können diese Techniken in Verbindung verwendet werden, um die Bindung von verschiedenen Konstrukte und bewerten die Auswirkungen der verschiedenen N-linked Glycoforms erhalten das Glykoprotein in verschiedenen Zelllinien zu äußern. Durchführung von BLI und ITC mit Glykoproteine produziert in HEK293F, HEK293S und behandelt mit Endo H bieten einen tiefen Einblick in die Rolle von Glykoproteinen in biologische Aktivität und therapeutisches Engagement.
Wir erfolgreich diese Protokolle zur Charakterisierung der extrazellulären Domäne (ECD) des menschlichen CD2228, ein Glykoprotein Mitglied der Sialinsäure Säure bindenden Ig-ähnliche Lektine (Siglecs) Familie, die unerlässlich für die Aufrechterhaltung der Homöostase der B-Zelle44 . Wir eingehende Konstrukt-Konzept für Kristallisation durchgeführt und das Röntgen-Dataset durch Einweichen mit Hg HA abgebaut. Wir auch getränkt CD22 Kristalle mit ihrer Liganden Sialinsäure Säure (α2-6 Sialyllactose), eine Struktur von der immun-Rezeptor-Ligand-Komplex zu erhalten und somit die Baupläne für das Struktur-geführte Design von Glycan Mimetika45,46vorgesehen. Darüber hinaus wir die Fragment Antigen-Bindung (Fab) von Anti-CD22 therapeutische Antikörper Epratuzumab - eine therapeutische Kandidaten derzeit in klinischen Phase-III-Studien für non-Hodgkin Lymphom47- generiert, um seine Bindungsaffinität von BLI bestimmen und ITC, differentiell glykosylierten baut CD22 ECD. Diese Studien zeigten eine entscheidende Rolle für N-linked Glykosylierung Epratuzumab Engagement, mit möglichen Folgen für die CD22 Anerkennung auf dysfunktionale B-Zellen.
Protokoll
(1) Konstrukt Design für Glykoprotein ECD
- Die Aminosäure-Sequenz des menschlichen CD22 zu bewerten (Uniprot) die InterPro und Phyre2 Server Ermittlung vorhergesagten Domänenelemente und Grenzen befinden sich innerhalb der Protein-48,-49.
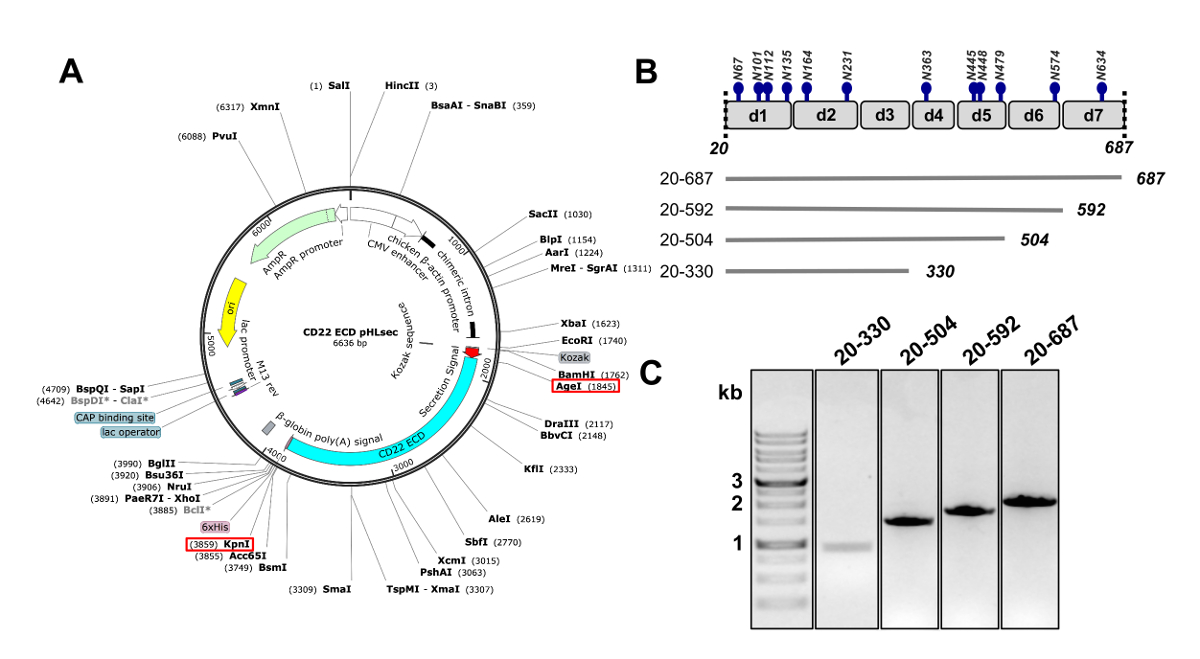
- Klonen Sie die Reihenfolge der menschlichen CD22, fehlt das Signalpeptid transmembranen und cytosolischen Domänen (Rückstände 20-687, nachstehend CD22 extrazellulären Domäne, CD22 ECD) in die pHLsec Säugetieren Ausdruck Vektor25 mit Restriktionsenzymen AgeI und KpnI ( Abbildung 2 ( A) 50.
Hinweis: Der pHLsec-Vektor ist optimiert für die Überexpression von löslichen, sekretierten Proteine in Säugerzellen25. Dieser Vektor enthält ein Sekret Signal für die extrazelluläre Sekretion von löslichen Glykoproteine ermöglichen. pHLsec enthält einen C-terminale (His)6 X Tag um Affinitätsreinigung von Zelle Überstände mit immobilisierten Metall Affinität Chromatographie Methoden zu erleichtern. - Abgeschnittene Konstrukte des CD22 ECD mit sequentiellen Deletionen der C-terminalen Ig Domains zu klonen: Domänen 1-6 (Rückstände 20-687), Domänen 1-5 (Rückstände 20-592), Domänen 1-4 (Rückstände 20-504) und Domänen 1-3 (Rückstände 20-330) (Abbildungen 2 b und 2 C)50 .
- Bewerten Sie die primäre Sequenz von CD22 ECD mit dem NetNGlyc-Server vorhergesagten N verbunden Glykosylierung Seiten präsentieren das Konstrukt51zu identifizieren.
- Mit Site-verwiesene Mutagenese, von Standardprotokollen52 oder durch Überlappung PCR53, mutieren Sie Standortes vorhergesagten N verbunden Glykosylierung (Asn, Gln und/oder Asn, Ala) um Konstrukte des CD22 ECD zu erstellen, die entweder ein einzelnes oder mehrere enthalten N-linked Glykosylierung Mutationen.
- Nach der Sequenz Überprüfung der geklonten Konstrukte, verwandeln Sie in kompetente E. Coli DH5α54 und Maxi-Prep Zellen die DNA (gemäß Herstellervorschrift) zur Vorbereitung der Transfektion.
2. HEK293F und HEK293S Zelle Einrichtung
Hinweis: Alle Manipulation von HEK293F oder HEK293S Zellen mit notwendigen Reagenzien und Geräte muss in einer Biosafety Level 2 Anlage in eine geeignete biologische Kabinett durchgeführt werden. Die äußere Oberfläche aller Elemente muss mit einem 70 % igen Ethanol Lösung oder gleichwertige Reagenz sterilisiert werden.
- HEK293F und HEK293S-Federung-Zellen (siehe Tabelle der Materialien) zu erhalten und bei-80 ° C bis zur Verwendung aufbewahren.
- Warmen Medien (siehe Tabelle der Materialien) für 1 h bei 37 ° C Wasserbad. Übertragen Sie 24 mL erwärmten Medien auf einem 125 mL ratlos Zelle Kultur Kolben mit einer belüftete Kappe.
- Erhalten Sie 1 mL Zelle aliquoten von-80 ° C und Transfer zu Eis.
- Inkubieren Sie Zellen in einem 37 ° C Wasserbad für ca. 1 min, um die Zellen teilweise Auftauen. 1 mL der Zellen aus dem Fläschchen in den 125 mL ratlos Zelle Kultur Kolben mit den Medien zu übertragen.
- Schließen Sie die Zelle Kultur Kolben mit der belüftete Kappe und Ort Kolben in einem Boston-Shaker set 130 u/min, 37 ° C, Luftfeuchtigkeit von 70 % und 8 % CO2.
3. HEK293 Zellen Wartung
Hinweis: Zelldichte und die Lebensfähigkeit der Zellen muss ca. 24 h nach dem Auftauen überprüft. Diese Schritt wird sichergestellt, dass Zellen nach Inokulation wiederherstellen; anfängliche Lebensfähigkeit sollte > 80 %.
- Vorsichtig 10 µL Zellen aus der 125 mL Flasche mit frischen Aussetzung Zellen zu entfernen und in eine sterile 1,5 mL reaktionscup übertragen. Schließen Sie die Küvette und wieder in den Inkubator.
- Pipette 10 µL Trypan blau-Lösung in die 1,5 mL reaktionscup mit den Zellen, gründlich mischen und 10 µL der Kammer der Zählung Folie übertragen.
- Eine automatische Zelle Zähler zählende Folie hineingesteckt und erhalten Werte für Zelldichte (in Einheiten von Zellen mL-1) und der Zellviabilität (in Prozent).
- Berechnen Sie das Volumen der Zellen, die erforderlich wären, um eine frische 200 mL Kultur bei einer endgültigen Dichte ~0.8 X 106 Zellen mL-1 mit den folgenden Gleichungen zu impfen:
 (1)
(1) (2)
(2)
Hinweis: Es kann dauern ~ 5D eine geeignete Zelle Dichte zum animpfen in eine 200 mL-Kultur zu erhalten. - Sobald die Zelldichte zum animpfen einer 200 mL-Kultur ausreicht, Warm-up Medien für 1 h bei 37 ° C Wasserbad und das erwärmte Medium in der Biosicherheit Schrank übertragen.
- Übertragen Sie das erforderliche Volumen der Medien (wie in Gleichung 2 berechnet) mit einer serologischen Pipette, sorgfältig in einem 500 mL ratlos Zelle Kultur Kolben mit einer belüftete Kappe werden.
- Mit einer serologischen Pipette übertragen Sie das erforderliche Volumen der Aussetzung Zellen (gerechnet in Gleichung 1) in den 500 mL ratlos Zelle Kultur Kolben mit den Medien.
- Kappe der neuen 200 mL Wartung Lager und wieder in den Inkubator. Wachsen Sie Zellen zu einer Dichte von ca. 3 x 106 Zellen mL-1. Die Passage Zellen bei einer Dichte von 0,8 x 106 Zellen mL-1 alle 2-3-d weiterhin eine stabile Kultur der Zellen (wie in Abschnitt 3.4 3.7 beschrieben). Lassen Sie sich nicht die Zellen zu eine Dichte von ~ 4 x 106 Zellen mL-1überschreiten.
(4) die Transfektion von HEK293 Zellen für Glykoprotein Ausdruck
- Berechnen Sie das Volumen der Zellen und Medien, die für eine 200 mL-Kultur zur Transfektion 0,8 x 10 benötigt6 Zellen mL-1 (mit Gleichungen 1 und 2 von Abschnitt 3.4).
Hinweis: Die Anzahl von 200 mL Transfektionen, die ausgeführt werden können hängt die Zelldichte der Wartung Aktie. - Übertragen Sie die gewünschte Lautstärke von Medien und Zellen zur Transfektion in eine neue 500 mL Zelle Kultur Flasche mit eine belüftete Kappe und Rückkehr den Zelle bestand in den Inkubator.
- Inkubieren Sie Zellen für 1 h vor Transfektion damit Zellen akklimatisieren folgt aufteilen.
- 50 µg DNA in einem sterilen 50 mL konische Rohr übertragen und mit 5 mL Medien verdünnen. Vakuum filter der verdünnte DNA mit Hilfe eine 0,22 µm-Wasserfilter-System in ein anderes steriles Röhrchen.
- Mischung verdünnt, DNA in ein 1:1 Masse: Volumen-Verhältnis mit Transfection Reagens gefiltert. Vorsichtig schwenken der DNA: Transfektion Reagenzlösung zu mischen und die Lösung bei Raumtemperatur für 10 min inkubieren.
- DNA: Transfektion Reagenzlösung direkt die Zellen einfügen. Inkubieren Sie transfizierte Zellen bei 130 u/min, 37 ° C, Luftfeuchtigkeit von 70 % und 8 % CO2 in einem Shaker für 5-7 d.
5. Optimierung der Zelle Transfection Bedingungen
Hinweis: Um die Zelle Transfektion Bedingungen für maximale Glykoprotein Ertrag optimieren, transfizieren Sie Zellen bei einer Vielzahl von anfänglichen Zelldichten und bewerten Sie Protein Rendite im Laufe der Zeit(Abbildung 3). Zellen transfizieren, wie in Abschnitt 4 beschrieben, am ersten Zelldichten von 0,5 x 106 bis 2 x 106 Zellen mL-1 55. Testversion Transfektionen können auf 25 mL Gesamtvolumen (in 125 mL ratlos Zelle Kultur Kolben) mit 6 µg DNA-spart Platz und Reagenzien verkleinert werden. Auch kann die Menge an DNA optimierte55.
- Jeden Tag nach Transfektion (1-7 Tage), übertragen eine Aliquote 500 µL aus der Zellkultur in einem sterilen 1,5 mL reaktionscup (in der Biosicherheits-Schrank).
- Regelmäñig Zellen bei 12.000 x g für 5 min in einem Microcentrifuge sofort nach der Entnahme zu spinnen. Übertragen Sie überstand auf eine neue 1,5 mL reaktionscup und speichern bei 4 ° C, bis alle Proben entnommen werden.
- Quantitate sekretierten Glykoprotein von Densitometrie
- Sobald alle Proben gewonnen werden, aliquoten 20 µL der einzelnen Probe in eine neue 1,5 mL reaktionscup und mischen mit 6 µL 4 X Laemmli Probenpuffer nicht reduzieren.
- Kochen Sie Proben für 5 min bei 95 ° C in einem Thermo-Block. Drehen Sie Proben 1 min bei 12.000 x g in einem Microcentrifuge.
- Laden Sie 20 µL jeder Probe pro Bohrloch in einem 10-gut 4-15 % Steigung SDS-PAGE Gel. Enthalten Sie eine Fahrspur für Protein Größe Marker. Laufen Sie Gel bei 250 V für 20 min in einem Puffer Tris/Glycin/SDS.
- Laufen Anschluss übertragen Gel, Coomassie Fleck (siehe Tabelle der Materialien) für 20 min. de-Flecken-Gel in DdH2O für 20 min. Bild Gel.
- Durchführen Sie Densitometrie mit ImageJ, Anschluss an standard-Protokolle57,58.
- Kompilieren und plot-Daten mit "Tage nach Transfektion" auf der X-Achse und "Densitometrie Werte" auf der Y-Achse(Abbildung 3).
Hinweis: Alternativ, wenn Protein-Expression zur Visualisierung von SDS-PAGE ausreicht, Techniken wie Western blotting verwendeten56möglicherweise.
- Quantitate sekretierten Glykoprotein von BLI
- Verwendung von Ni-NTA Biosensoren, die Menge der freigesetzten Glykoprotein mit BLI59quantitate.
- Kompilieren und plot-Daten mit "Tage nach Transfektion" auf der X-Achse und "Proteinkonzentration (µg/mL)" auf der Y-Achse(Abbildung 3).
6. Reinigung der löslichen Glykoprotein aus HEK293 überstand
- Ernten von Zellen durch Zentrifugation bei 6.371 x g für 20 min bei 4 ° C. Überstand mit sekretierten CD22 ECD zu behalten und mit einem 0,22 µm-Filter filtern.
- Überstand bei 4 mL min-1 auf einem Pre äquilibriert zu laden (20 mM Tris pH 9.0, 150 mM NaCl, 5 mM imidazol) Ni-NTA-Spalte (5 mL Volumen) mit einem Benchtop-Chromatographie-System.
Hinweis: Andere Affinität-basierte Reinigungstechniken können verwendet werden basierend auf die Affinität-Tags enthalten im Konstrukt Design in Abschnitt 1. - Nach überstand laden die Affinität Spalte mit 3-4 Spalte Volumen (CV) von Waschpuffer (20 mM Tris pH 9.0, 150 mM NaCl, 5 mM imidazol) waschen.
- Eluieren Sie gereinigten Glykoprotein aus der Spalte mit einer 4-100 % Gefälle (4 CVs) der Elution Buffer (20 mM Tris pH 9.0, 150 mM NaCl, 500 mM imidazol) beim Sammeln von Brüchen (Abbildung 3B).
- Pool-Fraktionen mit der eluierten Gipfel eine zentrifugale Filtration Gerät mit 10 kDa nominalen Molekulargewicht Limit (NMWL) und Konzentrat durch Zentrifugieren bei 4.000 x g bei 4 ° C für 15 Minuten oder bis die Probe ein Volumen von 500 µL erreicht.
- Injizieren Sie konzentrierten Glykoprotein in eine 500 µL Probenschleife und Belastung bei 0,5 mL min-1 auf einer Pre-äquilibriert (20 mM Tris, pH 9,0, 150 mM NaCl) Ausschluss Spalte Hochleistungs-Größe (ca. 24 mL Volumen) auf ein schnelles Protein Flüssigkeitschromatographie (FPLC) System bei 4 ° C beim Sammeln von Brüchen (Abb. 3C).
- Führen Sie SDS-PAGE Gel eluierten Bruchteile, Brüche, enthält das Glykoprotein zu identifizieren und bündeln Sie entsprechende Fraktionen zu. Die SDS-PAGE-Gel kann ausgeführt werden, wie in Abschnitt 560beschrieben.
7. Deglycosylation des gereinigten Glykoprotein
- Messen die Konzentration des gereinigten Proteins nach Größe Ausgrenzung Chromatographie mit Absorption bei 280 nm dividiert durch die vom Aussterben bedroht-Koeffizient (z. B. 1,418 M-1 cm-1 für CD22 ECD).
Hinweis: Der theoretische Aussterben Koeffizient der interessierenden Proteine kann mit Servern wie ExPASy ProtParam61berechnet werden. - Inkubieren Sie gereinigtes Protein mit Endo H für 1 h bei 37 ° C, bei einem Verhältnis von 1 mg gereinigtes Protein, 10 µL des kommerziellen Enzym in 1 X Endo H Puffer (lt. Herstellerangaben).
Hinweis: Endo H bindet hohe Mannose Glykane produziert einen einzigen GlcNAc Glyko-verlassen in HEK293S bei jeder Glykosylierung Seite21. Endo H nicht Glykane auf in HEK293F Zellen22, produzierten Proteine Spalten, jedoch andere Enzyme (z. B.PNGaseF24) dazu benutzt werden können. - Konzentrieren Sie Deglycosylated ECD, 500 µL zu und Gel Filtration Chromatographie auf eine Spalte der Ausgrenzung Hochleistungs-Größe (etwa 24 mL Volumen) bei 0,5 mL min-1 auf einem FPLC entfernen Endo H und daraus resultierende Aggregate zu trennen.
- Speichern Sie das Deglycosylated-Protein bei 4 ° C bis zur Verwendung in nachgeschalteten Experimente.
8. die Kristallisation von Glykoproteinen
Hinweis: Kristallisation Studien mit im Handel erhältlichen Bildschirmen führen und Drop Experimente mit Hilfe eines Roboters Kristallisation sitzen eingerichtet.
- Konzentrat rein, Deglycosylated ECD auf 10 mg mL-1 mit einem zentrifugale Filtration Gerät mit 10 kDa NMWL bei 4.000 x g (4 ° C), bis die gewünschte Konzentration erreicht ist.
- Bestimmen Sie die Proteinkonzentration mithilfe Absorption bei 280 nm und dividiert durch die vom Aussterben bedroht-Koeffizient.
- Zentrifuge Probe bei 12.000 x g für 5 min bei 4 ° C vor der Kristallisation Studien zu unerwünschten Staub oder andere Verunreinigungen aus der Probe zu entfernen.
- Füllen Sie den Tank Brunnen 96-Well-Sitzung fallen Kristallisation Platten mit 80 µL Kristallisation Lösung von einem kommerziellen Kristallisation Bildschirm.
Hinweis: Wir verwenden dünnbesetzte Matrix kommerzielle Bildschirme, die entworfen wurden, basierend auf den erfolgreichsten Kristallisation Bedingungen in Bezug auf Strukturen, die in die PDB-Datei hinterlegt. - Mit Hilfe eines Roboters Kristallisation verzichten Tropfen in den Brunnen der Kristallisation Platte mit einer Fallhöhe von 200 nL in einem Verhältnis von gereinigten Protein: Kristallisation Lösung von 1:1.
- Sobald die gesamte Platte verzichtet hat, Dichtplatte die mit Klebeband und Ort in einem Teller-Imager für Inspektion durch sichtbares und ultraviolettes Licht.
- Überprüfen Sie die Kristallisation Platten sofort nach der Einrichtung und in den folgenden Wochen mit sichtbares und ultraviolettes Licht um Bedingungen zu identifizieren, die anfängliche Glykoprotein Kristall Hits geben.
- Kristalle von ersten Kristallisation Hits mit feinrechen basierend auf der Bedingung des Kristall-Hit oder zufällige Matrix Mikro-säen Methoden62,63,64,65erhalten weiter zu optimieren.
- Cryo-schützen alle Kristalle fehlen ausreichende Cryo-Schutzmittel im Rahmen der Kristallisation durch Einweichen des Kristalls in Mutter Alkohol Lösung ergänzt mit 20 % (V/V) Glycerin-Lösung (oder gleichwertige Cryo-Schutzmittel, wie Ethylenglykol oder Polyethylenglykol 400).
- Berg-Kristalle in Cryoloops und Flash Einfrieren in flüssigem Stickstoff vor der Erhebung von Daten zu einer Heimat Quelle Diffraktometer oder mit Synchrotronstrahlung.
9. Abbau mit schweren Atom Derivatisierung
Hinweis: Vor jeder Manipulation von HA-Verbindungen müssen Sicherheitsaspekte berücksichtigt werden. HA Verbindungen in Proteinkristallographie verwendet werden für ihre starke Affinität zu biologischen Molekülen ausgewählt und Bergen Risiken für die menschliche Gesundheit aus über längere Zeit. Ergreifen Sie geeignete Sicherheitsmaßnahmen für HA-Verbindungen wie bereits in ihre Sicherheitsdatenblätter.
- Testen Sie verschiedene HA Verbindungen, reproduzieren Konzentrationen und Inkubationszeiten, gut diffracting Kristalle, die in Abschnitt 8 in einer 24-Well-Kristallisation-Platte mit den hängenden Tropfen Dampf Diffusion Methode66gewonnen.
- Entscheiden, welche HA werden für Kristall Derivatisierung verwendet. Server (z. B. das Heavy-Atom Datenbanksystem67) können helfen, mit HA zusammengesetzte Auswahl, sicherzustellen, dass sie für den Protein- und Kristallisation Zustand geeignet sind.
Hinweis: HA-Bildschirme sind auch für einfache Screening am effektivsten HA Verbindungen zur schrittweisen im Handel erhältlich. Eine Reihe von "magischen sieben" HA Verbindungen um hohe Erfolgswahrscheinlichkeit für HA Derivatisierung68haben zuvor beschrieben worden. - Workstation für HA Einweichen (Abb. 4A) eingerichtet. Übertragen Sie Kristalle mit einer Cryoloop, schnell auf eine 0,2 µL drop auf einem 22 mm-Deckglas mit HA-Lösung verdünnt in der Kristallisation Zustand, so dass die Endkonzentration von HA von 1-20 mM reicht werden. Versiegeln Sie den Tropfen zu und inkubieren Sie für unterschiedlich lange (Abbildung 4B). Ein guter Ausgangspunkt ist 5, 10, 60 und 90 min und Übernachtung.
- Sichtkontrolle Kristalle mit einem Lichtmikroskop, identifizieren mögliche Risse oder Veränderungen in der Farbe, die negative Auswirkungen auf das Glykoprotein Kristall oder Kristall Derivatisierung angeben können.
- Montieren Sie Kristalle in Cryoloops und Rücken-Einweichen Kristalle für 30 s in drei aufeinander folgenden 0,2 µL sinkt mit Mutter Alkohol Lösung ergänzt mit 20 % (V/V) Glycerin (oder Alternative Cryo-Schutzmittel)69. Rücken-Einweichen die Kristalle entfernt HA zusammengesetzte, der mußte unspezifisch und teilweise Belegung verursacht durch schwache Bindung HA reduziert. Flash-Kristalle Einfrieren in flüssigem Stickstoff (Abbildung 4B).
- Verwenden Sie für die Datenerhebung, Verarbeitung, Struktur-Lösung und Verfeinerung zuvor beschriebenen Protokolle26,70,71,72.
10. Einweichen Glykoprotein Kristalle mit seinen Liganden
- Reproduzieren Sie gut diffracting Kristalle, die in Abschnitt 8 in einer Kristallisation 24-Well-Platte mit der hängenden Tropfen Dampf-Diffusion-Methode erhalten.
- Bereiten Sie eine Stammlösung 50 mM Liganden in 20 mM Tris, pH 9,0, 150 mM NaCl.
Hinweis: Die Konzentration des Liganden sollte nach der Affinität zu seinen Glykoprotein vorbereitet werden. Wenn die Affinität unbekannt ist, kann es erforderlich, eine Methode wie ITC (Abschnitt 12.2) verwenden, um die Affinität vor Beginn Einweichen Experimente zu bestimmen. Sicherstellen Sie, dass die Liganden auf die gewünschte Konzentration in den erforderlichen Puffer löslich ist. - Die Tropfen mit ECD Kristalle unterschiedlicher Konzentration des Liganden hinzu und verschließen Sie die Tropfen für Inkubation bei Längen zwischen 5 min bis 5D.
- Verfolgen Sie visuell Kristalle mit einem Lichtmikroskop, mögliche Änderungen in der Morphologie zu identifizieren.
- Montieren Sie Kristalle in Cryoloops und Cryo-schützen sie in Mutter-Alkohol-Lösung ergänzt mit 20 % (V/V) Glycerin (oder andere Cryo-Schutzmittel wie Ethylenglykol oder niedermolekularen Polyethylenglykol 400)69.
- Verwenden Sie für die Datenerhebung, Verarbeitung, Struktur-Lösung und Verfeinerung zuvor beschriebenen Protokolle73,74,75.
11. Produktion von Fragment Antigen binden (Fab)
- Subclone Gene, die Fab schwere Kette (HC) und leichte Kette (LC) Sequenzen von Anti-ECD Antikörper, z. B.Epratuzumab entsprechen.
Hinweis: Alternativ kann IgG durch das Enzym Papain, Fab-Fragmente76generieren abgespalten werden. - Transfizieren Sie Zellen, wie beschrieben in Abschnitt 4, mit folgenden Änderungen:
- Verwenden Sie eine Gesamtmasse von DNA für die Transfektion von Fab-Fragmente von 90 µg pro 200 mL der Kultur.
- Transfizieren Sie HC und LC Plasmide im Verhältnis 2:1, die LC-Dimer-Bildung zu reduzieren.
- Nach 7 d der Inkubation Ernte Zellen, überstand zu behalten und mit einem Vakuum-driven Filtration Gerät 0,22 µm filtern.
- Equilibrate Anti-LC (Kappa oder Lambda) Affinität Spalten in PBS-Puffer mit einem Benchtop-Chromatographie-System.
Hinweis: Wenn LC-Dimer-Bildung ein Problem während der Reinigung, Protein G Affinitätschromatographie als Alternative zur Kappa/Lambda-LC Affinitätsreinigung einsetzbar. - Laden Sie überstand auf Affinität Spalte bei 4 mL min-1. Waschen Sie nach probenbeladung, Spalte mit 3-4 CVs von PBS.
- Eluieren Protein aus Spalte mit einer isokratischen Elution mit 100 mM Glycin, pH 2.2, sofort neutralisieren die eluierten Fraktionen mit 10 % (V/V) 1 M Tris, pH 9,0 in jeder Bruchteil.
Hinweis: Die eluierten Fab kann weiter durch Ionenaustausch-Chromatographie und/oder Größe Ausgrenzung Chromatographie mit einem FPLC bei 4 ° c gereinigt werden
12. die Charakterisierung von Fab und kleine Molekül binden an das Glykoprotein
- Biolayer Interferometrie
- 50 mL 1 x Kinetik Puffer (1 X PBS, 0,002 % (V/V) Tween-20, 0,01 % (w/V) BSA) vorzubereiten.
- Hydrat sechs Ni-NTA-Biosensoren in 200 µL 1 X Kinetik Puffer für 10 min in einer Platte vornässen.
- Verdünnen Sie sein-tagged ECD in 1 mL 1 X Kinetik Puffer auf eine Endkonzentration von 25 ng µL-1. Pipette Verdünnungsreihen des gereinigten Fab in 200 µL 1 x Kinetik Puffer, mit einer hohen Konzentration von 500 nM und spätere serielle Verdünnungen von 250 nM, 125 nM und 62,5 nM.
- Aliquoten Reagenzien in schwarzen flachen Boden aus Polypropylen 96-Well Mikrotestplatte wie in Abbildung 5A, wo jeder gut 200 µL der angegebenen Lösung enthält.
- Sammeln Sie Daten mithilfe der Kinetik-Assays in der Datenerfassung Software, wie zuvor beschrieben38,39,77 (Abb. 5A).
- Kurz, übertragen die Biosensoren in Vertiefungen mit 1 x Kinetik Puffer Baseline für 60 s vor dem Laden 25 ng µL-1 von Glykoprotein für 240 s (oder bis zu einer Schwelle von 1,0 nm erreicht ist) bei 1.000 u/min.
- Nach einem zweiten Ausgangswert von 60 s in 1 x Kinetik Puffer übertragen die Biosensoren in Vertiefungen, enthält die serielle Verdünnung der Fab. Die 180 s-Verein-Phase folgt anschließend eine 180 s Dissoziation Schritt 1 x Kinetik Puffer.
Hinweis: Biosensoren wiederverwendet werden wenn das obige Protokoll eine Regeneration Schritt besteht aus drei Zyklen folgt von Biosensoren im Puffer (PBS mit 500 mM imidazol) stripping waschen für 5 s gefolgt von 5 s in 1 x Kinetik Puffer für die Neutralisation. Biosensoren wiederverwendet werden, bis zu ~ 10-20 Mal am selben Tag oder bis schlechte Daten Qualität beobachtet.
- Analysieren Sie die Daten mit Hilfe der Analysesoftware (Abb. 5A):
- Unter der Registerkarte "1, Import und Daten auswählen.
- Tab 2, Schritt 1: Datenselektion, wählen Sie "Sensorauswahl" und Höhepunkt Referenz Wells (Zeilen E und F, Abb. 5A), rechter Mausklick und gut verweisen soll. Unter Schritt 2: Subtraktion, wählen Sie "Referenz-Brunnen". Unter Schritt 3: Y-Achse auszurichten, wählen Sie "Grundlinie" aus Zeitbereich von 0,1 bis 59,8 s. Unter Schritt 4: Inter Schritt Korrektur, wählen Sie "An Dissoziation ausrichten." Unter Schritt 5: Prozess, wählen Sie 'Savitzky-Golay Filter' und drücken Sie Prozessdaten.
- Tab 3 Wählen Sie unter "Assoziation und Dissoziation" unter Schritt zur Analyse mit einem 1:1 Modell. Wählen Sie "Globale Anpassung" und "Gruppe durch Farbe". Kurven der rechten Maustaste, wählen Sie "Farbe ändern", legen Sie alle Kurven auf die Farbe Ihrer Wahl. Wählen Sie "Fit Kurven". Wenn Daten gut ausgerüstet sind, kann ein Bericht exportiert werden, indem Sie "Bericht speichern" auswählen.
- Wiederholen Sie Experiment mit Glykoprotein in HEK293F und HEK293S Zellen (Abschnitt 5) hergestellt und nach der Endo H Behandlung (Abschnitt 7), die Wirkung, wenn überhaupt, von verschiedenen Glycoforms auf Fab Anerkennung. Darüber hinaus wiederholen Sie das Experiment mit dem ECD Trunkierungen, Einblick in die Domänen der Fab gebunden.
- Isothermen Titration Kalorimetrie der Fab-Glykoprotein Interaktion
Hinweis: ITC-Experimente, die hier beschriebenen erfolgt über eine automatisierte ITC-Instrument. Experimente sind in 1 mL rund 96-Well Unterflasche durchgeführt.- Dialyse ECD und Fab in einem einzigen 4 L Becher 20 mM Tris, pH 8.0, 150 mM NaCl bei 4 ° C über Nacht mit einer Stir Bar.
- Dialysierter ECD und Fab 5 µM und 50 µM bzw. zu konzentrieren, mit einem zentrifugalen Filter mit 10 kDa NMWL, 4.000 x g für 5 min bei 4 ° C vor dem Gebrauch Puffer gewährleistet die Konzentrator-Membranen dreimal mit 5 mL der Dialyse zu waschen.
Hinweis: Irgendeine Fehlanpassung im Puffer zwischen den Proben in der Zelle und Spritze verursachen unerwünschte Hitze während des ITC-Experiment und Daten von schlechter Qualität freigegeben werden. - Experiment 1: Fügen Sie 400 µL des ECD auf A1 in der Zelle geladen werden und 120 µL der Fab auf gut A2 auf der Spritze zu verladen. Auch A3 leer zurück Mischprobe Folgendes experiment abgeschlossen. Jedes nachfolgende Experiment zugesetzt werden, um die Platte in der gleichen Reihenfolge (d. h. experiment 2: Zelle - A4, Spritze - A5, leeren Brunnen - A6; Abbildung 5 ( B).
Hinweis: Enthalten Puffer in Puffer Steuerelemente (zu bestätigen, das Instrument gut benehmen) am Anfang und Ende jeder Lauf sowie Liganden (in Spritze) in Puffer (in Zelle) Kontrollen, die Wärme der Verdünnung der Probe in der Spritze zu berechnen. Der berechnete Wärme der Verdünnung dann experimentelle Rohdaten während der Datenanalyse (Abb. 5B) subtrahiert werden soll. - Führen Sie insgesamt 16 Injektionen mit einem Volumen von 2,5 μL für jede Injektion. Die einspritzdauer ist 5 s, mit 180 s Abstand zwischen den Injektionen. Stellen Sie die Zellentemperatur bis 25 ° C, mit einer mitreißenden Geschwindigkeit von 750 u/min und Filter beträgt 5 s.
Hinweis: Basierend auf der Affinität und Thermodynamik der ECD:Fab Interaktion, kann es erforderlich, die probenkonzentration, Anzahl der Injektionen oder Zellentemperatur zu ändern sein. - Analysieren Sie die Daten mit der Analysesoftware, wie oben beschrieben40,41,43 (Abb. 5B).
- Wiederhole das Experiment zumindest in Duplikate, Berechnung der mittleren K-D -Werte und Standardfehler. Experiment mit ECD von verschiedenen Glycoforms (§ § 5 und 6), die Wirkung, wenn überhaupt, von Glycoforms über die Thermodynamik der Fab: Glykoprotein Interaktion zu wiederholen.
- Richten Sie für Isothermen Titration Kalorimetrie der Liganden-Glykoprotein Interaktionen das ITC-Experiment, wie beschrieben in Abschnitt 12.2 mit folgenden Änderungen:
- Dialyse ECD in 4 L der Dialyse-Puffer über Nacht. Lösen Sie den Liganden mit Dialyse-Puffer nach Abschluss der Dialyse auf.
- Führen Sie ITC Experimente bei deutlich höheren Konzentrationen zu niedrig-Affinität Interaktionen erkennen können. Durchführen Sie für die ECD und Ligand Interaktion ITC Experimente bei Konzentrationen von 100 µM des ECD in der Zelle und 1 mM Liganden in der Spritze.
Ergebnisse
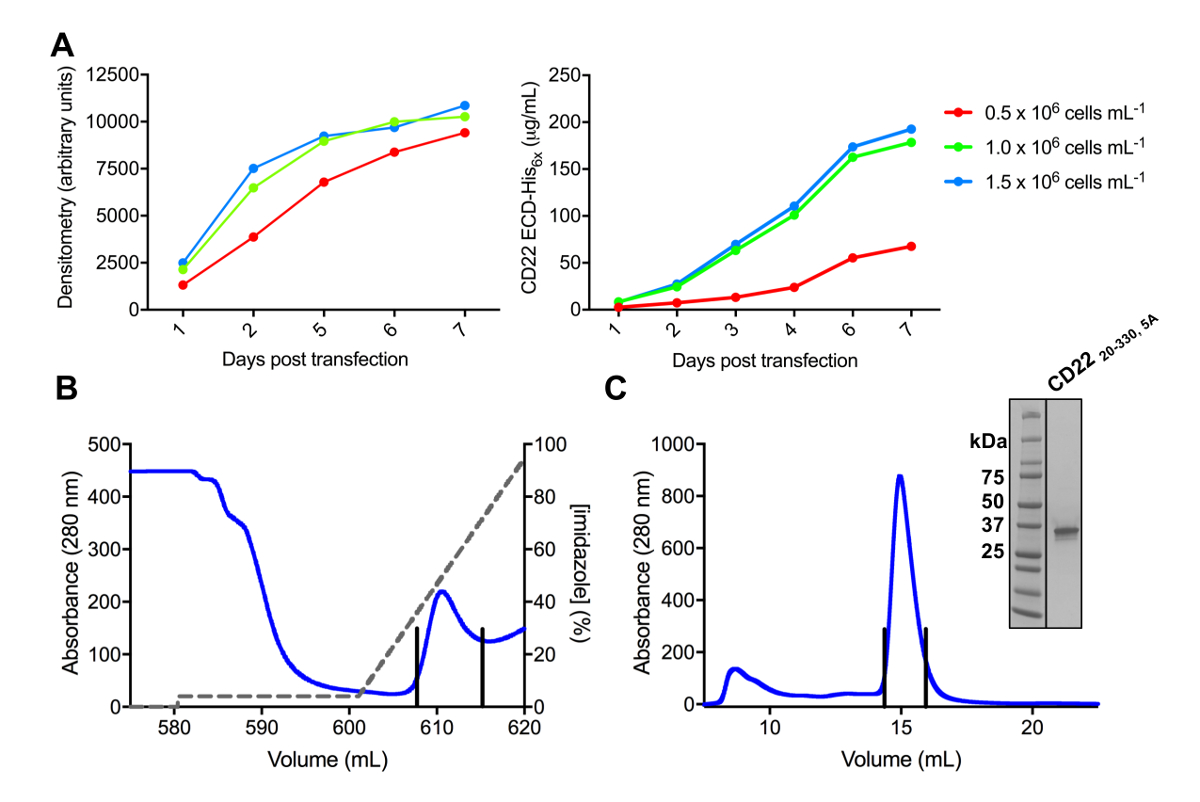
Verschiedene Konstrukte des CD22 ECD waren erfolgreich in der pHLsec Ausdruck Vektor kloniert und überexprimiert in Säugetieren HEK293F und HEK293S Zelllinien (Abbildung 2 und 3A). Alle Konstrukte wurden zur Größe Homogenität durch Größe Ausgrenzung Chromatographie gereinigt und ergab ein hochreines Muster zur Kristallisation Studien (Abb. 3 b und 3 C). Die CD22 Konstrukt, das zu gut diffracting Kristalle geführt war die d1-d3 abschneiden (Rückstände 20-330), mit fünf der sechs vorhergesagten N verbunden Glykosylierung Seiten mutierte vom Asn, Ala (N67A, N112A, N135A, N164A und N231A), in den HEK293S Zellen, so dass nur produziert die Glykosylierung Seite an Position N101 wurde beibehalten (dieses Konstrukt heißt CD2220-330, 5A). Kristalle wurden in mehreren Bedingungen des Bildschirms MCSG-1 dünnbesetzte Matrix erhalten, aber die besten Kristalle wurden von einem Zustand mit 30 % (w/V) Polyethylenglykol 4000, Lithiumchlorid 0,2 M und 0,1 M Tris, pH 8,5. Diese nativen kristalle gebeugt auf 2,1 Å Auflösung; mit bekannten Strukturen der Ig Domänen des zugehörigen Siglec-Proteine Ausbeute nicht keine Lösungen in den Herrn suchen.
Phasenabgleich Informationen erwerben, getränkt wir native Kristalle mit einem Panel von HA-Verbindungen, die Hg, Pt, Os, Ta und Br bei Konzentrationen von 1-20 mM HA Verbindung für eine Inkubationszeit von 5 min bis 1 d (Abbildung 4) enthalten. Wir Kristalle für Änderungen in der Morphologie überwacht, und fanden, dass Kristalle mit zusammengesetzten bei 20 mM HA getränkt führte die schnelle Rissbildung und Auflösen des Kristalls. Wir froren insgesamt 63 Kristalle, die ihrer Form nach festgelegten Inkubationszeiten, die mit Tantal Bromid Cluster, Platin chlorid, Quecksilberoxid Acetat und Quecksilberchlorid getränkt wurden beibehalten. Kristalle mit 7 mM von Quecksilberchlorid für 30 min zeigte anomale Signal auf einem Fluoreszenz-Scan bei der Canadian Light Source (CLS) 08-BM-Strahlrohr (Saskatoon, Kanada) getränkt und erlaubt für Multi-Wellenlänge anomalen Dispersion Röntgen Datenerhebung auf einer einzigen Kristall. Diese Datasets erlaubt uns, die Quecksilber-Unterkonstruktion der CD22 lösen20-330, 5A, die eine einzelne Quecksilber-Atom gebunden, eine kostenlose Cystein an Position C308 und letztendlich konnten wir die Struktur der CD22 bauen ergab20-330, 5A in der stufenweisen Elektron-Dichte-Karte mit AutoBuild78.
Sobald die Unliganded Struktur gelöst war, interessierte uns die Struktur der CD22 verpflichtet, seinen Liganden, α2-6 Siallylactose zu lösen. Wir berechnet zuerst die Affinität der CD22 α2-6 Sialyllactose mit ITC die Bindung Thermodynamik der Interaktion zu charakterisieren. Wir beobachteten eine Affinität von ~ 280 µM und verwendet diese Informationen, um eine Ausgangskonzentration identifizieren (~ 100 x KD) Liganden, zum Einweichen von unseren einheimischen CD2220-330, 5A -Kristalle zu verwenden. Wir CD2220-330, 5A Kristalle mit 25 mM Siallylactose für 5 min, 2 h, 14 h, 40 h und 5D getränkt und auf Änderungen im Kristall Morphologie überwacht. Insgesamt 75 ~ Kristalle wurden von verschiedenen Zeitpunkten eingefroren und an die CLS Synchrotron Strahlrohr 08-ID (Saskatoon, Kanada) für remote-Datenerfassung. Insgesamt sechs Röntgen-Datensätze wurden von gut diffracting Kristalle gesammelt. Die Struktur von jedem x-ray Dataset wurde durch Herrn mit Unliganded CD2220-330, 5A Struktur als eine anfängliche Suche Modell gelöst. Die daraus resultierende Elektronendichte für alle Datensätze wurde dann für positive Dichte in der Fo-Fc-Karte inspiziert, die α2-6 Sialyllactose innerhalb der Bindungsstelle des CD22 gebunden entsprechen würde. Bemerkenswert ist, alle Datensätze gesammelt, auch die von Kristallen getränkt nach nur 5 min von Inkubationszeit enthaltenen positiven Dichte entspricht der Liganden in die Bindungsstelle. Allgemeine Strukturen des Unliganded und der liganded CD22 waren sehr ähnlich mit minimalen Konformationsänderungen, die was den Erfolg der durchnässten Experimente mit α2-6 Sialyllactose erklären könnte.
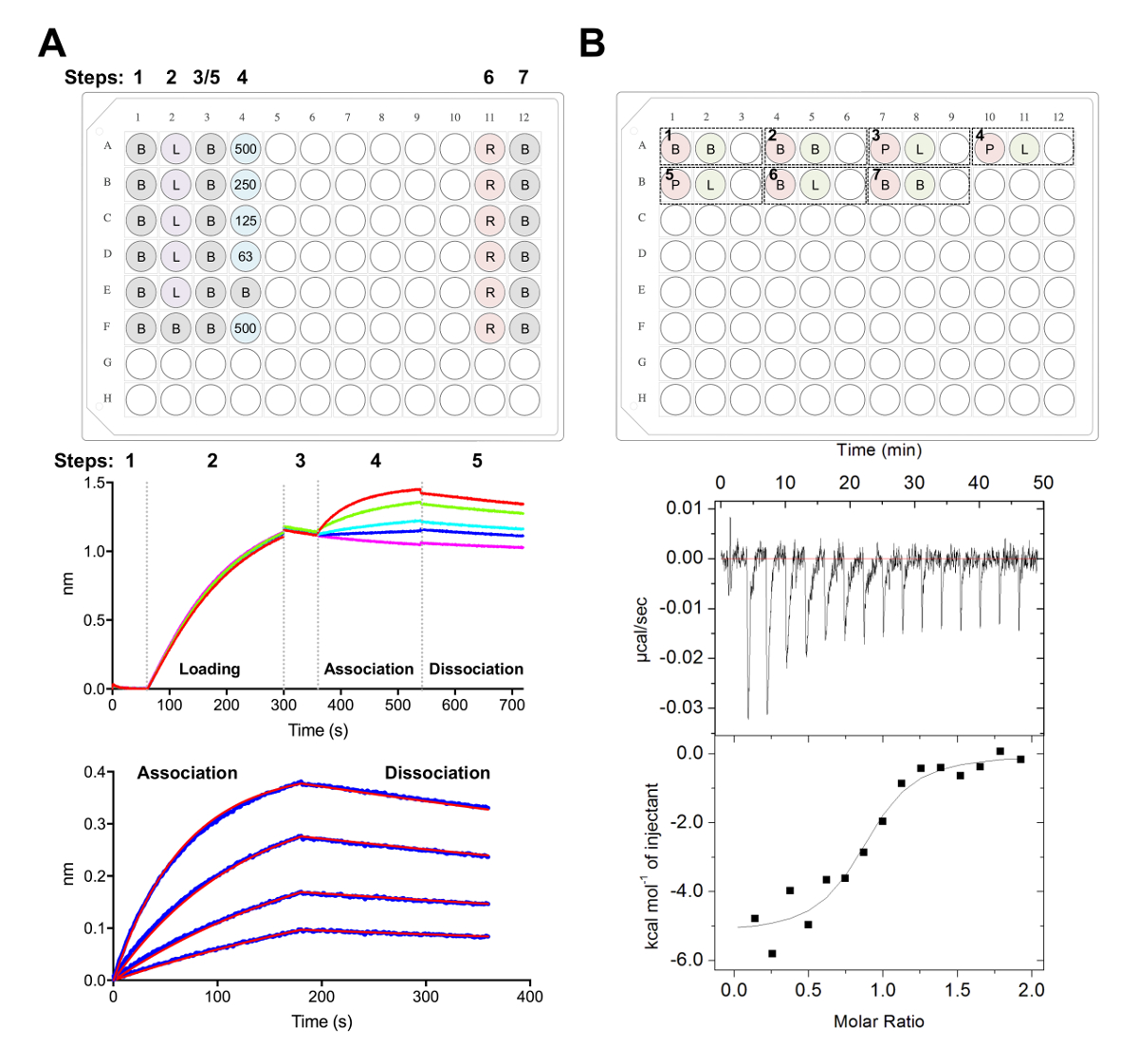
Wir zeichnen als nächstes die Antigene Oberfläche der CD22 von therapeutischen Antikörpern Epratuzumab in BLI und ITC Experimente (Abbildung 5) anerkannt. Kinetik und Thermodynamik profile Epratuzumab Fab Bindung an CD22 Konstrukte mit verschiedenen Glycoforms offenbart eine zunehmende Affinität zu CD22 mit reduzierten N verbunden Glycan Größe mit bis zu einer 14-fold Verbesserung der Affinität für kleinere Glykoproteinen (327 nM Vs 24 nM in BLI; 188 nM Vs 58 nM im ITC). Die CD22 N verbunden Glycan Beschränkung des Zugangs des Antikörpers zu sein Epitop wurde durch BLI mit Einpunkt-Mutanten der CD22 und lösen die Epratuzumab Fab-CD22 d1-d3 Co Kristall Struktur28gekennzeichnet.

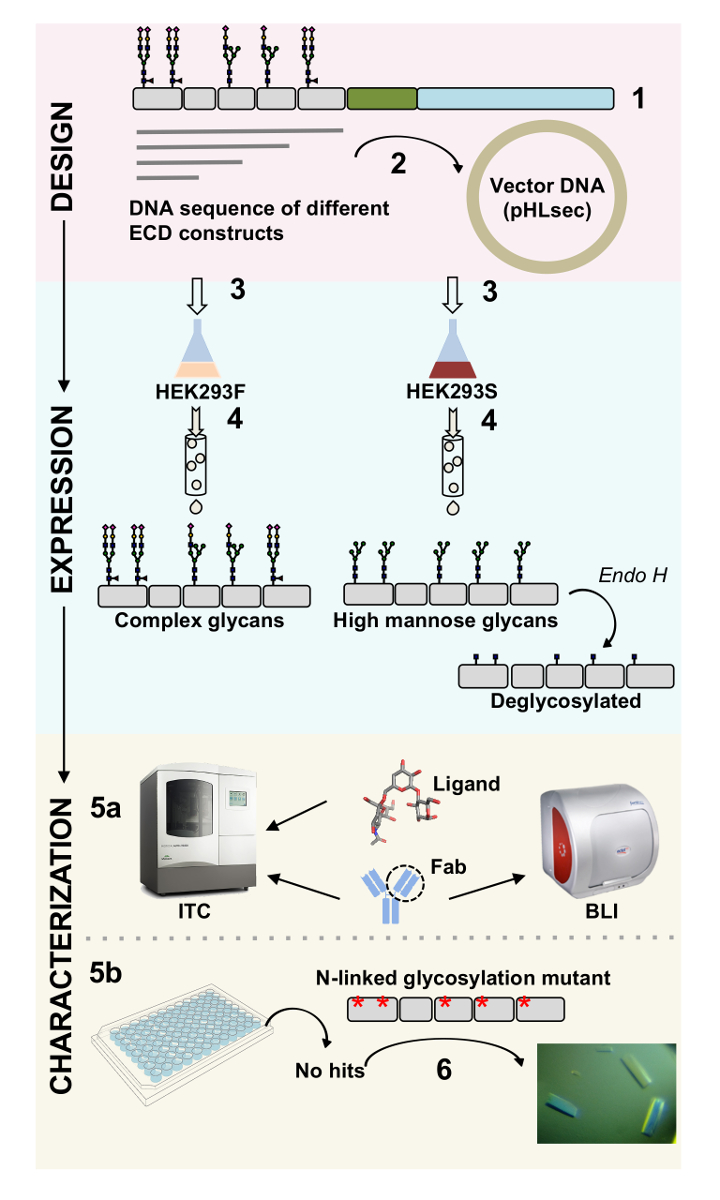
Abbildung 1 . Übersicht der Glykoprotein Charakterisierung von Konstrukt Design auf biophysikalische und strukturelle Charakterisierung. (1) primäre Sequenzanalyse der repräsentativen Glykoprotein. In grau, der extrazellulären Domäne (ECD); in grün, Segment transmembranen (TM); und blau, die cytosolische Domäne das Glykoprotein. Vorhergesagten N verbunden Glykane sind beschriftet. (2) Klonen ECD Konstrukte. (3) Ausdruck der ECD-Konstrukte in Säugerzellen. (4) Glykoprotein Reinigung. Während Proteine in HEK293F komplexen Glykoproteinen enthalten werden, werden Proteine in HEK293S hohe Mannose Glykanen haben. Enzymatische Behandlung von Glykoproteinen in HEK293S Zellen mit Endo H Ergebnisse in Glykoproteinen mit nur GlcNAc Glyko-an N-linked Glykosylierung Standorten produziert. (5a) Glykoproteine sind für ihre Bindung an Antikörper von Biolayer Interferometrie (BLI) und Isotherme Titration Kalorimetrie (ITC) getestet. Affinität zu kleinen Liganden kann auch durch ITC gemessen werden. (5 b) Kristallisation Studien von Glykoproteinen mit homogenen N verbunden Glykane, wie die in HEK293S und Deglycosylated mit Endo H. (6) In einigen Fällen ist Mutation des N-linked Glykosylierung Seiten notwendig, Kristalle zu erhalten. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 2 . Gestaltung von CD22 ektodomäne DNA für Ausdruck in Säugetierzellen baut. A) Darstellung des pHLsec Plasmids für Transiente Transfektion von CD22 ECD Konstrukte verwendet. AgeI und KpnI Sites zum Klonen verwendet werden mit roten Kästchen angezeigt. B) The CD22 ECD enthält sieben Ig Domänen (d1-d7) und 12 vorhergesagten N verbunden Glykosylierung Standorte (in blau). Vier Konstrukte wurden von CD22 ECD entwickelt. C) 1 % Agarose-Gel mit PCR Amplifikate der CD22 ECD baut für Klonen in der Säugetier-Expressionsvektor pHLsec. Erste Spur enthält 1 kb DNA-Marker. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 3 . Expression und Reinigung von Glykoproteinen. (A) Wirkung der Zelldichte Ausdruck Renditen. Glykoprotein Ausdruck in kleinen 25 mL Kultur HEK293F Aussetzung Zellen transfiziert mit drei unterschiedlichen Ausgangspunkt dichten von Zellen (0,5 x 106 Zellen mL-11.0 x 106 Zellen mL-1und 1, 5 x 106 Zellen mL -1). Quantifizierung von Densitometrie von SDS-PAGE im linken Panel und durch quantitative BLI im rechten Bereich durchgeführt. Werte sind repräsentativ für ein Glykoprotein Vorbereitung. B) Chromatogramm der ersten Reinigungsstufe für konstruieren CD2220-330, 5A von 600 mL überstand mit einer Ni-NTA-Affinität-Spalte. Das Glykoprotein war eluiert mit einem Farbverlauf von Imidazol (graue Linie), wo der Elution Puffer, der 500 mM Imidazol enthält 100 % entspricht. Gepoolte Brüche werden durch vertikale Linien dargestellt. C) Größe-Ausschluss Chromatogramm für konstruieren CD2220-330, 5A mit einem Hochleistungs-gel-Filtration-Spalte. Gepoolte Fraktionen aus der Elution Gipfel sind mit vertikalen Linien dargestellt. Einschub: Coomassie gefärbt SDS-PAGE Gel zeigt die Reinheit des das Glykoprotein. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 4 . Einweichen mit schweren Atomen Crystal. (A) Probe Workstation zum Einweichen native Kristalle mit HA-Verbindungen. Alle erforderlichen Werkzeuge gekennzeichnet sind. (B) Schritte befolgt, um Kristalle von Tränken konstruieren CD2220-330, 5A mit HA-Verbindungen. Schritt 1, geöffnet auch mit Kristallen und Transfer-Kristalle mit einer Schleife zu einem Rückgang von 0,2 µL auf einem Deckglas mit HA-Lösung verdünnt in der Kristallisation Zustand, so dass die Endkonzentration von HA von 1-10 mM reicht. Schritt2, versiegeln den Rückgang der Kristallisation Platte und inkubieren Sie Kristalle mit zusammengesetzten für verschiedene Zeiträume HA. Schritt 3, montieren Sie die eingeweichten Kristall in der Schleife und zurück-Tränken für 30 s in drei aufeinander folgenden 0,2 µl Tropfen mit der Mutter-Alkohol-Lösung ergänzt mit 20 % (V/V) Glycerin auf einem Deckglas verzichtet. Schritt 4, Flash den Kristall montiert auf einer Schleife mit flüssigem Stickstoff eingefroren und legen Sie sie in ein Puck für den Versand in das Synchrotron-Strahlrohr. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 5 . Biolayer Interferometrie und Isothermen Titration Kalorimetrie Messungen. (A) Vertreter BLI Experiment. Oberseite: Beispiel der Platte Setup für eine Kinetik-Experiment, wo sind die folgenden gekennzeichnet: 1 X Kinetik Puffer (B), seine6 X-tagged Glykoprotein laden (L), repräsentative Fab Konzentrationen (62,5, 125, 250, 500 nM), PBS + 500 mM Regeneration Puffer-Puffer (R) und 1 x Kinetik Neutralisation (B). Jedes enthält gut 200 µL der Lösung. Schritt wird für die Kinetik-Experiment am oberen Rand der Platte angezeigt. Mitte-Panel: Repräsentative Rohdaten von BLI Experiment mit Ni-NTA Biosensoren durchgeführt und die Platte im oberen Feld beschrieben. Schritt Zahlen entsprechen Grundlinie (1),6 X Glykoprotein laden (2), Basislinie (3), Verband in serielle Verdünnung der Fab (4) und Dissoziation (5). Regeneration-Schritte sind nicht vertreten (Schritte 6 und 7). Bodenplatte: Repräsentative Analysierte Daten zeigen rohen Assoziation und Dissoziation (blaue Linie) mit den entsprechenden 1:1 passen (rote Linie). B) Oberseite: repräsentative Platte Setup für eine einzelne ITC ausführen auf einem automatisierten ITC-Instrument mit sieben Experimente in einer 96-Well Runde Unterflasche. Jedes Experiment besteht aus drei Brunnen. Der erste Brunnen (rot) entspricht Probe für die Zelle (400 µL), der zweite Brunnen (grün) entspricht zum Beispiel für die Spritze (120 µL). Die dritte ist nun leer, und der Mischproben werden dazu auch nach dem Experiment Abschluss zurückgegeben werden. Experimente 1, 2 und 7 sind Puffer in Puffer Steuerelemente. Experimente von 3-5 sind dreifacher Experimente mit Glykoprotein (P) in der Zelle und Fab oder Liganden (L) in der Spritze. Experiment 6 stellt eine Ligand Wärme der Verdünnung Kontrolle und sollte während der Datenanalyse aus Experimenten 3-5 subtrahiert werden. Bodenplatte: Repräsentative roh (oben) und verarbeiteten (unten) ITC Daten zeigen Fab (Epratuzumab) binden an CD22 ECD produziert in HEK293F Zellen. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}
Diskussion
Membran verankerte Glykoproteine sind entscheidend für die Funktion der Zellen und sind attraktive therapeutische Ziele. Hier präsentieren wir ein Protokoll für die strukturelle und biophysikalischen Charakterisierung des ECD Membran Glykoproteine, sowohl allein als auch in komplexen mit niedermolekularen Liganden und Fab-Fragmente. Wir haben erfolgreich dieses Protokoll verwendet, um festzustellen, die Kristallstruktur der drei N-Terminal-die meisten Ig Domänen der extrazellulären Teil des menschlichen CD2228, eine kritische ko-Rezeptor auf B-Zellen, die humorale Immunität Check79zu halten. Wir haben auch die Bindungsstelle des CD22 mit seiner natürlichen Liganden α2-6 Sialyllactose geprägt und definiert die Art der Anerkennung eines therapeutischen Antikörpers gegen menschliche CD22. Diese Ergebnisse geben Einblicke in die Struktur-Funktions-Beziehung der ein wichtiges Mitglied der Siglecs Familie, die Ausdruck auf B-Zellen und eine molekulare Roadmap für die Entwicklung von neuen CD22 gezielte niedermolekularer und Antikörper-basierten beschränkt hat Therapeutika. Während dieses Protokoll für ein Ig-haltigen B-Zell-Rezeptor erfolgreich eingesetzt wurde, schlagen wir vor, dass unser Ansatz für die strukturelle und biophysikalischen Charakterisierung von jede Membran Glykoprotein mit einer unterschiedlichen Domäne Organisation angewendet werden kann. In einem solchen Fall zu konstruieren Design und kombinatorische Glycan N verbunden, die Mutationen (entweder auf Gln oder Ala) ausgewertet werden können, um ein Konstrukt für Kristallzüchtung und hochauflösende Beugung zu finden.
Eine homogene und reine Glykoprotein-Probe ist von entscheidender Bedeutung für Kristallzüchtung und x-ray Diffraction sowie für nachgeschaltete biophysikalische Charakterisierung. N-linked Glykane auf Glykoproteine sind von Natur aus heterogen und kann Konformationsänderungen und chemischen Heterogenität innerhalb der Glykoprotein, das Kristallbildung abhalten kann. Um diese Mikro-Heterogenität zu reduzieren, können Strategien, die Punktmutationen entfernen Asn Rückstände Glykane N verbunden oder mit mutierten Zelllinien (z. B. HEK293S), gefolgt von der Behandlung mit Endoglycosidases (z. B. EndoH) Hafen voraussichtlich einführen erheblich Verbesserung der Kristallisation Erfolg15,21,22. In diesem Protokoll besprechen wir die Reinigung des löslichen Glykoproteine und Fabriken, die in die Zelle überstand abgesondert werden. Glykoprotein Absonderung bietet einen relativ einfachen Weg zur Reinheit, ohne die Notwendigkeit einer Zelle Lyse oder die Zugabe von Chemikalien oder Reinigungsmitteln. Die Zelle überstand, erhalten folgende Zelle Ernte läuft dann direkt über eine Spalte, die Affinität für das Protein des Interesses (z. B. Ni-NTA für sein-tagged Glykoproteine oder LC Affinität für Fab-Fragmente) hat. Jedoch kann abhängig von der Spalte und die Bedingungen der Zelle überstand (z. B. pH-Wert), die verbindliche Fähigkeit des Proteins des Interesses auf die Spalte beeinträchtigt werden. Wenn dies der Fall ist, ist es möglicherweise erforderlich, zu konzentrieren und Austausch der Zelle überstand zur Verbesserung der Bindung an die Spalte zu puffern. Darüber hinaus ist es dringend empfohlen, Qualitätskontrolle Schritte während der Reinigung eingesetzt werden, um Protein Reinheit zu beurteilen. Ausführung einer SDS-PAGE Gel oder Western-Blot aller Proben (vor, während und nach Reinigungsschritte) kann Ausbeute Einblicke, ob die vorgeschlagenen Reinigung-Schema für das Protein des Interesses geeignet ist. Wenn kontaminierenden Bands auf SDS-PAGE zu sehen sind, oder wenn mehrere Arten während der Reinigung erzielt werden (z. B. mehrere Peaks auf Größe Ausschluss), zusätzliche Reinigungsschritte sollte berücksichtigt werden, z. B. Ionenaustausch-Chromatographie, um zu gewinnen in Reinheit und Erhöhung der Chancen der nachgeschalteten Kristallisation80.
Für Makromolekulare Kristallisation ist es oft wichtig, des Proteins des Interesses zuzulassen für das Screening von eine große Anzahl von möglichen Kristallisation Bedingungen bei hoher proteinkonzentrationen geeignet Kristall Treffer finden hohe Erträge zu erzielen. In der Regel die HEK293 Zelllinien hier diskutiert (HEK293F und HEK293S) sind robuste Expressionssysteme, und können leicht ausgeweitet werden, um mehr von der Probe nach Bedarf zu produzieren. Es ist jedoch möglich, dass das Protein des Interesses innerhalb dieser Zelllinien nicht ausreichend Ausdrücken kann. In diesen Fällen andere Zelllinien, wie Expi293 Zellen81,82, übergeordneten Ebenen der Proteinexpression zeigen gefunden worden und sollte als Alternative in Betracht.
Wenn wohlgeordneten, interferenzgitters Kristalle nicht nach Prüfung der mehrere Konstrukte des Proteins des Interesses trotz hoher Reinheit erhalten werden, es möglicherweise notwendig, Kristallisation Techniken zur Förderung der Kristallbildung zu erweitern. Es hat sich gezeigt, dass Fab-Fragmente von Antikörpern und Nanobodies ausgezeichnete Kristallisation Enhancer werden und wohlgeordneten Kristall Verpackung83,84,85fördern können. Diese Fragmente können zum Ausdruck und zur Homogenität gereinigt und in einem Komplex mit dem Protein des Interesses zur Kristallisation zu fördern. Wichtig ist, können Fab-Fragmente produziert wie in Abschnitt 10 beschrieben haben eine Tendenz zu nicht-funktionale LC Dimere86bilden. Diese Dimere sind Verunreinigungen und sollte bei der Reinigung entfernt werden. Nach unserer Erfahrung LC Dimere oft haben eine unterschiedliche Retentionsvolumen Größe Ausschluss, oder als eine unterschiedliche Spitze auf Ionenaustausch-Chromatographie eluieren, und so können aus der Fab Reinigung - entfernt werden, dies ist jedoch nicht immer der Fall. Wenn diese Techniken nicht ausreicht, um LC Dimere aus der Fab Reinigung zu entfernen sind, können zusätzliche Reinigungsverfahren wie Eiweiß G Affinitätsreinigung, eingesetzt werden, um Reinheit zu verbessern.
Alternative zu co-Komplexierung mit Fab-Fragmente, können gut dokumentierte Techniken wie zufällige Matrix Microseeding Chancen auf geordnete Kristalle63,70verbessern. Bei dieser Methode wird der Zusatz von geringen Mengen von zerkleinerten, suboptimale Kristalle in der Kristallisation Zustand, Bereitstellung eines Kristalls blasensiedens Kristallwachstum zu fördern. Dies kann mit Kristallen des Proteins des Interesses oder mit ähnlichen Domain-Architektur und tertiäre Struktur durchgeführt werden. Darüber hinaus kann zufällige Matrix Microseeding versucht, das Protein allein kristallisieren oder im Komplex mit einem Fab-Fragment oder kleine Molekül des Interesses durchgeführt werden. Jüngste Fortschritte in der Kryo-Elektronenmikroskopie machen diese Technik auch eine attraktive Alternative zum Röntgen-Kristallographie für hochauflösende strukturelle Informationsbeschaffung für Moleküle mit entsprechenden Features87,88, 89,90,91.
Beim Ausstieg der Röntgenbeugung Datasets von Herrn fehlschlägt, kann HA Einweichen zur Lösung des Problems der Phase von anomalen Dispersion oder isomorphous Ersatz benötigt werden. Inspektion der Aminosäure-Sequenz des Proteins liefern Hinweise auf die Strategie für HA Derivatisierung, einschließlich der optimale pH-Wert für die Bindung. Insbesondere können ungepaarte Cysteine innerhalb des Proteins speziell HA Verbindungen binden, die Quecksilber enthalten. Einweichen native Kristalle mit HA-Verbindungen ist ein iterativer Prozess, die Identität der optimale HA Verbindung, seine Konzentration und die erforderlichen Inkubationszeit zu bestimmen. Wenn erste Einweichen Versuche gut diffracting Kristalle, enthält eine HA für den Ausstieg nicht nachgeben, ist es möglicherweise erforderlich, Aminosäure Auswechslungen um die Wahrscheinlichkeit der HA Bindung verbessern und anomale Signal einführen. Beispiele dafür sind Mutationen zu umfassen einen kostenlosen Cystein Rückstand um effizient Hg, Au, Pt oder Pb. Expression von Proteinen binden für anomale phasing-in einem Seleno-Methionin ergänzt Medien in E. Coli anomale Phasing, ausgiebig genutzt wird jedoch eine gleichwertiges System, das zuverlässig Seleno-Methionin enthält ist nicht verfügbar für Säugetier-Zellen in Suspension92,93, und ist ein Bereich der zukünftigen Entwicklung.
Sobald die Unliganded Struktur der das Glykoprotein Interesse vorliegt, kann die Kristalle mit niedermolekularen Liganden Einweichen durchgeführt werden, um eine Struktur der immun-Rezeptor-Ligand-Komplex erhalten. Diese Daten liefern eine Blaupause für die rationale Gestaltung spezifischer und hoher Affinität Liganden, die eignet sich als kleine Molekül Therapeutika sowie hochauflösende Einblicke in die biologische Funktion von das Glykoprotein. Beim Versuch, Glykoprotein Kristalle mit niedermolekularen Liganden von Interesse zu genießen, kann Inspektion der Kristallstruktur Unliganded angeben, ob Einweichen sollte möglich sein. Wenn nahe Kristall-Verpackung Kontakte befinden sich rund um die Liganden-Bindungsstelle oder ganzen Regionen voraussichtlich Konformationsänderungen auf Liganden verbindlich zu unterziehen, einweichen dürfte problematisch sein. In diesem Fall sollten andere Methoden wie Co Kristallisation von der Protein-Ligand-komplex durchgeführt werden.
Offenlegungen
Die Autoren erklären keinen Interessenkonflikt.
Danksagungen
Röntgenbeugung Experimente, die in diesem Dokument beschriebenen wurden durchgeführt mit Strahllinien 08-ID und 08-BM am Canadian Light Source, die für Innovation, Naturwissenschaften und Engineering Research Council of Canada Kanada-Stiftung unterstützt wird, die Universität von Saskatchewan, die Regierung von Saskatchewan, westliche ökonomische Diversifikation Kanada, des National Research Council Canada und der Canadian Institute der Gesundheitsforschung. Wir möchten die Struktur- & biophysikalische Core Facility, The Hospital für kranke Kinder, für den Zugriff auf die ITC und BLI Instrumente erkennen. J.E.O. wurde von Banting Postdoctoral Fellowship BPF-144483 von Canadian Institutes of Health Research unterstützt. T.S ist ein Empfänger von Kanada Graduate Stipendium Master Award und einem Vanier Canada Graduate Stipendium Canadian Institutes of Health Research. Diese Arbeit wurde unterstützt durch Zuschuss PJT-148811 (J.-p.j.) von der Canadian Institutes of Health Research in Betrieb. Diese Forschung wurde unter anderem dank der Mittel aus dem Programm Kanada Forschung Stühle (J.-p.j.)durchgeführt.
Materialien
| Name | Company | Catalog Number | Comments |
| 0.22 μm Steritop filter | EMD Millipore | SCGPS02RE | |
| 10 well 4-15% gradient SDS-PAGE gel | Bio-Rad | 4561084 | |
| 10x glycobuffer 3 | New England Biolabs | P0702S | Comes with Endo H reagent |
| 10x Kinetics Buffer | PALL FortéBio | 18-1092 | |
| 10x Tris/Glycine/SDS Buffer | Bio-Rad | 1610732 | |
| 1 mL round bottom 96 well block | ThermoFisher | 260251 | |
| 22 mm cover slip | Hampton research | HR3-231 | |
| 4x Laemmli Sample Buffer | Bio-Rad | 1610747 | |
| 96-3 well INTELLIPLATE low volume reservior | Art Robbins Instruments | 102-0001-03 | |
| AgeI | New England Biolabs | R0552S | |
| ÄKTA Pure | GE Healthcare | ||
| ÄKTA Start | GE Healthcare | ||
| Amicon Ultra 15 centrifugal filtration device 10KDa MWCO | Millipore | UFC901008 | |
| Amicon Ultra 4 centrifugal filtration device 10KDa MWCO | Millipore | UFC801008 | |
| Auto-iTC200 | Malvern | ||
| Axygen MaxyClear Snaplock 1.5 mL microtubes | Fisher Scientific | MCT150C | |
| Countess Cell Counting Chamber Slides | Thermo Fisher Scientific | C10228 | |
| CryoLoop 18 x 0.05-0.1 mm | Hampton research | HR4-945 | |
| CryoLoop 18 x 0.1-0.2 mm | Hampton research | HR4-947 | |
| CryoLoop 18 x 0.2-0.3 mm | Hampton research | HR4-970 | |
| Digital Dry Bath | Bio-Rad | 1660562EDU | |
| E. coli DH5α | Invitrogen | 18258012 | |
| Endo H | New England Biolabs | P0702S | |
| Erlenmeyer flask (baffled base), polycarbonate, sterile, 500 mL, DuoCAP | TriForest Labware | FBC05000S | |
| Erlenmeyer flask 125 mL (baffled base), polycarbonate, sterile, 125 mL with vented cap | VWR | 89095-258 | |
| Falcon Disposable sterile serological pipet, non-pyrogenic, 10 mL | Greiner Bio-One | 607180 | |
| Falcon Disposable sterile serological pipet, non-pyrogenic, 25 mL | Greiner Bio-One | 760180 | |
| Falcon Disposable sterile serological pipet, non-pyrogenic, 5 mL | Greiner Bio-One | 606180 | |
| Falcon Disposable sterile serological pipet, non-pyrogenic, 50 mL | Greiner Bio-One | 768180 | |
| FectoPRO DNA Transfection Reagent, Polyplus | VWR | 10118-842 | |
| Freestyle 293F cells | Thermo Fisher Scientific | R79007 | |
| Freestyle Expression medium | Thermo Fisher Scientific | 12338001 | |
| Heavy Atom Screens Au | Hampton research | HR2-444 | |
| Heavy Atom Screens Hg | Hampton research | HR2-446 | |
| Heavy Atom Screens M1 | Hampton research | HR2-448 | |
| Heavy Atom Screens M2 | Hampton research | HR2-450 | |
| Heavy Atom Screens Pt | Hampton research | HR2-442 | |
| HEK 293S | ATCC | ATCC CRL-3022 | |
| HisTrap Affinity Column | GE Healthcare | 17525501 | |
| HiTrap KappaSelect Affinity Columns | GE Healthcare | 17545811 | |
| HiTrap LambdaSelect Affinity Columns | GE Healthcare | 17548211 | |
| KpnI | New England Biolabs | R0142S | |
| MCSG-1 Crystal Screen 1.7 mL block | Anatrace | MCSG-1 | |
| MCSG-2 Crystal Screen 1.7 mL block | Anatrace | MCSG-2 | |
| MCSG-3 Crystal Screen 1.7 mL block | Anatrace | MCSG-3 | |
| MCSG-4 Crystal Screen 1.7 mL block | Anatrace | MCSG-4 | |
| Mercuric chloride | Sigma | 1044170100 | |
| Microplate, 96 well, polypropelene, flat bottom, black | Greiner Bio-One | 655209 | |
| Minstrel DT UV | Formulatrix | ||
| Multitron Pro shaker | Infors HT | MP25-TA-CO2HB | |
| Nanodrop 2000/2000c Spectrophotometer | Thermo Fisher Scientific | ND-2000 | |
| Nanosep 3K Omega centrifugal device | PALL Life Science | OD003C33 | |
| Ni-NTA biosensors | PALL FortéBio | 18-5102 | |
| Octet RED96 | PALL ForteBio | ||
| Oryx 4 crystallizaiton robot | Douglas Instrument | ORY-4/1 | |
| Platinum chloride | Sigma | 520632-1g | |
| Precision Plus Protein Standard | Bio-Rad | 161-0374 | |
| PureLink HiPure Plasmid Maxiprep Kit | Invitrogen | K210006 | |
| Quick Coomassie Stain | Protein Ark | GEN-QC-STAIN-1L | |
| Steriflip Sterile 50 mL Disposable Vacuum Filtration System 0.22 µm Millipore Express | EMD Millipore | SCGP00525 | |
| Superdex 200 Increase 10/300 GL | GE Healthcare | 28990944 | |
| Superose 6 10/300 GL | GE Healthcare | 17517201 | |
| Tantalum bromide cluster | Jena bioscience | PK-103 | |
| Top96 Crystallization Screen | Rigaku Reagents | 1009846 | |
| Tryphan Blue | Thermo Fisher Scientific | T10282 | |
| VDX 24-well with sealant | Hampton research | HR3-172 | |
| α2-6 sialyllactose | Sigma Aldrich | A8556-1mg |
Referenzen
- Sachs, J. N., Engelman, D. M. Introduction to the membrane protein reviews: The interplay of structure, dynamics, and environment in membrane protein function. Annu Rev Biochem. 75 (1), 707-712 (2006).
- Cournia, Z., et al. Membrane protein structure, function, and dynamics: A perspective from experiments and theory. J Membr Biol. 248 (4), 611-640 (2015).
- Macauley, M. S., et al. Antigenic liposomes displaying CD22 ligands induce antigen-specific B cell apoptosis. J Clin Invest. 123 (7), 3074-3083 (2013).
- Hyde, C. A. C., et al. Targeting extracellular domains D4 and D7 of vascular endothelial growth factor receptor 2 reveals allosteric receptor regulatory sites. Mol Cell Biol. 32 (19), 3802-3813 (2012).
- Tai, W., Mahato, R., Cheng, K. The role of HER2 in cancer therapy and targeted drug delivery. J Control Release. 146 (3), 264-275 (2010).
- Zarei, O., Benvenuti, S., Ustun-Alkan, F., Hamzeh-Mivehroud, M., Dastmalchi, S. Strategies of targeting the extracellular domain of RON tyrosine kinase receptor for cancer therapy and drug delivery. J Cancer Res Clin Oncol. 142 (12), 2429-2446 (2016).
- Rosman, Z., Shoenfeld, Y., Zandman-Goddard, G. Biologic therapy for autoimmune diseases: an update. BMC Med. 11 (1), 88(2013).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Barclay, A. N. Membrane proteins with immunoglobulin-like domains - A master superfamily of interaction molecules. Semin Immunol. 15 (4), 215-223 (2003).
- Barclay, A. N. Ig-like domains: evolution from simple interaction molecules to sophisticated antigen recognition. Proc Natl Acad Sci. 96 (26), 14672-14674 (1999).
- Aebi, M. N-linked protein glycosylation in the ER. Biochim Biophys Acta - Mol Cell Res. 1833 (11), 2430-2437 (2013).
- Ohtsubo, K., Marth, J. D. Glycosylation in cellular mechanisms of health and disease. Cell. 126 (5), 855-867 (2006).
- Lodish, H., Berk, A., Zipursky, S., Al, E. Glycosylation in the ER and Golgi complex. Mol Cell Biol. (4), Section 17.7 (2000).
- Thomas, P., Smart, T. G. HEK293 cell line: A vehicle for the expression of recombinant proteins. J Pharmacol Toxicol Methods. 51 (3), 187-200 (2005).
- Lee, J. E., Fusco, M. L., Ollmann Saphire, E. An efficient platform for screening expression and crystallization of glycoproteins produced in human cells. Nat Protoc. 4 (4), 592-604 (2009).
- Betenbaugh, M. J., Tomiya, N., Narang, S., Hsu, J. T. A., Lee, Y. C. Biosynthesis of human-type N-glycans in heterologous systems. Curr Opin Struct Biol. 14 (5), 601-606 (2004).
- Yang, Z., et al. Engineered CHO cells for production of diverse, homogeneous glycoproteins. Nat Biotechnol. 33 (8), 842-844 (2015).
- Bláha, J., Kalousková, B., Skořepa, O., Pažický, S., Novák, P., Vaněk, O. High-level expression and purification of soluble form of human natural killer cell receptor NKR-P1 in HEK293S GnTI-cells. Protein Expr Purif. 140, 36-43 (2017).
- Bláha, J., Pachl, P., Novák, P., Vaněk, O. Expression and purification of soluble and stable ectodomain of natural killer cell receptor LLT1 through high-density transfection of suspension adapted HEK293S GnTI- cells. Protein Expr Purif. 109, 7-13 (2015).
- Chaudhary, S., Pak, J. E., Gruswitz, F., Sharma, V., Stroud, R. M. Overexpressing human membrane proteins in stably transfected and clonal human embryonic kidney 293S cells. Nat Protoc. 7 (3), 453-466 (2012).
- Chang, V. T., et al. Glycoprotein structural genomics: Solving the glycosylation problem. Structure. 15 (3), 267-273 (2007).
- Davis, S. J., Crispin, M. Solutions to the glycosylation problem for low- and high-throughput structural glycoproteomics. Funct Struct Proteomics Glycoproteins. , 127-158 (2011).
- Elbein, A. D., Tropea, J. E., Mitchell, M., Kaushal, G. P. Kifunensine, a potent inhibitor of the glycoprotein processing mannosidase I. J Biol Chem. 265 (26), 15599-15605 (1990).
- Zheng, K., Bantog, C., Bayer, R. The impact of glycosylation on monoclonal antibody conformation and stability. MAbs. 3 (6), 568-576 (2011).
- Aricescu, A. R., Lu, W., Jones, E. Y. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr Sect D Biol Crystallogr. 62 (10), 1243-1250 (2006).
- Adams, P. D., et al. The Phenix software for automated determination of macromolecular structures. Methods. 55 (1), 94-106 (2011).
- May, A. P., Robinson, R. C., Vinson, M., Crocker, P. R., Jones, E. Y. Crystal structure of the N-terminal domain of sialoadhesin in complex with 3' sialyllactose at 1.85 Å resolution. Mol Cell. 1 (5), 719-728 (1998).
- Ereño-Orbea, J., et al. Molecular basis of human CD22 function and therapeutic targeting. Nat Commun. 8 (1), 764(2017).
- Yu, X. -L., et al. Crystal structure of HAb18G/CD147: implications for immunoglobulin superfamily homophilic adhesion. J Biol Chem. 283 (26), 18056-18065 (2008).
- Garman, E., Murray, J. W. Heavy-atom derivatization. Acta Crystallogr - Sect D Biol Crystallogr. 59 (11), 1903-1913 (2003).
- Agniswamy, J., Joyce, M. G., Hammer, C. H., Sun, P. D. Towards a rational approach for heavy-atom derivative screening in protein crystallography. Acta Crystallogr Sect D Biol Crystallogr. 64 (4), 354-367 (2008).
- Rose, J. P., Wang, B. C., Weiss, M. S. Native SAD is maturing. IUCrJ. 2 (20), 431-440 (2015).
- Olieric, V., et al. Data-collection strategy for challenging native SAD phasing. Acta Crystallogr Sect D Struct Biol. 72 (3), 421-429 (2016).
- Rillahan, C. D., et al. Disubstituted sialic acid ligands targeting Siglecs CD33 and CD22 associated with myeloid leukaemias and B cell lymphomas. Chem Sci. 5 (6), 2398-2406 (2014).
- Mesch, S., et al. From a library of MAG antagonists to nanomolar CD22 ligands. ChemMedChem. 7 (1), 134-143 (2012).
- Chiu, M. L., Gilliland, G. L. Engineering antibody therapeutics. Curr Opin Struct Biol. 38, 163-173 (2016).
- Elgundi, Z., Reslan, M., Cruz, E., Sifniotis, V., Kayser, V. The state-of-play and future of antibody therapeutics. Adv Drug Deliv Rev. 122 (2016), 2-19 (2017).
- Yang, D., Singh, A., Wu, H., Kroe-Barrett, R. Determination of high-affinity antibody-antigen binding kinetics using four biosensor platforms. J Vis Exp. (122), e55659(2017).
- Kamat, V., Rafique, A. Designing binding kinetic assay on the bio-layer interferometry (BLI) biosensor to characterize antibody-antigen interactions. Anal Biochem. 536, 16-31 (2017).
- Brautigam, C. A., Zhao, H., Vargas, C., Keller, S., Schuck, P. Integration and global analysis of isothermal titration calorimetry data for studying macromolecular interactions. Nat Protoc. 11 (5), 882-894 (2016).
- Duff, M. R., Grubbs, J., Howell, E. E. Isothermal titration calorimetry for measuring macromolecule-ligand affinity. J Vis Exp. (55), e2796(2011).
- Livingstone, J. R. Antibody characterization by isothermal titration calorimetry. Nature. 384 (6608), 491-492 (1996).
- Freyer, M. W., Lewis, E. A. Isothermal titration calorimetry: Experimental design, data analysis, and probing macromolecule/ligand binding and kinetic interactions. Methods Cell Biol. 84, 79-113 (2008).
- Macauley, M. S., Crocker, P. R., Paulson, J. C. Siglec-mediated regulation of immune cell function in disease. Nat Rev Immunol. 14 (10), 653-666 (2014).
- Zaccai, N. R., et al. Structure-guided design of sialic acid-based Siglec inhibitors and crystallographic analysis in complex with sialoadhesin. Structure. 11 (5), 557-567 (2003).
- Pantophlet, R., et al. Bacterially derived synthetic mimetics of mammalian oligomannose prime antibody responses that neutralize HIV infectivity. Nat Commun. 8 (1), 1601(2017).
- Leonard, J. P., et al. Epratuzumab, a humanized anti-CD22 antibody, in aggressive non-Hodgkin's lymphoma: phase I/II clinical trial results. Clin Cancer Res. 10 (16), 5327-5334 (2004).
- Finn, R. D., et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 45, 190-199 (2017).
- Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., Sternberg, M. J. E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 10 (6), 845-858 (2015).
- Lessard, J. C. Molecular cloning. Methods Enzymol. 529, 85-98 (2013).
- Gupta, R., Jung, E., Brunak, S. NetNGlyc: Prediction of N-glycosylation sites in human proteins. , (2004).
- Liu, H., Naismith, J. H. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 8, 91(2008).
- Heckman, K. L., Pease, L. R. Gene splicing and mutagenesis by PCR-driven overlap extension. Nat Protoc. 2 (4), 924-932 (2007).
- Froger, A., Hall, J. E. Transformation of plasmid DNA into E. coli using the heat shock method. J Vis Exp. (6), e253(2007).
- Akula, I., Julien, J. -P. Optimization of glycoprotein expression by transient transfection in HEK293 F/S suspension cells. , Available from: https://www.polyplus-transfection.com/wp-content/uploads/2015/09/FectoPRO-Technical-Note-031716.pdf (2015).
- Taylor, S. C., Berkelman, T., Yadav, G., Hammond, M. A defined methodology for reliable quantification of western blot data. Mol Biotechnol. 55 (3), 217-226 (2013).
- Tan, H. Y., Ng, T. W. Accurate step wedge calibration for densitometry of electrophoresis gels. Opt Commun. 281 (10), 3013-3017 (2008).
- Gassmann, M., Grenacher, B., Rohde, B., Vogel, J. Quantifying Western blots: pitfalls of densitometry. Electrophoresis. 30 (11), 1845-1855 (2009).
- Jonnalgadda, K., Markley, L., Estes, S., Prajapati, S., Takkar, R., Kumaraswamy, S. Rapid, reliable quantitation of Fc-fusion protein in cell culture supernatants. , Available from: https://www.fortebio.com/documents/ForteBio_App_Note_13.pdf (2018).
- JoVE Science Education Database. Basic methods in cellular and molecular biology: Separating protein with SDS-PAGE. J Vis Exp. , (2018).
- Wilkins, M. R., et al. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol. 112, 531-552 (1999).
- Till, M., et al. Improving the success rate of protein crystallization by random microseed matrix screening. J Vis Exp. (78), e50548(2013).
- Obmolova, G., Malia, T. J., Teplyakov, A., Sweet, R., Gilliland, G. L. Promoting crystallization of antibody-antigen complexes via microseed matrix screening. Acta Crystallogr Sect D Biol Crystallogr. 66 (8), 927-933 (2010).
- D'Arcy, A., Bergfors, T., Cowan-Jacob, S. W., Marsh, M. Microseed matrix screening for optimization in protein crystallization: What have we learned. Acta Crystallogr Sect F, Struct Biol Commun. 70 (9), 1117-1126 (2014).
- Luft, J. R., et al. Efficient optimization of crystallization conditions by manipulation of drop volume ratio and temperature. Protein Sci. 16 (4), 715-722 (2007).
- Dessau, M. A., Modis, Y. Protein crystallization for X-ray crystallography. J Vis Exp. (47), e2285(2011).
- Sugahara, M., Asada, Y., Ayama, H., Ukawa, H., Taka, H., Kunishima, N. Heavy-atom Database System: A tool for the preparation of heavy-atom derivatives of protein crystals based on amino-acid sequence and crystallization conditions. Acta Crystallogr D Biol Crystallogr. 61 (9), 1302-1305 (2005).
- Boggon, T. J., Shapiro, L. Screening for phasing atoms in protein crystallography. Structure. 8 (7), 143-149 (2000).
- Vera, L., Stura, E. A. Strategies for protein cryocrystallography. Cryst Growth Des. 14 (2), 427-435 (2014).
- Pichlo, C., Montada, A. A., Schacherl, M., Baumann, U. Production, crystallization and structure determination of C. difficile PPEP-1 via microseeding and Zinc-SAD. J Vis Exp. (118), e55022(2016).
- Leslie, A. G. W., et al. Automation of the collection and processing of X-ray diffraction data - a generic approach. Acta Crystallogr Sect D Biol Crystallogr. 58 (11), 1924-1928 (2002).
- Pike, A. C. W., Garman, E. F., Krojer, T., Von Delft, F., Carpenter, E. P. An overview of heavy-atom derivatization of protein crystals. Acta Crystallogr Sect D Struct Biol. 72 (3), 303-318 (2016).
- Cooper, D. R., Porebski, P. J., Chruszcz, M., Minor, W. X-ray crystallography: Assessment and validation of protein-small molecule complexes for drug discovery. Expert Opin Drug Discov. 6 (8), 771-782 (2011).
- Hassell, A. M., et al. Crystallization of protein-ligand complexes. Acta Crystallogr Sect D Biol Crystallogr. 63 (1), 72-79 (2006).
- Muller, I., et al. Guidelines for the successful generation of protein-ligand complex crystals. Acta Crystallogr Sect D Struct Biol. 73 (2), 79-92 (2017).
- Zhao, Y., et al. Two routes for production and purification of Fab fragments in biopharmaceutical discovery research: Papain digestion of mAb and transient expression in mammalian cells. Protein Expr Purif. 67 (2), 182-189 (2009).
- Shah, N. B., Duncan, T. M. Bio-layer interferometry for measuring kinetics of protein-protein interactions and allosteric ligand effects. J Vis Exp. (84), e51383(2014).
- Terwilliger, T. C., et al. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr Sect D Biol Crystallogr. 64 (1), 61-69 (2008).
- Walker, J. A., Smith, K. G. C. CD22: An inhibitory enigma. Immunology. 123 (3), 314-325 (2008).
- Gräslund, S., et al. Protein production and purification. Nat Methods. 5 (2), 135-146 (2008).
- Jain, N. K., et al. A high density CHO-S transient transfection system: Comparison of ExpiCHO and Expi293. Protein Expr Purif. 134, 38-46 (2017).
- Fang, X. T., Sehlin, D., Lannfelt, L., Syvänen, S., Hultqvist, G. Efficient and inexpensive transient expression of multispecific multivalent antibodies in Expi293 cells. Biol Proced Online. 19 (1), 11(2017).
- Löw, C., et al. Nanobody mediated crystallization of an archeal mechanosensitive channel. PLoS One. 8 (10), 77984(2013).
- Hunte, C., Michel, H. Crystallization of membrane proteins mediated by antibody fragments. Curr Opin Struct Biol. 12 (4), 503-508 (2002).
- Ereño-Orbea, J., Sicard, T., Cui, H., Carson, J., Hermans, P., Julien, J. -P. Structural basis of enhanced crystallizability induced by a molecular chaperone for antibody antigen-binding fragments. J Mol Biol. 430 (3), 322-336 (2018).
- Spooner, J., et al. Evaluation of strategies to control Fab light chain dimer during mammalian expression and purification: A universal one-step process for purification of correctly assembled Fab. Biotechnol Bioeng. 112 (7), 1472-1477 (2015).
- Elmlund, D., Le, S. N., Elmlund, H. High-resolution cryo-EM: The nuts and bolts. Curr Opin Struct Biol. 46, 1-6 (2017).
- Merk, A., et al. Breaking cryo-EM resolution barriers to facilitate drug discovery. Cell. 165 (7), 1698-1707 (2016).
- Bai, X. C., McMullan, G., Scheres, S. H. W. How cryo-EM is revolutionizing structural biology. Trends Biochem Sci. 40 (1), 49-57 (2015).
- Wlodawer, A., Li, M., Dauter, Z. High-resolution cryo-EM maps and models: A crystallographer's perspective. Structure. 25 (10), 1589-1597 (2017).
- Bartesaghi, A., et al. 2.2 Å resolution cryo-EM structure of β-galactosidase in complex with a cell-permeant inhibitor. Science. 348 (6239), 1147-1151 (2015).
- Hendrickson, W. a, Horton, J. R., LeMaster, D. M. Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): A vehicle for direct determination of three-dimensional structure. EMBO J. 9 (5), 1665-1672 (1990).
- Walden, H. Selenium incorporation using recombinant techniques. Acta Crystallogr Sect D Biol Crystallogr. 66 (4), 352-357 (2010).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten