Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Combinación de hibridación in situ de fluorescencia múltiple con inmunohistoquímica fluorescente en secciones de cerebro de ratón frescas, congeladas o fijas
En este artículo
Resumen
Este protocolo describe un método para combinar la hibridación fluorescente in situ (FISH) y la inmunohistoquímica de fluorescencia (IHC) en secciones de cerebro de ratón frescas congeladas y fijas, con el objetivo de lograr una señal de FISH y IHC de fluorescencia multimarca. IHQ se dirigió a proteínas citoplasmáticas y unidas a la membrana.
Resumen
La hibridación fluorescente in situ (FISH) es una técnica molecular que identifica la presencia y distribución espacial de transcritos específicos de ARN dentro de las células. El fenotipado neuroquímico de neuronas identificadas funcionalmente suele requerir un marcaje concurrente con múltiples anticuerpos (proteína diana) mediante inmunohistoquímica (IHQ) y la optimización de la hibridación in situ (ARN dirigido), en conjunto. Se puede lograr una "firma neuroquímica" para caracterizar neuronas particulares, sin embargo, los factores que complican la situación incluyen la necesidad de verificar los objetivos de FISH e IHQ antes de combinar los métodos, y el número limitado de ARN y proteínas que pueden ser atacados simultáneamente dentro de la misma sección de tejido.
Aquí describimos un protocolo, utilizando preparaciones cerebrales de ratón frescas congeladas y fijadas, que detecta múltiples ARNm y proteínas en la misma sección cerebral utilizando RNAscope FISH seguido de inmunotinción de fluorescencia, respectivamente. Utilizamos el método combinado para describir el patrón de expresión de ARNm de baja abundancia (p. ej., receptor de galanina 1) y ARNm de alta abundancia (p. ej., transportador de glicina 2), en núcleos de tronco encefálico identificados inmunohistoquímicamente.
Las consideraciones clave para el etiquetado de proteínas aguas abajo del ensayo FISH van más allá de la preparación de tejidos y la optimización del etiquetado de la sonda FISH. Por ejemplo, descubrimos que la especificidad de unión y etiquetado de anticuerpos puede verse afectada negativamente por el paso de proteasa dentro del ensayo de sonda FISH. Las proteasas catalizan la escisión hidrolítica de los enlaces peptídicos, lo que facilita la entrada de la sonda FISH en las células, sin embargo, también pueden digerir la proteína objetivo del ensayo IHQ posterior, produciendo una unión fuera del objetivo. La localización subcelular de la proteína diana es otro factor que contribuye al éxito de la IHQ tras el ensayo de la sonda FISH. Observamos que la especificidad de la IHQ se conserva cuando la proteína diana está unida a la membrana, mientras que la IHQ dirigida a la proteína citoplasmática requirió una amplia resolución de problemas. Finalmente, encontramos que el manejo del tejido congelado fijo montado en portaobjetos es más desafiante que el tejido congelado fresco, sin embargo, la calidad de la IHQ fue en general mejor con el tejido congelado fijo, cuando se combinó con ARNscope.
Introducción
Las proteínas y los ARNm que definen neuroquímicamente las subpoblaciones de neuronas se identifican comúnmente con una combinación de inmunohistoquímica (IHQ) y/o hibridación in situ (ISH), respectivamente. La combinación de ISH con técnicas IHQ facilita la caracterización de patrones de colocalización exclusivos de las neuronas funcionales (codificación neuroquímica) al maximizar la capacidad de marcaje múltiple.
Los métodos de ISH fluorescentes (FISH), incluido el ARNoscopio, tienen una mayor sensibilidad y especificidad en comparación con los métodos de detección de ARN anteriores, como el ISH radiactivo y el ISH cromogénico no radiactivo. FISH permite la visualización de transcripciones individuales de ARNm como manchas teñidas punteadas1. Además, el ensayo RNAscope permite marcar un mayor número de dianas de ARN a la vez, utilizando diferentes etiquetas de fluoróforos. A pesar de estas ventajas, las limitaciones técnicas pueden afectar al número de fluoróforos/cromógenos que se pueden utilizar en un solo experimento. Estos incluyen la disponibilidad de juegos de filtros de microscopio; estas consideraciones se agravan cuando la identificación neuroquímica utiliza la combinación de FISH e IHQ, en comparación con el uso de cada técnica de forma aislada, ya que los pasos inherentes óptimos para un método pueden ser perjudiciales para el otro.
La aplicación previa de FISH combinada con IHQ ha demostrado la expresión de dianas celulares específicas en linfomas de células B humanas2, embriones de pollo3, embriones de pez cebra4, retina de ratón5 y células del oído interno de ratón6. En estos estudios, la preparación del tejido se realizó en parafina fijada en formol (FFPE)2,3,5 o en montura entera fresca 4,6. Otros estudios aplicaron el ARNoscopio cromogénico en preparaciones fijas de cerebro de ratón y rata 7,8,9. En particular, Baleriola et al.8 describieron dos preparaciones tisulares diferentes para la combinación de ISH-IHC; secciones fijas del cerebro del ratón y secciones del cerebro humano FFPE. En una publicación reciente, combinamos FISH e IHQ fluorescente en secciones frescas congeladas, para visualizar simultáneamente ARNm de baja abundancia (receptor de galanina 1, GalR1), ARNm de alta abundancia (transportador de glicina 2, GlyT2) y proteína10 transportadora vesicular de acetilcolina (vAChT) en la formación reticular del tronco encefálico.
El núcleo del tracto solitario (NTS, por sus siglas en inglés) es una de las principales regiones del cerebro implicada en la función autonómica. Situada en el cerebro posterior, esta población heterogénea de neuronas recibe e integra un gran número de señales autónomas, incluidas las que regulan la respiración. El NTS alberga varias poblaciones neuronales, que pueden caracterizarse fenotípicamente por el patrón de expresión de dianas de ARNm que incluyen GalR1 y GlyT2 y marcadores proteicos para la enzima tirosina hidroxilasa (TH) y el factor de transcripción Paired-like homeobox 2b (Phox2b).
El propietario de RNAscope recomienda preparaciones de tejido fresco congelado, pero el tejido preparado por fijación de perfusión transcárdica de animal entero, junto con la crioprotección a largo plazo (almacenamiento a -20 °C) de secciones fijas de tejido congelado, es común en muchos laboratorios. Por lo tanto, se buscó establecer protocolos para la FISH en combinación con IHQ utilizando preparaciones de tejido fresco congelado y congelado fijo. Aquí, proporcionamos secciones cerebrales frescas congeladas y congeladas fijas: (1) un protocolo para FISH combinado e IHQ fluorescente (2) una descripción de la calidad del ARNm y el marcaje de proteínas producido, cuando se utiliza cada preparación (3) una descripción de la expresión de GalR1 y GlyT2 en el NTS.
Nuestro estudio reveló que, cuando se combinó con la metodología de ARNoscopio, el éxito de la IHC varió en las preparaciones frescas congeladas y congeladas fijas y dependió de la localización de las proteínas diana dentro de la célula. En nuestras manos, el marcaje de proteínas unidas a la membrana siempre tuvo éxito. Por el contrario, la IHQ para la proteína citoplasmática requirió la resolución de problemas incluso en los casos en que la proteína citoplasmática estaba sobreexpresada en un animal transgénico (Phox2b-GFP)11. Finalmente, mientras que GalR1 se expresa en neuronas no catecolaminérgicas en el NTS, la expresión de GlyT2 está ausente en el NTS.
Protocolo
En la Figura 1 se puede encontrar un resumen de los pasos de preprocesamiento de tejidos. Todos los procedimientos se llevaron a cabo de conformidad con el Comité de Ética y Cuidado de los Animales de la Universidad de Nueva Gales del Sur de acuerdo con las directrices para el uso y cuidado de los animales con fines científicos (Consejo Nacional de Salud e Investigación Médica de Australia).
1. Preparación de muestras de tejido cerebral fresco congelado
- Perfusión transcárdica
- Preparar tampón fosfato (PB) 0,1 M heparinizado (2500 U/L), pH 7,5. Haga una suspensión de etanol de hielo seco mezclando hielo seco con etanol. Tendrá una temperatura de aproximadamente -72 °C y se utilizará para la congelación inmediata del tejido extraído.
- Eutanasia de ratones adultos C57BL/6 y Phox2b-GFP11 (ID de la base de datos de Informática del Genoma del Ratón MGI:5776545) anestesiando con pentobarbital sódico (70 mg/kg, i.p.), utilizando un calibre de aguja de 27,5 pulgadas.
PRECAUCIÓN: El pentobarbital es un barbitúrico. Es agudamente tóxico en dosis altas y puede causar la muerte por paro respiratorio. Consulte las pautas locales de salud, legales y de seguridad de materiales antes de usar. - Exponga el corazón y canule el ventrículo izquierdo con una aguja de extracción (calibre de 23 pulgadas). Realizar perfusión transcárdica con PB heparinizado 0,1 M hasta que la sangre se aclare (2-3 minutos) a un caudal de 11-13 mL/min. Determinar la depuración sanguínea mediante el control de la coloración del hígado y el efusado de la aurícula derecha12.
- Aísle el cerebro de la cavidad del cráneo, insértelo inmediatamente en un compuesto de temperatura de corte óptima (OCT) en un criomold o papel de aluminio y colóquelo en el baño de etanol de hielo seco. Guarde el tejido congelado incrustado en un recipiente hermético a -80 °C durante un máximo de 3 meses.
- Seccionamiento de tejido fresco congelado
- Ajuste la temperatura del criostato a -20 °C. Deje el tejido incrustado en OCT y un mandril de criostato en el criostato durante ~ 30 minutos para permitir el equilibrio a la nueva temperatura.
NOTA: Mantenga el pañuelo congelado en todo momento; transportar el tejido desde el congelador a -80 °C hasta el criostato en hielo seco. - Asegure el tejido al mandril de criostato preenfriado con compuesto OCT. En este protocolo, los bloques de tejido se montaron en el mandril en el plano coronal.
NOTA: Recorte el exceso de OCT del tejido, con una cuchilla de afeitar, para minimizar la cantidad de OCT que se corta con el criostato y posteriormente se transfiere al portaobjetos de vidrio. - Corte secciones coronales de 14 μm de espesor y móntelas en portaobjetos de microscopía de vidrio cargados.
- Caliente las guías a temperatura ambiente antes de montar las secciones. Una vez montada la sección, guarde los portaobjetos en una caja de portaobjetos en el criostato.
- Si es necesario montar más de una sección en un portaobjetos, caliente el área para la segunda sección colocando un dedo en el lado opuesto del portaobjetos durante 5 a 10 segundos para ayudar a la adherencia de la sección al portaobjetos. Una sección de tejido frío no se adhiere a un portaobjetos frío. Las secciones deben adherirse a las diapositivas planas; Doblarlos hará que se caigan de los portaobjetos durante los pasos de lavado.
- Si se observan grietas en las secciones, aumente la temperatura del criostato entre 1 y 5 °C para evitarlo. Es especialmente importante colocar secciones de tejido muy cerca unas de otras en el mismo portaobjetos. Esto evitará el desperdicio de sondas y reactivos FISH durante el ensayo.
- Almacene las secciones de papel tisú montadas en portaobjetos de vidrio en un recipiente hermético a -80 °C durante un máximo de 6 meses.
NOTA: Mantenga las secciones congeladas en todo momento y evite los ciclos de congelación y descongelación, para evitar la degradación del ARN. Transporte la caja portaobjetos desde el interior del criostato hasta el congelador a -80 °C con hielo seco.
- Ajuste la temperatura del criostato a -20 °C. Deje el tejido incrustado en OCT y un mandril de criostato en el criostato durante ~ 30 minutos para permitir el equilibrio a la nueva temperatura.
- Fijación de tejido fresco congelado
- El día en que se vaya a realizar el ensayo de sonda FISH, prepare paraformaldehído (PFA) al 4% en PB 0,1 M, pH 7,5 (solución de PFA al 4%). Filtre pasando a través de papel de filtro (Grado 1: 11 μm, Tabla de Materiales) en un embudo Buchner o filtro de crisol.
PRECAUCIÓN : El PFA es dañino y tóxico por contacto con la piel o inhalación. Todos los procedimientos con solución de PFA deben realizarse en un gabinete de campana extractora. Los residuos de la solución de PFA deben eliminarse con cuidado siguiendo los protocolos de seguridad institucionales. - Enfriar la solución de PFA al 4% a 4 °C. Transporta el pañuelo deslizante desde el congelador a -80 °C en hielo seco y sumérgelo inmediatamente en el fijador preenfriado durante 15 minutos.
NOTA: Es importante que este paso de fijación no supere los 15 minutos, ya que la sobrefijación dará lugar a un etiquetado de fondo no específico.
- El día en que se vaya a realizar el ensayo de sonda FISH, prepare paraformaldehído (PFA) al 4% en PB 0,1 M, pH 7,5 (solución de PFA al 4%). Filtre pasando a través de papel de filtro (Grado 1: 11 μm, Tabla de Materiales) en un embudo Buchner o filtro de crisol.
- Deshidratación de tejido fresco congelado
- Deshidratar las secciones de tejido sumergiendo los portaobjetos en concentraciones graduadas de etanol. En un frasco Coplin, primero sumérjalo al 50%, luego al 70% y finalmente al etanol absoluto, durante 5 minutos cada uno a temperatura ambiente. Repita la incubación final de etanol absoluto por segunda vez.
- Seque los portaobjetos al aire y delinee el grupo de secciones con un rotulador de barrera hidrofóbico, asegurándose de que el área interna se mantenga al mínimo.
NOTA: Asegúrese de que el portaobjetos de vidrio esté completamente seco antes de dibujar la barrera hidrofóbica. La barrera hidrofóbica debe rodear las secciones de tejido completamente sin espacios y debe estar seca antes de continuar con el procesamiento.
2. Preparación de muestras de tejido cerebral congelado fijo
- Fijación de perfusión transcárdica
- Sacrificar ratones mediante anestesia con pentobarbital sódico (70 mg/kg, i.p) seguido de perfusión transcárdica, primero con 0,1 M de PB y luego con solución de PFA al 4%. Fijar con 10 minutos de perfusión a 11-13 mL/min.
- Aislar el cerebro de la cavidad craneal después de la fijación por perfusión y sumergir durante la noche en una solución de PFA al 4%, a 4 °C.
- Seccionamiento tisular de tejido fijo
- Enjuague el cerebro con solución salina estéril tamponada con fosfato (PBS) 0,1 M antes de retirar las capas meníngeas, con la ayuda de un microscopio de disección, utilizando pinzas finas.
- Cortar el cerebro con precisión en bloques (separar el tronco encefálico del prosencéfalo antes de seccionar el vibratome) utilizando una matriz cerebral (Tabla de materiales). Específicamente, corte el tronco encefálico caudalmente en la decusación piramidal y diseccione el cerebelo. Del mismo modo, corte el prosencéfalo inmediatamente rostral al quiasma óptico.
- Fije el tejido en un mandril de micrótomo vibratorio con cianoacrilato e insértelo en una solución de agar al 2%.
- Corte secciones de tejido de 30 μm de espesor con un micrótomo vibratorio y almacene las secciones cortadas en una solución crioprotectora (sacarosa libre de RNasa al 30%, etilenglicol al 30%, polivinilpirrolidona (PVP-40), en PB 0,1 M, pH 7,4). Las secciones de tejido pueden almacenarse en crioprotector a -20 °C durante un máximo de 6 meses.
- Preparación de secciones fijas previas a FISH
- El día de la pesca, lave las secciones flotantes tres veces, durante 10 minutos por lavado, para eliminar la solución crioprotectora. Para lavar, coloque las secciones en PBS de 0,1 M en una placa de cultivo celular de 12 pocillos y agite en un agitador de plataforma giratoria (90 - 100 rpm).
- Después de los lavados, use un pincel para montar secciones en portaobjetos de microscopía de vidrio y seque al aire durante al menos 2 horas.
NOTA: Las secciones deben adherirse planas a los portaobjetos, ya que cualquier pliegue pronunciado hará que se desprendan durante los lavados. - Con un bolígrafo de barrera hidrofóbico, dibuje una barrera alrededor de las secciones para restringir los reactivos FISH a las secciones. Una vez más, es importante minimizar el área interna del contorno dibujado con el bolígrafo barrera.
POSIBLE PUNTO DE RUPTURA: Las secciones podrían almacenarse a temperatura ambiente, durante la noche, para continuar el ensayo al día siguiente.
3. Ensayo FISH
NOTA: El resto del protocolo se aplica tanto al tejido fresco congelado como al tejido congelado fijo.
- Preparar los reactivos e instrumentos para las etapas de hibridación y amplificación.
- Ajuste una incubadora de sobremesa y un baño de agua a 40 °C.
- Prepare una cámara humidificada y protegida de la luz para incubar los portaobjetos. La humidificación evita el secado de los tejidos: los portaobjetos se colocan de forma segura sobre un depósito húmedo. Idealmente, la cámara está hecha de poliestireno de alta resistencia, es resistente a la luz y hermética para mantener una atmósfera saturada de vapor de agua. El cierre de la cámara se basa en una fricción mínima para evitar el movimiento. Utilizamos una caja de portaobjetos forrada con toallitas húmedas de laboratorio (Tabla de Materiales) en la parte inferior. Coloque la caja portaobjetos dentro de la incubadora para precalentarla a 40 °C.
- Calentar el tampón de lavado 50x (tabla de materiales) y las sondas a 40 °C durante 10 minutos, utilizando el baño de agua, y luego enfriar a temperatura ambiente.
- Prepare 1 L de 1x Wash Buffer a partir de la concentración de caldo 50x.
- Prepare la mezcla de la sonda (Tabla de materiales): la sonda C1 está lista para usar a la concentración de stock, mientras que las sondas C2 y C3 se envían con una concentración de 50x y requieren dilución con el diluyente suministrado en el kit.
NOTA: Las mezclas de sonda pueden almacenarse a 4 °C durante un máximo de 6 meses.
- Tratamiento con proteasa
- Incubar secciones con Proteasa III (Tabla de Materiales) a temperatura ambiente durante 30 minutos.
NOTA: Asegúrese de que la proteasa III y los reactivos de incubación en los procesos posteriores (mezcla de sondas, soluciones de amplificación, tampón de bloqueo y sueros de anticuerpos) cubran las secciones por completo. Se puede usar una punta de pipeta para esparcir el reactivo sobre la sección para cubrir toda el área dentro de la barrera hidrofóbica. - Lave los portaobjetos dos veces con 0,1 M PBS, durante 2 minutos cada vez, en una placa de Petri cuadrada de plástico grande. Aquí se utilizó una placa de bioensayo cuadrada de 245 mm x 245 mm (Tabla de Materiales). Sosténgalo desde un lado del plato e inclínelo suavemente de 3 a 5 veces. Después de los lavados, saque el exceso de 0,1 M PBS del portaobjetos y agregue inmediatamente el siguiente reactivo. No deje que las secciones de tejido se sequen.
NOTA: Durante cada lavado, los portaobjetos se sumergen en solución a temperatura ambiente. Este es el flujo de trabajo para todos los pasos de lavado posteriores. Las secciones fijas de 30 μm de espesor se desprenden de los portaobjetos más fácilmente que las secciones de 14 μm de espesor, sea suave durante los lavados.
- Incubar secciones con Proteasa III (Tabla de Materiales) a temperatura ambiente durante 30 minutos.
- Hibridación y amplificación
- Después de lavar la solución de proteasa, coloque los portaobjetos en la cámara humidificada y precalentada. Incubar secciones con mezcla de sonda (Tabla de Materiales) durante 2 horas a 40 °C dentro de una incubadora de sobremesa.
NOTA: Asegúrese de que haya al menos 2 secciones reservadas para sondas de control positivo y negativo para evaluar la calidad del ARN de la muestra y la permeabilización óptima. Las sondas de control positivo se dirigen a los genes de limpieza; en este caso, se trataba de un cóctel de ARN dirigidos a la ubiquitina C (UBC; alta abundancia), la peptidilpropilisomerasa B (PPIB; moderada abundancia) y la ARN polimerasa 2a (POLR2A; baja abundancia). Las sondas de control negativo se dirigen al gen bacteriano de la 4-hidroxi-tetrahidrodipicolinato reductasa (DapB), que normalmente está ausente en las muestras de cerebro de ratón. Una señal DapB positiva indica una señal no específica y/o contaminación bacteriana de la muestra. - Después de la hibridación con la mezcla de sondas, los pasos de amplificación de la señal consisten en la incubación con Amp 1-FL (30 minutos), luego con Amp 2-FL (15 minutos), seguido de Amp 3-FL (30 minutos) y finalmente Amp 4-FL (15 minutos), cada uno a 40 °C. Con los frascos cuentagotas provistos, cubra las secciones de tejido con una solución de amplificación. Proceda al ensayo IHQ después del último paso de amplificación.
- Enjuague los portaobjetos con tampón de lavado dos veces durante 2 minutos entre la hibridación de la sonda y cada paso de amplificación.
- Después de lavar la solución de proteasa, coloque los portaobjetos en la cámara humidificada y precalentada. Incubar secciones con mezcla de sonda (Tabla de Materiales) durante 2 horas a 40 °C dentro de una incubadora de sobremesa.
4. Ensayo IHQ
- Paso de bloqueo IHQ
- Para evitar la unión inespecífica de anticuerpos, incubar las secciones durante 1 h a temperatura ambiente con una solución de bloqueo que contenga un 10 % de suero normal de caballo, 0,3 % de Tween20 en 1x TBSm (50 mM de Tris-Cl, pH 7,5, 150 mM de NaCl, 0,05 % de merthiolato) después del ensayo FISH. Preparar anticuerpos primarios en un tampón de dilución que contenga 1x TBSm, 5% de suero normal de caballo y 0,1% de Tween20. Los principales proveedores de anticuerpos se enumeran en la Tabla de materiales.
- Inmunohistoquímica
- Eliminar el exceso de tampón de bloqueo moviendo el portaobjetos e incubar secciones con anticuerpos primarios durante la noche a 4 °C.
- Lavar los portaobjetos 3 veces (5 minutos cada uno) con 1x TBSm e incubar con un anticuerpo secundario en un diluyente que contenga 1x TBSm, 1% de suero normal de caballo y 0,1% de Tween20 durante 2 horas a temperatura ambiente. Los anticuerpos secundarios utilizados en este protocolo se enumeran en la Tabla de Materiales.
- Lave los portaobjetos 3 veces con 1x TBSm (5 minutos cada uno) antes de cubrirlos con medio de montaje con o sin DAPI (Tabla de Materiales).
5. Imágenes
- Examine la inmunotinción bajo un microscopio de epifluorescencia equipado con una cámara (consulte la Tabla de materiales para obtener más detalles). Adquiera imágenes representativas con un aumento de 20x y guárdelas como archivos TIFF.
- Exporte imágenes representativas a un software de procesamiento de imágenes (Tabla de materiales) para ajustar el brillo/contraste para aumentar la claridad y reflejar la representación real.
6. OPCIONAL: Análisis cuantitativo de transcripciones objetivo
NOTA: Este es un artículo de métodos y no se proporcionan resultados cuantitativos. El método de cuantificación presentado aquí proviene de Dereli et al.10.
- Adquiera imágenes de las regiones de interés como se explica en 5.1 y aplique los mismos ajustes del microscopio y de la cámara (como el tiempo de exposición y la intensidad de la luz) a todas las imágenes del mismo fluoróforo.
- Trazar los perfiles neuronales utilizando un software de análisis de imágenes (Tabla de Materiales).
- Alinear las secciones con referencia al nivel de Bregma de acuerdo con un atlas cerebral estereotáxico13.
- Aplique el mismo brillo y contraste a todas las imágenes del mismo fluoróforo. Considere solo las neuronas con núcleos teñidos con DAPI.
- Cuente manualmente el número de células de ARNm, que expresan proteínas, ARNm/ARNm, proteínas/proteínas y ARNm/proteínas que coexpresan dentro de la región de interés.
- Para disminuir el sesgo en los resultados experimentales, haga que la persona que cuantifica los resultados experimentales no se ciegue a los grupos experimentales.
- Aplique la corrección de Abercrombie14 al total de recuentos de células utilizando la siguiente ecuación de Abercombie:
Recuento de células corregido = recuento manual de células x espesor de sección / (espesor de sección + tamaño nuclear)
Por ejemplo, para secciones de 14 μm de espesor, se calcula que el ancho nuclear promedio es de 7,7 ± 0,3 μm y el espesor promedio de la sección es de 14 ± 1 μm basado en 30 celdas y 10 secciones respectivamente en 5 animales10. De acuerdo con la ecuación de Abercrombie, el recuento de células corregido sería el recuento manual de células x 14/(14+7,7).

Figura 1: Flujo de trabajo paralelo de los pasos de preprocesamiento de tejidos para tejidos recién congelados y fijados con paraformaldehído. Los pasos de procesamiento para el tejido fresco congelado se muestran en los cuadros con contorno rojo, mientras que los del tejido fijo con paraformaldehído (PFA) se muestran en los cuadros con contorno azul. Haga clic aquí para ver una versión más grande de esta figura.
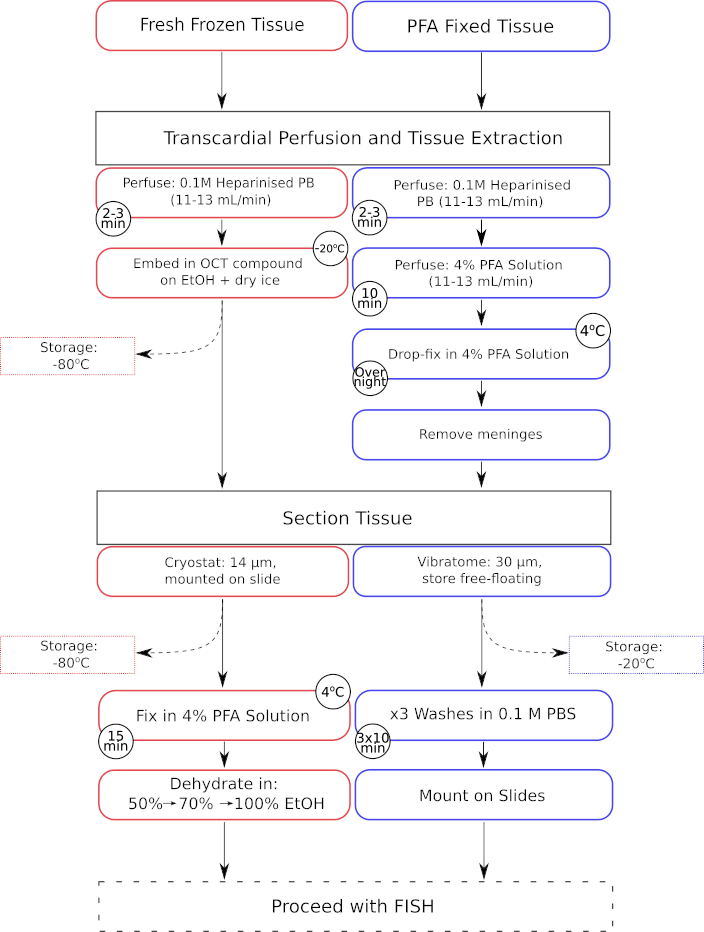{kind=link}

Figura 2: Resumen de la sonda FISH combinada y el procedimiento de inmunohistoquímica. Después del preprocesamiento del tejido, el tejido montado en portaobjetos se rodea con una pluma de barrera hidrofóbica, como se ve en el primer fotograma, y se incuba en una solución de proteasa a temperatura ambiente. Después de los lavados, el tejido se transfiere a una incubadora de sobremesa para la hibridación durante 2 horas antes de los pasos de amplificación secuencial. El sistema de hibridación in situ utiliza un diseño patentado de 'sonda Z', preamplificadores y amplificadores como se ve en los fotogramas 3-66. Una vez que el tejido se ha sometido al procesamiento de la sonda FISH, se lava antes de bloquearlo con suero de caballo normal. La incubación primaria de anticuerpos se lleva a cabo durante la noche a 4 °C para maximizar la unión anticuerpo-antígeno. La incubación secundaria de anticuerpos (2 horas) se realizó a temperatura ambiente. Haga clic aquí para ver una versión más grande de esta figura.
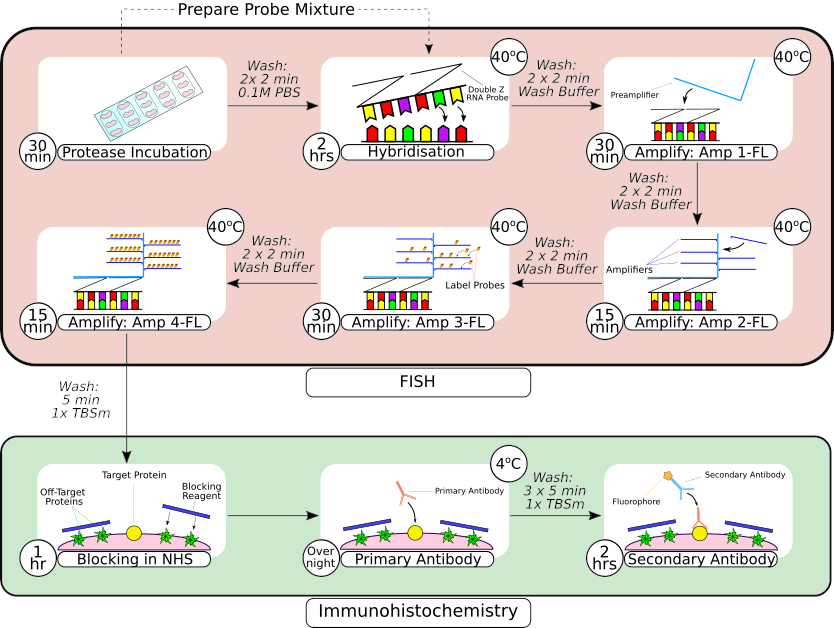{kind=link}
Resultados
Aquí, describimos un método para combinar FISH multiplex con IHQ fluorescente para localizar la expresión de ARNm para GalR1 y GlyT2 utilizando tejidos frescos congelados y fijados con paraformaldehído, respectivamente, en el NTS de ratón. En la Figura 1 y en la Figura 2 se muestra una línea de procesamiento de tejidos, FISH y procedimientos IHQ descritos en los métodos. La Tabla 1 proporciona un resumen de las combinaciones de sonda FISH...
Discusión
En las neurociencias, la FISH y la IHQ se utilizan de forma rutinaria para investigar la organización espacial y la importancia funcional del ARNm o las proteínas dentro de las subpoblaciones neuronales. El protocolo descrito en este estudio mejora la capacidad de detección simultánea de ARNm y proteínas en secciones cerebrales. Nuestro ensayo combinado multiplex FISH-IHC permitió la identificación fenotípica de distintas subpoblaciones neuronales en el NTS tanto en preparaciones cerebrales frescas congeladas com...
Agradecimientos
Este trabajo fue financiado por la subvención del Proyecto de Descubrimiento del Consejo de Investigación Australiano DP180101890 y la subvención del proyecto de la Fundación de Investigación Médica Rebecca L Cooper PG2018110
Materiales
| Name | Company | Catalog Number | Comments |
| ANIMALS | |||
| C57BL/6 mouse | Australian BioResources, Moss Vale | MGI: 2159769 | |
| Phox2b-eGFP mouse | Australian BioResources, Moss Vale | MGI: 5776545 | |
| REAGENTS | |||
| Cyanoacrylate | Loctite | ||
| Ethylene Glycol | Sigma-Aldrich | 324558 | |
| Heparin-Sodium | Clifford Hallam Healthcare | 1070760 | Consult local veterinary supplier or pharmacy. |
| Lethabarb (Sodium Pentabarbitol) Euthanasia Injection | Virbac (Australia) Pty Ltd | N/A | Consult a veterinarian for local pharmaceutical regulations regarding Sodium Pentabarbitol |
| Molecular grade agarose powder | Sigma Aldrich | 5077 | |
| OCT Compound, 118mL | Scigen Ltd | 4586 | |
| Paraformaldehyde, prilled, 95% | Sigma-Aldrich | 441244-1KG | |
| Polyvinylpyrrolidone, average mol wt 40,000 (PVP-40) | Sigma-Aldrich | PVP40 | |
| ProLong Gold Antifade Mountant | Invitrogen | P36930 | With or without DAPI |
| RNAscope Multiplex Fluorescent Reagent Kit (up to 3-plex capability) | Advanced Cell Diagnostics, Inc. (ACD Bio) | ADV320850 | Includes 50x Wash buffer and Protease III |
| RNase Away | Thermo-Fisher Scientific | 7003 | |
| Tris(hydroxymethyl)aminomethane | Sigma-Aldrich | 252859 | |
| Tween-20, for molecular biology | Sigma-Aldrich | P9416 | |
| EQUIPMENT | |||
| Benchtop incubator | Thermoline scientific micro incubator | Model: TEI-13G | |
| Brain Matrix, Mouse, 30g Adult, Coronal, 1mm | Ted Pella | 15050 | |
| Cryostat | Leica | CM1950 | |
| Drawing-up needle (23 inch gauge) | BD | 0288U07 | |
| Hydrophobic Barrier Pen | Vector labs | H-4000 | |
| Kimtech Science Kimwipes Delicate Task Wipes | Kimberley Clark Professional | 34120 | |
| Olympus BX51 | Olympus | BX-51 | |
| Peristaltic pump | Coleparmer Masterflex | L/S Series | |
| Retiga 2000R Digital Camera | QImaging | RET-2000R-F-CLR | colour camera |
| SuperFrost Plus Glass Slides (White) | Thermo-Fisher Scientific | 4951PLUS4 | |
| Vibrating Microtome (Vibratome) | Leica | VT1200S | |
| Whatman qualitative filter paper, Grade 1, 110 mm diameter | Merck | WHA1001110 | |
| SOFTWARES | |||
| CorelDRAW | Corel Corporation | Version 7 | |
| FIJI (ImageJ Distribution) | Open Source/GNU General Public Licence (GPL) | N/A | ImageJ 2.x: Rueden, C. T.; Schindelin, J. & Hiner, M. C. et al. (2017), "ImageJ2: ImageJ for the next generation of scientific image data", BMC Bioinformatics 18:529, PMID 29187165, doi:10.1186/s12859-017-1934-z and Fiji: Schindelin, J.; Arganda-Carreras, I. & Frise, E. et al. (2012), "Fiji: an open-source platform for biological-image analysis", Nature methods 9(7): 676-682, PMID 22743772, doi:10.1038/nmeth.2019 |
| PRIMARY ANTIBODIES | |||
| Anti-Tyrosine Hydroxylase Antibody | Millipore Sigma | AB1542 | Sheep polyclonal (1:1000 dilution), RRID: AB_90755 |
| Anti-Tyrosine Hydroxylase Antibody, clone LNC1 | Millipore Sigma | MAB318 | Mouse monoclonal (1:1000 dilution), RRID: AB_2201528 |
| Anti-Vesicular Acetylcholine Transporter (VAchT) Antibody | Sigma-Aldrich | ABN100 | Goat polyclonal (1:1000 dilution), RRID: AB_2630394 |
| GFP Antibody | Novus Biologicals | NB600-308 | Rabbit polyclonal (1:1000 dilution), RRID: AB_10003058 |
| Phox2b Antibody (B-11) | Santa Cruz Biotechnology | sc-376997 | Mouse monoclonal (1:1000 dilution), RRID: AB_2813765 |
| SECONDARY ANTIBODIES | |||
| Alexa Fluor 488 AffiniPure Donkey Anti-Rabbit IgG (H+L) (min X Bov, Ck, Gt, GP, Sy Hms, Hrs, Hu, Ms, Rat, Shp Sr Prot) | Jackson ImmunoResearch | 711-545-152 | Donkey anti-Rabbit (1:400 dilution), RRID: AB_2313584 |
| AMCA AffiniPure Donkey Anti-Sheep IgG (H+L) (min X Ck, GP, Sy Hms, Hrs, Hu, Ms, Rb, Rat Sr Prot) | Jackson ImmunoResearch | 713-155-147 | Donkey anti-Sheep (1:400 dilution), RRID: AB_AB_2340725 |
| Cy5 AffiniPure Donkey Anti-Goat IgG (H+L) (min X Ck, GP, Sy Hms, Hrs, Hu, Ms, Rb, Rat Sr Prot) | Jackson ImmunoResearch | 705-175-147 | Donkey anti-Goat (1:400 dilution), RRID: AB_2340415 |
| Cy5 AffiniPure Donkey Anti-Mouse IgG (H+L) (min X Bov, Ck, Gt, GP, Sy Hms, Hrs, Hu, Rb, Rat, Shp Sr Prot) | Jackson ImmunoResearch | 715-175-151 | Donkey anti-Mouse (1:400 dilution), RRID: AB_2619678 |
| Cy5 AffiniPure Donkey Anti-Sheep IgG (H+L) (min X Ck, GP, Sy Hms, Hrs, Hu, Ms, Rb, Rat Sr Prot) | Jackson ImmunoResearch | 713-175-147 | Donkey anti-Sheep (1:400 dilution), RRID: AB_2340730 |
| RNASCOPE PROBES | |||
| Galanin Receptor 1 oligonucleotide probe | ACDBio | 448821-C1 | targets bp 482 - 1669 (Genebank ref: NM_008082.2) |
| Glycine transporter 2 oligonucleotide probe | ACDBio | 409741-C3 | targets bp 925 - 2153 (Genebank ref: NM_148931.3) |
| Phox2b oligonucleotide probe | ACDBio | 407861-C2 | targets bp 1617 - 2790 (Genebank ref: NM_008888.3) |
Referencias
- Wang, F., et al. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. Journal of Molecular Diagnostics. 14 (1), 22-29 (2012).
- Annese, T., et al. RNAscope dual ISH-IHC technology to study angiogenesis in diffuse large B-cell lymphomas. Histochemistry and Cell Biology. 153 (3), 185-192 (2020).
- Morrison, J. A., McKinney, M. C., Kulesa, P. M. Resolving in vivo gene expression during collective cell migration using an integrated RNAscope, immunohistochemistry and tissue clearing method. Mechanisms of Development. 148, 100-106 (2017).
- Gross-Thebing, T., Paksa, A., Raz, E. Simultaneous high-resolution detection of multiple transcripts combined with localization of proteins in whole-mount embryos. BMC Biology. 12, 55 (2014).
- Stempel, A. J., Morgans, C. W., Stout, J. T., Appukuttan, B. Simultaneous visualization and cell-specific confirmation of RNA and protein in the mouse retina. Molecular Vision. 20, 1366-1373 (2014).
- Kersigo, J., et al. A RNAscope whole mount approach that can be combined with immunofluorescence to quantify differential distribution of mRNA. Cell and Tissue Research. 374 (2), 251-262 (2018).
- Grabinski, T. M., Kneynsberg, A., Manfredsson, F. P., Kanaan, N. M. A method for combining RNAscope in situ hybridization with immunohistochemistry in thick free-floating brain sections and primary neuronal cultures. PLoS One. 10 (3), 0120120 (2015).
- Baleriola, J., Jean, Y., Troy, C., Hengst, U. Detection of axonally localized mRNAs in brain sections using high-resolution in situ hybridization. Journal of Visualized Experiments. (100), e52799 (2015).
- Fe Lanfranco, M., Loane, D. J., Mocchetti, I., Burns, M. P., Villapol, S. Combination of fluorescent in situ hybridization (FISH) and immunofluorescence imaging for detection of cytokine expression in microglia/macrophage cells. Bio-Protocol. 7 (22), (2017).
- Dereli, A. S., Yaseen, Z., Carrive, P., Kumar, N. N. Adaptation of respiratory-related brain regions to long-term hypercapnia: focus on neuropeptides in the RTN. Frontiers in Neuroscience. 13, 1343 (2019).
- Lazarenko, R. M., et al. Acid sensitivity and ultrastructure of the retrotrapezoid nucleus in Phox2b-EGFP transgenic mice. Journal of Comparative Neurology. 517 (1), 69-86 (2009).
- Gage, G. J., Kipke, D. R., Shain, W. Whole animal perfusion fixation for rodents. Journal of Visualized Experiments. (65), e3564 (2012).
- Paxinos, G., Franklin, K. B. . The mouse brain in stereotaxic coordinates. , (2004).
- Abercrombie, M. Estimation of nuclear population from microtome sections. Anatomical Records. 94, 239-247 (1946).
- Kerr, N., et al. The generation of knock-in mice expressing fluorescently tagged galanin receptors 1 and 2. Molecular and Cellular Neurosciences. 68, 258-271 (2015).
- Kachidian, P., Pickel, V. M. Localization of tyrosine hydroxylase in neuronal targets and efferents of the area postrema in the nucleus tractus solitarii of the rat. Journal of Comparative Neurology. 329 (3), 337-353 (1993).
- Stornetta, R. L., et al. Expression of Phox2b by brainstem neurons involved in chemosensory integration in the adult rat. Journal of Neuroscience. 26 (40), 10305-10314 (2006).
- Gilmor, M. L., et al. Expression of the putative vesicular acetylcholine transporter in rat brain and localization in cholinergic synaptic vesicles. Journal of Neuroscience. 16 (7), 2179-2190 (1996).
- Fisher, J. M., Sossin, W., Newcomb, R., Scheller, R. H. Multiple neuropeptides derived from a common precursor are differentially packaged and transported. Cell. 54 (6), 813-822 (1988).
- Towle, A. C., Lauder, J. M., Joh, T. H. Optimization of tyrosine-hydroxylase immunocytochemistry in paraffin sections using pretreatment with proteolytic-enzymes. Journal of Histochemistry and Cytochemistry. 32 (7), 766-770 (1984).
- Biancardi, V., et al. Mapping of the excitatory, inhibitory, and modulatory afferent projections to the anatomically defined active expiratory oscillator in adult male rats. Journal of Comparative Neurology. 529 (4), 853-884 (2021).
- Matthews, D. W., et al. Feedback in the brainstem: an excitatory disynaptic pathway for control of whisking. Journal of Comparative Neurology. 523 (6), 921-942 (2015).
- Ramos-Vara, J. A. Principles and methods of immunohistochemistry. Methods in Molecular Biology. 1641, 115-128 (2017).
- Shi, S. R., Key, M. E., Kalra, K. L. Antigen retrieval in formalin-fixed, paraffin-embedded tissues: an enhancement method for immunohistochemical staining based on microwave oven heating of tissue sections. Journal of Histochemistry and Cytochemistry. 39 (6), 741-748 (1991).
- Yamashita, S., Katsumata, O. Heat-induced antigen retrieval in immunohistochemistry: mechanisms and applications. Methods in Molecular Biology. 1560, 147-161 (2017).
- Yamashita, S., Okada, Y. Mechanisms of heat-induced antigen retrieval: analyses in vitro employing SDS-PAGE and immunohistochemistry. Journal of Histochemistry and Cytochemistry. 53 (1), 13-21 (2005).
- Yamashita, S. Heat-induced antigen retrieval: mechanisms and application to histochemistry. Progress in Histochemistry and Cytochemistry. 41 (3), 141-200 (2007).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados