
Method Article
Desarrollo y pruebas de ensayos cuantitativos de PCR específicos de especies para aplicaciones de ADN ambiental
* Estos autores han contribuido por igual
En este artículo
Resumen
Los ensayos de ADN ambiental requieren un diseño riguroso, pruebas, optimización y validación antes de que pueda comenzar la recopilación de datos de campo. Aquí, presentamos un protocolo para llevar a los usuarios a través de cada paso del diseño de un ensayo qPCR específico de la especie, basado en sondas para la detección y cuantificación de un ADN de especies objetivo a partir de muestras ambientales.
Resumen
Se están desarrollando nuevos métodos no invasivos para detectar y monitorear la presencia de especies para ayudar en la pesca y la gestión de la conservación de la vida silvestre. El uso de muestras de ADN ambiental (eDNA) para detectar macrobiota es uno de esos grupos de métodos que se está volviendo rápidamente popular y que se está implementando en los programas de gestión nacionales. Aquí nos centramos en el desarrollo de ensayos específicos de especies para aplicaciones de PCR cuantitativas basadas en sondas (qPCR). El uso de qPCR basado en sonda ofrece una mayor especificidad de lo que es posible solo con imprimaciones. Además, la capacidad de cuantificar la cantidad de ADN en una muestra puede ser útil en nuestra comprensión de la ecología del eDNA y la interpretación de los patrones de detección de eDNA en el campo. Se necesita una cuidadosa consideración en el desarrollo y pruebas de estos ensayos para asegurar la sensibilidad y especificidad de detectar las especies objetivo a partir de una muestra ambiental. En este protocolo delinearemos los pasos necesarios para diseñar y probar ensayos basados en sondas para la detección de una especie objetivo; incluyendo la creación de bases de datos de secuencia, diseño de ensayos, selección y optimización de ensayos, rendimiento de ensayos de prueba y validación de campos. Seguir estos pasos ayudará a lograr un ensayo eficiente, sensible y específico que se puede usar con confianza. Demostramos este proceso con nuestro ensayo diseñado para poblaciones del mejillón(Actinonaias ligamentina),una especie de mejillón de agua dulce que se encuentra en el río Clinch, EE.UU.
Introducción
Los investigadores y gerentes están cada vez más interesados en el uso de ensayos de ADN ambiental para la detección de especies. Durante tres décadas, se ha utilizado pcr cuantitativo o en tiempo real (qPCR/rtPCR) en numerosos campos para la detección y cuantificación específica de secuencias de ácidos nucleicos1,2. Dentro del campo relativamente nuevo de la investigación de eDNA, el uso de estos ensayos con una curva estándar para la cuantificación de copias de ADN objetivo por volumen o peso de la muestra de eDNA se ha convertido en una práctica rutinaria. Las secuencias de ADN mitocondrial generalmente están dirigidas en ensayos de eDNA porque el genoma mitocondrial está presente en miles de copias por célula, pero también son posibles ensayos para secuencias nucleares de ADN o ARN. Es vital entender que los ensayos publicados para muestras de eDNA no siempre son iguales en rendimiento. La fiabilidad de un ensayo para detectar sólo el ADN de una especie objetivo (es decir, especificidad) y la detección de bajas cantidades de ADN objetivo (es decir, sensibilidad) puede variar considerablemente debido a diferencias en la forma en que se diseñó, seleccionó, optimizó y probó el ensayo. La presentación de informes de medidas cuantitativas de rendimiento de los ensayos se ha pasado por alto en gran medida, pero recientemente están surgiendo normas para mejorar la transparencia en el desarrollo de ensayos3,4,5,6,7,8.
Optimización e informes de ayudas al rendimiento de ensayos en el diseño del estudio y la interpretación de los resultados de la encuesta eDNA. Los ensayos que reaccionan cruzadamente con el ADN de especies no objetivo podrían conducir a detecciones de falsos positivos, mientras que los ensayos con poca sensibilidad pueden no detectar el ADN de las especies objetivo incluso cuando está presente en la muestra (falsos negativos). Una comprensión de la sensibilidad y la selectividad del ensayo ayudará a informar el esfuerzo de muestreo necesario para detectar especies raras. Dado que hay muchas fuentes naturales de variación en eDNA, los estudios deben limitar las fuentes de variación controlables tanto como sea posible, incluyendo la optimización completa y la caracterización del ensayo eDNA3.
Las condiciones que afectan directamente a la especificidad o sensibilidad de un ensayo cambiarán el rendimiento del ensayo. Esto puede ocurrir en diferentes condiciones de laboratorio (es decir, diferentes reactivos, usuarios, máquinas, etc.). Por lo tanto, este protocolo debe revisarse al aplicar un ensayo en condiciones nuevas. Incluso los ensayos bien caracterizados en la literatura deben ser probados y optimizados cuando se adoptan por un nuevo laboratorio o cuando se utilizan diferentes reactivos (por ejemplo, solución de mezcla maestra)5,9. La especificidad del ensayo puede cambiar cuando se aplica a una región geográfica diferente, ya que el ensayo se está aplicando a muestras de una nueva comunidad biótica que pueden incluir especies no objetivo contra las que no se ha probado el ensayo, y puede producirse una variación genética en las especies objetivo. Una vez más, el ensayo debe reevaluarse cuando se utiliza en una nueva ubicación. Las condiciones de campo difieren de las condiciones de laboratorio porque en el campo los inhibidores de la PCR son más propensos a estar presentes en las muestras. Los inhibidores de la PCR afectan directamente a la reacción de amplificación y, por lo tanto, afectan al rendimiento del ensayo. Por esta razón, se requiere un control positivo interno al desarrollar un ensayo eDNA.
Por último, las condiciones ambientales en el campo pueden afectar las moléculas de ADN de las especies objetivo y su captura a través de la degradación, el transporte y la retención del ADN. Además, diferentes protocolos para la recolección y extracción de ADN varían en su eficiencia y capacidad para retener el ADN. Sin embargo, es importante tener en cuenta que estos procesos afectan a la capacidad de detección de eDNA, pero no al rendimiento de los ensayos moleculares. Por lo tanto, la detección de ADN de las especies objetivo en muestras de campo es una función tanto del rendimiento técnico del ensayo qPCR como de las condiciones de campo y los protocolos de recolección, almacenamiento y extracción. Al utilizar un ensayo bien caracterizado y de alto rendimiento, los usuarios pueden sentirse seguros de las capacidades del ensayo; permitiendo a los investigadores centrarse ahora en la comprensión de los factores de ensayo externos (es decir, variables ambientales, diferencias en los protocolos de captura o extracción) que afectan a la detección de eDNA.
Aquí nos centramos específicamente en el rendimiento técnico del ensayo a través de un diseño y optimización rigurosos. Demostramos el protocolo utilizando un ensayo basado en sondas desarrollado para la detección de un mejillón de agua dulce, el mejillón(Actinonaias ligamentina),a partir del agua muestreada en el río Clinch, EE.UU. Recientemente Thalinger et al. (2020) presentaron directrices para la validación de ensayos eDNA específicos. El diseño del ensayo siguiendo nuestro protocolo traerá un ensayo al nivel 4 de Thalinger et al., además de un paso adicional hacia el nivel 56. En este punto, se optimizará el rendimiento técnico de un ensayo y estará listo para su uso regular en aplicaciones de laboratorio y campo. El uso posterior del ensayo en experimentos de laboratorio, mesocosmo y campo puede abordar preguntas relacionadas con la detección de eDNA y los factores que influyen en la capacidad de detección, los pasos finales para la validación de nivel 56.
Protocolo
1. Generación de una base de datos de secuencias de secuencias de ADN mitocondrial de especies de interés objetivo y no objetivo
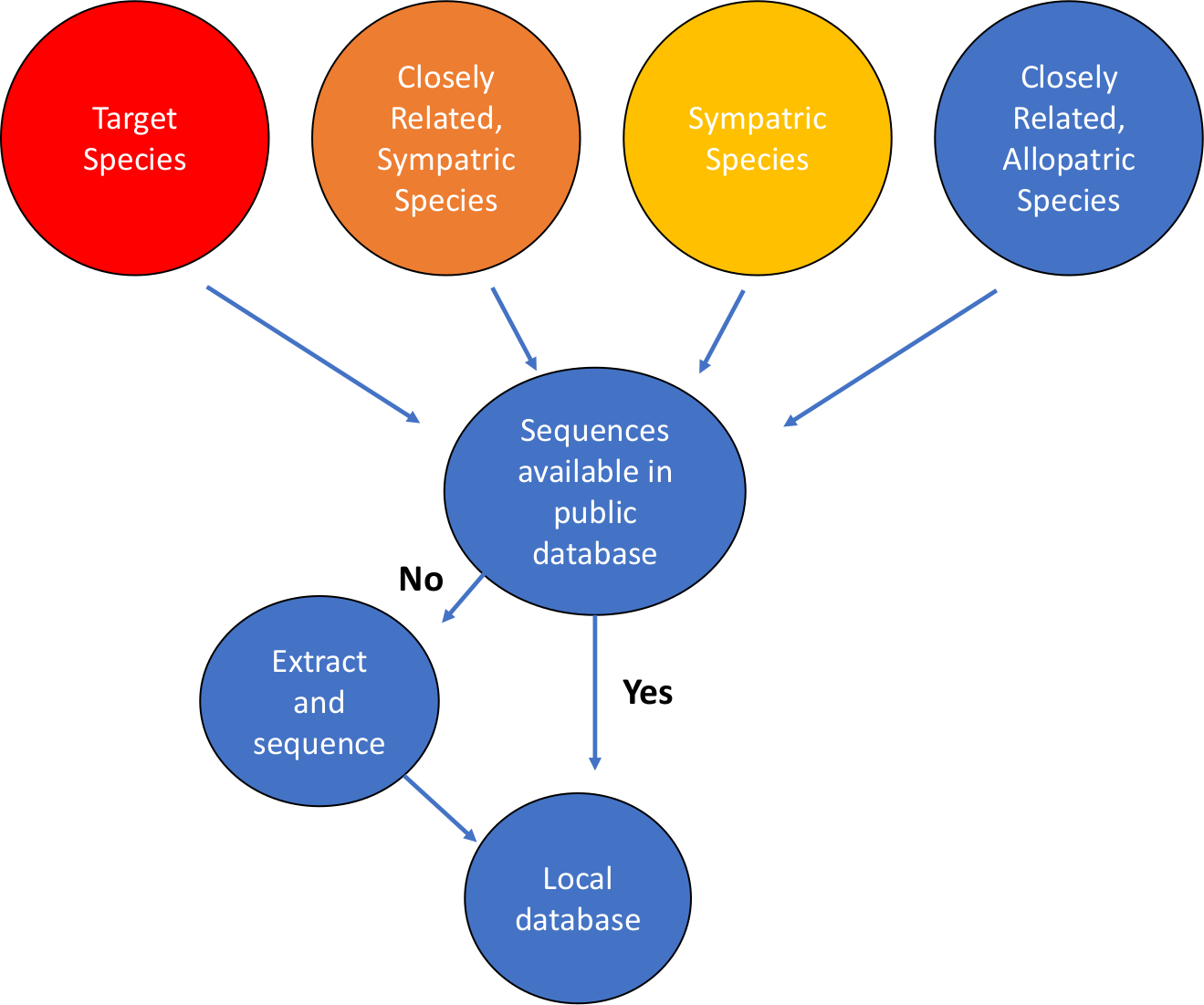
- Defina la pregunta, los objetivos y el sistema que se están abordando. Identificar las especies objetivo para la detección de eDNA. Identifique el sistema geográfico en el que se utilizará el ensayo. Hacer una lista de especies de interés, incluyendo las especies objetivo, especies simátricas (co-ocurrencia) dentro del mismo taxa (generalmente nivel de orden o familia), y especies alopastrices estrechamente relacionadas, aquellas que pueden no estar en la misma ubicación geográfica que el objetivo (Figura 1).
NOTA: Aquí, las poblaciones del río Clinch de la especie A. ligamentina fueron blanco de ataques. - Buscar y descargar secuencias de múltiples regiones genéticas para especies en la lista del Paso 1. Se pueden utilizar bases de datos de secuencia como NCBI (Centro Nacional de Información Biotecnológica), BOLD (Código de Barras de Base de Datos de Vida), EMBL (Laboratorio Europeo de Biología Molecular) y DDBJ (Dna Data Bank of Japan). NCBI, EMBL y DDBJ comparten información de secuencia.
- Utilizando la base de datos de nucleótidos del NCBI, busque el organismo objetivo (por ejemplo, Actinonaias ligamentina)y la región genética (por ejemplo, citocromo c oxidasa I (COI) o NADH-dehydrogenasa 1 (ND1); Ejemplo de cadena de búsqueda: Actinonaias ligamentina Y ND1)
- A continuación, seleccione todas las secuencias que coincidan con las especificaciones y seleccione Enviar a. Elija Complete record, File y download format as GenBank o FASTA y, a continuación, Create File. Estas secuencias ahora se guardan en el equipo.
- Repita estos pasos para todas las especies de la lista definida en el paso 1. Mantenga secuencias para cada región genética en un archivo independiente, ya que se analizarán por separado.
- Descargue todas las secuencias relevantes (o una proporción grande y representativa de secuencias) para las especies objetivo identificadas en el Paso 1. Incluya variantes geográficas si es posible.
- Repita las secuencias de búsqueda y descarga de especies no objetivo relacionadas y simpatrices del mismo grupo taxonómico que se identificaron en el Paso 1 (por ejemplo, si la especie objetivo es el mejillón (A. ligamentina) secuencias de descarga para todas las demás especies de mejillón de agua dulce en la Unión Familiar que se producen en el sistema de interés).
- Repita la búsqueda y descarga de especies estrechamente relacionadas pero alopastrices (geográficamente separadas) enumeradas en el Paso 1.1.
NOTA: No todas las especies (objetivos y no objetivos) estarán disponibles en las bases de datos públicas. Aumente la base de datos de referencia local amplificando y secuenciando especímenes de interés interno verificados taxonómicamente. Si se trabaja con una especie que tiene una alta diversidad genética dentro de las especies o que trabaja en un área geográficamente grande donde se pueden esperar variantes geográficas, reúna secuencias de toda la gama.
2. Diseño de ensayo
- Alinee secuencias de cada región genética por separado utilizando software de alineación que se puede encontrar en varios programas bioinformáticas y de edición de secuencias genéticas. Haga esta alineación para cada una de las diferentes regiones genéticas.
- Por ejemplo, utilizando el software Geneious Prime (https://www.geneious.com) importa los archivos de secuencia descargados en el programa.
- Cree carpetas independientes para cada región genética.
- Dentro de una carpeta que contiene secuencias de una región genética, seleccione todas las secuencias.
- Utilice la herramienta Alineación múltiple para crear una alineación de nucleótidos de las secuencias seleccionadas. Puede haber varias opciones para el tipo de alineación, utilizando las alineaciones Geneious o MUSCLE y los parámetros predeterminados funcionan bien.
- Elija regiones prometedoras para el diseño de ensayos mediante la visualización de datos de secuencia alineados. Una región que tiene una gran cantidad de datos de secuencia disponibles para las especies de interés, es altamente divergente entre las especies, y muestra que la baja variación dentro de las especies es un buen candidato. Esto aumentará la probabilidad de que las imprimaciones y las sondas diseñadas sean capaces de discriminar el objetivo de especies no objetivo, al tiempo que garantizarán que las variantes intraespecíficas se amplíen con el ensayo.
- Diseño de imprimaciones de ensayo y sonda.
- Utilice el software de diseño de ensayos qPCR y siga las instrucciones. La herramienta PrimerQuest (https://www.idtdna.com/) de IDT para diseñar 5 conjuntos de ensayos qPCR se utilizó aquí.
- Pegue la secuencia seleccionada en el paso 2.2 en el cuadro de entrada Secuencia. Si la alineación crea espacios, elimínelos de la secuencia.
- Seleccione qPCR 2 Imprimaciones + Sondeo en la opción Elegir su diseño.
- Descargue los ensayos recomendados.
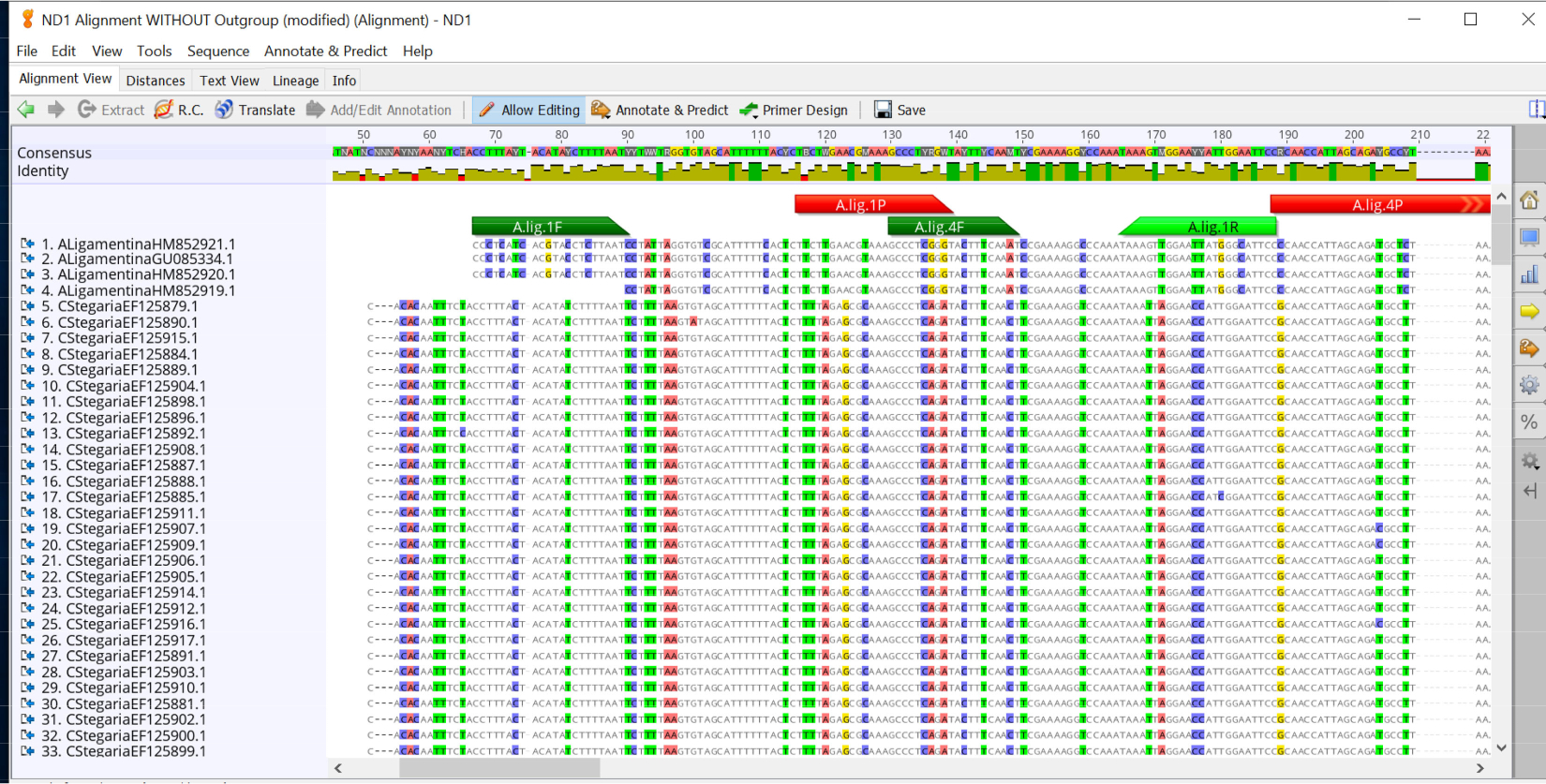
- Copie las secuencias de la imprimación hacia delante del primer ensayo y busque esta secuencia de imprimación en la alineación creada en el paso 2.1.4. Si utiliza Geneious Prime, utilice la herramienta Anotar y predecir para agregar la región de imprimación a la alineación. Haga esto para todas las combinaciones de imprimación y sonda (Figura 2).
- Inspeccione estas regiones de la alineación en busca de variación dentro de las especies objetivo, así como dentro de las especies que coexisten.
- Si hay variación genética intraespecífica, busque ensayos donde las imprimaciones y la sonda no se encuentran dentro de estas regiones.
- Para evitar la amplificación de especies no objetivo, busque desajustes con especies no objetivo. Elija los ensayos con más discrepancias para una mayor validación. (2018) sugieren elegir conjuntos con al menos dos de las tres regiones (las dos imprimaciones, o una imprimación y una sonda) que tengan al menos dos desajustes con todas las especies no objetivo. Sin embargo, tenga en cuenta que los desajustes en la sonda contribuyen menos a la especificidad10.
NOTA: Las diferencias dentro de 3 pares base del extremo de 3' de cada imprimación aumentan la especificidad mejor que las diferencias en el extremo de 5' de las imprimaciones10.
- Tenga en cuenta los siguientes parámetros importantes en el diseño del ensayo.
- Determine las temperaturas de fusión y recocido de las imprimaciones y la sonda. Idealmente, la temperatura de fusión (Tm) de las imprimaciones debe ser entre 60-64 °C y dentro de 2 °C entre sí, y el Tm de la sonda debe ser 6-8 grados más alto que el Tm de las imprimaciones. Ajuste la temperatura de recocido (Ta) de la reacción qPCR 5 °C por debajo de la temperatura de fusión, alrededor de 55-60 °C11.
- Examine el contenido de GC. Elija entre un 35 % y un 65% de contenido GC y evite regiones con 4 O más Gs consecutivos. Tener 1 o 2 Gs o Cs en las 5 últimas bases del extremo de 3' de la imprimación (abrazadera GC) podría aumentar la especificidad, ya que ayudaría a la imprimación a hacer un vínculo más fuerte12.
- Busca estructuras de horquilla y dimer. Ensayo de imprimaciones y sonda para estructuras de horquillas y atenuadores predichos utilizando un programa de análisis de oligonucleótidos (por ejemplo, OligoAnalyzer -IDT13; OligoCalculator14). Estas estructuras pueden causar amplificación no objetivo y menor eficiencia. Evite los ensayos que se predicen para formar estas estructuras.
- Determine la longitud de imprimación. Apunta a imprimaciones entre 18-25 bases de longitud y longitud de la sonda entre 20 –25 bases. Las imprimaciones y sondas más largas pueden tener una menor eficiencia de amplificación.
- Determine la longitud del amplificador. Debe estar entre aproximadamente 100 y 250 pares base. Este rango es generalmente lo suficientemente corto como para una alta eficiencia de PCR, pero lo suficientemente largo como para facilitar la verificación por sanger secuenciación4,15.
- Sondas de diseño. Asegúrese de que las sondas no tengan una base G en el extremo de 5', ya que podría amortiguar la señal de los colorantes verdes y amarillos11. Diseñamos sondas de doble enfriamiento, con saciados IDT 3IABkFQ y ZEN y fluoróforos FAM o HEX.
NOTA: Determine las sondas MGB: Las sondas TaqMan MGB (aglutinante de ranura menor) se utilizan a menudo para estudios de eDNA. Sin embargo, dado que estas sondas son muy cortas, pueden enlazarse a no destinos incluso con un desajuste de 2 o 3 pares base10. - Determine la sonda Tm. La temperatura de fusión de la sonda debe ser 6-8 °C más alta que las imprimaciones. Las temperaturas más bajas disminuyen el éxito de unión de la sonda.
- Determine la longitud y la ubicación de la sonda. La sonda debe tener entre 20 y 25 bp de longitud e idealmente situada cerca del sitio de encuadernación de imprimación en la misma hebra sin solaparla.
3. Evaluación y optimización de ensayos
- En el desarrollo y pruebas de ensayos silico. Antes de ordenar conjuntos de primer sondeo, evalúe la especificidad (amplificación potencial no objetivo) probando la amplificación de imprimación en silico.
- Imprimaciones de prueba a través de la Primera Explosión16 de NCBI o programas similares que pueden identificar posibles no objetivos en la base de datos NCBI nt/nr que podrían amplificarse con el ensayo. Si utiliza imprimaciones de pasta de primer plano en el cuadro Usar mi propia imprimación en Parámetros de imprimación. En las opciones Parámetros de comprobación de especificidad del par de imprimación, seleccione nr como base de datos y escriba la Orden del organismo de interés (por ejemplo, "Unionida" o "Unionoida") en el cuadro Organismo.
- Continúe evaluando conjuntos de imprimación/sondeo visualmente en datos de secuencia alineados.
- Para evaluar imprimaciones y sondeos al mismo tiempo en silico, cree una cadena de texto de la imprimación delantera, 12 N, la sonda, 12 N y el complemento inverso de la imprimación inversa. Si la secuencia de sondeo está dentro de 12 pares base de una de las imprimaciones, utilice el número de N correspondiente al número de pares base entre la imprimación y la sonda.
- Utilice la búsqueda de nucleótidos explosión (Blastn) de NCBI para buscar en la base de datosnr 17. Utilice la pestaña Taxonomía para buscar especies no objetivo con pocos desajustes; estos deben ser probados en el laboratorio durante la optimización del ensayo.
NOTA: En las pruebas silico ayuda a descartar ensayos no específicos, pero los ensayos potencialmente específicos deben ser probados empíricamente (in vitro), ya que no todas las especies tienen secuencias en las bases de datos genéticas y la imprimación y las sondas todavía pueden unirse a no objetivos, incluso si el software lo considera poco probable.
- Elija de tres a cinco combinaciones de imprimación/sonda para probar en el laboratorio.
- Ordene imprimaciones, sondas y un estándar de ADN sintético, así como imprimaciones adicionales de cola M13 para la secuenciación de amplificadores.
- Pida imprimaciones sintéticas de oligonucleótidos y sondas de una empresa que fabrica oligos. Las sondas están etiquetadas con un tinte fluorescente y un quencher. Se deben seleccionar diferentes fluoróforos para ensayos que necesitan ser multiplexados. Compruebe si su instrumento qPCR busca una lista de los fluoróforos que el instrumento puede detectar.
- Diseñe y ordene imprimaciones M13-tailed para la verificación de las detecciones qPCR con secuenciación Sanger agregando la secuencia M13 Forward (-20), GTA AAA CGA CGG CCA GT, al final de 5' de la imprimación delantera, y la secuencia M13 Reverse (-27), CAG GAA ACA GCT ATG AC, al final de la imprimación inversa de 5'.
- El estándar de ADN sintético contiene la secuencia de destino (incluidas las regiones de imprimación) a una concentración conocida en copias/μL. Cuantificar muestras desconocidas basadas en una curva realizada por concentraciones conocidas de esta norma (es decir, la curva estándar). Adquirir el estándar sintético de la misma empresa que fabrica las imprimaciones y la sonda. Siga las recomendaciones del fabricante para la resuspensión y el almacenamiento. Diluya los estándares en el tampón TE con un portador de ARN utilizando utensilios plásticos de baja retención para reducir la hidrólisis y la unión a las superficies.
NOTA: Si la curva estándar no funciona bien (mala eficiencia de PCR, consulte el paso 3.4.2), intente volver a suspender el estándar en agua o Tris-HCl. - Suspenda las imprimaciones y las sondas en agua libre de nucleasa, Tris-HCl o Tampón TE a concentraciones convenientes para su uso en ensayos. Generalmente, diluya las existencias de trabajo 20 veces en la mezcla maestra para lograr la concentración de ensayo final optimizada. Almacene los oligos suspendidos a una constante de -20 °C cuando no esté en uso.
- La optimización y las pruebas de ensayos in vitro (en el laboratorio). Rechazar los ensayos que tienen poca eficiencia, reacción cruzada con especies co-ocurrencias, o tienen poca sensibilidad18. Incluya el uso de un control positivo interno (IPC) durante el desarrollo del ensayo, así como al ejecutar muestras reales.
- En primer lugar, encuentre los valores óptimos de temperatura y concentración de imprimación/sonda para el ensayo. Una vez optimizados estos parámetros para la eficiencia de la PCR (Paso 3.4.2), reactividad cruzada (Paso 3.4.3) y sensibilidad (Paso 3.4.4), proceda a probar el ensayo con un IPC multiplexado (Paso 3.4.5).
- Pruebe la temperatura óptima de recocido (Ta) para la imprimación y las sondas utilizando un gradiente de temperatura PCR centrado 5 ° C por debajo de la imprimación media prevista Tm.
- Pruebe las concentraciones óptimas de imprimación y sonda. Típicamente, 200 nM, 400 nM, y 800 nM concentraciones de imprimación y 75 nM, 125 nM, y 200 nM concentraciones de sonda se prueban.
- Cree una curva estándar y determine la eficiencia y el rango lineal. Pruebe al menos seis diluciones de 10 veces de un estándar de ADN sintético que contenga la secuencia objetivo, a aproximadamente 100 copias/reacción a 105 copias/reacción (Figura 3A).
- Utilice el software qPCR para trazar el valor Cq (umbral para el ciclo en la cuantificación) de cada estándar en el eje y y la base de registro 10 de la concentración estándar inicial en copias/reacción en el eje X. El software qPCR debe ejecutar automáticamente una regresión lineal (Figura 3B).
- Calcular la eficiencia desde la pendiente de la regresión, E = -1 + 10(-1/pendiente). Por ejemplo, si la pendiente es -3.4, E= -1 + 10(0.29) = 0.97 o 97%. Compruebe también los valores r2 que indican qué tan bien encajan las réplicas estándar en la curva. El software qPCR debe calcular esto automáticamente también (Figura 3B). Aspirar a valores de eficiencia del 100% (±10 %) y r2 valores de ≥0,989,15,19,20,21,22.
- Inspeccione visualmente la curva estándar en busca de sesgo, es decir, desviaciones de la regresión en una dirección coherente o para un rendimiento de curva estándar deficiente medido por la eficiencia y los valores r2 (Figura 3C y 3D).
- Especificidad: Evaluar la reactividad cruzada con especies no objetivo para disminuir la probabilidad de falsos positivos. Cuando las detecciones de eDNA puedan dar lugar a costosas decisiones de gestión, verifique las detecciones positivas mediante secuenciación de amplificadores.
- No objetivos: Ejecutar el ensayo contra las extracciones genómicas de ADN de especímenes taxonómicamente verificados de especies relacionadas y de especies geográficamente co-ocurrencias; con la máxima prioridad es probar contra especies estrechamente relacionadas, co-ocurrencia. Utilice concentraciones totales de ADN similares para muestras objetivo y no objetivo. La concentración elegida debe producir amplificación a partir de muestras de especies objetivo cerca del medio del rango lineal de la curva estándar. La amplificación debe observarse únicamente con las especies objetivo.
- Si se observa una amplificación no objetivo, limpie y secuencia el producto para verificar su identidad. No es raro observar la contaminación de las especies objetivo en muestras de tejido de especies no objetivo, por lo que todas las amplificaciones en esta etapa deben verificarse mediante secuenciación. Reamplifique los amplificadores limpios de las pruebas de especificidad utilizando las imprimaciones de cola M13 y la secuencia con imprimaciones M13.
- En el laboratorio post-PCR, transfiera los productos qPCR que se secuenciarán a tubos frescos. Retire las imprimaciones residuales y los componentes de reacción con un kit de limpieza (por ejemplo, MinElute PCR Purification Kit).
- Hacer 1:100 diluciones de las eluciones y amplificar 1 μL de cada uno durante 30 ciclos en una reacción PCR de 50 μL con las imprimaciones de cola M13 y una polimerasa de alta fidelidad (por ejemplo, Phusion High-Fidelity DNA Polymerase).
- Ejecute 10 μL de cada reacción en un gel de agarose del 1% para comprobar si hay una sola banda del tamaño esperado. Si no se observa ninguna banda, aumente el número de ciclos o la cantidad de muestra. Si se observan varias bandas, el gel purifica la banda del tamaño esperado.
- Retire las imprimaciones residuales y los componentes de reacción con un kit de limpieza como el anterior y mida las concentraciones de ADN de las eluciones.
- Configure las reacciones de secuenciación con las imprimaciones M13 de acuerdo con las instrucciones de la instalación de secuenciación.
NOTA: Nunca abra muestras amplificadas en el laboratorio qPCR. Prepare muestras para la secuenciación en un laboratorio dedicado a muestras post-PCR.
- Sensibilidad: La sensibilidad afecta la posibilidad de falsos negativos, o fallas en la detección del ADN de las especies objetivo cuando está presente. Evalúe el límite de detección (LOD) y el límite de cuantificación (LOQ) para cada ensayo. Por último, incluya un control positivo interno (IPC) para evaluar la inhibición de la PCR de las muestras. Multiplex y pruebe este ensayo IPC con el ensayo diseñado para asegurarse de que los dos ensayos no interfieren entre sí.
- LOD: Haz seis diluciones en serie 4 veces del estándar de ADN sintético, con 8-24 réplicas por dilución estándar(Figura 4). Calcule la concentración inicial más baja con un 95% de detección. Las gráficas LOD y LOQ se pueden generar con un script R de calculadora LOD/LOQ5.
NOTA: Los datos debajo de la LOD no deben ser censurados. Debido a la especificidad de la PCR, no hay un límite inferior para los verdaderos positivos. La LOD es la concentración más alta por debajo de la cual se puede esperar que ocurran falsos negativos. - LOQ: A partir de la misma serie de dilución, calcule la concentración estándar inicial de ADN más baja cuantificable con un coeficiente de variación (CV) por debajo del 35%.
NOTA: Lod y LOQ deben notificarse en copias/reacción. Cuando se utiliza un ensayo validado y las muestras de campo se amplifican por debajo del LOQ, los resultados deben notificarse como detecciones % en lugar de concentraciones de eDNA, porque la concentración exacta no se puede medir con confianza5.
- LOD: Haz seis diluciones en serie 4 veces del estándar de ADN sintético, con 8-24 réplicas por dilución estándar(Figura 4). Calcule la concentración inicial más baja con un 95% de detección. Las gráficas LOD y LOQ se pueden generar con un script R de calculadora LOD/LOQ5.
- Utilice un control positivo interno (IPC) para probar la inhibición de la PCR. La inhibición puede conducir a una disminución de la sensibilidad y falsos negativos. Pruebe la capacidad del ensayo IPC que se va a multiplexar con el ensayo de destino.
- Un ensayo IPC se puede multiplexar con el ensayo de destino utilizando una sonda con un tinte de reportero diferente al ensayo de destino. Este ensayo IPC consiste en una secuencia corta de ADN sintético de una especie no relacionada con el taxón objetivo, incorporado en la mezcla maestra qPCR a una baja concentración de aproximadamente 102 copias/reacción, junto con imprimación y sondas que lo detectan. Esta concentración más baja es necesaria para evitar la competencia con la secuencia objetivo para la polimerasa y los nucleótidos23.
- Compare el valor Cq de la plantilla IPC del ejemplo con el de la plantilla IPC en el control sin plantilla. En este control sin plantilla (NTC), la única entrada de ADN es la de la plantilla IPC. La plantilla IPC en esta reacción debe amplificarse como se esperaba. Si la plantilla IPC de una muestra amplifica a 2 o más ciclos diferentes de la de la plantilla IPC en el NTC, se inhibe la muestra eDNA. Las muestras que muestran inhibición se pueden diluir 1:10 y volver a analizarse. Si una muestra permanece inhibida, esa muestra debe eliminarse del análisis.
- En primer lugar, encuentre los valores óptimos de temperatura y concentración de imprimación/sonda para el ensayo. Una vez optimizados estos parámetros para la eficiencia de la PCR (Paso 3.4.2), reactividad cruzada (Paso 3.4.3) y sensibilidad (Paso 3.4.4), proceda a probar el ensayo con un IPC multiplexado (Paso 3.4.5).
- Desarrollo y pruebas in situ del ensayo
- En el laboratorio: Si se dispone de acceso al organismo en el laboratorio, así como a especies simátricas; tomar muestras de agua de los recintos con estas especies, procesar las muestras y probar el ensayo contra estas muestras de eDNA. Secuenciar productos como se ha mencionado anteriormente para verificar la amplificación del objetivo previsto utilizando las imprimaciones de cola M13.
- En el campo:
- Identificar los sitios donde se sabe que ocurre el organismo objetivo y se sabe que no ocurren. Es preferible tener alguna medida de abundancia en cada sitio donde se produce la especie objetivo.
- Decida qué volúmenes de muestra y métodos de recolección de muestras (por ejemplo, filtración, centrifugación, etc.) se utilizarán.
- Incluya un control en blanco o negativo en cada sitio, se trata de agua limpia que se ha traído al sitio de campo y luego se recoge y prepara con el mismo equipo de campo y protocolos utilizados para el muestreoeDNA 24. El propósito del campo en blanco es detectar la contaminación del equipo de muestreo y el equipo de campo traídos al sitio. Lleve el campo en blanco antes de procesar muestras de agua de campo.
- Tome varias muestras de agua por sitio, preferiblemente 3 muestras por sitio.
- De vuelta en el laboratorio, procesar y extraer muestras.
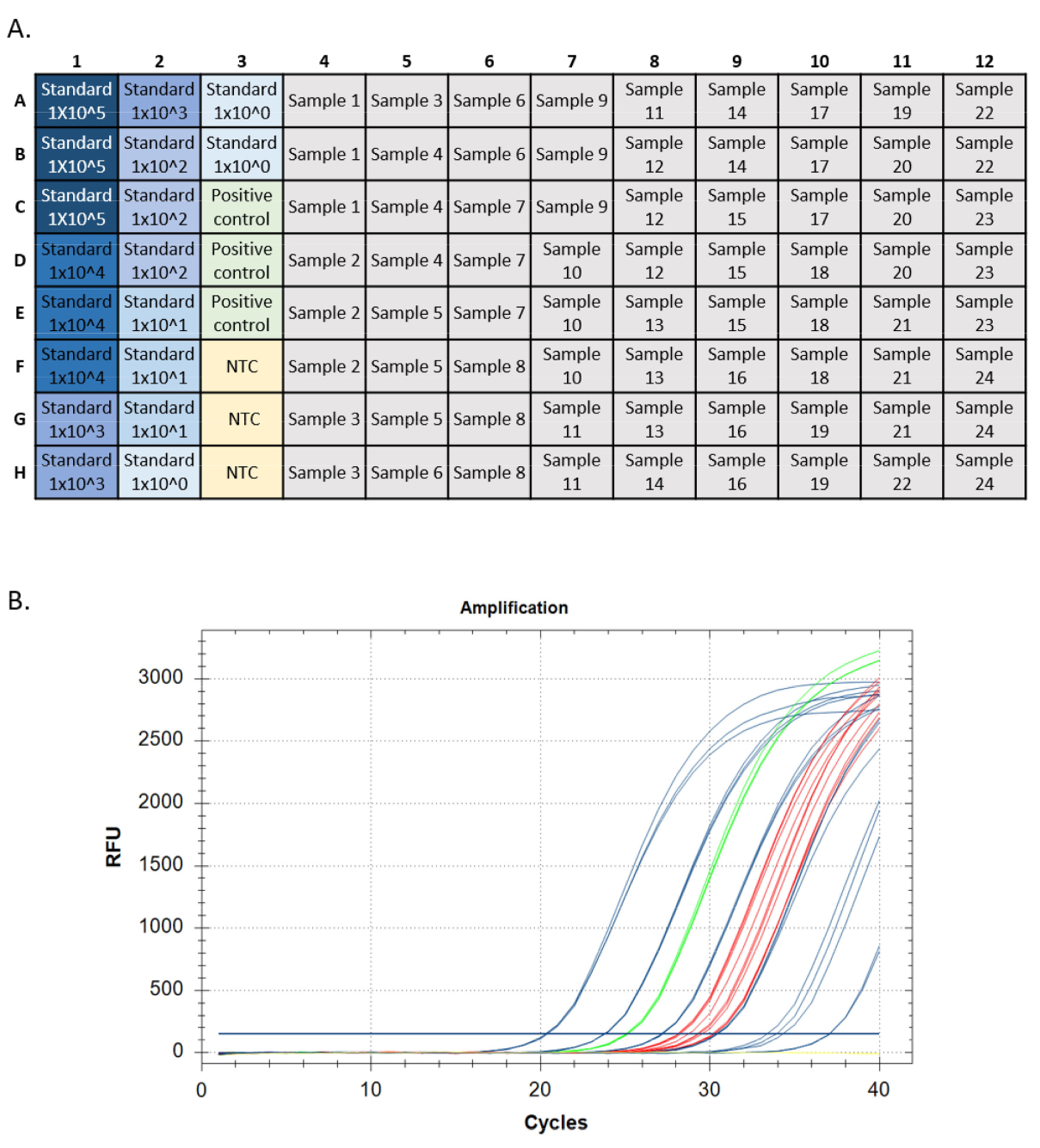
- Ejecute el ensayo utilizando una placa configurada de forma similar a la Figura 5A y compare la frecuencia de concentración y detección de eDNA con las diferencias de sitio conocidas en ocurrencia y abundancia. Confirme todas las detecciones secuenciando24,25.
NOTA: Lo anterior validaría un ensayo a través del nivel 4 de la escala6 (optimización del rendimiento técnico del ensayo) y comenzaría a recopilar datos compatibles con la validación de ensayos de nivel 5. El nivel 5 incorpora el modelado de probabilidad y el uso del ensayo para estudios de ecología eDNA. Creemos que esto está fuera del alcance del desarrollo básico de ensayos, pero alentamos estas aplicaciones de ensayos de laboratorio y de campo investigados para mejorar el diseño de ensayos y la interpretación de datos.
Resultados
En el diseño de un ensayo qPCR específico de la especie para el mucket(A. ligamentina),se descargaron secuencias disponibles de todas las especies de Unionidae en el río Clinch. Especies estrechamente relacionadas como Lampsilis siliquoidea también fueron incluidas en la base de datos de referencia a pesar de que no se encuentran en el mismo río. No todas las especies en el sistema fluvial de interés se encontraron en GenBank, por lo que se secuenciaron especies adicionales en casa. Las secuencias se alinearon utilizando el software Geneious y el software Primer Quest (IDT) se utilizó para diseñar múltiples ensayos. Se agregaron cinco conjuntos de imprimaciones y sonda a la alineación para la evaluación visual (Figura 2). Luego fueron probados en silico usando Primer-Blast, después de lo cual se les ordenó para más pruebas invitro. En el laboratorio, todos los ensayos fueron probados utilizando extracciones de ADN de 27 especies disponibles para verificar la especificidad. Un ensayo (A.lig.1) amplificó con éxito sólo las especies objetivo (Cuadro 1; Tabla 2). Este ensayo avanzó para nuevas pruebas de eficiencia de ensayos, LOD y LOQ. Tiene una longitud de amplificador de 121 pares base. La Tabla 3 muestra la secuencia utilizada para el estándar de ADN sintético A. ligamentina. La Figura 3A y la Figura 3B muestran los resultados de un ensayo exitoso con buena eficiencia y valores r2. La Figura 3C y la Figura 3D muestran un ensayo cuya curva estándar tiene una eficiencia deficiente; este ensayo fue descartado. Se constató que la LOD y la LOQ para el ensayo seleccionado (A.lig.1) eran 5.00 copias/reacción utilizando el método discreto descrito en Klymus et al5. El IPC que se mintió con el ensayo(Tablas 3-6)no afectó la curva estándar del ensayo A. ligamentina. El IPC que usamos es un fragmento de la transcripción hemT del ratón. IDT prediseñado este ensayo para otra aplicación, pero modificamos su uso como IPC para las aplicaciones eDNA de nuestro laboratorio.
Una ejecución qPCR exitosa debe cumplir ciertos criterios para cada medida de rendimiento (es decir, amplificación de curva estándar, control positivo de ADN genómico, sin control de plantilla y control positivo interno). Los estándares de ensayo de destino deben tener curvas de amplificación exponenciales. Estas curvas deben alcanzar una meseta de punto final si se permite correr suficientes ciclos. Esto es indicativo de que la sonda fluorescente se consume completamente durante la reacción, y los niveles de fluorescencia alcanzan un límite máximo. Los estándares de amplificación posteriores pueden no alcanzar una meseta en 40 ciclos. Los controles positivos (ADN genómico e IPC) deben tener el mismo patrón. Las incógnitas pueden amplificarse o no, pero la amplificación en incógnitas también debe tener un patrón exponencial y una meseta de punto final (Figura 5).
En un qPCR de calidad, las diluciones estándar amplifican a Cq espaciado uniformemente de aproximadamente cada 3,3 ciclos por cada diferencia de 10 veces en la concentración. Cada réplica de una dilución estándar amplifica de una manera estrechamente agrupada que tiene casi el mismo Cq (representado por los valores r2). Todas las diluciones estándar deben exhibir amplificación (Figura 3A). En un qPCR deficiente, los estándares pueden exhibir forma no exponencial, variación desigual en los valores Cq entre diluciones, no llegar a una meseta de punto final, o algunas diluciones pueden no amplificarse en absoluto (Figura 3D).
Los parámetros importantes para una curva estándar son la eficiencia, r2,pendiente e intercepción y. La eficiencia debe caer entre 90%-110% con valores ideales cercanos al 100% y r2 los valores deben estar por encima de 0,98 con resultados ideales que se acerquen a 1,015,22. Los valores de pendiente deben estar entre -3.2 y -3.5 con resultados ideales cercanos a -3.322. Los valores de e-interceptar deben caer entre un Cq de 34-41 con resultados ideales que tienen un Cq de 37.0. La intercepción y es el Cq predicho de una reacción con 1 copia de la secuencia de destino, la unidad más pequeña que se puede medir en un solo qPCR. Es probable que se inhiban las incógnitas con Cq que la interceptación y. Ejecutar más de 40 ciclos de PCR puede ser necesario para detectar el objetivo en caso de inhibición o un conjunto de imprimación ineficiente, sin embargo la cuantificación no es posible en estas circunstancias y se deben ejecutar controles negativos adicionales sin la secuencia objetivo, pero que contenga ADN total similar a las incógnitas, para descartar la amplificación de fuentes no específicas.
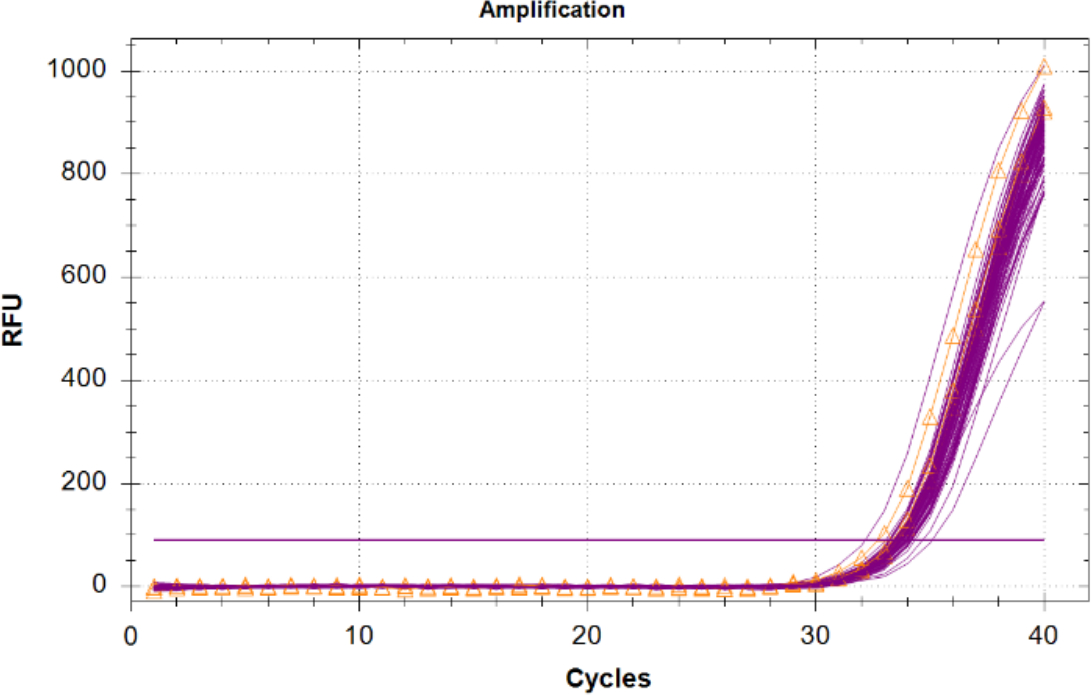
La amplificación del Control Positivo Interno (IPC) en muestras desconocidas debe compararse con los resultados del ipc de control de plantilla negativo, ya que no hay competencia para reactivos y no hay inhibidores presentes. Las incógnitas con un IPC que tienen un Cq de 2 ciclos o superior al valor promedio de Cq de la NTC, o que no amplifican deben considerarse inhibidos. Si no hay inhibidores presentes en las muestras, toda amplificación IPC debe tener una agrupación estrecha en la gráfica con valores Cq casi los mismos que la NTC (Figura 6).
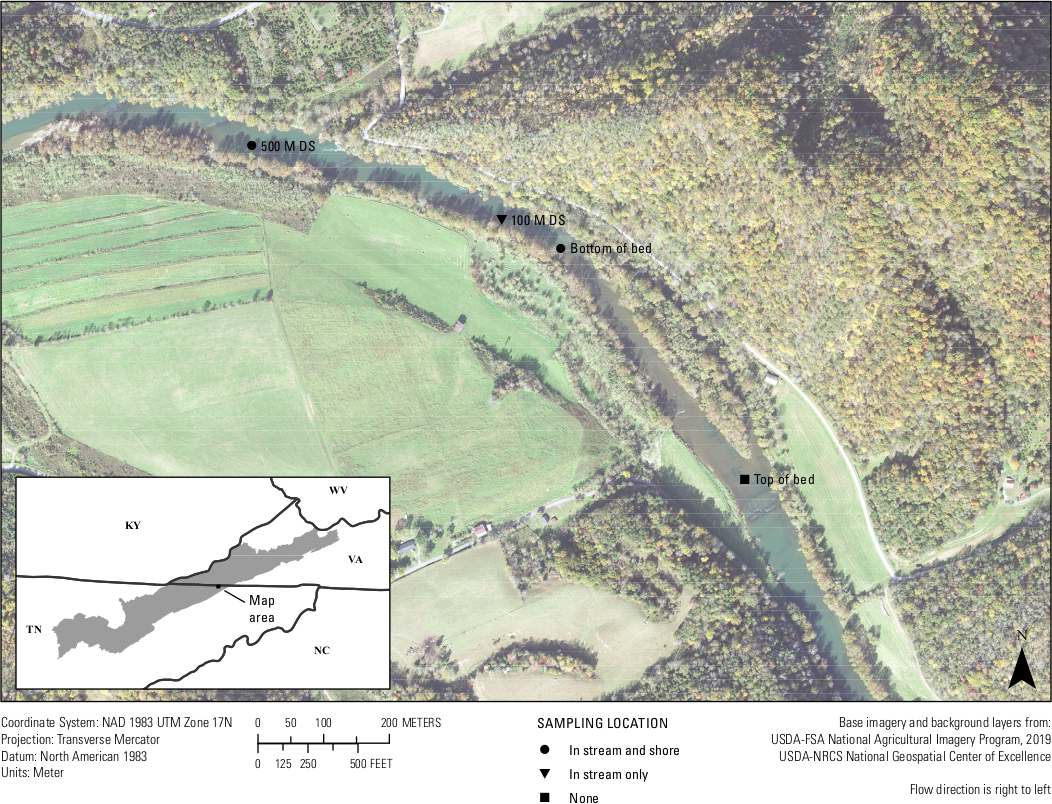
Finalmente, se realizaron pruebas in situ del ensayo. Entre el 25 y el 26 de septiembre de 2019 se filtraron veinte muestras de agua del río Clinch y tres muestras en blanco de campo a menos de 500 metros de un lecho de mejillón conocido por tener A. ligamentina. Se filtraron aproximadamente cuatro muestras de agua de 1 L por lugar de muestreo. Lugares incluidos en la parte inferior del lecho de mejillón en corriente, parte inferior del lecho de mejillón cerca de la orilla, 100 m aguas abajo de la cama en corriente, 500 m aguas abajo de la cama en corriente y 500 m aguas abajo de la cama cerca de la orilla (Figura 7). De vuelta en el laboratorio, cada filtro se cortó por la mitad y el ADN se extrajo de sólo la mitad de un filtro. La mitad restante del filtro para cada muestra se almacenó en un congelador de -80 °C. A continuación, las muestras se ejecutaron utilizando el ensayo A.lig.1 multiplexado con el IPC. De las 23 muestras, cinco fueron encontradas inhibidas. Estas muestras se diluyeron 1:10 y las diluciones se volvieron a ejecutar. Diecinueve de las 20 muestras de campo amplificadas utilizando el ensayo diseñado. De estas 19 muestras, cinco estaban por encima de la LOD y loq del ensayo de 5 copias/reacción; lo que significa que la mayoría de las muestras tenían una detección de eDNA, pero en un nivel donde es probable que se produzcan resultados negativos falsos y que el ensayo no podría cuantificar con confianza el número de copia de esas 14 muestras. Sin embargo, entre el 75 y el 100% de las cuatro réplicas biológicas amplificadas en cada lugar de muestreo. Dos de los tres espacios en blanco de campo fueron negativos, mientras que un campo en blanco mostró amplificación, haciendo hincapié en la importancia de la técnica limpia en el campo.

Figura 1: Flujo de trabajo para la construcción de bases de datos de secuencias de ADN mitocondriales. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Alineaciones de secuencia para especies de mejillón del río Clinch con posibles imprimaciones y sondas para el ensayo Actinonaias ligamentina ND1. Imprimaciones delanteras en verde oscuro, sonda en rojo y imprimación inversa en verde claro. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Ejemplos de curva estándar y regresión lineal. A. Ejemplo de una curva estándar aceptable derivada de la amplificación de tres réplicas cada una de las seis diluciones estándar. Una serie de dilución estándar de 10 veces con la mayor concentración del estándar a la izquierda, con concentraciones decrecientes moviéndose hacia la derecha. La línea horizontal que cruza todos los trazados es el umbral para el ciclo en quantitation (Cq). Donde cada seguimiento cruza este umbral es donde se determina el Cq. B. Regresión lineal hecha de las réplicas estándar de la Figura 3A. Las réplicas de las diluciones estándar se trazan en círculos y las incógnitas (muestras) se trazan con x. La eficiencia es del 98,9%, r2 acercándose a 1,0, y pendiente de -3.349. C. Ejemplo de una curva estándar deficiente derivada de la amplificación de tres réplicas cada una de las seis diluciones estándar. D. Una regresión lineal que forma la curva estándar para las réplicas estándar amplificadas en el ejemplo 3C. Tenga en cuenta la mala eficiencia y los valores r2. También tenga en cuenta que sólo 4 de los 6 estándares amplificados. Si después de repetir las corridas, la curva estándar no mejora, el problema puede ser con un conjunto de imprimación/sonda deficiente que no amplifica el ADN objetivo como se esperaba en cuyo caso, este ensayo no debe considerarse. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: Ejemplos de configuraciones de placas para las ejecuciones qPCR estándar LOD y LOQ. Los estándares utilizados en la curva son en azul, la concentración estándar disminuye de oscuro a azul claro. Control positivo del ADN en verde y sin control de plantilla (NTC) en amarillo. Concentraciones estándar experimentales en gris que muestran 24 réplicas para cada dilución estándar. La serie de dilución se basó en dos placas (A, B), cada una con una curva estándar, control positivo y NTC. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: Trazas de configuración y amplificación de placas de una ejecución qPCR. A. Configuración de placa, estándares que se muestran en color azul, más oscuro que indica la mayor concentración del estándar. Control positivo del ADN en verde, sin controles de plantilla en amarillo (NTC), objetivos de muestra en gris. B. Trazas de amplificación de una ejecución qPCR. Estándares mostrados en azul, control positivo del ADN en verde, sin controles de plantilla en amarillo, y desconocidos en rojo. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 6: Trazas de amplificación para el Control Positivo Interno (IPC). Trazas IPC para todas las muestras desconocidas en magenta y el IPC de los controles sin plantilla (NTC) mostrados en naranja con triángulos. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 7: Mapa que muestra los sitios de recolección de eDNA de un lecho de mejillón en el río Clinch a lo largo de la frontera entre Virginia y Tennessee. Se recogieron muestras en Wallens Bend en la parte inferior de la cama, a 100 m aguas abajo de la cama y a 500 m aguas abajo de la cama. Los sitios fueron recogidos en medio del arroyo (en arroyo) o aproximadamente 1 – 2 metros de la costa (costa). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| componente | nombre | Secuencia 5' – 3' | Etiqueta fluorescente | |
| Primer delantero | A.lig.1-f | CCCTCATCACGTACCTCTTAATC | ||
| Introducción inversa | A.lig.1-r | GGAATGCCCATAATTCCAACTTTA | ||
| sonda | Sonda A.lig.1 | TTCTTGAACGTAAAGCCCTCGGGT | Fam | |
Tabla 1: El ensayo actinonaias ligamentina qPCR diseñado (A.lig.1) que incluye secuencias para las imprimaciones hacia delante e inversas y la sonda.
| especie | amplificado | En el río Clinch |
| 1. Actinonaias ligamentina | Sí | Sí |
| 2. Actinonaias pectorosa | No | Sí |
| 3. Amblema plicata | No | Sí |
| 4. Corbicula spp. | No | Sí |
| 5. Cumberlandia monodonta | No | Sí |
| 6. Ciclonaias tuberculata | No | Sí |
| 7. Cyprogenia stegaria | No | Sí |
| 8. Elliptio dilatata | No | Sí |
| 9. Epioblasma brevidens | No | Sí |
| 10. Epioblasma capsaeformis | No | Sí |
| 11. Epioblasma florentina aureola | No | Sí |
| 12. Epioblasma triquetra | No | Sí |
| 13. Fusconaia cor | No | Sí |
| 14. Fusconaia subrotunda | No | Sí |
| 15. Lampsilis ovata | No | Sí |
| 16. Lampsilis siliquoidea | No | No |
| 17. Lasmigona costata | No | Sí |
| 18. Lemiox rimosus | No | Sí |
| 19. Lexingtonia dolabelloides | No | Sí |
| 20. Medionidus conradicus | No | Sí |
| 21. Plethobasus cyphyus | No | Sí |
| 22. Pleurobema plenum | No | Sí |
| 23. Ptychobranchus fasciolaris | No | Sí |
| 24. Ptychobranchus subtentus | No | Sí |
| 25. Quadrula pustulosa | No | Sí |
| 26. Strophitus ondulatus | No | Sí |
| 27. Iris Villosa | No | Sí |
Tabla 2: Una lista de especies utilizadas para las pruebas de especificidad in vitro del ensayo A.lig.1. El ensayo amplió el ADN genómico del objetivo(Actinonaias ligamentina)y no amplió ninguna de las especies no objetivo.
| componente | Secuencia 5'-3' | ||||
| Norma Actinonaias ligementina | CCCTCATCACGTAC CTCTTAATCCTATTAGGTGTCGCATTTTTCACTCTTCTTGAACGTA | ||||
| AAGCCCTCGGGT ACTTTCAAATCCGAAAAGGCCCAAATAAAGTTGGAATTATGGGCATTC | |||||
| CCCAACCATTAGCAGATGCTCTAAAGCTCTTCGTAAAGAATGAGTAACACCAACCTCCT | |||||
| CAAACTACCTACCCTTCATCTTAACCCCACTATGTTAATTTTAGCACTTAGACTTT | |||||
| GACAATTATTTCCATCCTTTATANTATCATCCAAATANTTTTTGGTGCTCCTATTCT | |||||
| TGTGTATCTCCCCCCTAGCTGTTTATACAACACTTATAACAGGCTGAGCCTCAAACTCCA | |||||
| AATATGCCCTTTTAGGAGCTATTCGAGCCATAGCCCAAACCATTTCTTATGAGGTTACAA | |||||
| TAAC | |||||
| Plantilla IPC (Hem-T) | CTACATAAGTAACACCTTCTCATGTCCAAAGCTCTCTGAGTGTCCCCCACATCTCAGACGCT | ||||
| GTATGACAGTCTCCTTTCGTGTGAACATTCGGCTCTCTATGTTCTCAAGGACTGCAC | |||||
Cuadro 3: Secuencia (5'-3') de la norma de ligamentina Actinonaias y la plantilla IPC (Hem-T) utilizada para este ensayo. La secuencia de las imprimaciones hacia delante e inversas está en negrita e cursiva, y la de la sonda está subrayada.
| componente | nombre | Secuencia 5' – 3' | Etiqueta fluorescente | |
| Primer delantero | HemT-F | TCTGAGTGTCCCCCGAATCT | ||
| Introducción inversa | HemT-R | GCAGTCCTTGAGAACATAGAGC | ||
| sonda | HemT-P | TGACAGTCTCCTTTCGTGTGAACATTCG | Cy5 | |
Tabla 4: El ensayo del Control Positivo Interno (IPC), incluidas las secuencias para las imprimaciones hacia delante e inverso y la sonda.
| Volumen por muestra (μL) | componente |
| 10 | Mezcla de maestros ambientales |
| 1 | 20uM A. lig.1 Mezcla de F/R |
| 1 | Sonda 2.5uM A. lig.1 |
| 1 | Mezcla de imprimación IPC de 5uM (HemT-F/ R) |
| 0.75 | Sonda IPC de 2,5uM (HemT-P) |
| 1.5 | 1 X 103 concentración de la plantilla IPC |
| 2.75 | H20 |
| 2 | muestra |
| 20 | Volumen total |
Tabla 5: La mezcla de PCR utilizada para el ensayo A.lig.1 multiplexado con el ensayo IPC.
| paso | Temperatura (°C) | Hora | |
| 1 | Denatura inicial | 95 | 10 min |
| 2 | desnaturalizar | 95 | 15 seg |
| 3 | recocido | 60 | 1 min |
| 4 | Vaya al paso 2, repita 39X |
Tabla 6: Condiciones de reacción para el ensayo A.lig.1.
Discusión
Al igual que con cualquier estudio, definir la cuestión a abordar es el primer paso y el diseño del ensayo eDNA depende del alcance del estudio26. Por ejemplo, si el objetivo de la investigación o encuesta es detectar una o unas pocas especies, un ensayo basado en sondas objetivo es el mejor. Sin embargo, si el objetivo es evaluar una suite más grande o un conjunto de especies, los ensayos de metabarcodificación de secuenciación de alto rendimiento son más adecuados. Una vez que se determina qué enfoque tomar, se recomienda un estudio piloto que incluya diseño de ensayos, pruebas y optimización24. El diseño de ensayos comienza con una lista de especies como se describe en Figura 1. Esta lista será la base para entender qué tan bien funciona un ensayo en términos de especificidad y el rango geográfico al que podría aplicarse6,10. Se recomienda diseñar el ensayo para un área geográfica específica, permitiendo al diseñador probar mejor un ensayo de reactividad cruzada contra otras especies en esa área, y ser consciente de las limitaciones que esto tiene en la extensión de un ensayo a otras áreas donde puede ocurrir una especie objetivo24. Una vez completada la lista, las secuencias se pueden descargar de bases de datos genéticas públicas. Dado que estas bases de datos están incompletas27, uno debe secuenciar tantas especies en la lista como sea posible en casa para completar la base de datos de referencia local de secuencias que se utilizarán en el diseño de ensayos. Priorice las especies estrechamente relacionadas, ya que estas son las no metas más probables que amplificarán. Centrarse en todas las especies dentro del mismo género o familia que la especie objetivo es un buen lugar para comenzar. Las comparaciones con especies estrechamente relacionadas ayudarán a identificar regiones de secuencia únicas para las especies objetivo. Esto puede ayudar a informar cómo puede funcionar el ensayo en otros sistemas o ubicaciones. Las regiones mitocondriales son la opción habitual para el desarrollo de ensayos, porque hay más información de secuencia de una variedad más amplia de especies disponibles en genes mitocondriales que se han utilizado en proyectos de código de barras de vida, y porque el ADN mitocondrial está presente a una concentración mucho mayor en copias/células que el ADN nuclear24,28,29. Se deben evaluar varias regiones genéticas para un mayor desarrollo de ensayos, ya que la cobertura de secuencia varía entre taxones en las bases de datos de repositorio genéticos. Después de crear esta base de datos local de secuencias de referencia, se utiliza una combinación de visualización manual de datos de secuencia alineados y programas de software informático para diseñar los ensayos de imprimación/sondeo. Uno no debe confiar estrictamente en el software para determinar qué ensayos probar. Es importante verificar visualmente sobre alineaciones donde las imprimaciones y sondas se sientan en los objetivos y no objetivos para obtener una mejor comprensión de cómo podrían actuar en un PCR. Por último, la detección y optimización del ensayo incluye tres niveles (in silico, in vitro e in situ)6,7,24,25. En el diseño y pruebas silico son importantes para producir una breve lista de ensayos con una buena probabilidad de éxito, pero las pruebas empíricas (in vitro) son cruciales para seleccionar el ensayo con el mejor rendimiento real. La optimización in vitro y las pruebas de ensayos incluyen medir la eficiencia de la reacción y definir la sensibilidad y especificidad del ensayo. Los límites de detección y cuantificación son dos parámetros que a menudo se pasan por alto en el desarrollo de ensayos, pero importantes para la interpretación de datos. Al ejecutar varias réplicas de las curvas estándar para un ensayo, lod y LOQ se pueden medir fácilmente1,5,30. Pocos estudios discuten los resultados con respecto a la LOD o LOQ del ensayo, pero Sengupta et al. (2019) incorporan la LOD y loQ de su ensayo en su interpretación de datos y gráficos para una comprensión más clara de sus resultados31. Los controles positivos internos también deben multiplexarse en el ensayo diseñado. Sin pruebas de inhibición de pcr en las muestras, pueden ocurrir falsos negativos24,32. Proponemos el uso de un ensayo IPC multiplexado con el ensayo objetivo como el método más fácil para las pruebas de inhibición de PCR23. Por último, es necesario realizar pruebas in situ del ensayo a partir de muestras recogidas en campo y laboratorio para garantizar que la amplificación objetivo se produzca en muestras ambientales24.
Existen limitaciones para el uso de ensayos qPCR específicos de especies basados en sondas con muestras de eDNA. Por ejemplo, el diseño de varios ensayos para pruebas puede estar limitado por la disponibilidad de la secuencia, y puede ser necesario comprometerse en aspectos del rendimiento del ensayo. Estas opciones deben guiarse por los objetivos del estudio y deben ser notificadas con los resultados26. Por ejemplo, si el objetivo es la detección de una especie rara y se esperan pocos positivos, se podría utilizar un ensayo con especificidad imperfecta (es decir, amplificación de especies no objetivo) si todas las detecciones se verifican mediante secuenciación. Si el objetivo es monitorear el rango geográfico de una especie y no se necesitan datos de concentración de eDNA, se podría utilizar un ensayo con eficiencia imperfecta y los datos reportados sólo como detección porcentual. Además, a menos que todos los conespecíficos potenciales se prueben en el laboratorio, lo que rara vez es posible, no se puede conocer con absoluta certeza la verdadera especificidad de un ensayo. Por ejemplo, el ensayo fue diseñado y probado contra varias especies de mejillón de agua dulce en el río Clinch. Para utilizar este ensayo en un sistema fluvial diferente, tendríamos que probarlo contra un conjunto de especies en la nueva ubicación. La variación genética dentro de la especie o población que no se prueba durante el desarrollo del ensayo también podría afectar la especificidad. Por último, incluso si se ha verificado que un ensayo tiene un alto rendimiento técnico; condiciones cambian cuando se trabaja en el campo. Las condiciones no relacionadas con el ensayo, como el flujo de agua, el pH y el comportamiento animal, pueden cambiar la capacidad de detección de eDNA, al igual que el uso de diferentes protocolos de recolección y extracción de eDNA. El uso de ensayos optimizados y bien descritos ayudará a facilitar la comprensión de la influencia que tales parámetros tienen en la detección de eDNA.
El campo de la eDNA está madurando más allá de la etapa de análisis exploratorio para aumentar la estandarización de métodos y técnicas. Estos desarrollos mejorarán nuestra comprensión de las técnicas, habilidades y limitaciones de eDNA. El proceso de optimización que delineamos anteriormente mejora la sensibilidad, especificidad y reproducibilidad de un ensayo. El objetivo final de este refinamiento y estandarización de los métodos eDNA es mejorar las capacidades de los investigadores para hacer inferencias basadas en datos eDNA, así como aumentar la confianza del usuario final y del titular de la estaca en los resultados.
Divulgaciones
Los autores no declaran conflicto de intereses. Los patrocinadores de la financiación no tuvieron ningún papel en el diseño del estudio; en la recopilación, análisis o interpretación de datos; en la escritura del manuscrito; o en la decisión de publicar los resultados.
Agradecimientos
Agradecemos a Alvi Wadud y Trudi Frost que ayudaron en el desarrollo y pruebas de imprimación. La financiación para el diseño de ensayos reportado en este estudio fue proporcionada por el Programa Estratégico de Investigación y Desarrollo Ambiental del Departamento de Defensa (RC19-1156). Cualquier uso del comercio, producto o nombres de empresas es sólo para fines descriptivos y no implica la aprobación por parte del Gobierno de los Estados Unidos. Los datos generados durante este estudio están disponibles como una versión de datos del USGS https://doi.org/10.5066/P9BIGOS5.
Materiales
| Name | Company | Catalog Number | Comments |
| 96 Place Reversible Racks with Covers | Globe Scientific | 456355AST | |
| Clean gloves (ie. latex, nitrile, etc.) | Kimberly-Clark | 43431, 55090 | |
| CFX96 Touch Real-Time PCR Detection System | Bio-Rad | 1855196 | |
| Fisherbrand Premium Microcentrifuge Tubes: 1.5mL | Fisher Scientific | 5408129 | |
| Fisherbrand Premium Microcentrifuge Tubes: 2.0mL | Fisher Scientific | 2681332 | |
| Hard-Shell 96-Well PCR Plates, low profile, thin wall, skirted, white/clear | Bio-Rad | #HSP9601 | |
| IPC forward and reverse primers | Integrated DNA Technologies, Inc. | none | custom product |
| IPC PrimeTime qPCR Probes | Integrated DNA Technologies, Inc. | none | custom product |
| IPC Ultramer DNA Oligo synthetic template | Integrated DNA Technologies, Inc. | none | custom product |
| Labnet MPS 1000 Compact Mini Plate Spinner Centrifuge for PCR Plates | Labnet | C1000 | |
| Microcentrifuge machine | Various | - | Any microcentrifuge machine that hold 1.5mL and 2.0mL tubes is typically okay. |
| Microseal 'B' PCR Plate Sealing Film, adhesive, optical | Bio-Rad | MSB1001 | |
| Nuclease-Free Water (not DEPC-Treated) | Invitrogen | AM9932 | |
| Pipette Tips GP LTS 1000 µL F 768A/8 | Rainin | 30389272 | |
| Pipette Tips GP LTS 20 µL F 960A/10 | Rainin | 30389274 | |
| Pipette Tips GP LTS 200 µL F 960A/10 | Rainin | 30389276 | |
| Pipettes | Rainin | Various | Depending on lab preference, manual or electronic pipettes can be used at various maximum volumes. |
| TaqMan Environmental Master Mix 2.0 | Thermo Fisher Scientific | 4396838 | |
| Target forward and reverse primers | Integrated DNA Technologies, Inc. | none | custom product |
| Target PrimeTime qPCR Probes | Integrated DNA Technologies, Inc. | none | custom product |
| Target synthetic gBlock gene fragment | Integrated DNA Technologies, Inc. | none | custom product. used for qPCR standard dilution series |
| TE Buffer | Invitrogen | AM9849 | |
| VORTEX-GENIE 2 VORTEX MIXER | Fisher Scientific | 50728002 |
Referencias
- Kubista, M., et al. The real-time polymerase chain reaction. Mol Aspects Med. 27 (2-3), 95-125 (2006).
- Higuchi, R. D., Walsh, P. S., Griffith, R. Simultaneous amplification and detection of specific DNA sequences. Biotechnology. 10, 5(1992).
- Mauvisseau, Q., et al. Influence of accuracy, repeatability and detection probability in the reliability of species-specific eDNA based approaches. Scientific Reports. 9 (1), 580(2019).
- Hernandez, C., et al. 60 specific eDNA qPCR assays to detect invasive, threatened, and exploited freshwater vertebrates and invertebrates in Eastern Canada. Environmental DNA. , (2020).
- Klymus, K. E., et al. Reporting the limits of detection and quantification for environmental DNA assays. Environmental DNA. , (2019).
- Thalinger, B., et al. A validation scale to determine the readiness of environmental DNA assays for routine species monitoring. bioRxiv. , (2020).
- Helbing, C. C., Hobbs, J. Environmental DNA Standardization Needs for Fish and Wildlife Population Assessments and Monitoring. CSA Group. , (2019).
- Sepulveda, A. J., Nelson, N. M., Jerde, C. L., Luikart, G. Are Environmental DNA Methods Ready for Aquatic Invasive Species Management. Trends in Ecology & Evolution. , (2020).
- Svec, D., Tichopad, A., Novosadova, V., Pfaffl, M. W., Kubista, M. How good is a PCR efficiency estimate: Recommendations for precise and robust qPCR efficiency assessments. Biomolecular Detection and Quantification. 3, 9-16 (2015).
- Wilcox, T. M., et al. Robust detection of rare species using environmental DNA: the importance of primer specificity. PLoS One. 8 (3), 59520(2013).
- Prediger, E. How to design primers and probes for PCR and qPCR. IDT. , Available from: http://www.idtdna.cco/pages/education/decoded/article/designing-pcr-primers-and-probes (2020).
- Thornton, B., Basu, C. Real-time PCR (qPCR) primer design using free online software. Biochemistry and Molecular Biology Education. 39, 145-154 (2011).
- Owczarzy, R., et al. IDT SciTools: a suite for analysis and design of nucleic acid oligomers. Nucleic Acids Research. 36, Web Server issue 163-169 (2008).
- Kibbe, W. A. OligoCalc: an online oligonucleotide properties calculator. Nucleic Acids Research. 35, Web Server issue 43-46 (2007).
- Taylor, S. C., et al. The Ultimate qPCR Experiment: Producing Publication Quality, Reproducible Data the First Time. Trends in Biotechnology. 37 (7), 761-774 (2019).
- Ye, J., et al. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 13 (134), 11(2012).
- Altschul, S. F., Gish, W., Miller, W., Myers, E. W., Lipman, D. J. Basic Local Alignment Search Tool. Journal of Molecular Biology. 215, 403-410 (1990).
- Bustin, S. A., et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clinical Chemistry. 55 (4), 611-622 (2009).
- Bio-Rad. Bio-Rad Vol. 5279. , ed Bio-Rad (2020).
- Bio-Rad. Bio-Rad Vol. 6894. , Bio-Rad (2020).
- Eurogentec. Eurogentec. Vol. 0708-V2. , ed Eurogentec (2020).
- Bustin, S., Huggett, J. qPCR primer design revisited. Biomolecular Detection and Quantification. 14, 19-28 (2017).
- Hoorfar, J., et al. Practical considerations in design of internal amplification controls for diagnostic PCR assays. Journal of Clinical Microbiology. 42 (5), 1863-1868 (2004).
- Goldberg, C. S., et al. Critical considerations for the application of environmental DNA methods to detect aquatic species. Methods in Ecology and Evolution. 7 (11), 1299-1307 (2016).
- Guan, X., et al. Environmental DNA (eDNA) Assays for Invasive Populations of Black Carp in North America. Transactions of the American Fisheries Society. 148 (6), 1043-1055 (2019).
- Mosher, B. A., et al. Successful molecular detection studies require clear communication among diverse research partners. Frontiers in Ecology and the Environment. 18 (1), 43-51 (2019).
- Kwonga, S., Srivathsana, A., Meier, R. An update on DNA barcoding: low species coverage and numerous unidentified sequences. Cladistics. 28, 6(2012).
- Rees, H. C., et al. REVIEW: The detection of aquatic animal species using environmental DNA - a review of eDNA as a survey tool in ecology. Journal of Applied Ecology. 51 (5), 1450-1459 (2014).
- Evans, N. T., Lamberti, G. A. Freshwater fisheries assessment using environmental DNA: A primer on the method, its potential, and shortcomings as a conservation tool. Fisheries Research. 197, 60-66 (2018).
- Forootan, A., et al. Methods to determine limit of detection and limit of quantification in quantitative real-time PCR (qPCR). Biomolecular Detection and Quantification. 12, 1-6 (2017).
- Sengupta, M. E., et al. Environmental DNA for improved detection and environmental surveillance of schistosomiasis. Proceedings of the National Academy of Sciences of the United States of America. 116 (18), 8931-8940 (2019).
- Klymus, K. E., Richter, C. A., Chapman, D. C., Paukert, C. Quantification of eDNA shedding rates from invasive bighead carp Hypophthalmichthys nobilis and silver carp Hypophthalmichthys molitrix. Biological Conservation. 183, 77-84 (2015).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados