
Method Article
Développement et essais d’analyses quantitatives pcr spécifiques aux espèces pour les applications de l’ADN environnemental
* Ces auteurs ont contribué à parts égales
Dans cet article
Résumé
Les analyses d’ADN environnemental nécessitent une conception, des tests, une optimisation et une validation rigoureux avant que la collecte de données sur le terrain puisse commencer. Ici, nous présentons un protocole pour emmener les utilisateurs à travers chaque étape de la conception d’une espèce spécifique, basée sur la sonde qPCR analyse pour la détection et la quantification d’un ADN d’espèces cibles à partir d’échantillons environnementaux.
Résumé
De nouvelles méthodes non invasives de détection et de surveillance de la présence d’espèces sont en cours d’élaboration pour aider à la gestion des pêches et de la conservation de la faune. L’utilisation d’échantillons d’ADN environnemental (ADN électronique) pour détecter le macrobiote est l’une de ces méthodes qui devient rapidement populaire et qui est mise en œuvre dans les programmes nationaux de gestion. Ici, nous nous concentrons sur le développement d’analyses ciblées spécifiques aux espèces pour les applications quantitatives pcr (qPCR) basées sur des sondes. L’utilisation de qPCR à base de sonde offre une plus grande spécificité que ce qui est possible avec les amorces seules. En outre, la capacité de quantifier la quantité d’ADN dans un échantillon peut être utile dans notre compréhension de l’écologie de l’ADN électronique et de l’interprétation des modèles de détection de l’ADN électronique sur le terrain. Un examen attentif est nécessaire dans l’élaboration et l’essai de ces essais afin d’assurer la sensibilité et la spécificité de la détection des espèces cibles à partir d’un échantillon environnemental. Dans ce protocole, nous délimités les étapes nécessaires à la conception et à l’essai d’analyses basées sur des sondes pour la détection d’une espèce cible; y compris la création de bases de données de séquences, la conception d’analyses, la sélection et l’optimisation des analyses, les performances d’analyse des tests et la validation sur le terrain. Suivre ces étapes aidera à réaliser un test efficace, sensible et spécifique qui peut être utilisé en toute confiance. Nous démontrons ce processus avec notre essai conçu pour les populations de la boue (Actinonaias ligamentina), une espèce de moules d’eau douce trouvée dans la rivière Clinch, Etats-Unis.
Introduction
Les chercheurs et les gestionnaires s’intéressent de plus en plus à l’utilisation d’analyses d’ADN environnementale pour la détection des espèces. Depuis trois décennies, le PCR quantitatif ou en temps réel (qPCR/rtPCR) est utilisé dans de nombreux domaines pour la détection et la quantification spécifiques à la séquence des acides nucléiques1,2. Dans le domaine relativement nouveau de la recherche sur l’ADN, l’utilisation de ces essais avec une courbe standard pour la quantification des copies de l’ADN cible par volume ou poids de l’échantillon d’ADN est maintenant devenue une pratique courante. Les séquences d’ADN mitochondrial sont généralement ciblées dans les analyses d’ADN électronique parce que le génome mitochondrial est présent en milliers d’exemplaires par cellule, mais des analyses pour les séquences d’ADN nucléaire ou d’ARN sont également possibles. Il est essentiel de comprendre que les analyses publiées pour les échantillons d’ADN électronique ne sont pas toujours égales en termes de performances. La fiabilité d’un essai dans la détection de l’ADN d’une espèce cible (c.-à-d. la spécificité) et la détection de faibles quantités d’ADN cible (c.-à-d. sensibilité) peuvent varier considérablement en raison de différences dans la façon dont l’essai a été conçu, sélectionné, optimisé et testé. Les mesures quantitatives de déclaration des performances d’essai ont été largement négligées auparavant, mais récemment, des normes visant à améliorer la transparence dans le développement des analyses émergent3,4,5,6,7,8.
Optimisation et rapports des aides à la performance d’analyse dans la conception et l’interprétation des résultats de l’enquête eDNA. Les analyses qui réagissent croiséement avec l’ADN des espèces non cibles pourraient mener à de fausses détections positives, tandis que les analyses avec une faible sensibilité peuvent ne pas détecter l’ADN des espèces cibles même lorsqu’il est présent dans l’échantillon (faux négatifs). Une compréhension de la sensibilité et de la sélectivité des analyses aidera à éclairer l’effort d’échantillonnage nécessaire pour détecter les espèces rares. Étant donné qu’il existe de nombreuses sources naturelles de variation dans l’ADN, les études doivent limiter autant que possible les sources contrôlables de variation, y compris l’optimisation et la caractérisation complètes de l’analyseeDNA 3.
Les conditions qui affectent directement la spécificité ou la sensibilité d’un essai modifieront les performances de l’essai. Cela peut se produire dans différentes conditions de laboratoire (c.-à-d. différents reagents, utilisateurs, machines, etc.). Par conséquent, ce protocole devrait être revu lors de l’application d’un essai dans de nouvelles conditions. Même les essais bien caractérisés dans la littérature devraient être testés et optimisés lorsqu’ils sont adoptés par un nouveau laboratoire ou lors de l’utilisation de différents réacgages (p. ex., solution master-mix)5,9. La spécificité de l’analyse peut changer lorsqu’elle est appliquée à une région géographique différente, parce que l’analyse est appliquée à des échantillons provenant d’une nouvelle communauté biotique qui peuvent inclure des espèces non cibles contre qui l’essai n’a pas été testé, et des variations génétiques dans les espèces cibles peuvent se produire. Encore une fois, l’analyse devrait être réévaluée lorsqu’elle est utilisée dans un nouvel emplacement. Les conditions sur le terrain diffèrent des conditions de laboratoire parce que sur le terrain, les inhibiteurs du PCR sont plus susceptibles d’être présents dans les échantillons. Les inhibiteurs du PCR affectent directement la réaction d’amplification et affectent ainsi les performances de l’analyse. Pour cette raison, un contrôle positif interne est nécessaire lors de l’élaboration d’un essai eDNA.
Enfin, les conditions environnementales sur le terrain peuvent affecter les molécules d’ADN de l’espèce cible et leur capture par la dégradation, le transport et la rétention de l’ADN. De plus, les différents protocoles de collecte et d’extraction de l’ADN varient en fonction de leur efficacité et de leur capacité à conserver l’ADN. Cependant, il est important de noter que ces processus affectent la détectabilité de l’ADN, mais pas la performance d’un test moléculaire. Ainsi, la détection de l’ADN des espèces cibles dans les échantillons de champ est fonction à la fois de la performance technique de l’essai qPCR ainsi que des conditions sur le terrain et des protocoles de collecte, d’entreposage et d’extraction. Lors de l’utilisation d’un essai bien caractérisé et très performant, les utilisateurs peuvent se sentir confiants dans les capacités de l’essai; permettre aux chercheurs de se concentrer maintenant sur la compréhension des facteurs externes d’analyse (c.-à-d. les variables environnementales, les différences dans les protocoles de capture ou d’extraction) affectant la détection de l’ADN.
Ici, nous nous concentrons spécifiquement sur l’analyse des performances techniques grâce à une conception rigoureuse et l’optimisation. Nous démontrons le protocole à l’aide d’un test à base de sonde développé pour la détection d’une moule d’eau douce, le mucket (Actinonaias ligamentina), à partir de l’eau échantillonnée dans la rivière Clinch, Etats-Unis. Récemment, Thalinger et coll. (2020) ont présenté des lignes directrices pour la validation des essais ciblés de l’ADN. La conception d’essai suivant notre protocole apportera un essai au niveau 4 de Thalinger et autres plus une étape supplémentaire vers le niveau 56. À ce stade, les performances techniques d’un essai seront optimisées et il sera prêt pour une utilisation régulière dans les applications de laboratoire et de terrain. Une utilisation plus approfondie de l’analyse en laboratoire, en mésocosme et sur le terrain peut alors répondre aux questions concernant la détection de l’ADN et les facteurs influençant la détectabilité, les dernières étapes de la validation de niveau 56.
Protocole
1. Génération d’une base de données de séquences de séquences d’ADN mitochondriales provenant d’espèces cibles et non cibles d’intérêt
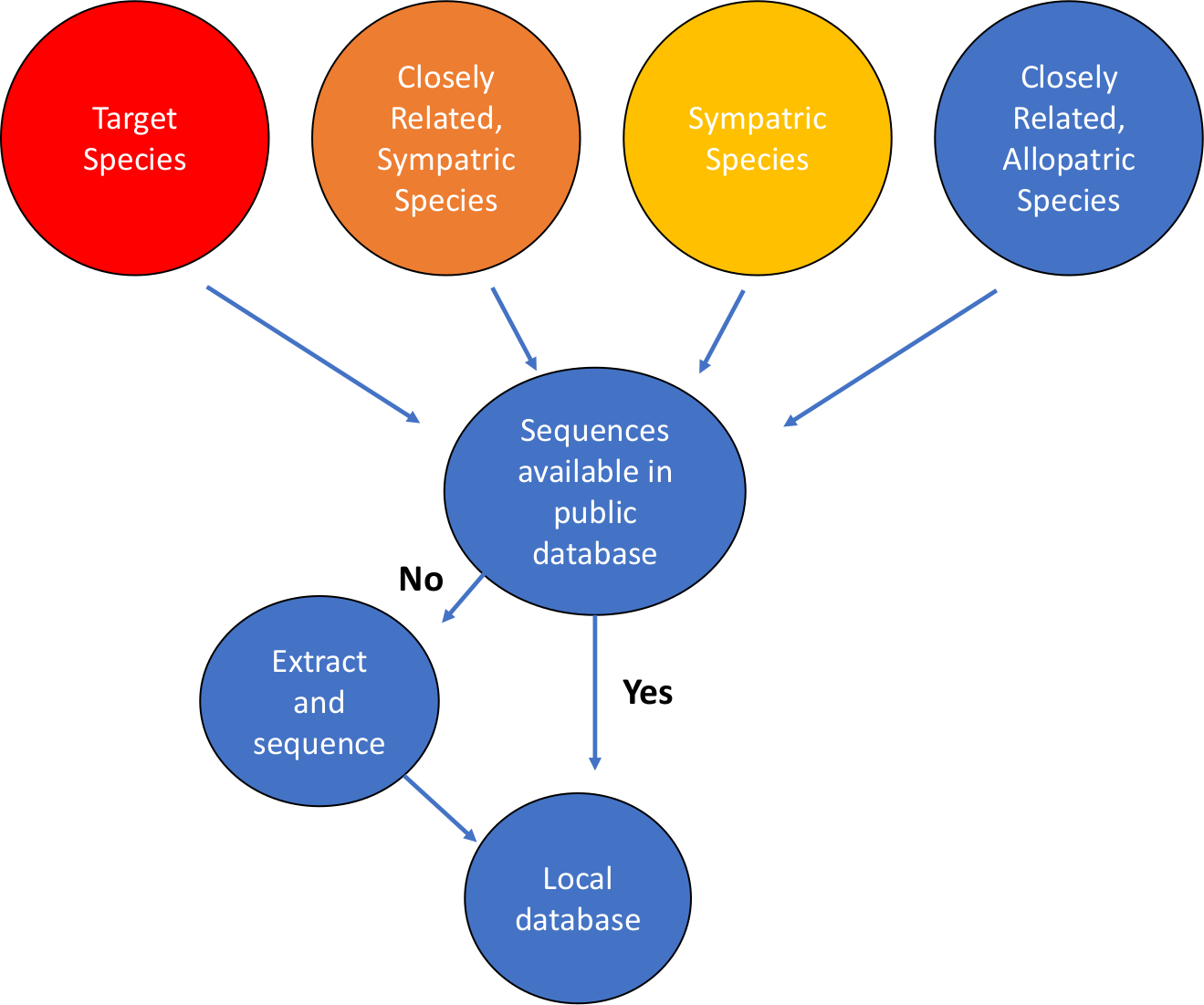
- Définissez la question, les cibles et le système à traiter. Identifier les espèces cibles pour la détection de l’ADN. Identifiez le système géographique dans lequel l’analyse sera utilisée. Dressez une liste des espèces d’intérêt, y compris les espèces cibles, les espèces sympatriques (co-présentes) dans le même taxa (habituellement ordre ou niveau familial), et les espèces allopatriques étroitement apparentées, celles qui ne se trouvent peut-être pas au même endroit géographique que la cible (figure 1).
REMARQUE : Ici, les populations de l’espèce A. ligamentina de la rivière Clinch ont été ciblées. - Rechercher et télécharger des séquences à partir de plusieurs régions génétiques pour les espèces sur la liste de l’étape 1. Des bases de données de séquences telles que NCBI (National Center for Biotechnology Information), BOLD (Barcode of Life Database), EMBL (European Molecular Biology Laboratory) et DDBJ (DNA Data Bank of Japan) peuvent être utilisées. NCBI, EMBL et DDBJ partagent tous des informations de séquence.
- À l’aide de la base de données nucléotide de ncbi, recherchez l’organisme cible (p. ex., Actinonaias ligamentina)et la région génétique (p. ex., cytochrome c oxidase I (COI) ou NADH-déshydrogénase 1 (ND1); Exemple chaîne de recherche: Actinonaias ligamentina ET ND1)
- Ensuite, sélectionnez toutes les séquences qui correspondent aux spécifications et sélectionnez Envoyer à. Choisissez l’enregistrement complet, le format de fichier et de téléchargement comme GenBank ou FASTA et créez ensuite le fichier. Ces séquences sont maintenant enregistrées sur l’ordinateur.
- Répétez ces étapes pour toutes les espèces de la liste définie à l’étape 1. Conservez les séquences pour chaque région génétique dans un fichier distinct car celles-ci seront analysées séparément.
- Téléchargez toutes les séquences pertinentes (ou une grande proportion représentative de séquences) pour les espèces cibles identifiées à l’étape 1. Inclure des variantes géographiques si possible.
- Répétez les séquences de recherche et de téléchargement pour les espèces non cibles apparentées et sympatriques du même groupe taxonomique qui ont été identifiées à l’étape 1 (p. ex., si l’espèce cible est le mucket (A. ligamentina) télécharger des séquences pour toutes les autres espèces de moules d’eau douce dans les Unionidae familiales qui se produisent dans le système d’intérêt).
- Répétez la recherche et téléchargez les espèces étroitement apparentées mais allopâtres (géographiquement distinctes) énumérées à l’étape 1.1.
REMARQUE : Toutes les espèces (cibles et non cibles) ne seront pas disponibles dans les bases de données publiques. Augmentez la base de données de référence locale en amplifiant et séquençant des spécimens taxonomiquement vérifiés d’espèces d’intérêt à l’interne. Si vous travaillez avec une espèce qui a une grande diversité génétique au sein de l’espèce ou si vous travaillez dans une zone géographiquement grande où l’on peut s’attendre à des variantes géographiques, rassemblez des séquences de l’ensemble de l’aire de répartition.
2. Conception d’essai
- Alignez les séquences de chaque région génétique séparément à l’aide d’un logiciel d’alignement que l’on retrouve dans divers programmes d’édition de séquences génétiques et de bioinformatiques. Faites cet alignement pour chacune des différentes régions génétiques.
- Par exemple, à l’aide du logiciel Geneious Prime (https://www.geneious.com) importez les fichiers de séquences téléchargés dans le programme.
- Créez des dossiers distincts pour chaque région génétique.
- Dans un dossier qui contient des séquences d’une région génétique, sélectionnez toutes les séquences.
- Utilisez l’outil d’alignement multiple pour créer un alignement nucléotide des séquences sélectionnées. Il peut y avoir plusieurs options pour le type d’alignement, en utilisant les alignements Geneious ou MUSCLE et les paramètres par défaut fonctionne bien.
- Choisissez des régions prometteuses pour la conception d’analyse grâce à la visualisation de données de séquence alignées. Une région qui a beaucoup de données de séquence disponibles pour les espèces d’intérêt, est très divergente entre les espèces, et montre faible variation à l’intérieur de l’espèce est un bon candidat. Cela augmentera la probabilité que les amorces et les sondes conçues soient en mesure de discriminer la cible des espèces non cibles, tout en veillant à ce que les variantes intraspécifiques s’amplifient avec l’analyse.
- Conception des amorces d’essai et de la sonde.
- Utilisez le logiciel de conception d’analyse qPCR et suivez les instructions. L’outil PrimerQuest (https://www.idtdna.com/) d’IDT pour concevoir 5 ensembles d’analyses qPCR a été utilisé ici.
- Coller la séquence sélectionnée à l’étape 2.2 dans la boîte d’entrée séquence. Si l’alignement créait des espaces, supprimez-les de la séquence.
- Sélectionnez qPCR 2 Primers + Probe dans l’option Choisissez votre conception.
- Téléchargez les analyses recommandées.
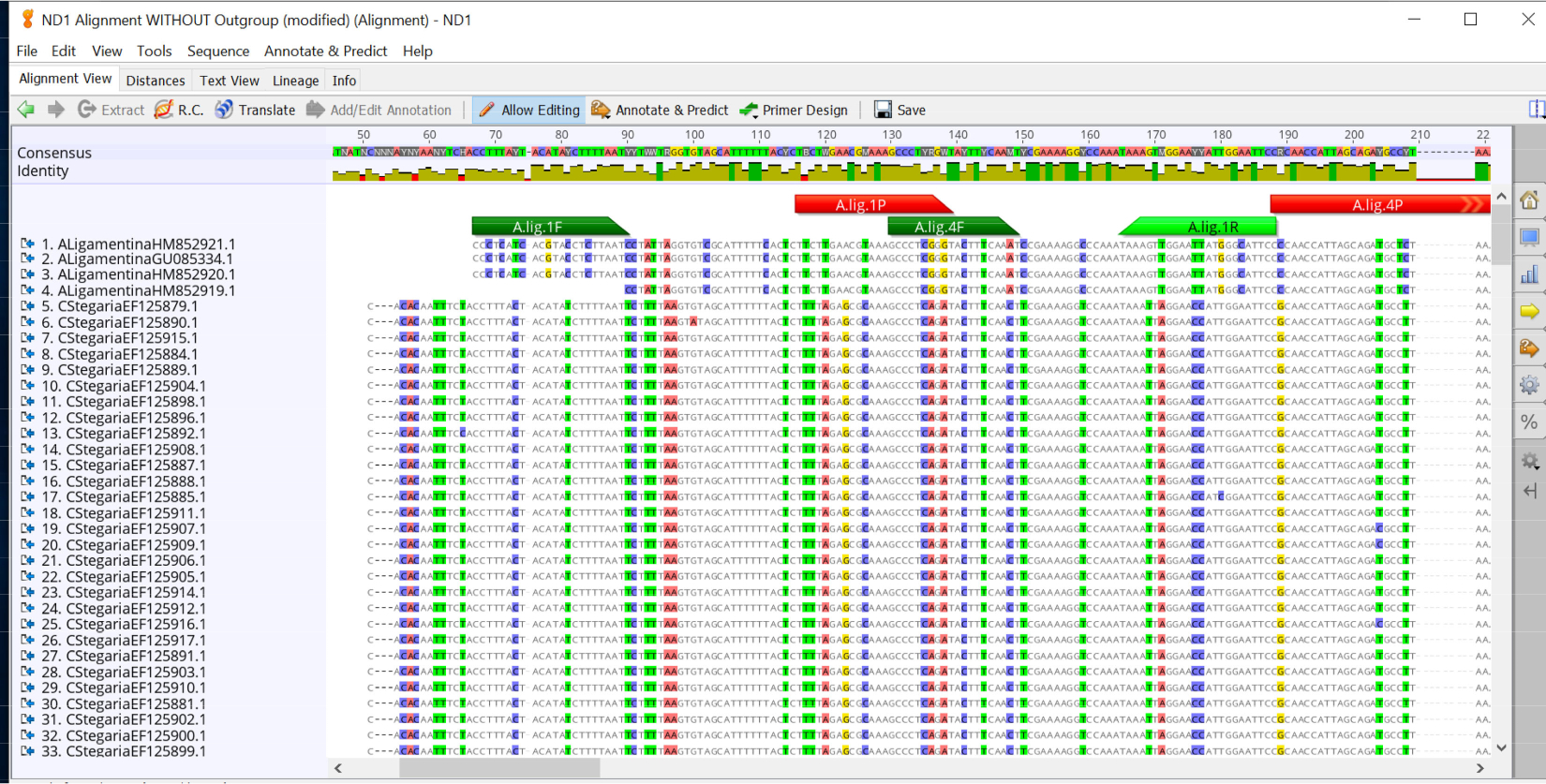
- Copiez les séquences de l’amorce avant du premier essai et recherchez cette séquence d’amorce dans l’alignement créé à l’étape 2.1.4. Si vous utilisez Geneious Prime, utilisez l’outil Annotate et Predict pour ajouter la région d’amorce à l’alignement. Faites ceci pour toutes les combinaisons d’amorce et de sonde( figure 2).
- Inspecter ces régions de l’alignement pour décexer les variations à l’intérieur de l’espèce cible ainsi qu’au sein des espèces co-présentes.
- S’il y a variation génétique intraspécifique, recherchez des analyses où les amorce et la sonde ne tombent pas dans ces régions.
- Pour prévenir l’amplification des espèces non cibles, recherchez les inadéquations avec les espèces non cibles. Choisissez des analyses avec le plus de décalages pour une validation ultérieure. Currier et coll. (2018) suggèrent de choisir des ensembles avec au moins deux des trois régions (les deux amorces, ou une amorce et une sonde) ayant au moins deux inadéquations avec toutes les espèces non cibles. Cependant, gardez à l’esprit que les inadéquations à la sonde contribuent moins à la spécificité10.
REMARQUE: Les différences dans les 3 paires de base de l’extrémité 3' de chaque amorce augmentent la spécificité mieux que les différences à la fin de 5' des amorces10.
- Considérez les paramètres importants suivants dans la conception d’essai.
- Déterminer les températures de fusion et d’annelation des amorce et de la sonde. Idéalement, la température de fusion (Tm) des amorces devrait être comprise entre 60-64 °C et à moins de 2 °C l’une de l’autre, et le Tm de la sonde devrait être 6-8 degrés plus élevé que le Tm des amorces. Réglez la température d’annealing (Ta) de la réaction qPCR 5 °C en dessous de la température de fusion, autour de 55-60 °C11.
- Examinez le contenu GC. Choisissez entre 35 et 65% de contenu GC, et évitez les régions avec 4 G consécutifs ou plus. Avoir 1 ou 2 G ou Cs dans les 5 dernières bases de la fin de 3' de l’amorce (pince GC) pourrait augmenter la spécificité car il aiderait l’amorce à faire un lien plus fort12.
- Recherchez des structures d’épingle à cheveux et de dimère. Testez les amorces et la sonde pour les structures et les dimers en épingle à cheveux prévus à l’aide d’un programme d’analyse des oligonucléotides (p. ex., OligoAnalyzer -IDT13; OligoCalculator14). Ces structures peuvent provoquer une amplification non cible et une efficacité moindre. Évitez les analyses qui sont prévues pour former ces structures.
- Déterminez la longueur de l’amorce. Visez des amorces de 18 à 25 bases de longueur et de longueur de sonde entre 20 et 25 bases. Les amorces et les sondes plus longues peuvent avoir une efficacité d’amplification plus faible.
- Déterminer la longueur de l’amplicon. Il devrait être entre environ 100 et 250 paires de base. Cette plage est généralement assez courte pour une efficacité PCR élevée, mais suffisamment longue pour faciliter la vérification par sanger séquençage4,15.
- Conception de sondes. Assurez-vous que les sondes n’ont pas de base G à l’extrémité de 5', car il pourrait amortir le signal des teintes vertes etjaunes 11. Nous avons conçu des sondes doubles trempées, avec des étanches IDT 3IABkFQ et ZEN et des fluorophores FAM ou HEX.
REMARQUE : Déterminez les sondes MGB : Les sondes TaqMan MGB (classeur de rainures mineures) sont souvent utilisées pour des études sur l’ADN. Cependant, parce que ces sondes sont très courtes, elles peuvent se lier à des non-cibles même avec un décalage de paire de base de 2 ou3 10. - Déterminer la sonde Tm. La température de fusion de la sonde devrait être de 6 à 8 °C supérieure à celle des amorces. Des températures plus basses diminuent le succès de liaison de la sonde.
- Déterminez la longueur et l’emplacement de la sonde. La sonde doit avoir entre 20 et 25 bp de longueur et idéalement située à proximité du site de liaison de l’amorce sur le même brin sans la chevaucher.
3. Analyse de dépistage et d’optimisation
- Dans le développement et les tests d’analyse silico. Avant de commander des ensembles d’amorce-sonde, évaluez la spécificité (amplification non-cible potentielle) en testant l’amplification d’amorce dans le silicic.
- Testez les amorces par l’intermédiaire de Primer-Blast16 de NCBI ou de programmes similaires qui peuvent identifier des non-cibles potentielles dans la base de données NCBI nt/nr qui pourraient amplifier avec l’analyse. Si vous utilisez des amorces de pâte Primer-Blast sur la boîte d’amorce Utilisez ma propre boîte d’amorce sous paramètres Primer. Dans les options de vérification des paramètres de spécification de la paire d’amorce, sélectionnez nr comme base de données et tapez l’ordre de l’organisme d’intérêt (par exemple, « Unionida » ou « Unionoida ») dans la boîte organisme.
- Continuez d’évaluer visuellement les ensembles d’amorce/sonde sur des données de séquence alignées.
- Afin d’évaluer les amorces et les sondes en même temps en silice, créez une chaîne de texte de l’amorce avant, 12 N, la sonde, 12 N, et le complément inverse de l’amorce inverse. Si la séquence de sonde se situe à moins de 12 paires de base de l’une des amorces, utilisez le nombre de N correspondant au nombre de paires de base entre l’amorce et la sonde.
- Utilisez la recherche Nucleotide Blast (Blastn) de NCBI pour rechercher contre la base de données nr17. Utilisez l’onglet Taxonomie pour rechercher des espèces non cibles avec peu d’inadéquations; ceux-ci doivent être testés en laboratoire lors de l’optimisation des analyses.
REMARQUE : Dans le cas du silico, les tests aident à exclure les analyses non spécifiques, mais les analyses potentiellement spécifiques doivent être testées empiriquement (in vitro), car toutes les espèces n’ont pas de séquences dans les bases de données génétiques et l’amorce et les sondes peuvent toujours se lier à des non-cibles même si elles sont jugées peu probables par le logiciel.
- Choisissez trois à cinq combinaisons d’amorce/sonde à tester en laboratoire.
- Commandez des amorces, des sondes et une norme d’ADN synthétique ainsi que des amorces supplémentaires à queue M13 pour le séquençage amplicon.
- Commandez des amorces et des sondes synthétiques d’oligonucléotide d’une entreprise qui fabrique des oligos. Les sondes sont étiquetées avec un colorant fluorescent et un quencher. Différents fluorophores doivent être sélectionnés pour les analyses qui doivent être multiplexées. Vérifiez votre instrument qPCR pour obtenir une liste des fluorophores que l’instrument peut détecter.
- Concevoir et commander des amorces à queue M13 pour la vérification des détections qPCR avec séquençage Sanger en ajoutant la séquence M13 Forward (-20), GTA AAA CGA CGG CCA GT, à la fin de 5' de l’amorce avant, et la séquence M13 Reverse (-27), CAG GAA ACA GCT ATG AC, à la fin de 5' de l’amorce inverse.
- La norme d’ADN synthétique contient la séquence cible (y compris les régions d’amorce) à une concentration connue en copies/μL. Quantifier des échantillons inconnus en fonction d’une courbe faite par des concentrations connues de cette norme (c.-à-d. la courbe standard). Acquérir la norme synthétique de la même entreprise qui fabrique les amorces et la sonde. Suivez les recommandations du fabricant en matière de réanimation et de stockage. Diluer les normes dans le tampon TE avec un transporteur d’ARN à l’aide de plasticware à faible rétention pour réduire l’hydrolyse et la liaison aux surfaces.
REMARQUE : Si la courbe standard ne fonctionne pas bien (faible efficacité du PCR, voir l’étape 3.4.2), essayez de suspendre à nouveau la norme dans l’eau ou tris-HCl. - Suspendez les amorces et les sondes dans l’eau exempte de nucléase, tris-HCl, ou tampon TE à des concentrations pratiques pour une utilisation d’essai. En général, diluer les stocks de travail 20 fois dans le mélange principal pour atteindre la concentration d’analyse finale optimisée. Conserver les oligos suspendus à une température constante de -20 °C lorsqu’ils ne sont pas utilisés.
- Analyse et essais in vitro (en laboratoire). Rejeter les analyses qui ont une faible efficacité, qui réagissent de façon croisée avec les espèces co-présentes ou qui ont une faible sensibilité18. Inclure l’utilisation d’un contrôle positif interne (CIP) pendant le développement de l’analyse ainsi que lors de l’exécution d’échantillons réels.
- Tout d’abord, trouvez les valeurs optimales de concentration de la température et de l’amorce/sonde pour l’essai. Une fois que ces paramètres ont été optimisés pour l’efficacité du PCR (étape 3.4.2), la réactivité croisée (étape 3.4.3) et la sensibilité (étape 3.4.4), procéder à tester l’essai avec un IPC multiplexé (étape 3.4.5).
- Testez la température optimale d’annealing (Ta) pour l’amorce et les sondes à l’aide d’un gradient de température PCR centré à 5 ° C en dessous de l’amorce moyenne prévue Tm.
- Testez les concentrations optimales d’amorce et de sonde. En règle générale, des concentrations d’amorce de 200 nM, 400 nM et 800 nM et 75 nM, 125 nM et 200 nM de sonde sont testées.
- Créez une courbe standard et déterminez l’efficacité et la plage linéaire. Testez au moins six dilutions 10 fois d’une norme d’ADN synthétique contenant la séquence cible, à environ 100 copies/réaction à 105 copies/réaction (Figure 3A).
- Utilisez le logiciel qPCR pour tracer la valeur Cq (seuil de cycle à la quantification) de chaque norme sur l’axe y et la base de journal 10 de la concentration standard initiale en copies/réaction sur l’axe x. Le logiciel qPCR doit exécuter automatiquement une régression linéaire (Figure 3B).
- Calculer l’efficacité de la pente de la régression, E = -1 + 10(-1/pente). Par exemple, si la pente est de -3,4, E = -1 + 10(0,29) = 0,97 ou 97 %. Vérifiez également les valeurs r2 qui indiquent dans quelle mesure les répliques standard s’adaptent à la courbe. Le logiciel qPCR devrait également calculer automatiquement cette mesure (figure 3B). Viser des valeurs d’efficacité de 100% (±10%) et r2 valeurs de ≥0,989,15,19,20,21,22.
- Inspecter visuellement la courbe standard pour décelér les écarts par rapport à la régression dans une direction cohérente ou pour les mauvaises performances de la courbe standard mesurées par l’efficacité et les valeurs r2 (figure 3C et 3D).
- Spécificité : Évaluer la réactivité croisée avec les espèces non cibles afin de réduire le risque de faux positifs. Lorsque les détections eDNA peuvent entraîner des décisions de gestion coûteuses, vérifiez les détections positives par séquençage amplicon.
- Non cibles : Faire l’analyse contre les extractions d’ADN génomique de spécimens taxonomiquement vérifiés d’espèces apparentées et d’espèces géographiquement co-présentes; la plus haute priorité étant de tester contre des espèces étroitement apparentées et co-présentes. Utilisez des concentrations totales d’ADN similaires pour les échantillons cibles et non cibles. La concentration choisie devrait donner l’amplification à partir d’échantillons d’espèces cibles près du milieu de l’aire de répartition linéaire de la courbe standard. L’amplification ne doit être observée qu’avec les espèces cibles.
- Si l’amplification non cible est observée, nettoyez et séquencez le produit pour vérifier son identité. Il n’est pas rare d’observer la contamination de l’espèce cible dans des échantillons de tissus d’espèces non cibles, de sorte que toutes les amplifications à ce stade doivent être vérifiées par séquençage. Réamplifier les amplicons nettoyés à partir de tests de spécificité à l’aide des amorces à queue M13 et séquencer avec des amorces M13.
- Dans le laboratoire post-PCR, transférer les produits qPCR à séquencer dans des tubes frais. Retirez les amorces résiduelles et les composants de réaction à l’l’intérieur d’un kit de nettoyage (p. ex., kit de purification pcr MinElute).
- Effectuer 1:100 dilutions des élitutions et amplifier 1 μL de chacun pendant 30 cycles dans une réaction PCR de 50 μL avec les amorces à queue M13 et une polymélase haute fidélité (p. ex., polymélas d’ADN Haute Fidélité phusion).
- Exécutez 10 μL de chaque réaction sur un gel agarose de 1 % pour vérifier la taille d’une seule bande. Si aucune bande n’est observée, augmentez le nombre de cycles ou la quantité d’échantillon. Si plusieurs bandes sont observées, gel purifier la bande de la taille prévue.
- Retirez les amorces résiduelles et les composants de réaction avec un kit de nettoyage comme ci-dessus et mesurez les concentrations d’ADN des élitutions.
- Configurer les réactions de séquençage avec les amorces M13 selon les instructions de l’installation de séquençage.
REMARQUE : N’ouvrez jamais d’échantillons amplifiés dans le laboratoire qPCR. Préparer des échantillons pour le séquençage dans un laboratoire dédié aux échantillons post-PCR.
- Sensibilité : La sensibilité affecte le risque de faux négatifs ou de défauts de détection de l’ADN des espèces cibles lorsqu’il est présent. Évaluer la limite de détection (LOD) et la limite de quantification (LOQ) pour chaque essai. Enfin, inclure un contrôle positif interne (CIP) pour évaluer l’inhibition pcr des échantillons. Multiplex et tester ce test IPC avec l’analyse conçue pour s’assurer que les deux essais n’interfèrent pas les uns avec les autres.
- LOD : Effectuer six dilutions sérielles quadruples de la norme d’ADN synthétique, avec 8 à 24 répliques par dilution standard (figure 4). Calculez la concentration initiale la plus faible avec une détection de 95 %. Les parcelles LOD et LOQ peuvent être générées avec une calculatrice LOD/LOQ R script5.
REMARQUE : Les données inférieures au LOD ne doivent pas être censurées. En raison de la spécificité du PCR, il n’y a pas de limite inférieure pour les vrais positifs. Le LOD est la concentration la plus élevée au-dessous de laquelle on peut s’attendre à ce que de faux négatifs se produisent. - LOQ : À partir de la même série de dilution, calculer la concentration standard initiale d’ADN la plus faible quantifiable avec un coefficient de variation (CV) inférieur à 35 %.
REMARQUE : Le LOD et le LOQ doivent être rapportés en copies/réactions. Lors de l’utilisation d’un test validé et les échantillons sur le terrain amplifient en dessous de la LOQ, les résultats doivent être signalés comme des détections en % plutôt que des concentrations d’ADN, parce que la concentration exacte ne peut pas êtremesurée avec confiance 5.
- LOD : Effectuer six dilutions sérielles quadruples de la norme d’ADN synthétique, avec 8 à 24 répliques par dilution standard (figure 4). Calculez la concentration initiale la plus faible avec une détection de 95 %. Les parcelles LOD et LOQ peuvent être générées avec une calculatrice LOD/LOQ R script5.
- Utilisez un contrôle positif interne (CIP) pour tester l’inhibition du PCR. L’inhibition peut entraîner une diminution de la sensibilité et des faux négatifs. Testez la capacité de l’analyse IPC à être multiplexé avec l’analyse cible.
- Un test IPC peut être multiplexé avec l’analyse cible à l’aide d’une sonde avec un colorant reporter différent de l’analyse cible. Cet essai de CIP consiste en une courte séquence d’ADN synthétique d’une espèce sans rapport avec le taxa cible, incorporée dans le mélange principal qPCR à une faible concentration d’environ 102 copies/réaction, ainsi que des amorces et des sondes qui le détectent. Cette concentration plus faible est nécessaire pour éviter la concurrence avec la séquence cible de polymése et de nucléotides23.
- Comparez la valeur Cq du modèle IPC de l’échantillon à celle du modèle IPC dans le contrôle sans modèle. Dans ce modèle sans contrôle (CNT), la seule entrée d’ADN est celle du modèle IPC. Le modèle IPC dans cette réaction devrait amplifier comme prévu. Si le modèle IPC d’un échantillon amplifie à 2 cycles ou plus différents de celui du modèle IPC du CNT, l’échantillon eDNA est inhibé. Les échantillons qui montrent une inhibition peuvent être dilués 1:10 et re-testés. Si un échantillon reste inhibé, cet échantillon doit être retiré de l’analyse.
- Tout d’abord, trouvez les valeurs optimales de concentration de la température et de l’amorce/sonde pour l’essai. Une fois que ces paramètres ont été optimisés pour l’efficacité du PCR (étape 3.4.2), la réactivité croisée (étape 3.4.3) et la sensibilité (étape 3.4.4), procéder à tester l’essai avec un IPC multiplexé (étape 3.4.5).
- Développement et essais d’analyse in situ
- En laboratoire : Si l’accès à l’organisme en laboratoire ainsi qu’aux espèces sympatriques est disponible; prélever des échantillons d’eau dans les enclos avec ces espèces, traiter les échantillons et tester l’analyse par rapport à ces échantillons d’ADN. Séquencez les produits comme ci-dessus pour vérifier l’amplification de la cible prévue à l’aide des amorces à queue M13.
- Sur le terrain :
- Identifier les sites où l’organisme cible est connu pour se produire et connu pour ne pas se produire. Il est préférable d’avoir une certaine mesure de l’abondance à chaque site où l’espèce cible se trouve.
- Décider quels volumes d’échantillons et méthodes de collecte d’échantillons (p. ex., filtration, centrifugation, etc.) seront utilisés.
- Inclure un champ vierge ou négatif de contrôle à chaque site, c’est de l’eau propre qui a été apportée sur le site du terrain, puis recueillie et préparée avec le même équipement de terrain et les mêmes protocoles utilisés pour l’échantillonnage eDNA24. Le but du champ vide est de détecter la contamination de l’équipement d’échantillonnage et de l’équipement de terrain apportés sur le site. Prendre le champ vide avant de traiter les échantillons d’eau du champ.
- Prélevez plusieurs échantillons d’eau par site, de préférence 3 échantillons par site.
- Retour en laboratoire, traitement et extraction d’échantillons.
- Exécutez l’analyse à l’aide d’une plaque mise en place semblable à la figure 5A et comparez la fréquence de concentration et de détection de l’ADN électronique avec les différences connues de l’occurrence et de l’abondance sur le site. Confirmez toutes les détections en séquençant24,25.
REMARQUE : Ce qui précède validerait un essai au niveau 4 de l’échelle6 de Thalinger et coll. (2020) (optimisation des performances techniques de l’essai) et commencerait à recueillir des données à l’appui de la validation de test de niveau 5. Le niveau 5 intègre la modélisation des probabilités et l’utilisation de l’analyse des études d’écologie de l’ADN. Nous estimons que cela dépasse le cadre du développement de base de l’analyse, mais nous encourageons ces applications d’essais en laboratoire et sur le terrain afin d’améliorer la conception des analyses et l’interprétation des données.
Résultats
Lors de la conception d’un essai qPCR spécifique à chaque espèce pour le mucket (A. ligamentina), des séquences disponibles de toutes les espèces d’Unionidae dans la rivière Clinch ont été téléchargées. Des espèces étroitement apparentées comme Lampsilis siliquoidea ont également été incluses dans la base de données de référence, même si elles ne se trouvent pas dans la même rivière. Toutes les espèces du réseau fluvial d’intérêt n’ont pas été trouvées à GenBank, de sorte que d’autres espèces ont été séquencées dans la maison. Les séquences ont été alignées à l’aide du logiciel Geneious et le logiciel Primer Quest (IDT) a été utilisé pour concevoir plusieurs analyses. Cinq ensembles d’amorce et de sonde ont été ajoutés à l’alignement pour l’évaluationvisuelle ( figure 2). Ils ont ensuite été testés en silico à l’aide de Primer-Blast, après quoi ils ont été commandés pour d’autres tests in vitro. En laboratoire, tous les essais ont été testés à l’aide d’extractions d’ADN de 27 espèces disponibles pour vérifier la spécificité. Un essai (A.lig.1) n’a réussi à amplifier que l’espèce cible (tableau 1; Tableau 2). Cet essai a avancé pour d’autres essais de l’efficacité d’essai, LOD et LOQ. Il a une longueur amplicon de 121 paires de base. Le tableau 3 montre la séquence utilisée pour la norme a. ligamentina d’ADN synthétique. La figure 3A et la figure 3B montrent les résultats d’un essai réussi avec une bonne efficacité et des valeurs r2. La figure 3C et la figure 3D montrent un test dont la courbe standard a une faible efficacité; cet essai a été jeté. Le LOD et loq pour l’essai choisi (A.lig.1) se sont tous deux révélés être 5.00 copies/réaction utilisant la méthode discrète décrite dans Klymus et autres5. Le CIP qui a été multiplexé avec l’essai (tableaux 3-6) n’a pas affecté la courbe standard de l’essai de A. ligamentina. L’IPC que nous utilisons est un fragment de la transcription hemt souris. Cet essai a été préconceptionné par IDT pour une autre application, mais nous avons modifié son utilisation en tant qu’IPC pour les applications eDNA de notre laboratoire.
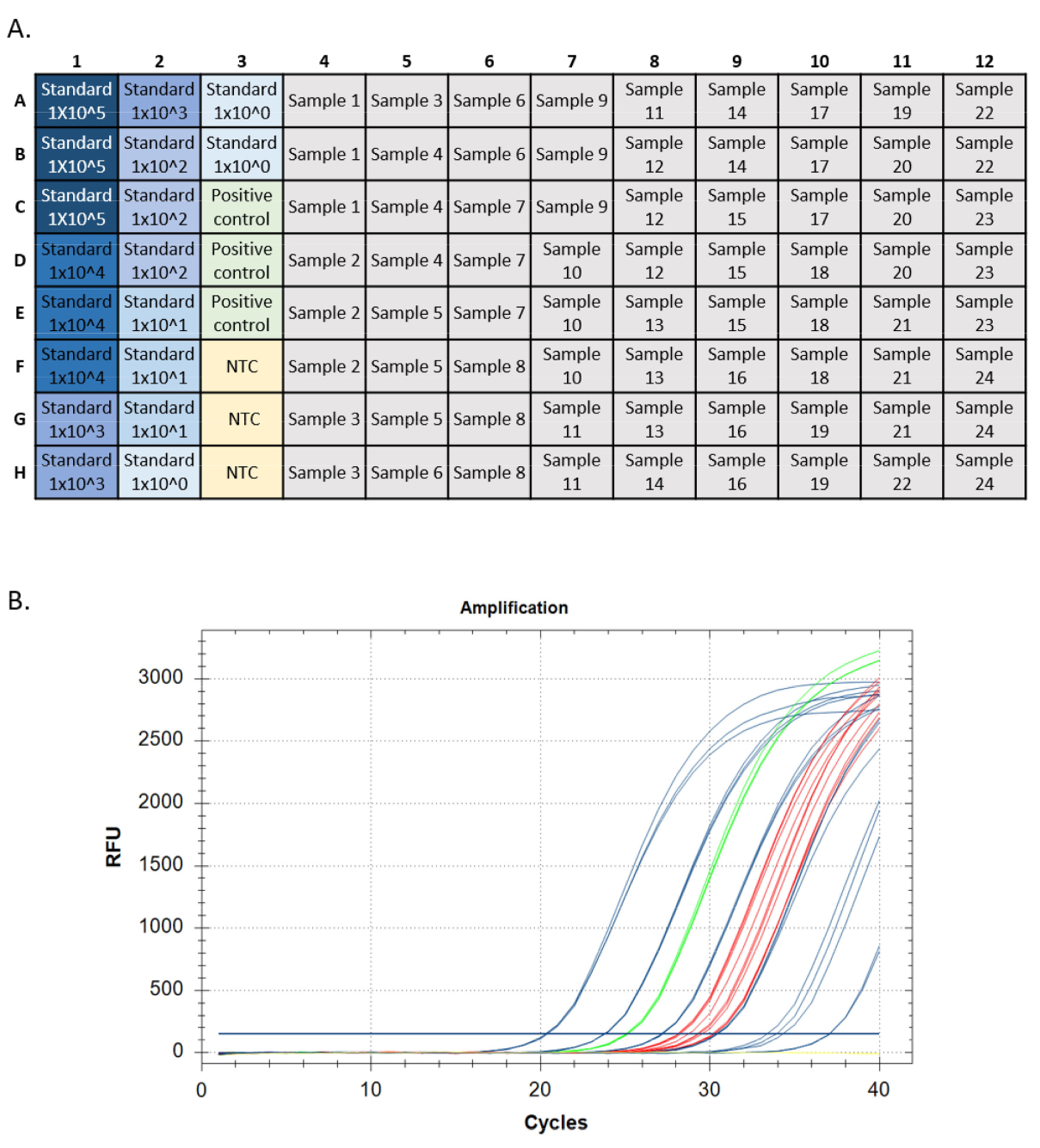
Une exécution qPCR réussie devrait répondre à certains critères pour chaque mesure de performance (c.-à-d. amplification standard de courbe, contrôle positif de l’ADN génomique, aucun contrôle de modèle et contrôle positif interne). Les normes d’analyse cible devraient avoir des courbes d’amplification exponentielles. Ces courbes devraient atteindre un plateau de point final si elles sont autorisées à exécuter suffisamment de cycles. Ceci indique que la sonde fluorescente est complètement consommée pendant la réaction, et les niveaux de fluorescence atteignant une limite maximale. Les normes d’amplification ultérieures peuvent ne pas atteindre un plateau en 40 cycles. Les contrôles positifs (ADN génomique et CIP) devraient avoir le même modèle. Les inconnues peuvent ou non amplifier, mais l’amplification en inconnus devrait également avoir un modèle exponentiel et un plateau de points de terminaison (figure 5).
Dans un qPCR de qualité, les dilutions standard s’amplifient à Cq uniformément espacé d’environ tous les cycles de 3,3 pour chaque différence de concentration de 10 fois. Chaque réplique d’une dilution standard amplifie d’une manière étroitement groupée ayant presque le même Cq (représenté par les valeurs r2). Toutes les dilutions standard s’exposent à l’amplification( Figure 3A). Dans un qPCR pauvre, les normes peuvent présenter une forme non exponentielle, une variation inégale des valeurs cq entre les dilutions, ne pas arriver à un plateau de point de terminaison, ou certaines dilutions peuvent ne pas amplifier du tout (Figure 3D).
Les paramètres importants pour une courbe standard sont l’efficacité, r2,pente, et y-intercept. L’efficacité devrait tomber entre 90%-110% avec des valeurs idéales proches de 100% et r2 valeurs devraient être au-dessus de 0,98 avec des résultats idéaux approchant 1,015,22. Les valeurs de pente devraient se situer entre -3,2 et -3,5 avec des résultats idéaux proches de -3,322. Les valeurs y-intercept devraient tomber entre un Cq de 34-41 avec des résultats idéaux ayant un Cq de 37,0. L’interception y est le Cq prévu d’une réaction avec 1 copie de la séquence cible, la plus petite unité qui peut être mesurée en un seul qPCR. Les inconnues avec Cq plus grand que l’interception y sont susceptibles d’être inhibées. L’exécution de plus de 40 cycles de PCR peut être nécessaire pour détecter la cible en cas d’inhibition ou d’un ensemble d’amorce inefficace, mais la quantification n’est pas possible dans ces circonstances et des contrôles négatifs supplémentaires sans la séquence cible, mais contenant l’ADN total semblable aux inconnus, devraient être exécutés pour exclure l’amplification des sources non spécifiques.
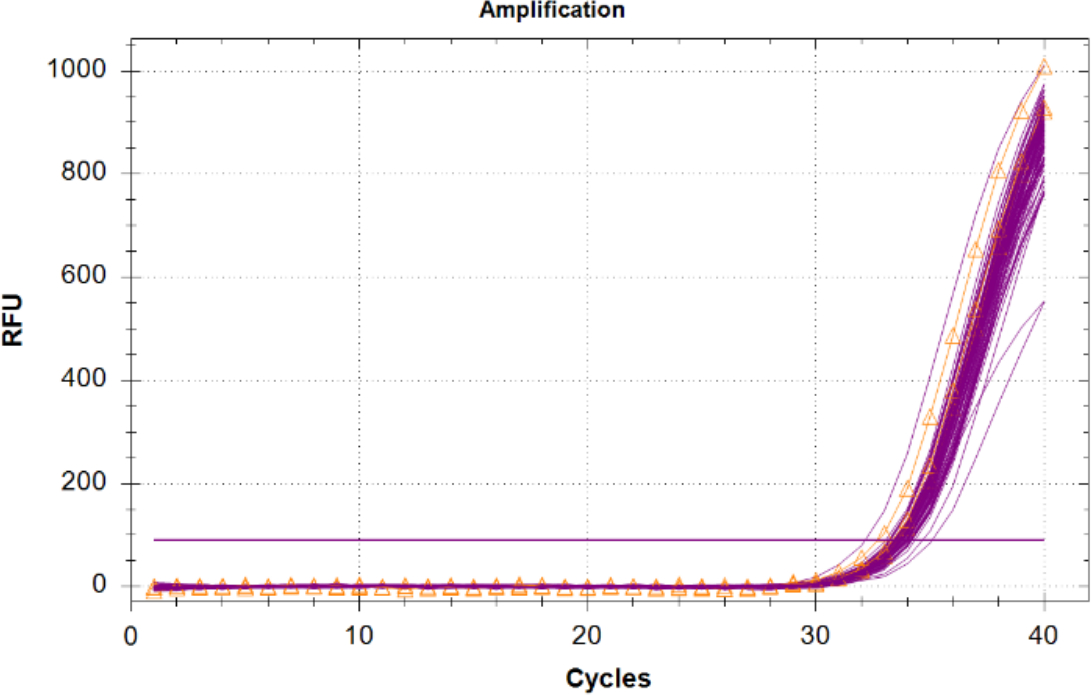
L’amplification du contrôle positif interne (CIP) dans des échantillons inconnus doit être comparée aux résultats du CIP de contrôle de modèle négatif, car il n’y a pas de concurrence pour les réa évaluateurs et aucun inhibiteur n’est présent. Les inconnues avec un CIP ayant un Cq de 2 cycles ou supérieure à la valeur moyenne cq du CNT, ou qui n’amplifient pas devraient être considérées comme inhibées. Si aucun inhibiteur n’est présent dans les échantillons, alors toute amplification iPC devrait avoir un regroupement serré dans la parcelle avec des valeurs Cq proches de la même que le CNT (Figure 6).
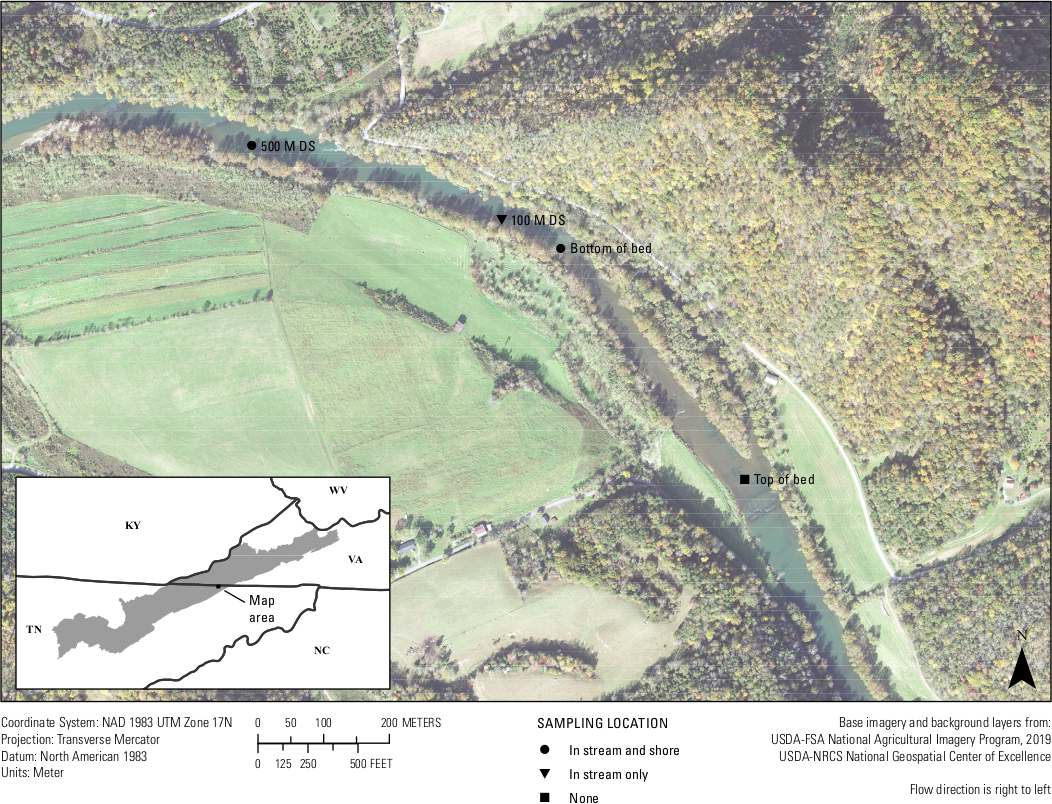
Enfin, des tests in situ de l’essai ont eu lieu. Vingt échantillons d’eau de la rivière Clinch et trois échantillons vierges ont été filtrés entre le 25 et le 26 septembre 2019 à moins de 500 mètres d’un lit de moule connu pour avoir A. ligamentina. Environ quatre échantillons d’eau de 1 L ont été filtrés par lieu d’échantillonnage. Sites d’emplacement inclus au fond du lit de moules dans le ruisseau, au fond du lit de moules près du rivage, à 100 m en aval du lit dans le ruisseau, à 500 m en aval du lit dans le ruisseau et à 500 m en aval du lit près de la rive (figure 7). De retour en laboratoire, chaque filtre a été coupé en deux et l’ADN n’a été extrait que de la moitié d’un filtre. Le reste de la moitié du filtre pour chaque échantillon a été conservé dans un congélateur de -80 °C. Des échantillons ont ensuite été exécutés à l’aide du multiplexé d’analyse A.lig.1 avec le CIP. Sur les 23 échantillons, cinq se sont révélés inhibés. Ces échantillons ont été dilués 1:10 et les dilutions ont été réédgées. Dix-neuf des 20 échantillons sur le terrain amplifiés à l’aide de l’analyse conçue. De ces 19 échantillons, cinq étaient au-dessus du LOD de l’essai et du LOQ de 5 copies/réaction ; ce qui signifie que la plupart des échantillons avaient une détection de l’AND, mais à un niveau où de faux résultats négatifs sont susceptibles de se produire et que l’analyse ne pouvait pas quantifier avec confiance le numéro de copie de ces 14 échantillons. Néanmoins, 75 à 100 % des quatre sites biologiques se répliquent amplifiés à chaque lieu d’échantillonnage. Deux des trois blancs de champ étaient négatifs, tandis qu’un vide de champ a montré l’amplification, soulignant l’importance de la technique propre dans le domaine.

Figure 1 : Flux de travail pour la construction de bases de données de séquences d’ADN mitochondriales. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 2 : Alignements de séquences pour les espèces de moules de rivière Clinch avec des amorces et des sondes prospectives pour l’analyse Actinonaias ligamentina ND1. Amorce avant en vert foncé, sonde en rouge et amorce inversée en vert clair. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 3 : Exemples de courbe standard et de régression linéaire. Unexemple d’une courbe standard acceptable dérivée de l’amplification de trois répliques chacune des six dilutions standard. Une série de dilution standard 10 fois avec la plus forte concentration de la norme sur la gauche, avec des concentrations décroissantes se déplaçant vers la droite. La ligne horizontale franchissant toutes les traces est le seuil de cycle à la quantitation (Cq). Là où chaque trace franchit ce seuil, c’est là que le Cq est déterminé. B. Régression linéaire faite à partir des répliques standard de la figure 3A. Les répliques des dilutions standard sont tracées en cercles et les inconnues (échantillons) sont tracées avec des x. L’efficacité est de 98,9%, r2 approchant 1,0, et la pente de -3,349. C. Exemple d’une courbe standard médiocre dérivée de l’amplification de trois répliques chacune des six dilutions standard. D. Une régression linéaire formant la courbe standard pour les répliques standard amplifiées dans l’exemple 3C. Notez la faible efficacité et les valeurs r2. Notez également que seulement 4 des 6 normes amplifiées. Si après les répétitions, la courbe standard ne s’améliore pas, le problème peut être avec un ensemble d’amorce/sonde pauvre qui n’amplifie pas l’ADN cible comme prévu dans ce cas, cet essai ne devrait pas être considéré. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 4 : Exemples de configurations de plaques pour les courses QPCR standard LOD et LOQ. Les normes utilisées dans la courbe sont en bleu, la concentration standard diminue de bleu foncé à bleu clair. Contrôle positif de l’ADN en vert et aucun contrôle de modèle (CNT) en jaune. Concentrations standard expérimentales en gris montrant 24 répliques pour chaque dilution standard. La série de dilution a été plaquée sur deux plaques (A, B), chacune avec une courbe standard, un contrôle positif et un CNT. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 5 : Traces de configuration et d’amplification de la plaque d’une course qPCR. Uneconfiguration de plaque. , normes montrées en bleu, couleur plus foncée indiquant la plus haute concentration de la norme. Contrôle positif de l’ADN en vert, pas de contrôles de modèle en jaune (CNT), cibles d’échantillon en gris. B. Amplification des traces d’une course qPCR. Normes indiquées en bleu, contrôle positif de l’ADN en vert, aucun contrôle de modèle en jaune, et inconnus en rouge. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 6 : Traces d’amplification pour le contrôle positif interne (CIP). Traces iPC pour tous les échantillons inconnus en magenta et le CIP à partir des contrôles sans modèle (CNT) montrés en orange avec des triangles. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 7 : Carte montrant les sites de collecte eDNA d’un lit de moules dans la rivière Clinch le long de la frontière entre la Virginie et le Tennessee. Des échantillons ont été prélevés à Wallens Bend, au fond du lit, à 100 m en aval du lit et à 500 m en aval du lit. Les sites ont été recueillis au milieu du cours d’eau (en cours d’eau) ou à environ 1 à 2 mètres du rivage (rivage). S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}
| composant | nom | Séquence 5' – 3' | Étiquette fluorescente | |
| Amorce avant | A.lig.1-f | CCCTCATCACGTACCTCTTAATC | ||
| Amorce inversée | A.lig.1-r | GGAATGCCCATAATTCCAACTTTA GGAATGCCCATAATTCCAACTTTA | ||
| sonde | Sonde A.lig.1 | TTCTTGAACGTAAAGCCCTCGGGT TTCTTGAACGTAAAGCCCTCGGGT TTCTTGAACGTAAAGCCCTCGGGT TTC | Fam | |
Tableau 1 : Essai qPCR actinonaias ligamentina conçu (A.lig.1), y compris des séquences pour les amorces avant et arrière et la sonde.
| espèce | amplifié | Dans la rivière Clinch |
| 1. Actinonaias ligamentina | oui | oui |
| 2. Actinonaias pectorosa | Non | oui |
| 3. Plicata Amblema | Non | oui |
| 4. Corbicula spp. | Non | oui |
| 5. Cumberlandia monodonta | Non | oui |
| 6. Cyclonaias tuberculata | Non | oui |
| 7. Stegaria Cyprogenia | Non | oui |
| 8. Elliptio dilatata | Non | oui |
| 9. Brevidens Epioblasma | Non | oui |
| 10. Epioblasma capsaeformis | Non | oui |
| 11. Epioblasma florentina aureola | Non | oui |
| 12. Epioblasma triquetra | Non | oui |
| 13. Cor Fusconaia | Non | oui |
| 14. Subrotunda Fusconaia | Non | oui |
| 15. Lampsilis ovata | Non | oui |
| 16. Lampsilis siliquoidea | Non | Non |
| 17. Lasmigona costata | Non | oui |
| 18. Rimosus lemiox | Non | oui |
| 19. Dolabelloides Lexingtonia | Non | oui |
| 20. Conradicus Medionidus | Non | oui |
| 21. Plethobasus cyphyus | Non | oui |
| 22. Plénum pleurobema | Non | oui |
| 23. Fasciolaris Ptychobranchus | Non | oui |
| 24. Ptychobranchus sous-ténor | Non | oui |
| 25. Pustulosa Quadrula | Non | oui |
| 26. Strophitus undulatus | Non | oui |
| 27. Iris de Villosa | Non | oui |
Tableau 2 : Liste des espèces utilisées pour le test de spécificité in vitro de l’essai A.lig.1. L’analyse a amplifié l’ADN génomique de la cible (Actinonaias ligamentina) et n’a pas amplifié l’une des espèces non cibles.
| composant | Séquence 5'-3' | ||||
| Actinonaias ligementina standard | CCCTCATCACGTAC (en) CTCTTAATCCTATTAGGTGTCGCATTTTTCACTCTTCTTGAACGTA | ||||
| AAGCCCTCGGGT ACTTTCAAATCCGAAAAGGCCCAAATAAAGTTGGAATTATGGGCATTC ACTTTCAAATCCGAAAAGGCC CAAATAAAGTTGGAATTATGGGCATTC ACTTTCAAATCCAAAGGGGGGGGCCAAAAAGGGGCCAAAGGCC CAAATA | |||||
| CCCAACCATTAGCAGATGCTCTAAAGCTCTTCGTAAAAGAATGAGTAACACCAACCTCCT | |||||
| CAAACTACCTACCCTTCATCTTAACCCCAACCACTATGTTATTATTAGCACTTAGACTTT | |||||
| GACAATTATTTCCATCCTTTATANTATCATCCCAAATANTTTTTGGTATGCTCCTATTCT | |||||
| TGTGTATCTCCTCCCTAGCTGTTTATATAACACTTATAACAGGCTGAGCCTCAAACTCCA | |||||
| AATATGCCCTTTTAGGAGCTATTCGAGCCATAGCCCAAACCATTTCTTATGAGGTTACAA | |||||
| TAAC (TAAC) | |||||
| Modèle IPC (Hem-T) | CTACATAAGTAACACCTTCTCATGTCCAAAGCTCTCTGAGTGTCCCTCGAATCTCAGACGCT | ||||
| GTATGACAGTCTCCTTGTGTGAACATTCGGCTCTATGTTCTCAAGGACTGCAC | |||||
Tableau 3 : Séquence (5'-3') de la norme Actinonaias ligamentina et du modèle IPC (Hem-T) utilisé pour cet essai. La séquence des amorces avant et arrière est en gras et italique, et celle de la sonde est soulignée.
| composant | nom | Séquence 5' – 3' | Étiquette fluorescente | |
| Amorce avant | HemT-F | TCTGAGTGTCCCTCGAATCT | ||
| Amorce inversée | HemT-R | GCAGTCCTTGAGAACATAGAGC | ||
| sonde | HemT-P | TGACAGTCTCCTTGTGTGAACATTCG | Cy5 (en) | |
Tableau 4 : L’analyse du contrôle positif interne (CIP), y compris les séquences pour les amorces avant et arrière et la sonde.
| Volume par échantillon (μL) | composant |
| 10 | Mix Maître de l’environnement |
| 1 | 20uM A. lig.1 F/R mix |
| 1 | Sonde 2.5uM A. lig.1 |
| 1 | Mélange d’amorce IPC 5uM (HemT-F/ R) |
| 0.75 | Sonde IPC de 2,5 uM (HemT-P) |
| 1.5 | 1 X 103 concentration du modèle IPC |
| 2.75 | H20 |
| 2 | échantillon |
| 20 | Total Volume |
Tableau 5 : Le mélange PCR utilisé pour le multiplex d’analyse A.lig.1 avec l’essai IPC.
| pas | Température (°C ) | Heure | |
| 1 | Dénoature initiale | 95 | 10 min |
| 2 | dénaturer | 95 | 15 sec |
| 3 | recuit | 60 | 1 min |
| 4 | Passez à l’étape 2, répétez 39X |
Tableau 6 : Conditions de réaction pour l’analyse A.lig.1.
Discussion
Comme pour toute étude, la définition de la question à traiter est la première étape et la conception de l’analyse eDNA dépend de la portée de l’étude26. Par exemple, si le but de la recherche ou de l’enquête est de détecter une ou quelques espèces, un test ciblé basé sur une sonde est le meilleur. Toutefois, si l’objectif est d’évaluer une suite plus grande ou un assemblage d’espèces, les analyses de métabarcoding de séquençage à haut débit sont mieux adaptées. Une fois qu’il est déterminé quelle approche prendre, une étude pilote comprenant la conception, les essais et l’optimisation des analyses est recommandée.24. La conception des analyses commence par une liste d’espèces décrites dans Figure 1. Cette liste sera à la base pour comprendre comment un test fonctionne en termes de spécificité et de l’étendue géographique à qui il pourrait être appliqué6,10. Il est encouragé à concevoir l’essai d’une zone géographique spécifique, permettant au concepteur de mieux tester un essai de réactivité croisée contre d’autres espèces dans cette région, et d’être conscient des limites que cela a sur l’extension d’un essai à d’autres zones où une espèce cible peut se produire24. Une fois la liste terminée, les séquences peuvent être téléchargées à partir de bases de données génétiques publiques. Étant donné que ces bases de données sont incomplètes27, il faut séquencer autant d’espèces sur la liste que possible à l’interne pour compléter la base de données de référence locale des séquences qui seront utilisées dans la conception d’analyse. Priorisez les espèces étroitement apparentées, car ce sont les non-cibles les plus probables qui s’amplifieront. Se concentrer sur toutes les espèces du même genre ou de la même famille que l’espèce cible est un bon point de départ. Les comparaisons avec des espèces étroitement apparentées aideront à identifier les régions séquence uniques aux espèces cibles. Cela peut aider à informer comment l’analyse peut effectuer dans d’autres systèmes ou emplacements. Les régions mitochondriales sont le choix habituel pour le développement d’analyse, parce que plus d’informations de séquence d’une plus grande variété d’espèces sont disponibles aux gènes mitochondriques qui ont été utilisés dans le code à barres des projets de vie, et parce que l’ADN mitochondrique est présent à une concentration beaucoup plus grande dans les copies/cellule que l’ADN nucléaire24,28,29. Plusieurs régions génétiques devraient être évaluées pour le développement ultérieur de l’analyse, car la couverture des séquences varie d’un taxa à l’autre dans les bases de données des référentiels génétiques. Une fois cette base de données locale de séquences de référence créée, une combinaison de visualisation manuelle des données de séquence alignées et des logiciels informatiques est utilisée pour concevoir les analyses d’amorce/sonde. Il ne faut pas se fier strictement au logiciel pour déterminer quels essais tester. Il est important de vérifier visuellement sur les alignements où les amorces et les sondes sont assises sur les cibles et les non-cibles pour mieux comprendre comment elles pourraient agir dans un PCR. Enfin, le dépistage et l’optimisation des analyses comprennent trois niveaux (in silico, in vitro et in situ)6,7,24,25. Dans la conception et les tests silico sont importants pour produire une courte liste d’essais avec de bonnes chances de succès, mais empirique (in vitro) test est crucial pour choisir l’essai avec les meilleures performances réelles. L’optimisation in vitro et l’essai des essais comprennent la mesure de l’efficacité de la réaction et la définition de la sensibilité et de la spécificité de l’essai. Les limites de détection et de quantification sont deux paramètres souvent négligés dans le développement d’analyses, mais importants pour l’interprétation des données. En exécutant plusieurs répliques des courbes standard pour un essai, LOD et LOQ peuvent facilement être mesurés1,5,30. Peu d’études discutent des résultats en ce qui concerne le LOD ou loq de l’essai, mais Sengupta et coll. (2019) intègrent le LOD et le LOQ de leur analyse dans leur interprétation des données et leurs graphiques pour une meilleure compréhension de leurs résultats.31. Les contrôles positifs internes doivent également être multiplexés dans l’analyse conçue. Sans test d’inhibition de la PCR dans les échantillons, de faux négatifs peuvent se produire24,32. Nous proposons l’utilisation d’un essai multiplexé d’IPC avec l’essai cible comme méthode la plus facile pour l’essai d’inhibition de PCR23. Enfin, des essais in situ de l’essai à partir d’échantillons prélevés sur le terrain et en laboratoire sont nécessaires pour s’assurer que l’amplification cible se produit dans les échantillons environnementaux.24.
Il existe des limites à l’utilisation d’analyses qPCR spécifiques aux espèces et basées sur des sondes avec des échantillons d’ADN. Par exemple, la conception de plusieurs essais pour les tests peut être limitée par la disponibilité de la séquence, et des compromis peuvent être nécessaires sur certains aspects des performances d’analyse. Ces choix doivent être guidés par les objectifs de l’étude et doivent être rapportés avec les résultats26. Par exemple, si l’objectif est la détection d’une espèce rare et que peu de points positifs sont attendus, un essai avec une spécificité imparfaite (c.-à-d. l’amplification d’espèces non cibles) pourrait être utilisé si toutes les détections étaient vérifiées par séquençage. Si l’objectif est de surveiller l’aire de répartition géographique d’une espèce et que les données sur la concentration de l’ADN sont pas nécessaires, un essai avec une efficacité imparfaite pourrait être utilisé et les données ne seraient déclarées qu’en pourcentage de détection. En outre, à moins que tous les conspécifiques potentiels ne soient testés en laboratoire, ce qui est rarement possible, on ne peut pas connaître avec une certitude absolue la véritable spécificité d’un essai. Par exemple, l’essai a été conçu et testé contre plusieurs espèces de moules d’eau douce dans la rivière Clinch. Pour utiliser cet essai dans un système fluvial différent, nous aurions besoin de le tester contre une suite d’espèces dans le nouvel emplacement. Les variations génétiques au sein de l’espèce ou de la population qui ne sont pas testées lors du développement de l’analyse peuvent également affecter la spécificité. Enfin, même si un essai a été vérifié pour avoir des performances techniques élevées; les conditions changent lorsque vous travaillez sur le terrain. Les conditions non liées à l’analyse telles que le débit d’eau, le pH et le comportement des animaux peuvent modifier la détectabilité de l’ADNE, tout comme l’utilisation de différents protocoles de collecte et d’extraction de l’ADN. L’utilisation d’analyses optimisées et bien décrites facilitera la compréhension de l’influence de ces paramètres sur la détection de l’ADN.
Le domaine de l’ADN est en train de mûrir au-delà du stade de l’analyse exploratoire pour accroître la normalisation des méthodes et des techniques. Ces développements amélioreront notre compréhension des techniques, des capacités et des limitations de l’ADN électronique. Le processus d’optimisation que nous décrivons ci-dessus améliore la sensibilité, la spécificité et la reproductibilité d’un essai. Le but ultime de ce raffinement et de cette normalisation des méthodes eDNA est d’améliorer les capacités des chercheurs à faire des inférences basées sur les données de l’ADN électronique ainsi que d’accroître la confiance des utilisateurs finaux et des détenteurs de pieu dans les résultats.
Déclarations de divulgation
Les auteurs ne déclarent aucun conflit d’intérêts. Les commanditaires du financement n’ont joué aucun rôle dans la conception de l’étude; dans la collecte, les analyses ou l’interprétation des données; dans l’écriture du manuscrit; ou dans la décision de publier les résultats.
Remerciements
Nous remercions Alvi Wadud et Trudi Frost qui ont aidé au développement et aux essais d’amorçage. Le financement de la conception de l’essai rapporté dans cette étude a été fourni par le Programme stratégique de recherche et de développement environnementaux du ministère de la Défense (RC19-1156). Toute utilisation de noms commerciaux, de produits ou d’entreprises est à des fins descriptives seulement et n’implique pas l’approbation par le gouvernement des États-Unis. Les données générées au cours de cette étude sont disponibles sous forme de communiqué de données de l’USGS https://doi.org/10.5066/P9BIGOS5.
matériels
| Name | Company | Catalog Number | Comments |
| 96 Place Reversible Racks with Covers | Globe Scientific | 456355AST | |
| Clean gloves (ie. latex, nitrile, etc.) | Kimberly-Clark | 43431, 55090 | |
| CFX96 Touch Real-Time PCR Detection System | Bio-Rad | 1855196 | |
| Fisherbrand Premium Microcentrifuge Tubes: 1.5mL | Fisher Scientific | 5408129 | |
| Fisherbrand Premium Microcentrifuge Tubes: 2.0mL | Fisher Scientific | 2681332 | |
| Hard-Shell 96-Well PCR Plates, low profile, thin wall, skirted, white/clear | Bio-Rad | #HSP9601 | |
| IPC forward and reverse primers | Integrated DNA Technologies, Inc. | none | custom product |
| IPC PrimeTime qPCR Probes | Integrated DNA Technologies, Inc. | none | custom product |
| IPC Ultramer DNA Oligo synthetic template | Integrated DNA Technologies, Inc. | none | custom product |
| Labnet MPS 1000 Compact Mini Plate Spinner Centrifuge for PCR Plates | Labnet | C1000 | |
| Microcentrifuge machine | Various | - | Any microcentrifuge machine that hold 1.5mL and 2.0mL tubes is typically okay. |
| Microseal 'B' PCR Plate Sealing Film, adhesive, optical | Bio-Rad | MSB1001 | |
| Nuclease-Free Water (not DEPC-Treated) | Invitrogen | AM9932 | |
| Pipette Tips GP LTS 1000 µL F 768A/8 | Rainin | 30389272 | |
| Pipette Tips GP LTS 20 µL F 960A/10 | Rainin | 30389274 | |
| Pipette Tips GP LTS 200 µL F 960A/10 | Rainin | 30389276 | |
| Pipettes | Rainin | Various | Depending on lab preference, manual or electronic pipettes can be used at various maximum volumes. |
| TaqMan Environmental Master Mix 2.0 | Thermo Fisher Scientific | 4396838 | |
| Target forward and reverse primers | Integrated DNA Technologies, Inc. | none | custom product |
| Target PrimeTime qPCR Probes | Integrated DNA Technologies, Inc. | none | custom product |
| Target synthetic gBlock gene fragment | Integrated DNA Technologies, Inc. | none | custom product. used for qPCR standard dilution series |
| TE Buffer | Invitrogen | AM9849 | |
| VORTEX-GENIE 2 VORTEX MIXER | Fisher Scientific | 50728002 |
Références
- Kubista, M., et al. The real-time polymerase chain reaction. Mol Aspects Med. 27 (2-3), 95-125 (2006).
- Higuchi, R. D., Walsh, P. S., Griffith, R. Simultaneous amplification and detection of specific DNA sequences. Biotechnology. 10, 5(1992).
- Mauvisseau, Q., et al. Influence of accuracy, repeatability and detection probability in the reliability of species-specific eDNA based approaches. Scientific Reports. 9 (1), 580(2019).
- Hernandez, C., et al. 60 specific eDNA qPCR assays to detect invasive, threatened, and exploited freshwater vertebrates and invertebrates in Eastern Canada. Environmental DNA. , (2020).
- Klymus, K. E., et al. Reporting the limits of detection and quantification for environmental DNA assays. Environmental DNA. , (2019).
- Thalinger, B., et al. A validation scale to determine the readiness of environmental DNA assays for routine species monitoring. bioRxiv. , (2020).
- Helbing, C. C., Hobbs, J. Environmental DNA Standardization Needs for Fish and Wildlife Population Assessments and Monitoring. CSA Group. , (2019).
- Sepulveda, A. J., Nelson, N. M., Jerde, C. L., Luikart, G. Are Environmental DNA Methods Ready for Aquatic Invasive Species Management. Trends in Ecology & Evolution. , (2020).
- Svec, D., Tichopad, A., Novosadova, V., Pfaffl, M. W., Kubista, M. How good is a PCR efficiency estimate: Recommendations for precise and robust qPCR efficiency assessments. Biomolecular Detection and Quantification. 3, 9-16 (2015).
- Wilcox, T. M., et al. Robust detection of rare species using environmental DNA: the importance of primer specificity. PLoS One. 8 (3), 59520(2013).
- Prediger, E. How to design primers and probes for PCR and qPCR. IDT. , Available from: http://www.idtdna.cco/pages/education/decoded/article/designing-pcr-primers-and-probes (2020).
- Thornton, B., Basu, C. Real-time PCR (qPCR) primer design using free online software. Biochemistry and Molecular Biology Education. 39, 145-154 (2011).
- Owczarzy, R., et al. IDT SciTools: a suite for analysis and design of nucleic acid oligomers. Nucleic Acids Research. 36, Web Server issue 163-169 (2008).
- Kibbe, W. A. OligoCalc: an online oligonucleotide properties calculator. Nucleic Acids Research. 35, Web Server issue 43-46 (2007).
- Taylor, S. C., et al. The Ultimate qPCR Experiment: Producing Publication Quality, Reproducible Data the First Time. Trends in Biotechnology. 37 (7), 761-774 (2019).
- Ye, J., et al. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 13 (134), 11(2012).
- Altschul, S. F., Gish, W., Miller, W., Myers, E. W., Lipman, D. J. Basic Local Alignment Search Tool. Journal of Molecular Biology. 215, 403-410 (1990).
- Bustin, S. A., et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clinical Chemistry. 55 (4), 611-622 (2009).
- Bio-Rad. Bio-Rad Vol. 5279. , ed Bio-Rad (2020).
- Bio-Rad. Bio-Rad Vol. 6894. , Bio-Rad (2020).
- Eurogentec. Eurogentec. Vol. 0708-V2. , ed Eurogentec (2020).
- Bustin, S., Huggett, J. qPCR primer design revisited. Biomolecular Detection and Quantification. 14, 19-28 (2017).
- Hoorfar, J., et al. Practical considerations in design of internal amplification controls for diagnostic PCR assays. Journal of Clinical Microbiology. 42 (5), 1863-1868 (2004).
- Goldberg, C. S., et al. Critical considerations for the application of environmental DNA methods to detect aquatic species. Methods in Ecology and Evolution. 7 (11), 1299-1307 (2016).
- Guan, X., et al. Environmental DNA (eDNA) Assays for Invasive Populations of Black Carp in North America. Transactions of the American Fisheries Society. 148 (6), 1043-1055 (2019).
- Mosher, B. A., et al. Successful molecular detection studies require clear communication among diverse research partners. Frontiers in Ecology and the Environment. 18 (1), 43-51 (2019).
- Kwonga, S., Srivathsana, A., Meier, R. An update on DNA barcoding: low species coverage and numerous unidentified sequences. Cladistics. 28, 6(2012).
- Rees, H. C., et al. REVIEW: The detection of aquatic animal species using environmental DNA - a review of eDNA as a survey tool in ecology. Journal of Applied Ecology. 51 (5), 1450-1459 (2014).
- Evans, N. T., Lamberti, G. A. Freshwater fisheries assessment using environmental DNA: A primer on the method, its potential, and shortcomings as a conservation tool. Fisheries Research. 197, 60-66 (2018).
- Forootan, A., et al. Methods to determine limit of detection and limit of quantification in quantitative real-time PCR (qPCR). Biomolecular Detection and Quantification. 12, 1-6 (2017).
- Sengupta, M. E., et al. Environmental DNA for improved detection and environmental surveillance of schistosomiasis. Proceedings of the National Academy of Sciences of the United States of America. 116 (18), 8931-8940 (2019).
- Klymus, K. E., Richter, C. A., Chapman, D. C., Paukert, C. Quantification of eDNA shedding rates from invasive bighead carp Hypophthalmichthys nobilis and silver carp Hypophthalmichthys molitrix. Biological Conservation. 183, 77-84 (2015).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.