
Method Article
Estrategia de análisis bioaformático y bioinformática basada en biosensores para la validación global de interacciones entre fármacos y proteínas
En este artículo
Resumen
Este estudio tenía como objetivo presentar una estrategia para identificar las interacciones entre fármacos y péptidos. La estrategia consiste en la biopanning de péptidos cortos que reconocen fármacos basados en un biosensor de micropeso de cristal de cuarzo (QCM), seguido de análisis bioinformática para evaluar cuantitativamente la información obtenida para el reconocimiento de fármacos y la anotación de los sitios de unión a fármacos sobre proteínas.
Resumen
Los receptores y las proteínas enzimáticas son biomoléculas importantes que actúan como objetivos de unión para moléculas pequeñas bioactivas. Por lo tanto, la validación rápida y global de las interacciones fármaco-proteínas es altamente deseable no sólo para entender los mecanismos moleculares subyacentes a la eficacia terapéutica, sino también para evaluar las características de los fármacos, tales como adsorción, distribución, metabolismo, excreción, y toxicidad (ADMET) para uso clínico. Aquí, presentamos una estrategia de alto rendimiento basada en biosensores para el biopanning de péptidos cortos con exhibición de fago T7 que se pueden mostrar fácilmente en el fósforo capsid. También se muestra el análisis posterior de las secuencias de aminoácidos de péptidos que contienen segmentos cortos, como "reliquias rotas", de los sitios de unión a fármacos que utilizan programas de bioinformática en la suite de contacto de ligando receptor (RELIC). Mediante la aplicación de este método a dos fármacos clínicamente aprobados, un irinotecán antitumoral y un oseltamivir anti-gripe, se explica en este artículo el proceso detallado para recoger las secuencias de péptidos que reconocen fármacos y resaltar los sitios de unión farmacológica de las proteínas diana. La estrategia descrita en este documento se puede aplicar para cualquier molécula pequeña de interés.
Introducción
La identificación de objetivos farmacorres unidos es esencial para el desarrollo de fármacos, así como para comprender los mecanismos moleculares de las enfermedades. En particular, las proteínas receptoras y enzimáticas son los objetivos moleculares más importantes de las moléculas pequeñas bioactivas1. Aunque la captura de afinidad es una técnica bien establecida para identificar las proteínas que unen fármacos2,las limitaciones técnicas, como la baja solubilidad de las proteínas, a menudo obstaculizan la validación de los objetivos farmacológicos2. Lo más importante es que las moléculas pequeñas inmovilizadas pierden el grado de libertad necesario para el acoplamiento y pueden ser inaccesibles para los sitios de unión internamente ubicados en proteínas objetivo más grandes. Además, el misfolding proteico, la incapacidad para analizar las condiciones de co-cristalización y las limitaciones debidas al tamaño molecular a menudo dificultan el uso de cristalografía por rayos X, resonancia magnética nuclear (RMN) y otros análisis experimentales para estudiar las interacciones entre fármacos y proteínas.
El uso de la pantalla de fago T7 biopanning es una manera eficiente para determinar el sitio de unión en proteínas para cebos de moléculas pequeñas3. En particular, una biblioteca de péptidos aleatorios con foda T7, que se puede construir insertando ADN sintético en un sitio de clonación múltiple, es eficaz. En comparación con la biblioteca de fagos T7 que muestra proteínas, los péptidos cortos se pueden diseñar fácilmente para ser mostrados en el fago T7 capsid sin restricciones físicas, que puede entrar en contacto estetéricamente con cualquier fármaco de molécula pequeña fijado en un soporte sólido2. Además, la introducción de un biosensor de micropeso de cristal de cuarzo (QCM) en la plataforma de biopanning de exhibición de fago T7 permite la identificación de tales interacciones débiles pero específicas de péptidos cortos con fármacos mediante el seguimiento de la reducción en la frecuencia QCM4,5. El fago T7 enlazado se recupera directamente infectando el huésped Escherichia coli (BLT5615), y se determina la secuencia de ADN de la región que codifica el péptido seleccionado por afinidad que alberga segmentos cortos que reconocen fármacos. Análisis posterior de la secuencia de aminoácidos de la población de péptidos proporciona información sobre el reconocimiento de drogas. En silico alineación emparejada de las secuencias de aminoácidos rescatados se puede utilizar para obtener información sobre el objetivo biológico de la droga dentro de un proteoma seleccionado. Esta identificación de alto rendimiento de fragmentos de proteínas con afinidad hacia un fármaco se puede utilizar para reconstruir heurísticamente el sitio de unión a fármacos de una manera similar a la de reconstruir un artefacto antiguo a partir de fragmentos de cerámica6. En particular, este enfoque único puede ser útil cuando los enfoques proteómicos convencionales fallan.
Aquí, presentamos una estrategia basada en biosensores para el biopanning de péptidos exhibidos por fagos T7 y análisis bioinformática para la validación objetivo de las moléculas pequeñas. Más allá de las limitaciones técnicas de los métodos convencionales, esta estrategia permite la identificación de sitios de unión a fármacos en proteínas objetivo para cualquier molécula pequeña de interés bajo el protocolo idéntico.
Protocolo
NOTA: Los siguientes son los pasos para examinar los fagos T7 que reconocen fármacos utilizando un biosensor QCM y recuperar los fagos examinados a través de la infección por E. coli (BLT5615). Los protocolos para la síntesis de un derivado de una molécula pequeña que forma una monocapa autoensamblada (SAM) y para la construcción de la biblioteca de péptidos aleatorios de 15 mer exhibida por fagos T7 se pueden encontrar en otros lugares6,7.
1. Preparación del chip del sensor QCM
- Conecte un chip de sensor cerámico en el oscilador de un aparato QCM de 27 MHz y registre la frecuencia intrínsica (Hz) en la fase de aire antes de la inmovilización de moléculas pequeñas.
- Separar el chip y soltar 20 μL de una solución (1 mM en 70% etanol) de un pequeño derivado de molécula que forma SAM en el electrodo de oro del chip del sensor usando una pipeta.
PRECAUCIÓN: El cristal del chip del sensor donde se encuentra el electrodo de oro (Au, 0,1 mm de espesor, 2,5 mm i.d., 4,9 mm2)es extremadamente delgado y puede agrietarse fácilmente (SiO2,0,06 mm de espesor, 9 mm i.d.). Por lo tanto, pipeta cuidadosamente. - Dejar reposar 1 h a temperatura ambiente (alrededor de 20 °C) en una placa de Petri con tejido humedecido y protegido de las luces de la habitación.
- Lave la superficie del electrodo suavemente con agua ultrapura; a continuación, retire las gotas de agua soplando aire con una jeringa o un plumero de aire.
- Configure el chip del sensor para el aparato QCM y registre la reducción de la frecuencia en la fase de aire para medir la cantidad de la molécula pequeña que ha sido inmovilizada.
NOTA: Al menos, 100 Hz de frecuencia intrínsica es necesario para la inmovilización exitosa de moléculas pequeñas (1 Hz inmoviliza 30 pg).
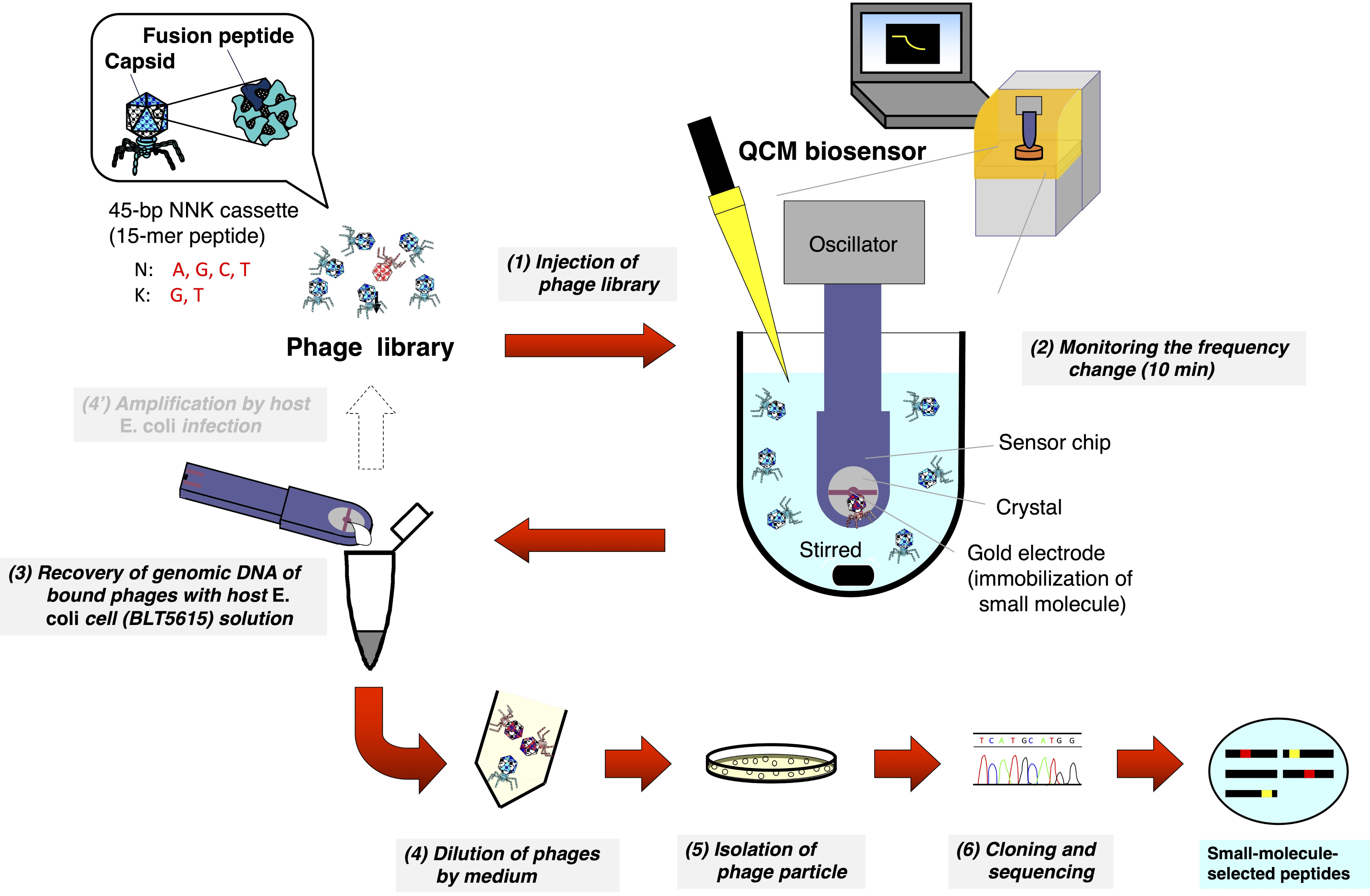
2. Biopanning de la biblioteca de fagos T7 utilizando un biosensor QCM (Figura 1)
- Ajuste una cubeta con un agitador magnético dedicado en el biosensor QCM y vierta 8 ml del búfer de reacción (Tris-HCl de 10 mM, NaCl de 200 mM, pH 7-8) en la cubeta.
- Conecte el chip del sensor QCM al oscilador y tire hacia abajo del brazo del oscilador para sumergir el chip en el búfer que se agita a 1000 rpm.
- Comience a monitorear la frecuencia QCM en el ordenador personal (PC) y espere hasta que el sensorgrama se equilible a alrededor de 3 Hz/min de la deriva de frecuencia.
- Inyecte 8 μL de una biblioteca de fagos T7 (1—2 × 1010 pfu/ml) en la cubeta (concentración final: 1-2 × 107 pfu/mL) y marque el punto de inyección en el sensor.
- Monitoree la reducción de frecuencia causada por la unión de fagos T7 a la molécula pequeña inmovilizada en la superficie del electrodo de oro durante 10 min.
- Detenga el monitor de frecuencia QCM, desaloje el chip del sensor del oscilador y retire el búfer soplando aire y/o limpiando con toallitas.
- Coloque el chip del sensor en una placa petri húmeda y deje caer los 20 μL de la suspensión de E. coli (BLT5615) (OD600 = 0.5—1.0 después de temblar a 37 °C añadiendo células huésped IPTG a 1 mM) en la fase de registro en el electrodo de oro.
- Cierre la tapa del plato y cúbralo con papel de aluminio para bloquear la luz.
- Incubar el plato a 37 °C durante 30 minutos en un mezclador de microplacas de 96 pozos (1000-1500 rpm) para mejorar la recuperación de los fagos T7 enlazados.
- Recupere los 20 μL de la solución y suspenda en 200 μL de media LB.
NOTA: Las muestras obtenidas en este paso se pueden conservar a 4 °C por una semana. - Preparar una serie de dilución de la solución de fago para el aislamiento de la placa y la secuenciación del ADN de acuerdo con el procedimiento general descrito en las instrucciones del fabricante8,9.
- Limpie la superficie del electrodo dorado con un hisopo de algodón empapado con un 1% solución de sulfato de dodecilo sódico.
- Lave la superficie de oro con agua ultrapura de la botella de lavado y luego retire las gotas de agua soplando aire con una jeringa o un plumero de aire.
- Gota 5 μL de solución de piraña (Conc. H2SO4: 30% H2O2 = 3:1) en la superficie de oro y dejar durante 5 min.
- Lave la superficie de oro de nuevo con agua y luego séquela soplando aire y/o limpiando con toallitas.
- Repita los pasos 2.14 y 2.15.
PRECAUCIÓN: Preparar la solución de pirañas inmediatamente antes de su uso. Utilice este líquido con cuidado, ya que es un ácido muy fuerte. El tratamiento durante más de 5 minutos erosiona el chip del sensor.
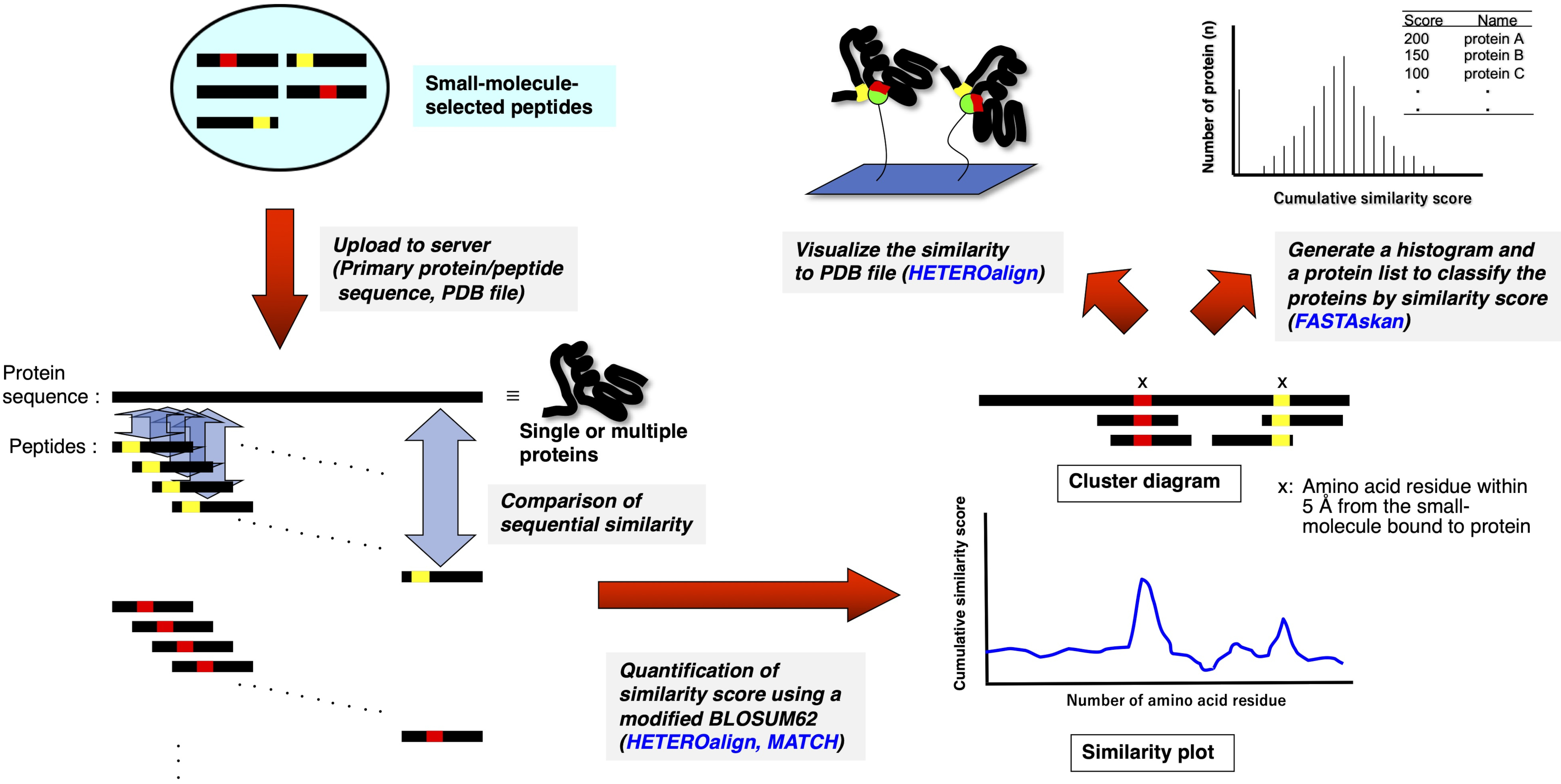
3. Análisis de bioinformática utilizando la suite del programa Receptor Ligand Contact (RELIC) (Figura 2)10,11
- Descomprima el programa RELIC independiente en un PC con un sistema operativo MS Windows.
- Alinee las secuencias de aminoácidos de los péptidos de 15 mer seleccionados con el medicamento o seleccionados aleatoriamente de la biblioteca primaria sin pantalla en cada archivo de formato de texto (nombre.txt).
- Escriba la secuencia de aminoácidos de una o varias proteínas en cada archivo de texto con formato FASTA, o descargue los archivos de texto de la base de datos en formato FASTA desde cualquier base de datos de proteínas (por ejemplo, UniProt (http://www.uniprot.org/) o DrugBank (https://www.drugbank.ca/).
- Coloque los archivos de texto (y los archivos PDB para HETEROalign) en la carpeta necesaria para ejecutar cada programa RELIC.
- Haga clic en el archivo ejecutable (programa.exe) de AADIV, INFO, MOTIF, MATCH, HETEROalign, FASTAcon y FASTAskan en la carpeta independiente para abrir la versión personal de FTN95.
- Escriba el nombre de archivo adecuado, junto con la extensión (nombre.txt), en el mensaje de comando para ejecutar cada programa y obtener el archivo de formato de texto necesario.
- Exporte el archivo de texto resultante a un software de hoja de cálculo (por ejemplo, Excel) para generar una gráfica de contenido de información (INFO) o puntuaciones de similitud acumulativas calculadas utilizando un BLOSUM62 (MATCH, HETEROalign).
NOTA: El servidor RELIC original (http://relic.bio.anl.gov) ya no está disponible y algunos programas RELIC de tipo independiente que funcionan en EQUIPOS con una plataforma Windows se pueden obtener del autor correspondiente (tkksg@rs.noda.tus.ac.jp).
Resultados
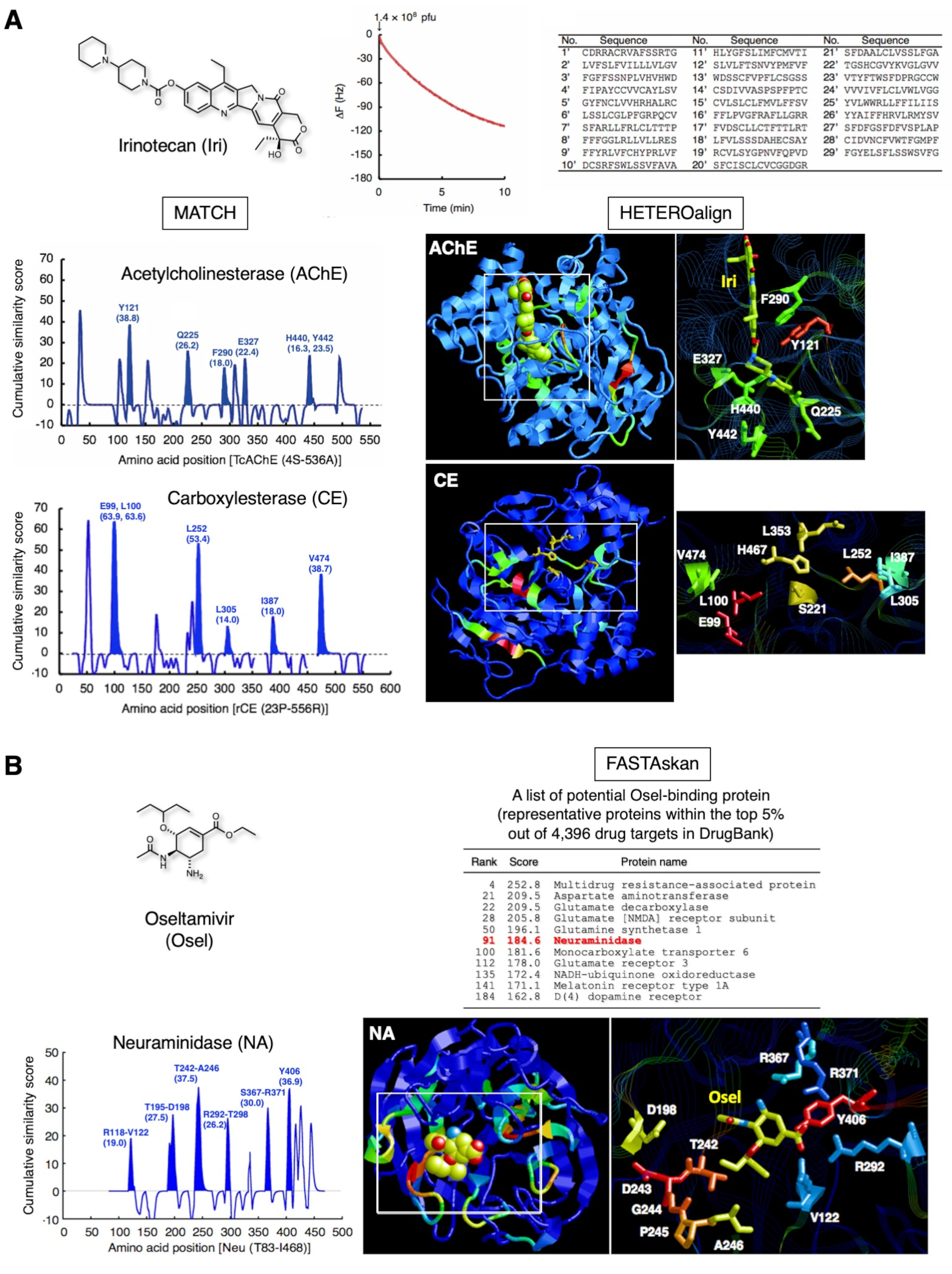
Los resultados representativos de dos medicamentos clínicamente aprobados se muestran en la Figura 3. Irinotecán(Figura 3A),un prodrug soluble en agua de camptotecina natural utilizado para el tratamiento del cáncer colorrectal avanzado y cáncer de pulmón de células no pequeñas, se convierte en SN-38 en el hígado, que inhibe la topoisomerasa I en las células cancerosas12. Además, este compuesto inhibe directamente acetilcolinesterasa (AChE)13,14. A través de la estrategia, 29 péptidos que reconocieron a Iri inmovilizado como SAM fueron identificados por subconjuntos de biopanning basados en biosensores QCM. La posterior alineación por pares de los 29 péptidos y AChE produjo puntuaciones máximas para Y121, Q225, F290, E327, H440 y Y442 y destacó la porción en la estructura tridimensional. Estos residuos de aminoácidos eran consistentes con aquellos que componen el sitio de unión iri de AChE. El mismo subconjunto de péptidos identificó con éxito E99, L100, L252, L305, I387 y V474 en las proximidades de la tríada catalítica (S221, E353 y H467) en carboxesterasa (CE), indicando que estos aminoácidos forman un andamio para el reconocimiento Iri durante la des-esterificación de Iri15. Estos residuos de aminoácidos en el sitio catalítico no se pueden identificar directamente utilizando cristalografía convencional de rayos X o análisis NMR, ya que la reacción enzimática procede sin problemas y no forma el complejo estático establemente en condiciones experimentales generales. Por lo tanto, que la detección combinatoria de sitios de unión a fármacos de múltiples proteínas, incluyendo aquellos en los complejos intermedios posiblemente formados con enzimas durante las reacciones metabólicas para un medicamento en particular, es posible utilizando los péptidos seleccionados por afinidad determinados para un fármaco.
La Figura 3B muestra los demás resultados obtenidos para oseltamivir (Osel), un fármaco antigripal que se activa al carboxilato oseltamivir, que a su vez inhibe la neuraminidasa (NA) del virus de la gripe16. Los 27 péptidos que reconocían a Osel cubriendo la superficie del electrodo de oro del chip del sensor QCM detectaron con éxito el sitio de unión al osel en NA16. Este sitio de enlace consta de bucles de péptidos no estructurados que potencialmente experimentan movimiento dinámico al acoplarse con Osel. Los péptidos que reconocen el Osel en el tapoide de fago T7 podrían imitar este acoplamiento dinámico cuando se une al Osel fijado en la superficie de electrodo de oro del chip del sensor QCM. Se han identificado eventos adversos neuropsiquiátricos (ANP) en pacientes jóvenes con gripe, cuyo efecto en los pacientes están posiblemente relacionados con la enfermedad en sí en lugar del medicamento. Los estudios han demostrado que Osel se exporta activamente desde el sistema nervioso central (SNC) de roedores a través de la proteína de resistencia multirresistente (MDR) en la barrera hematoencefálica (BBB)17. De hecho, una de las proteínas de la clase de este MDR mostró una puntuación alta (top 5% de 4.396 en la base de datos de proteínas DrugBank 1.018),además de otros transportadores, enzimas relacionadas con neurotransmisores y receptores, en nuestro estudio. Se está investigando la importancia farmacológica de estas proteínas con respecto a la aparición de efectos adversos de Osel.
Hasta ahora, se han identificado con éxito sitios de unión de moléculas pequeñas y múltiples en las proteínas objetivo para seis fármacos de moléculas pequeñas que utilizan nuestra estrategia(Figura 4). Para Brz2001 y roxithromiccin (RXM), sitios idénticos de unión a fármacos en una proteína objetivo se identificaron utilizando diferentes grupos de péptidos, los números y secuencias de aminoácidos para los que varió completamente7,19. Además, las piscinas de péptidos únicos obtenidas para Iri, RXM y Osel condujeron a la identificación de múltiples sitios de unión en diferentes proteínas para cada fármaco, como AChE y CE para Iri (Figura 3A)20,angiomotina y CYP3A4 para RXM19,21,y NA y proteína asociada a MDR para Osel. Se identificó un objetivo molecular desconocido para el compuesto antitumoral doxorubicina (FANCF)22,y el anti-angiogénico macrólido RXM (angiomotina)19.

Figura 1: Representación esquemática del biopanning basado en biosensores QCM de la biblioteca de péptidos mostrados por fagos T7. Una biblioteca de fagos T7 que muestra péptidos aleatorios se inyecta en la cubeta que contiene el búfer (bajo agitación) donde el chip biosensor QCM está sumergido y la frecuencia se estabiliza. Después de monitorear la reducción de frecuencia debido a la unión de fagos T7 a moléculas pequeñas inmovilizadas en la superficie de electrodo de oro del chip del sensor, el chip del sensor se separa del oscilador. El ADN del fago T7 enlazado se recupera directamente después de la infección por E. coli (BLT5615). Los fagos T7 resultantes se aíslan a través de la formación de placa y, finalmente, la secuencia de aminoácidos del péptido seleccionado por afinidad de fármaco que se muestra en el cápside de fago T7 se determina de acuerdo con el método general de exhibición de fagos. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Representación esquemática de la evaluación cuantitativa de la comparación de secuencias entre péptidos seleccionados por fármacos y proteínas simples o múltiples. Las secuencias de péptidos seleccionadas por fármacos están alineadas respectivamente con las secuencias primarias de aminoácidos de proteínas simples y múltiples, y la similitud en cada conjunto de aminoácidos 3-5 se anota acumulativamente a través de la alineación emparejada de acuerdo con una matriz BLOSUM62 modificada. La gráfica o diagrama resultante indica los residuos o porciones que constituyen un sitio potencial de unión a fármacos en la proteína. Un análisis adicional utilizando un programa RELIC apropiado resalta el sitio de encuadernación en la estructura tridimensional (si el archivo PDB está disponible) o clasifica proteínas enteras que posiblemente son el objetivo de unión (el programa HETEROalign no está disponible actualmente). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3:Resultados representativos de la recopilación de péptidos y posterior análisis bioinformática. (A) Irinotecán (prorrédug antitumoral, inhibidor de la topoisomerasa I). La interacción de fago T7 fue monitoreada durante 10 minutos como una reducción en la frecuencia QCM. El ADN del fago T7 enlazado fue recuperado y secuenciado para determinar la secuencia de aminoácidos correspondiente. Las secuencias de aminoácidos de 29 péptidos de 15 mer, recogidos con un subconjunto de biopanning de un ciclo, destacaron los aminoácidos que componen el sitio de unión Iri de AChE [PDB ID 1U65]. Una evaluación adicional utilizando los mismos 29 péptidos destacó los residuos vecinos de aminoácidos (residuos de andamios para la des-esterificación) de la tríada catalítica en CE, una enzima hepática que convierte Iri a SN-38 (forma activa) [PDB ID: 1K4Y]. Las puntuaciones de similitud de 103 péptidos seleccionados aleatoriamente de la biblioteca primaria no seleccionada7,19 se han restado de estas puntuaciones para eliminar el sesgo de biblioteca. Estas cifras se reproducen a partir de la referencia 20, con permiso de Elsevier. (B) Oseltamivir (medicamento antigrimoso). Los 27 péptidos que contenían aminoácidos que reconocen el Osel destacaron porciones desordenadas del sitio de unión al Osel para la neuraminidasa (NA) (enzima del virus) [PDB ID: 2HT7]. La validación global de la similitud de secuencia entre 27 péptidos y 4.396 proteínas en DrugBank 1.018 reveló que NA está dentro del rango superior del 5%, además de las proteínas humanas huésped asociadas con las funciones del sistema nervioso central. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4:Resumen delas moléculas pequeñas, cuyo objetivo de unión se identificó utilizando esta estrategia. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Discusión
Aquí se ha presentado una estrategia para la biopanning basada en biosensores QCM de péptidos que reconocen fármacos, seguida de análisis bioinformática para validar interacciones fármaco-proteínas utilizando los péptidos identificados. El diseño de los derivados de moléculas pequeñas para la inmovilización en el electrodo de oro del biosensor es un paso importante, ya que el vinculador introducido puede obstaculizar la unión y la recolección del péptido que reconoce el fármaco. Para evitar esto, los derivados con diferentes posiciones del vinculador introducido se preparan23. Alternativamente, para inmovilizar moléculas pequeñas hidrofóbicas, el chip del sensor está sumergido en agua a granel en una placa Petri de 10 cm, y una solución de 20 μL de la molécula pequeña (solución de 10 mM en sulfóxido de dimetil) se cae sobre el electrodo de oro del biosensor, para cubrir su superficie, e incubada durante 5 minutos. Esto permite la retención de una frecuencia intrínseco de sub-cien hercios de moléculas pequeñas, que se mantiene durante al menos 10 minutos durante el biopanning. De hecho, utilizando dicha inmovilización, los péptidos seleccionados por afinidad de Osel destacaron claramente el sitio de enlace de Osel en NA (Figura 3).
El fago T7 utilizado para preparar la biblioteca de péptidos aquí está genéticamente diseñado utilizando el casete NNK15 que codifica 32 codón para los 20 aminoácidos estándar y reprime la aparición de 2 codos stop (UAA, UGA) y emerge sólo UAG (Figura 1)6,7. Esto es importante para mostrar péptidos de larga duración de 15 mer y aumentar la diversidad de la biblioteca. El sistema de visualización de fagos T7 tiene un límite de visualización técnica de 107—109 fagos T7. Sin embargo, la diversidad de la biblioteca de péptidos de 15 mer es teóricamente 2015 (3,27 × 1019); por lo tanto, no se puede utilizar para la construcción completa de la biblioteca. Sin embargo, la búsqueda de similitudes o la minería de motivos conservados permite la detección de los aminoácidos que comprenden los sitios de unión farmacológica de proteínas incluso con esta limitada diversidad de péptidos en la biblioteca. Además, 3-5 aminoácidos se extiende dentro del péptido de la biblioteca (la tasa de aparición está entre 1/203 y 1/ 205,que se puede realizar utilizando el sistema de visualización de fago T7) están involucrados en el reconocimiento de fármacos de moléculas pequeñas; por lo tanto, no se requiere una coincidencia del 100% de las secuencias de péptidos con secuencias de aminoácidos de 15 mer que constituyen el sitio de unión de fármacos de la proteína objetivo. De hecho, aproximadamente 30 péptidos seleccionados por afinidad destacaron con éxito el sitio de unión de la proteína objetivo para cada fármaco probado(Figura 4). Por lo tanto, la diversidad de la biblioteca de fago T7 padre utilizado (1.7 × 107 pfu/ mL) se puede utilizar para reconstruir heurísticamente el sitio de unión a fármacos.
Por lo general, 3-5 copias de fagos T7 que mostraban las mismas secuencias que las de las secuencias de aminoácidos de 15 mer que albergan estiramientos de aminoácidos que reconocen drogas surgieron dentro de las 16 placas aisladas arbitrariamente, lo que indica el éxito de la selección de afinidad bajo nuestro protocolo. Esto indica que entre 18 y 30 secuencias de péptidos diferentes que reconocen fármacos se recogen dentro de las 96 placas aisladas (el número está asociado con el formato de microplaca), que se identifican posteriormente mediante secuenciación del ADN y obteniendo la secuencia de aminoácidos correspondiente. En la estrategia actual, la inyección de 8 μL de la biblioteca de fagos T7 en la cubeta que contiene 8 ml de amortiguador (dilución de 1000 veces de la biblioteca) es adecuada para reducir la unión no específica de los fagos T7. Aumentar la diversidad de péptidos seleccionados por afinidad, repetir la selección de un ciclo varias veces y usar 16 o 32 aislamientos de placa por cribado demostró ser más eficaz que el aislamiento de una sola solución a la vez. Por ejemplo, para recopilar eficazmente aproximadamente 30 péptidos seleccionados por afinidad secuenciados de forma diferente, se llevaron a cabo entre 3 y 6 conjuntos de selección de un ciclo, 16 o 32 placas aisladas en cada experimento. La aparición de secuencias idénticas en las placas de fago 16 o 32 T7 se indica detección accidental de antecedentes o podría contaminación como arrastre. Por el contrario, la ausencia de fagos T7 con la misma secuencia o apariencia de muchos fagos T7 con péptidos más cortos que la longitud de 15 mer indica que los fagos T7 en la población no emergieron específicamente con alta probabilidad. Como la reducción de la frecuencia qcm se produce en la misma medida incluso en tales casos, el éxito de la selección debe evaluarse exhaustivamente secuenciando el ADN del fago aislado T7, seguido de análisis bioinformática de las secuencias de aminoácidos de los péptidos. Además, a diferencia del protocolo convencional de visualización de fagos T7, repetir rondas de selección es menos eficaz, ya que la variación y el número de los fagos T7 son pequeños y no se concentran incluso después de repetir los pasos de amplificación y selección23.
Es importante destacar que este método es aplicable para la minería de pequeños sitios de unión a moléculas en los proteomas de los seres humanos, virus patógenos, e incluso plantas. Curiosamente, la longitud de visualización corta posiblemente no estructurada de péptidos en el cástago de fago T7 puede imitar la dinámica molecular de péptidos de proteínas durante el acoplamiento con una molécula pequeña; esto puede reflejar el enlace dinámico24. Más allá de las limitaciones técnicas de los métodos convencionales, esta estrategia, aplicable a protocolos idénticos para moléculas pequeñas, puede ampliar el proteoma farmacológico, así como proporcionar más granularidad con respecto al análisis de interacción fármaco-proteína.
Deben tenerse en cuenta ciertas limitaciones técnicas de este enfoque. La síntesis orgánica es necesaria para la inmovilización de moléculas pequeñas en la superficie del electrodo de oro del chip del biosensor. Para los no expertos en química orgánica, algunos reactivos de inmovilización están disponibles comercialmente para fijar mecánicamente la molécula pequeña mediante acoplamiento. Además, ciertas porciones sin sentido de los péptidos podrían resultar en la detección de una porción de la proteína no relevante para el acoplamiento de fármacos como falsos positivos. Esto corresponde empíricamente a los dominios ricos en beta-hoja o leucina ricos en leucina o valina, que están codificados por más codón que otros aminoácidos estándar, cuando se producen copias del fago T7. Controlar la longitud del péptido de biblioteca podría controlar la aparición de falsos positivos. Por el contrario, puede haber casos en los que no se detecten residuos de aminoácidos en el sitio de unión a fármacos que participan en el acoplamiento, como se demuestra mediante cristalografía de rayos X o análisis nmr. Esto puede resolverse recogiendo un mayor número de péptidos que reconocen fármacos o cambiando la dirección de fijación de las moléculas pequeñas en el electrodo de oro.
Muchas interacciones farmaco-proteínas relacionadas con los principales y secundarios efectos del consumo de drogas todavía pueden no ser identificadas en el proteoma; además, las enzimas y transportadores responsables de la absorción de fármacos, distribución, metabolismo, excreción y toxicidad, también podrían no ser identificados. La unión a proteínas no siempre es responsable de la bioactividad de un medicamento. Por lo tanto, una combinación de otra información de ensayos biológicos mejorará la identificación de objetivos esenciales de fármacos responsables de los principales y adversos efectos de los fármacos. Otras adaptaciones de esta técnica concisa aumentarán la practicidad y el rendimiento para la minería de los sitios de unión a proteínas de una amplia gama de fármacos de moléculas pequeñas. El método presentado en este documento contribuirá en gran medida no sólo a llevar a cabo investigaciones básicas en los campos relacionados, sino también a aclarar los mecanismos moleculares subyacentes a la eficacia terapéutica u otros efectos biológicos de los fármacos en el uso clínico.
Divulgaciones
Los autores no tienen conflictos de intereses que revelar.
Agradecimientos
El autor agradece a los Drs. Yujiro Hayashi y Hayato Ishikawa por proporcionar oseltamivir, y al Dr. Lee Makowski por proporcionar los programas independientes RELIC. El autor también reconoce a Tetsuya Kawagoe por la asistencia técnica del experimento QCM. Este trabajo fue parcialmente apoyado por JSPS KAKENHI Grant Number 17K01363 (Y.T.).
DECLARACIÓN DE DISPONIBILIDAD DE DATOS
Los programas RELIC independientes y los datos de secuencia de péptidos seleccionados por afinidad para fármacos, así como las secuencias proteicas de la base de datos de proteomas utilizadas en este artículo están disponibles en este autor bajo petición (tkksg@rs.noda.tus.ac.jp).
Materiales
| Name | Company | Catalog Number | Comments |
| AFFINIXQN | ULVAC, Inc. (Tokyo, Japan) | QCM2008-STKIT | Contains Glass cuvette, stir magnet, operation and analysis software with a Windows PC |
| AADIV | Northeastern University (Lee Makowski) | AADIV.exe | Calculates the frequency of occurrence of each of the 20 amino acids at each recombinant insert position, as well as the overall position-independent frequency of each amino acid within that set of peptide sequences. Also roughly estimates the sequence diversity of a display library by statistical sampling method based upon sequences obtained from a limited number of randomly sampled members of the library. |
| Ceramic Sensor Chip | ULVAC, Inc. (Tokyo, Japan) | QCMST27C | 4 sensor chips/package |
| Dimethyl sulfoxide | Sigma-Aldrich (St. Louis, MO, USA) | D8418 | |
| Ethanol | Merck (Kenilworth, NJ, USA) | 09-0850 | |
| FASTAcon | Northeastern University (Lee Makowski) | FASTAcon.exe | Identifies proteins from a population with short consensus sequences. |
| FASTAskan | Northeastern University (Lee Makowski) | FASTAskan.exe | Lists proteins with high similarity to a peptide population. |
| Immiblization kit for AFFINIX | ULVAC, Inc. (Tokyo, Japan) | QCMIMKT | SAM reagent and amine coupling reagent |
| INFO | Northeastern University (Lee Makowski) | INFO.exe | Provides mathematical measure of the probability of observing a particular peptide sequence by random chance (i.e., nonspecific binding) as opposed to by selection for a specific property (affinity to small molecule). |
| Liquid LB medium | Sigma-Aldrich (St. Louis, MO, USA) | L3522 | Autoclave for 20 min |
| MATCH | Northeastern University (Lee Makowski) | MATCH.exe | Identifies any stretches of amino acid residues within a particular protein that exhibit significant similarity to a group of affinity-selected peptides. Outputs as cluster dia- gram and cumulative similarity plot calculated from a modified BLOSUM62 matrix with a short window (5–6 amino acids in length). |
| MOTIF1 | Northeastern University (Lee Makowski) | MOTIF1.exe | Searches for three continuous amino acid sequence motifs within a peptide population. |
| MOTIF2 | Northeastern University (Lee Makowski) | MOTIF2.exe | Searches for patterns of three amino acids and does not allow conservative amino acid substitutions, but does allow identical gap lengths. |
| NaCl | Merck (Kenilworth, NJ, USA) | S3014 | |
| Receptor ligand contacts (RELIC) | Argonne National Laboratory (Lemont, IL, USA) | https://www.relic.anl.gov | Currently unavailable (Stand-alone program can be used from correspondence author upon request) |
| Tris | Merck (Kenilworth, NJ, USA) | 252859 |
Referencias
- Santos, R., et al. A comprehensive map of molecular drug targets. Nature Reviews Drug Discovery. 16 (1), 19-34 (2017).
- Ziegler, S., Pries, V., Hedberg, C., Waldmann, H. Target identification for small bioactive molecules: finding the needle in the haystack. Angewandte Chemie International Edition (English). 52 (10), 2744-2792 (2013).
- Piggott, A. M., Karuso, P. Identifying the cellular targets of natural products using T7 phage display. Natural Product Reports. 33 (5), 626-636 (2016).
- Takakusagi, Y., Takakusagi, K., Sakaguchi, K., Sugawara, F. Phage display technology for target determination of small-molecule therapeutics: an update. Expert Opinion on Drug Discovery. 15 (10), 1199-1211 (2020).
- Takakusagi, Y., Takakusagi, K., Sugawara, F., Sakaguchi, K. Use of phage display technology for the determination of the targets for small-molecule therapeutics. Expert Opinion on Drug Discovery. 5 (4), 361-389 (2010).
- Takakusagi, Y., Takakusagi, K., Sugawara, F., Sakaguchi, K. Using the QCM Biosensor-Based T7 Phage Display Combined with Bioinformatics Analysis for Target Identification of Bioactive Small Molecule. Methods in Molecular Biology. 1795, 159-172 (2018).
- Takakusagi, Y., et al. Mapping a disordered portion of the Brz2001-binding site on a plant monooxygenase, DWARF4, using a quartz-crystal microbalance biosensor-based T7 phage display. ASSAY and Drug Devevelopment Technologies. 11 (3), 206-215 (2013).
- Novagen. T7 Select® System Manual. Novagen. , TB178 1009JN (2009).
- Novagen. OrientExpressTM cDNA Manual. Novagen. , TB247 1109JN (2009).
- Mandava, S., Makowski, L., Devarapalli, S., Uzubell, J., Rodi, D. J. RELIC--a bioinformatics server for combinatorial peptide analysis and identification of protein-ligand interaction sites. Proteomics. 4 (5), 1439-1460 (2004).
- Makowski, L. Phage Nanobiotechnology. Petrenko, V. A., Smith, G. P. , RSC Publishing. Ch. 3 33-54 (2011).
- Garcia-Carbonero, R., Supko, J. G. Current perspectives on the clinical experience, pharmacology, and continued development of the camptothecins. Clinical Cancer Research. 8 (3), 641-661 (2002).
- Harel, M., et al. The crystal structure of the complex of the anticancer prodrug 7-ethyl-10-[4-(1-piperidino)-1-piperidino]-carbonyloxycamptothecin (CPT-11) with Torpedo californica acetylcholinesterase provides a molecular explanation for its cholinergic action. Molecular Pharmacology. 67 (6), 1874-1881 (2005).
- Dodds, H. M., Rivory, L. P. The mechanism for the inhibition of acetylcholinesterases by irinotecan (CPT-11). Molecular Pharmacology. 56 (6), 1346-1353 (1999).
- Bencharit, S., et al. Structural insights into CPT-11 activation by mammalian carboxylesterases. Nature Structural Biology. 9 (5), 337-342 (2002).
- Kim, C. U., et al. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. Journal of the American Chemical Society. 119 (4), 681-690 (1997).
- Hoffmann, G., et al. Nonclinical pharmacokinetics of oseltamivir and oseltamivir carboxylate in the central nervous system. Antimicrobial Agents and Chemotherapy. 53 (11), 4753-4761 (2009).
- Wishart, D. S., et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Research. 46 (1), 1074-1082 (2018).
- Takakusagi, K., et al. Multimodal biopanning of T7 phage-displayed peptides reveals angiomotin as a potential receptor of the anti-angiogenic macrolide Roxithromycin. European Journal of Medicinal Chemistry. 90, 809-821 (2015).
- Takakusagi, Y., et al. Efficient one-cycle affinity selection of binding proteins or peptides specific for a small-molecule using a T7 phage display pool. Bioorganic and Medicinal Chemistry. 16 (22), 9837-9846 (2008).
- Takakusagi, Y., Suzuki, A., Sugawara, F., Kobayashi, S., Sakaguchi, K. Self-assembled monolayer (SAM) of small organic molecule for efficient random-peptide phage display selection using a cuvette type quartz-crystal micobalance (QCM) device. World Journal of Engineering. 5, 1005-1006 (2009).
- Kusayanagi, T., et al. The antitumor agent doxorubicin binds to Fanconi anemia group F protein. Bioorganic and Medicinal Chemistry. 20 (21), 6248-6255 (2012).
- Takakusagi, Y., et al. Identification of C10 biotinylated camptothecin (CPT-10-B) binding peptides using T7 phage display screen on a QCM device. Bioorganic and Medicinal Chemistry. 15 (24), 7590-7598 (2007).
- Rodi, D. J., et al. Identification of small molecule binding sites within proteins using phage display technology. Combinatorial Chemistry and High Throughput Screening. 4 (7), 553-572 (2001).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados