
Method Article
Biosensor-based High Throughput Biopanning and Bioinformatics Analysis Strategy for the Global Validation of Drug-protein Interactions
In This Article
Summary
This study aimed to present a strategy for identifying drug-peptide interactions. The strategy involves the biopanning of drug-recognizing short peptides based on a quartz-crystal microbalance (QCM) biosensor, followed by bioinformatics analysis for quantitatively assessing the information obtained for the drug recognition and annotation of the drug-binding sites on proteins.
Abstract
Receptors and enzyme proteins are important biomolecules that act as binding targets for bioactive small molecules. Thus, the rapid and global validation of the drug-protein interactions is highly desirable for not only understanding the molecular mechanisms underlying therapeutic efficacy but also for assessing drug characteristics, such as adsorption, distribution, metabolism, excretion, and toxicity (ADMET) for clinical use. Here, we present a biosensor-based high throughput strategy for the biopanning of T7 phage-displayed short peptides that can be easily displayed on the phage capsid. Subsequent analysis of the amino acid sequences of peptides containing short segments, as "broken relics", of the drug-binding sites using bioinformatics programs in receptor ligand contact (RELIC) suite, is also shown. By applying this method to two clinically approved drugs, an anti-tumor irinotecan, and an anti-flu oseltamivir, the detailed process for collecting the drug-recognizing peptide sequences and highlighting the drug-binding sites of the target proteins are explained in this paper. The strategy described herein can be applied for any small molecules of interest.
Introduction
Identification of drug-binding targets is an essential for the development of drugs as well as for understanding the molecular mechanisms of diseases. In particular, receptor and enzyme proteins are the most important molecular targets of bioactive small molecules1. Although affinity capture is a well-established technique for identifying the drug-binding proteins2, technical limitations, such as low solubility of proteins, often hamper the validation of drug targets2. Most importantly, the immobilized small molecules lose the degree of freedom necessary for docking and may be inaccessible to the internally located binding sites on larger target proteins. Furthermore, protein misfolding, inability to analyze co-crystallization conditions, and limitations due to molecular size often hamper the use of X-ray crystallography, nuclear magnetic resonance (NMR), and other such experimental analyses for studying drug-protein interactions.
The use of the T7 phage display biopanning is an efficient way for determining the binding site on proteins for small molecule baits3. In particular, a T7 phage-displayed random peptide library, which can be constructed by inserting synthetic DNA into a multi-cloning site, is effective. Compared to the T7 phage library displaying proteins, the short peptides can be easily engineered to be displayed on the T7 phage capsid without physical restrictions, which can sterically contact with any small molecule drug fixed on a solid support2. Furthermore, the introduction of a quartz-crystal microbalance (QCM) biosensor into the T7 phage display biopanning platform allows the identification of such weak but specific interactions of short peptides with drugs by monitoring the reduction in QCM frequency4,5. The bound T7 phage is then directly recovered by infecting the host Escherichia coli (BLT5615), and the DNA sequence of the region that encodes the affinity-selected peptide harboring drug-recognizing short segments is determined. Subsequent analysis of the amino acid sequence of the peptide population provides information regarding drug recognition. In silico pairwise alignment of the rescued amino acid sequences can be used to obtain information regarding the biological target of the drug within a selected proteome. This high throughput identification of protein fragments with affinity towards a drug can be used to heuristically reconstruct the drug-binding site in a manner similar to that of reconstructing an ancient artefact from pottery shards6. In particular, this unique approach can be useful when conventional proteomics approaches fail.
Here, we present a biosensor-based strategy for the biopanning of T7 phage-displayed peptides and bioinformatics analysis for the target validation of the small molecules. Beyond technical limitations on conventional methods, this strategy enables the identification of drug-binding sites on target proteins for any small molecule of interest under the identical protocol.
Protocol
NOTE: The following are the steps for screening drug-recognizing T7 phages using a QCM biosensor and recovering the screened phages via E. coli (BLT5615) infection. The protocols for the synthesis of a derivative of a small molecule that forms a self-assembled monolayer (SAM) and for the construction of the T7 phage-displayed 15-mer random peptide library can be found elsewhere6,7.
1. Preparation of the QCM sensor chip
- Attach a ceramic sensor chip on the oscillator of a 27-MHz QCM apparatus, and record the intrinsic frequency (Hz) in the air phase before small molecule immobilization.
- Detach the chip and drop 20 μL of a solution (1 mM in 70% ethanol) of a small molecule derivative that forms SAM onto the gold electrode of the sensor chip using a pipette.
CAUTION: The sensor chip crystal where the gold electrode (Au, 0.1 mm thick, 2.5 mm i.d., 4.9 mm2) is located is extremely thin and may crack easily (SiO2, 0.06 mm thick, 9 mm i.d.). Hence, pipette carefully. - Leave for 1 h at room temperature (around 20 °C) in a Petri dish with moistened tissue and shielded from room lights.
- Wash the electrode surface gently with ultrapure water; then, remove the water drops by blowing air with a syringe or air duster.
- Set up the sensor chip for the QCM apparatus and record the reduction in frequency in the air phase to measure the amount of the small molecule that has been immobilized.
NOTE: At least, 100 Hz of intrinsic frequency is necessary for successful small molecule immobilization (1 Hz immobilizes 30 pg).
2. Biopanning of the T7 phage library using a QCM biosensor (Figure 1)
- Set a cuvette with a dedicated magnetic stirrer on the QCM biosensor and pour 8 mL of the reaction buffer (10 mM Tris-HCl, 200 mM NaCl, pH 7-8) into the cuvette.
- Attach the QCM sensor chip to the oscillator and pull down the arm of oscillator to immerse the chip into the buffer being stirred at 1000 rpm.
- Start monitoring the QCM frequency on the personal computer (PC) and wait until the sensorgram equilibrates to around 3 Hz/min of the frequency drift.
- Inject 8 μL of a T7 phage library (1—2 × 1010 pfu/mL) into the cuvette (final concentration: 1—2 × 107 pfu/mL) and mark the injection point on the sensor.
- Monitor the frequency reduction caused by the binding of T7 phages to the small molecule immobilized on the gold electrode surface for 10 min.
- Stop the QCM frequency monitor, dislodge the sensor chip from the oscillator, and remove the buffer by blowing air and/or wicking away with wipes.
- Put the sensor chip into a humid Petri dish and drop the 20 μL of the suspension of E. coli (BLT5615) (OD600 = 0.5—1.0 after shaking at 37 °C by adding IPTG to 1 mM) host cells in the log phase onto the gold electrode.
- Close the lid of the dish and cover it with aluminum foil to block out light.
- Incubate the dish at 37 °C for 30 min on a 96-well microplate mixer (1000-1500 rpm) for enhancing the recovery of the bound T7 phages.
- Recover the 20 μL of the solution and suspend it into 200 μL of LB medium.
NOTE: The samples obtained in this step can be preserved at 4 °C by one week. - Prepare a dilution series of the phage solution for plaque isolation and DNA sequencing according to the general procedure described in the manufacturer's instructions8,9.
- Wipe the gold electrode surface with a cotton swab soaked with 1% sodium dodecyl sulfate solution.
- Wash the gold surface with ultrapure water from the washing bottle and then remove water drops by blowing air with a syringe or air duster.
- Drop 5 μL of piranha solution (Conc. H2SO4: 30% H2O2 = 3:1) on the gold surface and leave for 5 min.
- Wash the gold surface again with water and then dry by blowing air and/or wicking away with wipes.
- Repeat steps 2.14 and 2.15.
CAUTION: Prepare the piranha solution immediately prior to use. Use this liquid carefully, as it is a very strong acid. Treatment for longer than 5 min erodes the sensor chip.
3. Bioinformatics analysis using Receptor Ligand Contact (RELIC) program suite (Figure 2)10,11
- Unzip the stand-alone RELIC program on a PC with an MS Windows operating system.
- Align the amino acid sequences of the 15-mer peptides affinity-selected using the drug or randomly selected from unscreened parent library in each text format file (name.txt).
- Type the amino acid sequence of single or multiple protein(s) in each text file with FASTA format, or download the database text files in FASTA format from any protein database (e.g. UniProt (http://www.uniprot.org/) or DrugBank (https://www.drugbank.ca/).
- Place the text files (and PDB files for HETEROalign) in the folder necessary for running each RELIC program.
- Click the executable file (program.exe) for AADIV, INFO, MOTIF, MATCH, HETEROalign, FASTAcon, and FASTAskan in the independent folder to open the Personal Version of FTN95.
- Type the appropriate filename, along with the extension (name.txt), in the command message to execute each program and obtain the required text format file.
- Export the resulting text file to a spreadsheet software (e.g., Excel) to generate a plot of information content (INFO) or cumulative similarity scores calculated using a BLOSUM62 (MATCH, HETEROalign).
NOTE: The original RELIC server (http://relic.bio.anl.gov) is no longer available and some stand-alone type RELIC programs that work on PCs with a Windows platform can be obtained from the corresponding author (tkksg@rs.noda.tus.ac.jp).
Results
The representative results for two clinically approved drugs are shown in Figure 3. Irinotecan (Figure 3A), a water-soluble prodrug of natural camptothecin used for treating advanced colorectal cancer and non-small cell lung cancer, is converted to SN-38 in the liver, which inhibits topoisomerase I in cancer cells12. Furthermore, this compound directly inhibits acetylcholinesterase (AChE)13,14. Through the strategy, 29 peptides that recognized Iri immobilized as an SAM was identified by QCM biosensor-based one-cycle biopanning subsets. Subsequent pairwise alignment of the 29 peptides and AChE yielded maximal scores for Y121, Q225, F290, E327, H440, and Y442 and highlighted the portion in the three-dimensional structure. These amino acid residues were consistent with those making up the Iri-binding site of AChE. The same subset of peptides successfully identified E99, L100, L252, L305, I387, and V474 in the vicinity of the catalytic triad (S221, E353, and H467) in carboxylesterase (CE), indicating that these amino acids form a scaffold for Iri recognition during de-esterification of Iri15. Such amino acid residues in the catalytic site cannot be identified directly using conventional X-ray crystallography or NMR analysis, as the enzyme reaction proceeds smoothly and does not form the static complex stably under general experimental conditions. Thus, that combinatorial detection of drug-binding sites of multiple proteins, including those in the intermediate complexes possibly formed with enzymes during metabolic reactions for a particular drug, is possible using the affinity-selected peptides determined for one drug.
Figure 3B shows the other results obtained for oseltamivir (Osel), an anti-flu drug that is activated to oseltamivir carboxylate, which in turn inhibits the neuraminidase (NA) of the influenza virus16. The 27 of peptides that recognized Osel covering the QCM sensor chip gold electrode surface successfully detected the the Osel-binding site in NA16. This binding site consists of unstructured peptide loops that potentially undergo dynamic movement while docking with Osel. The Osel-recognizing peptides on the T7 phage capsid might mimic this dynamic docking when binding to the Osel fixed on the gold electrode surface of the QCM sensor chip. Neuropsychiatric adverse events (NPAEs) have been identified in young patients with influenza, which effect on patients are possibly related to the disease itself rather than the drug. Studies have demonstrated that Osel is actively exported from the central nervous system (CNS) of rodents via multidrug resistance (MDR) protein in the blood-brain barrier (BBB)17. Indeed, one of the proteins of the class of this MDR showed a high score (top 5% out of 4,396 in the DrugBank 1.0 protein database18), in addition to other transporters, neurotransmitter-related enzymes, and receptors, in our study. The pharmacological significance of these proteins with regard to the appearance of adverse effects of Osel is being investigated.
So far, single and multiple small molecule-binding sites on the target proteins have been successfully identified for six small molecule drugs been using our strategy (Figure 4). For Brz2001 and roxithromycin (RXM), identical drug-binding sites on a target protein were identified using different pools of peptides, the numbers and amino acid sequences for which varied completely7,19. Furthermore, single peptide pools obtained for Iri, RXM and Osel led to the identification of multiple binding sites on different proteins for each drug, such as AChE and CE for Iri (Figure 3A)20, angiomotin and CYP3A4 for RXM19,21, and NA and MDR-associated protein for Osel. An unknown molecular target was identified for the anti-tumor compound doxorubicin (FANCF)22, and the anti-angiogenic macrolide antibiotic RXM (angiomotin)19.

Figure 1: Schematic representation of the QCM biosensor-based biopanning of the T7 phage-displayed peptide library. A T7 phage library that displays random peptides is injected into the cuvette containing the buffer (under stirring) where the QCM biosensor chip is immersed and the frequency is stabilized. After monitoring the frequency reduction due to the binding of T7 phages to small molecules immobilized on the gold electrode surface of the sensor chip, the sensor chip is detached from the oscillator. DNA from the bound T7 phage is then directly recovered after host E. coli (BLT5615) infection. The resulting T7 phages are isolated via plaque formation and, finally, the amino acid sequence of the drug affinity-selected peptide displayed on the T7 phage capsid is determined according to the general phage display method. Please click here to view a larger version of this figure.
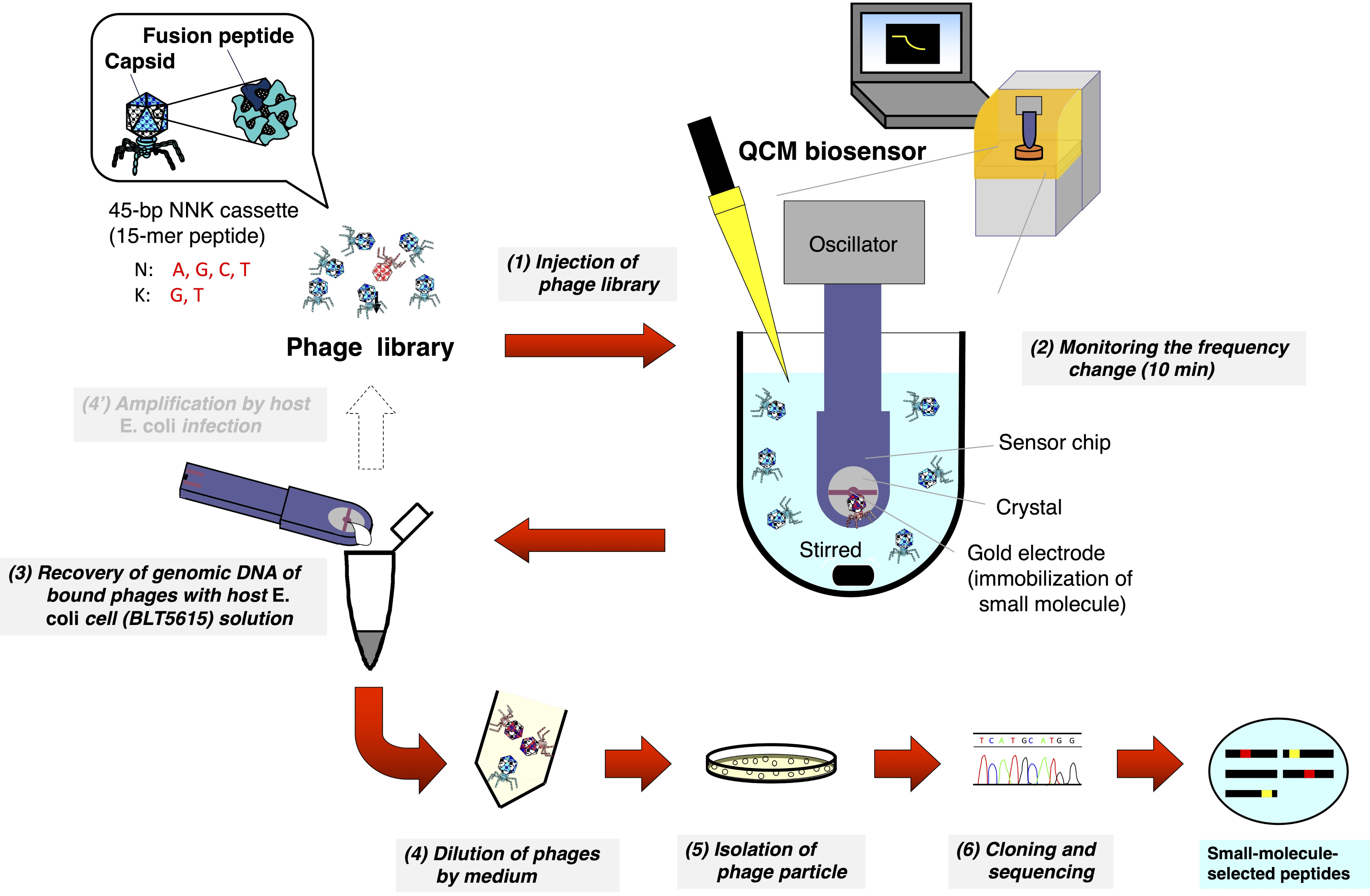{kind=link}

Figure 2: Schematic representation of the quantitative assessment of the sequence comparison between drug-selected peptides and single or multiple proteins. Drug-selected peptide sequences are respectively aligned with the primary amino acid sequences of single and multiple proteins, and the similarity in each 3—5 amino acid sets is cumulatively scored via pairwise alignment according to a modified BLOSUM62 matrix. The resulting plot or diagram indicates the residues or portions that constitute a potential drug-binding site on the protein. Further analysis using an appropriate RELIC program highlights the binding site on the three-dimensional structure (if PDB file is available) or ranks entire proteins that are possibly the binding target (HETEROalign program is currently unavailable). Please click here to view a larger version of this figure.
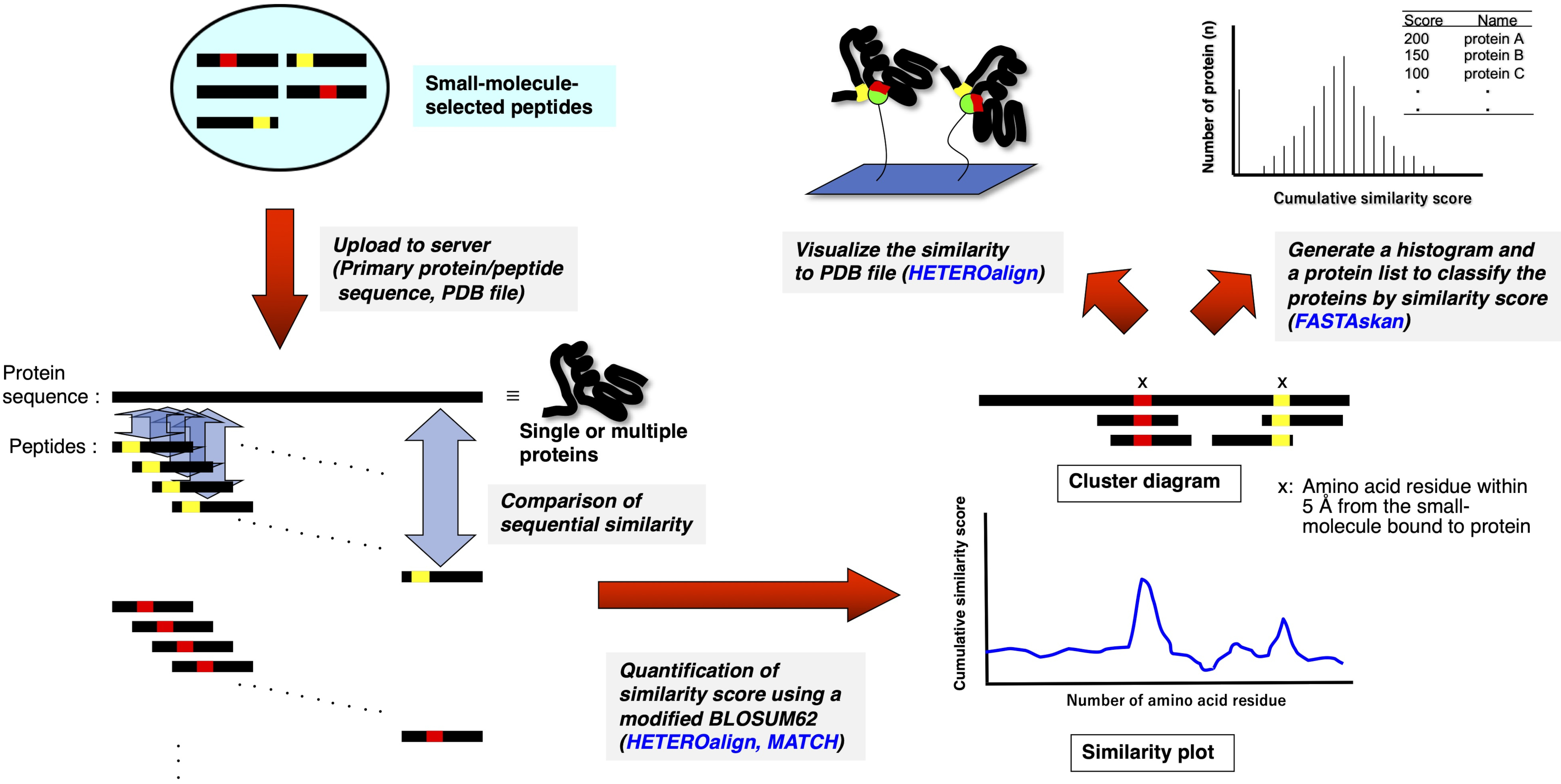{kind=link}

Figure 3: Representative results of peptide collection and subsequent bioinformatics analysis. (A) Irinotecan (anti-tumor prodrug, topoisomerase I inhibitor). T7 phage interaction was monitored for 10 min as a reduction in QCM frequency. The DNA of the bound T7 phage was recovered and sequenced to determine the corresponding amino acid sequence. The amino acid sequences of 29 15-mer peptides, collected using a subset of one-cycle biopanning, highlighted the amino acids that make up the Iri-binding site of AChE [PDB ID 1U65]. Further assessment using the same 29 peptides highlighted the neighboring amino acid residues (scaffold residues for de-esterification) of the catalytic triad in CE, a liver enzyme that converts Iri to SN-38 (active form) [PDB ID: 1K4Y].Similarity scores of 103 randomly selected peptides from the unscreened parent library7,19 have been subtracted from these scores to remove library bias. These figures are reproduced from Ref. 20, with permission from Elsevier. (B) Oseltamivir (anti-flu drug). The 27 peptides containing Osel-recognizing amino acids highlighted disordered portions of the Osel-binding site for neuraminidase (NA) (virus enzyme) [PDB ID: 2HT7]. Global validation of the sequence similarity between 27 peptides and 4,396 proteins in DrugBank 1.018 revealed NA to be within the top 5% range, in addition to the host human proteins associated with the functions of the central nervous system. Please click here to view a larger version of this figure.
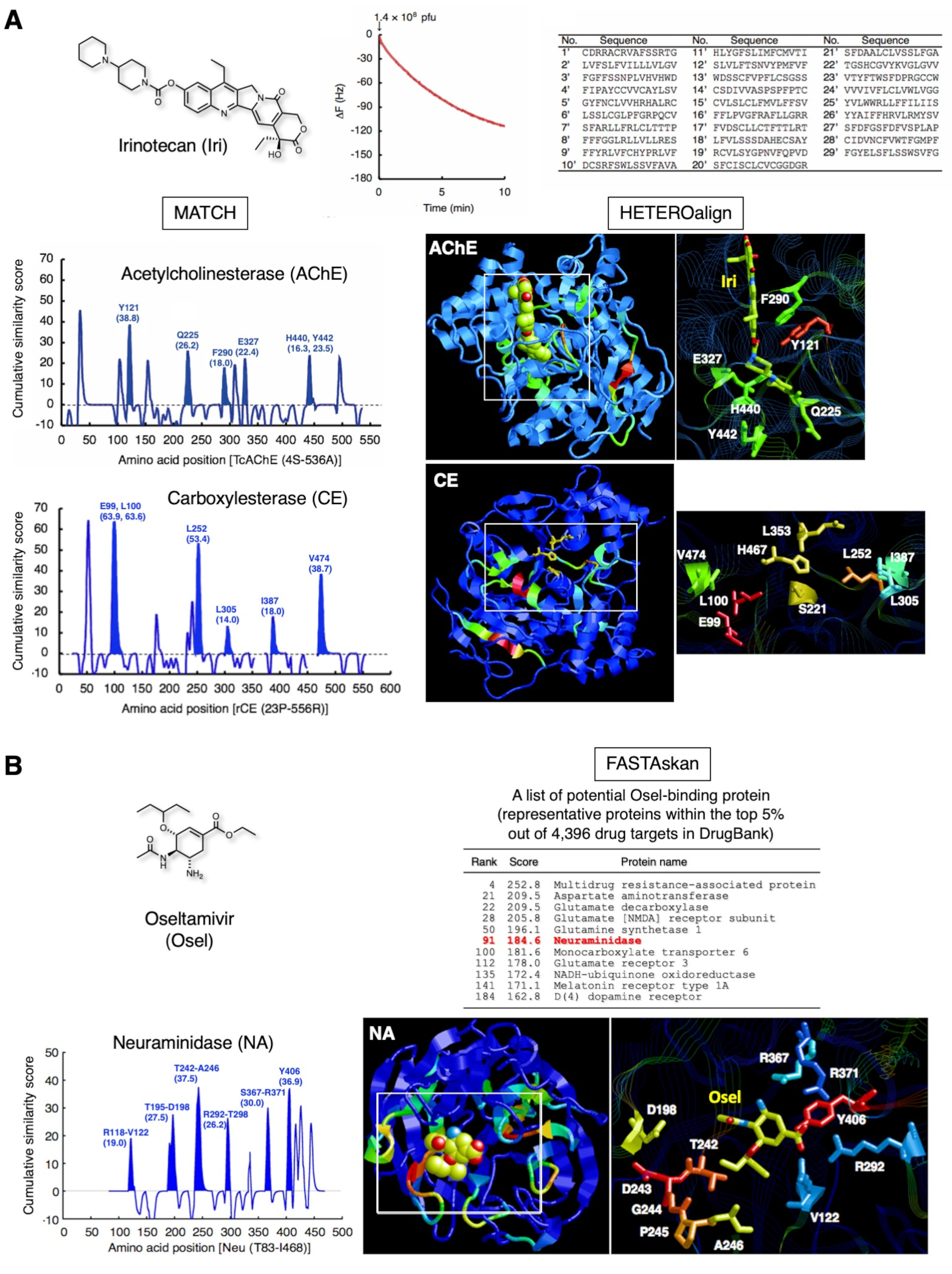{kind=link}

Figure 4: Summary of the small molecules, the binding targets of which were identified using this strategy. Please click here to view a larger version of this figure.
{kind=link}
Discussion
Here, a strategy for the QCM biosensor-based biopanning of drug-recognizing peptides, followed by bioinformatics analysis for validating drug-protein interactions using the identified peptides, has been presented. Designing of the small molecule derivatives for immobilization on the gold electrode of the biosensor is an important step, as the introduced linker may hinder the binding and collection of the peptide that recognizes the drug. To avoid this, derivatives with different positions of the introduced linker are prepared23. Alternatively, for immobilizing hydrophobic small molecules, the sensor chip is immersed in bulk water in a 10 cm Petri dish, and a 20 μL solution of the small molecule (10 mM solution in dimethyl sulfoxide) is dropped onto the gold electrode of the biosensor, to cover its surface, and incubated for 5 min. This allows retention of a sub-hundred hertz intrinsic frequency of small molecules, which is held for at least 10 min during the biopanning. Indeed, using such immobilization, the Osel affinity-selected peptides clearly highlighted the Osel-binding site in NA (Figure 3).
The T7 phage used for preparing the peptide library here is genetically engineered using the NNK15 cassette that encodes 32 codons for all 20 of standard amino acids and represses the emergence of 2 stop codons (UAA, UGA) and emerges only UAG (Figure 1)6,7. This is important for displaying 15-mer full-length peptides and increasing the diversity of the library. The T7 phage display system has a technical display limit of 107—109 T7 phages. However, the diversity of the 15-mer peptide library is theoretically 2015 (3.27 × 1019); thus, it cannot be used for complete library construction. Nevertheless, similarity search or mining of conserved motifs allows the detection of the amino acids comprising the drug-binding sites of proteins even with this limited diversity of peptides in the library. In addition, 3—5 amino acid stretches within the library peptide (the appearance rate is between 1/203 and 1/ 205, which can be realized using the T7 phage display system) are involved in the recognition of small molecule drugs; therefore, a 100% match of the peptide sequences with 15-mer amino acid sequences constituting the drug binding site of the target protein is not required. Indeed, approximately 30 affinity-selected peptides successfully highlighted the binding site of the target protein for each drug tested (Figure 4). Thus, the diversity of the parent T7 phage library used (1.7 × 107 pfu/mL) can be used to heuristically reconstruct the drug-binding site.
Typically, 3—5 copies of T7 phages that displayed the same sequences as those of the 15-mer amino acid sequences harboring drug-recognizing amino acid stretches emerged within the 16 plaques arbitrarily isolated, indicating the success of the affinity selection under our protocol. This indicates that 18—30 different drug-recognizing peptide sequences are collected within the 96 plaques isolated (the number is associated with the microplate format), which are identified subsequently using sequencing of the DNA and obtaining the corresponding amino acid sequence. In the present strategy, injection of 8 μL of the T7 phage library into the cuvette containing 8 mL buffer (1000-fold dilution of the library) is suitable for reducing the non-specific binding of T7 phages. To increase the diversity of the affinity-selected peptides, repeating one-cycle selection several times and using 16 or 32 plaque isolations per screening proved to be more effective than isolation from a single solution at a time. For example, to effectively collect approximately 30 differently sequenced affinity-selected peptides, 3—6 sets of one-cycle selection were conducted, 16 or 32 plaques were isolated in each experiment. Appearance of identical sequences in all 16 or 32 T7 phage plaques are indicated accidental detection of background or might contamination as a carryover. In contrast, the absence of T7 phages with the same sequence or appearance of many T7 phages with shorter peptides than the 15-mer length indicates that T7 phages in the population non-specifically emerged with high probability. As QCM frequency reduction occurs to the same extent even in such cases, the success of the selection should be comprehensively evaluated by sequencing the DNA of the isolated T7 phage, followed by bioinformatics analysis of the amino acid sequences of the peptides. Furthermore, unlike the conventional T7 phage display protocol, repeating rounds of selection is less effective, as the variation and number of the T7 phages are small and are not concentrated even after repeating the amplification and selection steps23.
Importantly, this method is applicable for the mining of small molecule-binding sites in the proteomes of humans, pathogenic viruses, and even plants. Interestingly, the possibly unstructured short display length of peptides on the T7 phage capsid can mimic the molecular dynamics of peptides of proteins during docking with a small molecule; this can reflect dynamic binding24. Beyond the technical limitations of conventional methods, this strategy, applicable to identical protocols for small molecules, may expand the druggable proteome as well as provide more granularity regarding drug-protein interaction analysis.
Certain technical limitations of this approach should be considered. Organic synthesis is necessary for small molecule immobilization on the gold electrode surface of the biosensor chip. For the non-experts in organic chemistry, some immobilization reagents are commercially available to mechanically fix the small molecule by coupling. Furthermore, certain nonsense portions of the peptides might result in the detection of a portion of the protein not relevant for drug docking as false positives. This corresponds empirically to beta-sheet or leucine-rich domains rich in leucine or valine residues, which are encoded by more codons than other standard amino acids, when copies of the T7 phage are produced. Controlling the library peptide length might control the occurrence of false positives. In contrast, there may be cases where amino acid residues in the drug-binding site that are involved in docking, as demonstrated using X-ray crystallography or NMR analysis, are not detected. This may be solved by collecting a larger number of the drug-recognizing peptides or changing the direction of fixing of the small molecules on the gold electrode.
Many drug-protein interactions that are related to the main and secondary effects of drug use may yet be unidentified in the proteome; in addition, enzymes and transporters responsible for drug absorption, distribution, metabolism, excretion, and toxicity, might also be still unidentified. Protein binding is not always responsible for the bioactivity of a drug. Thus, a combination of other information from biological assays will improve the identification of essential drug targets responsible for the main and adverse effects of drugs. Further adaptations of this concise technique will increase the practicality and throughput for mining of the protein-binding sites of a wide range of small molecule drugs. The method presented herein will largely contribute to not only conduct basic researches in the related fields but also clarify the molecular mechanisms underlying therapeutic efficacy or other biological effects of drugs in clinical use.
Disclosures
The authors have no conflicts of interest to disclose.
Acknowledgements
The author thanks Drs. Yujiro Hayashi and Hayato Ishikawa for providing oseltamivir, and Dr. Lee Makowski for providing the stand-alone RELIC programs. The author also acknowledges Tetsuya Kawagoe for the technical assistance of the QCM experiment. This work was partially supported by JSPS KAKENHI Grant Number 17K01363 (Y.T.).
DATA AVAILABILITY STATEMENT
The stand-alone RELIC programs and the sequence data of affinity-selected peptides for drugs as well as the protein sequences from proteome database used in this paper are available from this author upon request (tkksg@rs.noda.tus.ac.jp).
Materials
| Name | Company | Catalog Number | Comments |
| AFFINIXQN | ULVAC, Inc. (Tokyo, Japan) | QCM2008-STKIT | Contains Glass cuvette, stir magnet, operation and analysis software with a Windows PC |
| AADIV | Northeastern University (Lee Makowski) | AADIV.exe | Calculates the frequency of occurrence of each of the 20 amino acids at each recombinant insert position, as well as the overall position-independent frequency of each amino acid within that set of peptide sequences. Also roughly estimates the sequence diversity of a display library by statistical sampling method based upon sequences obtained from a limited number of randomly sampled members of the library. |
| Ceramic Sensor Chip | ULVAC, Inc. (Tokyo, Japan) | QCMST27C | 4 sensor chips/package |
| Dimethyl sulfoxide | Sigma-Aldrich (St. Louis, MO, USA) | D8418 | |
| Ethanol | Merck (Kenilworth, NJ, USA) | 09-0850 | |
| FASTAcon | Northeastern University (Lee Makowski) | FASTAcon.exe | Identifies proteins from a population with short consensus sequences. |
| FASTAskan | Northeastern University (Lee Makowski) | FASTAskan.exe | Lists proteins with high similarity to a peptide population. |
| Immiblization kit for AFFINIX | ULVAC, Inc. (Tokyo, Japan) | QCMIMKT | SAM reagent and amine coupling reagent |
| INFO | Northeastern University (Lee Makowski) | INFO.exe | Provides mathematical measure of the probability of observing a particular peptide sequence by random chance (i.e., nonspecific binding) as opposed to by selection for a specific property (affinity to small molecule). |
| Liquid LB medium | Sigma-Aldrich (St. Louis, MO, USA) | L3522 | Autoclave for 20 min |
| MATCH | Northeastern University (Lee Makowski) | MATCH.exe | Identifies any stretches of amino acid residues within a particular protein that exhibit significant similarity to a group of affinity-selected peptides. Outputs as cluster dia- gram and cumulative similarity plot calculated from a modified BLOSUM62 matrix with a short window (5–6 amino acids in length). |
| MOTIF1 | Northeastern University (Lee Makowski) | MOTIF1.exe | Searches for three continuous amino acid sequence motifs within a peptide population. |
| MOTIF2 | Northeastern University (Lee Makowski) | MOTIF2.exe | Searches for patterns of three amino acids and does not allow conservative amino acid substitutions, but does allow identical gap lengths. |
| NaCl | Merck (Kenilworth, NJ, USA) | S3014 | |
| Receptor ligand contacts (RELIC) | Argonne National Laboratory (Lemont, IL, USA) | https://www.relic.anl.gov | Currently unavailable (Stand-alone program can be used from correspondence author upon request) |
| Tris | Merck (Kenilworth, NJ, USA) | 252859 |
References
- Santos, R., et al. A comprehensive map of molecular drug targets. Nature Reviews Drug Discovery. 16 (1), 19-34 (2017).
- Ziegler, S., Pries, V., Hedberg, C., Waldmann, H. Target identification for small bioactive molecules: finding the needle in the haystack. Angewandte Chemie International Edition (English). 52 (10), 2744-2792 (2013).
- Piggott, A. M., Karuso, P. Identifying the cellular targets of natural products using T7 phage display. Natural Product Reports. 33 (5), 626-636 (2016).
- Takakusagi, Y., Takakusagi, K., Sakaguchi, K., Sugawara, F. Phage display technology for target determination of small-molecule therapeutics: an update. Expert Opinion on Drug Discovery. 15 (10), 1199-1211 (2020).
- Takakusagi, Y., Takakusagi, K., Sugawara, F., Sakaguchi, K. Use of phage display technology for the determination of the targets for small-molecule therapeutics. Expert Opinion on Drug Discovery. 5 (4), 361-389 (2010).
- Takakusagi, Y., Takakusagi, K., Sugawara, F., Sakaguchi, K. Using the QCM Biosensor-Based T7 Phage Display Combined with Bioinformatics Analysis for Target Identification of Bioactive Small Molecule. Methods in Molecular Biology. 1795, 159-172 (2018).
- Takakusagi, Y., et al. Mapping a disordered portion of the Brz2001-binding site on a plant monooxygenase, DWARF4, using a quartz-crystal microbalance biosensor-based T7 phage display. ASSAY and Drug Devevelopment Technologies. 11 (3), 206-215 (2013).
- Novagen. T7 Select® System Manual. Novagen. , TB178 1009JN (2009).
- Novagen. OrientExpressTM cDNA Manual. Novagen. , TB247 1109JN (2009).
- Mandava, S., Makowski, L., Devarapalli, S., Uzubell, J., Rodi, D. J. RELIC--a bioinformatics server for combinatorial peptide analysis and identification of protein-ligand interaction sites. Proteomics. 4 (5), 1439-1460 (2004).
- Makowski, L. Phage Nanobiotechnology. Petrenko, V. A., Smith, G. P. , RSC Publishing. Ch. 3 33-54 (2011).
- Garcia-Carbonero, R., Supko, J. G. Current perspectives on the clinical experience, pharmacology, and continued development of the camptothecins. Clinical Cancer Research. 8 (3), 641-661 (2002).
- Harel, M., et al. The crystal structure of the complex of the anticancer prodrug 7-ethyl-10-[4-(1-piperidino)-1-piperidino]-carbonyloxycamptothecin (CPT-11) with Torpedo californica acetylcholinesterase provides a molecular explanation for its cholinergic action. Molecular Pharmacology. 67 (6), 1874-1881 (2005).
- Dodds, H. M., Rivory, L. P. The mechanism for the inhibition of acetylcholinesterases by irinotecan (CPT-11). Molecular Pharmacology. 56 (6), 1346-1353 (1999).
- Bencharit, S., et al. Structural insights into CPT-11 activation by mammalian carboxylesterases. Nature Structural Biology. 9 (5), 337-342 (2002).
- Kim, C. U., et al. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. Journal of the American Chemical Society. 119 (4), 681-690 (1997).
- Hoffmann, G., et al. Nonclinical pharmacokinetics of oseltamivir and oseltamivir carboxylate in the central nervous system. Antimicrobial Agents and Chemotherapy. 53 (11), 4753-4761 (2009).
- Wishart, D. S., et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Research. 46 (1), 1074-1082 (2018).
- Takakusagi, K., et al. Multimodal biopanning of T7 phage-displayed peptides reveals angiomotin as a potential receptor of the anti-angiogenic macrolide Roxithromycin. European Journal of Medicinal Chemistry. 90, 809-821 (2015).
- Takakusagi, Y., et al. Efficient one-cycle affinity selection of binding proteins or peptides specific for a small-molecule using a T7 phage display pool. Bioorganic and Medicinal Chemistry. 16 (22), 9837-9846 (2008).
- Takakusagi, Y., Suzuki, A., Sugawara, F., Kobayashi, S., Sakaguchi, K. Self-assembled monolayer (SAM) of small organic molecule for efficient random-peptide phage display selection using a cuvette type quartz-crystal micobalance (QCM) device. World Journal of Engineering. 5, 1005-1006 (2009).
- Kusayanagi, T., et al. The antitumor agent doxorubicin binds to Fanconi anemia group F protein. Bioorganic and Medicinal Chemistry. 20 (21), 6248-6255 (2012).
- Takakusagi, Y., et al. Identification of C10 biotinylated camptothecin (CPT-10-B) binding peptides using T7 phage display screen on a QCM device. Bioorganic and Medicinal Chemistry. 15 (24), 7590-7598 (2007).
- Rodi, D. J., et al. Identification of small molecule binding sites within proteins using phage display technology. Combinatorial Chemistry and High Throughput Screening. 4 (7), 553-572 (2001).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved