
Method Article
Ensamblaje híbrido del genoma de novo para la generación de genomas completos de bacterias urinarias utilizando tecnologías de secuenciación de lectura corta y larga
En este artículo
Resumen
Este protocolo detalla un enfoque integral para el cultivo, la secuenciación y el ensamblaje del genoma híbrido de novo de las bacterias urinarias. Proporciona un procedimiento reproducible para la generación de secuencias completas y circulares del genoma útiles en el estudio de elementos genéticos cromosómicos y extracromosómicos que contribuyen a la colonización urinaria, la patogénesis y la diseminación de la resistencia a los antimicrobianos.
Resumen
Las secuencias completas del genoma proporcionan datos valiosos para la comprensión de la diversidad genética y los factores únicos de colonización de los microbios urinarios. Estos datos pueden incluir elementos genéticos móviles, como plásmidos y fagos extracromosómicos, que contribuyen a la diseminación de la resistencia a los antimicrobianos y complican aún más el tratamiento de la infección del tracto urinario (ITU). Además de proporcionar una resolución fina de la estructura del genoma, los genomas completos y cerrados permiten la genómica comparativa detallada y los análisis evolutivos. La generación de genomas completos de novo ha sido durante mucho tiempo una tarea desafiante debido a las limitaciones de la tecnología de secuenciación disponible. La secuenciación de próxima generación (NGS) de extremo pareado produce lecturas cortas de alta calidad que a menudo resultan en ensamblajes de genoma precisos pero fragmentados. Por el contrario, la secuenciación de nanoporos proporciona lecturas largas de menor calidad que normalmente conducen a ensamblajes completos propensos a errores. Tales errores pueden obstaculizar los estudios de asociación de todo el genoma o proporcionar resultados engañosos del análisis de variantes. Por lo tanto, los enfoques híbridos que combinan lecturas cortas y largas han surgido como métodos confiables para lograr genomas bacterianos cerrados altamente precisos. Aquí se informa un método integral para el cultivo de diversas bacterias urinarias, la identificación de especies mediante la secuenciación del gen 16S rRNA, la extracción de ADN genómico (gDNA) y la generación de lecturas cortas y largas por las plataformas NGS y Nanopore, respectivamente. Además, este método describe una línea bioinformática de algoritmos de control de calidad, ensamblaje y predicción de genes para la generación de secuencias completas del genoma anotadas. La combinación de herramientas bioinformáticas permite la selección de datos de lectura de alta calidad para el ensamblaje del genoma híbrido y el análisis posterior. El enfoque simplificado para el ensamblaje del genoma híbrido de novo descrito en este protocolo puede adaptarse para su uso en cualquier bacteria cultivable.
Introducción
El microbioma urinario es un área emergente de investigación que ha roto una idea errónea de décadas de que el tracto urinario es estéril en individuos sanos. Los miembros de la microbiota urinaria pueden servir para equilibrar el ambiente urinario y prevenir la infección del tracto urinario (ITU)1,2. Las bacterias uropatógenas invaden el tracto urinario y emplean diversos mecanismos de virulencia para desplazar la microbiota residente, colonizar el urotelio, evadir las respuestas inmunes y contrarrestar las presiones ambientales3,4. La orina es un medio relativamente limitado en nutrientes caracterizado por una alta osmolaridad, disponibilidad limitada de nitrógeno y carbohidratos, baja oxigenación y bajo pH5,6,7. La orina también se considera antimicrobiana, compuesta de altas concentraciones de urea inhibitoria y péptidos antimicrobianos como la catelicidina humana LL-378. La investigación de los mecanismos empleados tanto por las bacterias residentes como por los uropatógenos para colonizar el tracto urinario es fundamental para comprender mejor la salud del tracto urinario y desarrollar nuevas estrategias para el tratamiento de las infecciones urinarias. Además, a medida que el fracaso de las terapias antimicrobianas de primera línea se vuelve más común, es cada vez más importante monitorear la diseminación de elementos genéticos móviles portadores de determinantes de resistencia a los antimicrobianos dentro de las poblaciones de bacterias urinarias9,10.
Para investigar los genotipos y fenotipos de las bacterias urinarias, es imperativo su cultivo exitoso y la posterior secuenciación del genoma completo (WGS). Los métodos dependientes del cultivo son necesarios para detectar e identificar microbios viables en muestras de orina11. El cultivo clínico de orina estándar consiste en enchapar la orina en agar de sangre de oveja al 5% (BAP) y agar MacConkey e incubar aeróbicamente a 35 ° C durante 24 h12. Sin embargo, con un umbral de detección de ≥105 UFC/ml13,muchos miembros de la microbiota urinaria no se informan por este método. Las técnicas de cultivo mejoradas, como el cultivo cuantitativo mejorado de orina (EQUC)11, emplean varias combinaciones de diferentes volúmenes de orina, tiempos de incubación, medios de cultivo y condiciones atmosféricas para identificar microbios que comúnmente se omiten en el cultivo de orina estándar. Descrito en este protocolo es una versión modificada de EQUC, denominado aquí protocolo de cultivo de orina mejorado modificado, que permite el cultivo de diversas bacterias urinarias y uropatógenos utilizando medios selectivos y condiciones atmosféricas óptimas, pero no es inherentemente cuantitativo. El aislamiento exitoso de bacterias urinarias permite la extracción de ADN genómico (gDNA) para el WGS aguas abajo y el ensamblaje del genoma.
Los ensamblajes de genomas, ensamblajes completos en particular, permiten el descubrimiento de factores genéticos que pueden contribuir a la colonización, el mantenimiento del nicho y la virulencia tanto entre la microbiota residente como entre las bacterias uropatógenas. Los ensamblajes de genoma preliminares contienen un número diverso de secuencias contiguas (contigs) que pueden contener errores de secuenciación y carecen de información de orientación. En un ensamblaje completo del genoma, se ha verificado tanto la orientación como la precisión de cada par de bases14. Además, la obtención de secuencias completas del genoma proporciona información sobre la estructura del genoma, la diversidad genética y los elementos genéticos móviles15. Las lecturas cortas por sí solas pueden identificar la presencia o ausencia de genes importantes, pero pueden no identificar su contexto genómico16. Con tecnologías de secuenciación de lectura larga como Oxford Nanopore y PacBio, la generación de ensamblajes cerrados de novo de genomas bacterianos ya no requiere métodos extenuantes como el cierre manual de ensamblajes de novo mediante PCR multiplex17,18. La combinación de las tecnologías de secuenciación de lectura corta de próxima generación y secuenciación de lectura larga nanoporos permite la generación fácil de ensamblajes de genomas bacterianos precisos, completos y cerrados a costos relativamente bajos19. La secuenciación de lectura corta produce ensamblajes de genoma precisos pero fragmentados que generalmente consisten en un promedio de 40-100 contigs, mientras que la secuenciación de nanoporos genera lecturas largas de aproximadamente 5-100 kb de longitud que son menos precisas pero pueden servir como andamios para unir contigs y resolver la sintenia genómica. Los enfoques híbridos que utilizan tecnologías de lectura corta y larga pueden producir genomas bacterianos precisos y completos19.
Aquí se describe un protocolo integral para el aislamiento e identificación de bacterias de la orina humana, la extracción de ADN genómico, la secuenciación y el ensamblaje completo del genoma utilizando un enfoque de ensamblaje híbrido. Este protocolo proporciona un énfasis especial en los pasos necesarios para modificar adecuadamente las lecturas generadas por la secuenciación de lectura corta y larga para el ensamblaje preciso de un cromosoma bacteriano cerrado y elementos extracromosómicos como los plásmidos.
Protocolo
Las bacterias se cultivaron a partir de orina recolectada de mujeres que consintieron como parte de los estudios de revisión institucional aprobados por la junta 19MR0011 (UTD) y STU 032016-006 (UTSW).
1. Cultivo de orina mejorado modificado
NOTA: Todos los pasos de cultivo deben llevarse a cabo en condiciones estériles. Esterilizar todos los instrumentos, soluciones y medios. Limpie el área de trabajo con etanol al 70%, luego instale un quemador Bunsen y trabaje cuidadosamente cerca de la llama para reducir las posibilidades de contaminación. Alternativamente, se puede utilizar un gabinete de bioseguridad de clase II para mantener un ambiente estéril. Use el equipo de protección personal (EPP) adecuado para evitar la exposición a microbios potencialmente patógenos.
- Aislamiento de colonias y orina sembrada de glicerol
- Descongele la orina sembrada de glicerol a temperatura ambiente (RT). Una vez descongelada, vórtice la muestra durante 5 s para mezclar. En tubos de microcentrífuga estériles, prepare diluciones 1:3 y 1:30 de la orina en solución salina estéril 1x tamponada con fosfato (PBS) a un volumen final de 100 μL.
NOTA: La orina sembrada de glicerol se prepara mezclando 500 μL de orina sin diluir y 500 μL de glicerol estéril al 50% en crioviales y almacenándola a -80 °C. - Placas de agar precalentadas a 37 °C durante 15 min antes de su uso. Consulte la Figura 1 para conocer los tipos de medios y las condiciones de cultivo adecuadas para los géneros bacterianos urinarios comunes. Mezcle bien la orina diluida mediante pipeteo antes del recubrimiento, placa 100 μL de la orina diluida en la placa de agar deseada y extienda la muestra con perlas de vidrio estériles. Placa 100 μL del diluyente 1x PBS en una placa separada como un control sin crecimiento.
NOTA: Si se intenta cultivar especies uropatógenas comunes (por ejemplo, Escherichia coli, Klebsiella spp., Enterococcus faecalis, etc.), se recomienda utilizar agar cromogénico(Tabla de Materiales)ya que permite una fácil identificación de especies bacterianas uropatógenas(Figura 1). El ácido nalidíxico de colistina (CNA) o el agar MRS son útiles para aislar especies grampositivas fastidiosas (por ejemplo, Lactobacillus spp.)de orina conocida por contener uropatógenos gramnegativos, que pueden competir con las especies fastidiosas en agares no selectivos. - Incubar la placa invertida en la condición atmosférica deseada a 35 °C durante un período de 24 h para los uropatógenos y de 3-5 días para las bacterias fastidiosas (Figura 1).
- Después del período de incubación, retire las placas de la incubadora. De cada placa, elija las colonias que exhiben un color, morfología o patrones hemolíticos únicos.
- Vuelva a rayar la colonia bacteriana utilizando un bucle estéril sobre el agar correspondiente e incube la placa invertida durante 2-5 días en la atmósfera deseada para obtener colonias bien aisladas.
NOTA: Si se utiliza BAP para el cultivo primario, el parche de colonias en agar cromogénico puede proporcionar información útil sobre la heterogeneidad de la población bacteriana en la muestra.
- Descongele la orina sembrada de glicerol a temperatura ambiente (RT). Una vez descongelada, vórtice la muestra durante 5 s para mezclar. En tubos de microcentrífuga estériles, prepare diluciones 1:3 y 1:30 de la orina en solución salina estéril 1x tamponada con fosfato (PBS) a un volumen final de 100 μL.
- Cultivo en caldo líquido y aislados bacterianos de glicerol
- Una vez obtenidas las colonias aisladas que coincidan con la morfología de la colonia madre, escoja una sola colonia e inocule en 3 ml de caldo líquido utilizando un bucle de inoculación estéril. Consulte la Figura 1 para el caldo capaz de apoyar el crecimiento de géneros comunes de microbiota urinaria. Selle las placas de agar con parafilm y guárdelas a 4 °C durante 2-4 días. Incubar cultivos líquidos en las condiciones atmosféricas deseadas durante 1-5 días hasta que el cultivo esté visiblemente turbio.
- Después de observar el crecimiento, vórtice el cultivo y luego agregue 1 ml del cultivo nocturno a 500 μL de glicerol estéril al 50% en un criovial de 2 ml; sellar y mezclar suavemente por inversión. Prepare dos existencias de glicerol para cada colonia (una sirve como respaldo) y guárdelas a -80 °C.
2. Identificación de especies bacterianas mediante secuenciación del gen Sanger del ARNr 16S
NOTA: La identidad microbiana se puede confirmar alternativamente utilizando la espectrometría de masas de tiempo de vuelo de ionización por láser asistida por matriz (MALDI-TOF)20.
- Reacción en cadena de la colonia-polimerasa (PCR)
- Preparar 25 μL de la reacción de PCR en tubos de PCR añadiendo 12,5 μL de 2x Taq Polymerase Master Mix, 0,5 μL de 10 μM de imprimación 8F, 0,5 μL de imprimación 10 μM 1492R(Tabla de Materiales),y 11,5 μL de agua libre de nucleasas21.
NOTA: Si realiza PCR para múltiples muestras, haga una mezcla maestra de reacción de mezcla de Taq polimerasa, imprimaciones y agua estéril libre de nucleasas. Luego alícuota 25 μL en cada tubo de PCR. - Para realizar la PCR de colonia, deslice una colonia bien aislada de la nueva raya con un palillo de dientes estéril o una punta de pipeta. Resuspend la colonia en la mezcla de reacción de PCR preparada en el paso 2.1.1. Mezclar suavemente. Recoger el líquido en la parte inferior del tubo mediante un giro rápido a 2000 x g.
NOTA: Asegúrese de que la muestra esté libre de burbujas de aire. Incluya una muestra de control sin plantilla (NTC) que contenga solo la mezcla de reacción de PCR. - Coloque los tubos de muestra en el termociclador y ejecute el siguiente programa: 95 °C durante 3 min; 40 ciclos de: 95 °C durante 30 s, 51 °C durante 30 s y 72 °C durante 1 min 30 s; 72 °C durante 10 min; mantener a 10 °C.
- Preparar 25 μL de la reacción de PCR en tubos de PCR añadiendo 12,5 μL de 2x Taq Polymerase Master Mix, 0,5 μL de 10 μM de imprimación 8F, 0,5 μL de imprimación 10 μM 1492R(Tabla de Materiales),y 11,5 μL de agua libre de nucleasas21.
- Extracción en gel e identificación de especies
- Al finalizar la ejecución de PCR, verifique el producto de PCR en un gel de agarosa al 1% preparado en un tampón Tris-Borate-EDTA (TBE) 0.5x. Antes de fundir el gel, agregue bromuro de etidio (EtBr). Luego, funda el gel usando peines para pozos que contengan al menos 20 μL de volumen de muestra.
PRECAUCIÓN: EtBr es un agente intercalante sospechoso de ser cancerígeno. Siempre use guantes y EPP cuando lo manipule y deseche los materiales que contengan EtBr de acuerdo con las pautas de la institución. - Cuando el gel esté listo, coloque el gel en el tanque de electroforesis lleno con un tampón TBE 0.5x y retire el peine. Cargue la escalera de 1 kb en el primer pozo y 10-20 μL de la reacción de PCR en los pozos posteriores. Ejecute a 100-140 V hasta que se resuelva. Visualice el gel bajo luz UV y confirme la presencia de una banda claramente definida a ~ 1.5 kb que está ausente en el pozo NTC.
PRECAUCIÓN: Los rayos UV son dañinos para la piel y los ojos, use un protector apropiado al visualizar el gel y use el EPP apropiado.
NOTA: La PCR de colonias puede no tener éxito para algunas bacterias; proceder con PCR a partir de gDNA aislado es una opción alternativa22. - Elimine las bandas de ~ 1.5 kb con una maquinilla de afeitar y transfiera los esquejes de gel a tubos de microcentrífuga limpios. Proceda con el protocolo de extracción de gel según las instrucciones del fabricante(Tabla de materiales). Medir la concentración del ADN purificado mediante espectrofotómetro de microvolumen.
NOTA: Es deseable una concentración >10 ng/μL, y A260/280 entre 1.7-2.0 es aceptable. - Prepare dos reacciones de secuenciación de Sanger para cada muestra, una utilizando el 8F y la otra utilizando el cebador 1492R en agua libre de nucleasas de acuerdo con las pautas de cualquier servicio de secuenciación de Sanger elegido.
- Una vez que se reciben los datos de secuenciación, cargue las secuencias de ADN en el sitio web de ncbi Basic Local Alignment Search Tool (BLAST) (blast.ncbi.nlm.nih.gov/Blast.cgi), elija Nucleotide BLAST (blastn), seleccione la base de datos rRNA / ITS secuencias de ARN ribosómico 16S (bacterias y arqueas) y ejecute el programa Megablast. El aislado puede ser identificado por la más alta calidad de acierto a una referencia de la base de datos.
NOTA: Algunas especies bacterianas exhiben una alta identidad en sus secuencias de ARNr 16S y pueden ser indistinguibles solo por este método. La especiación requerirá homología de ADN y análisis bioquímicos para distinguir con confianza a los miembros del mismo género23.
- Al finalizar la ejecución de PCR, verifique el producto de PCR en un gel de agarosa al 1% preparado en un tampón Tris-Borate-EDTA (TBE) 0.5x. Antes de fundir el gel, agregue bromuro de etidio (EtBr). Luego, funda el gel usando peines para pozos que contengan al menos 20 μL de volumen de muestra.
3. Extracción de ADN genómico (gDNA)
NOTA: Esta sección utiliza reactivos y columnas de espín proporcionados en el kit de extracción de gDNA al que se hace referencia en la Tabla de materiales para la extracción de alto rendimiento de ADN genómico de calidad de diversas especies bacterianas. A continuación se proporcionan modificaciones e instrucciones recomendadas.
- Prepare los reactivos del kit según las instrucciones del fabricante.
- Preparar cultivos de 3-10 ml en caldo estéril apropiado(Figura 1)inoculando bacterias de colonias bien aisladas en el medio e incubando a la temperatura y presión atmosférica observadas en la Figura 1 hasta que se observe un crecimiento suficiente.
- Después de la incubación, medir la densidad óptica a 600 nm (OD600)del cultivo utilizando un espectrofotómetro24.
- Prepare la muestra para la cuantificación diluyendo cultivos nocturnos en una proporción de 1:10. Incluya también un espacio en blanco de los medios de cultivo estériles para la medición. Calcule la densidad óptica restando la lectura en blanco de la lectura de la muestra y multiplicando por el factor de dilución de diez.
- Utilizando la medición OD600 y una relación preestablecida OD600 a UFC/ml para la especie, calcule cuántos mililitros de cultivo son necesarios para obtener 2 x 109 células.
- Centrifugar el volumen de cultivo requerido durante 5 min a 5000 x g a pellet. Aspire el sobrenadante y vuelva a suspender el pellet en tampón TE frío de 200 μL (preenfriamiento sobre hielo al comienzo del procedimiento).
- Centrifugar la muestra durante 2 min a 5000 x g. Retire el sobrenadante y luego vuelva a suspender el gránulo en 180 μL de Tampón de lisis enzimática (ELB) y agregue 20 μL de RNasa A prehervida (10 mg / ml). Para una lisis eficiente de bacterias Gram-positivas, agregue 18 μL de mutanolisina (25 kU / ml). Vórtice bien, y luego incube las muestras a 37 ° C en el rotador durante 2 h.
NOTA: Se recomienda utilizar el ELB descrito en el protocolo del fabricante para las bacterias Gram-positivas y Gram-negativas. - Proceda de acuerdo con las instrucciones del fabricante.
NOTA: Repita los pasos de elución una o dos veces más para obtener un rendimiento adicional de gDNA, si lo desea. - Evalúe la calidad del gDNA extraído según las instrucciones de la sección 4 y almacene el gDNA a 4 °C si se va a utilizar en un plazo de 1 semana. Alternativamente, mantenga el gDNA a -20 °C para el almacenamiento a largo plazo.
4. Evaluación de la calidad del aDNb extraído
- Para evaluar la calidad por electroforesis en gel, prepare gel de agarosa al 1% como se describe en la subsección 2.2. Prepare la muestra en un tubo limpio: mezcle 1-2 μL de gDNA extraído y 3 μL de tinte de carga 2x en parafilm. Ejecute el gel una vez cargado y luego visualícelo bajo luz UV.
NOTA: La extracción exitosa de gDNA será evidente por una banda discreta en la parte superior del gel y un frotis mínimo(Figura 2A). La mancha es indicativa de cizalladura. Si no hay ninguna banda de gDNA es evidente y/o el frotis es sustancial, repita la extracción de gDNA. Considere reducir los tiempos de incubación en la RNasa A y la Proteinasa K. Si se observan dos bandas alrededor de 1.5-3 kb, esto sugiere contaminación por ARN(Figura 2B). Preparar RNasa A fresca y repetir la extracción. - Para evaluar la calidad por espectrofotómetro de microvolumen, mida la concentración de gDNA y la relación de absorbancia A260/280 por espectrofotómetro de microvolumen. Las concentraciones >50 ng/μL y A260/280 entre 1,7-2,0 son aceptables.
NOTA: El bajo rendimiento de gDNA puede deberse a una entrada baja, una entrada alta, contaminación de nucleasas, lisis insuficiente. Las relaciones de absorbancia por encima del rango indican contaminación por ARN. Repita la extracción si la calidad del gDNA es deficiente. - Para evaluar la calidad por fluorómetro, siga las instrucciones del fabricante para cuantificar la concentración de gDNA utilizando el kit de ensayo de alta sensibilidad y el instrumento de fluorómetro(Tabla de materiales). La concentración >50 ng/μL es deseable.
5. Secuenciación de lectura corta de próxima generación emparejada y preparación de bibliotecas
NOTA: La secuenciación de lectura corta se puede realizar en varios instrumentos en distintas longitudes y orientaciones de lectura. Se recomienda la secuenciación de extremo pareado de 150 pb (ciclo de 300) para el WGS bacteriano. Tanto la preparación de la biblioteca como la secuenciación pueden subcontratarse a instalaciones centrales o laboratorios comerciales.
- Prepare la biblioteca de secuenciación de acuerdo con las instrucciones del fabricante (Tabla de materiales). Siga la concentración de la biblioteca de carga final recomendada por el fabricante; sin embargo, una modificación recomendada es cargar la biblioteca agrupada a 1,8 pM para una generación de lectura óptima en los instrumentos NextSeq.
- Aunque es opcional, utilice un bioanalizador(Tabla de materiales)para evaluar la distribución de fragmentos de biblioteca agrupados y asegurarse de que el tamaño del fragmento sea de 600 pb en promedio.
6. Preparación de la biblioteca de secuenciación Nanopore MinION
- Preparar la biblioteca de secuenciación de acuerdo con el protocolo del fabricante(Tabla de Materiales). El uso de dos kits de expansión de código de barras permite la multiplexación de hasta 24 muestras en una sola celda de flujo. Se recomienda realizar la preparación de la biblioteca en dos partes, 12 muestras a la vez al multiplexar 24 muestras. Las 24 muestras se pueden agrupar como se describe a continuación.
NOTA: Las muestras se pueden almacenar a 4 ° C durante la noche al finalizar la ligadura de código de barras nativa, lo que proporciona un punto de parada en el protocolo, si es necesario. Al final de la sección de ligadura de código de barras nativa del protocolo de preparación de la biblioteca, se recomienda agrupar cantidades equimolares de cada muestra hasta la masa máxima de ADN (ng) posible.- Para ello, cuantifique todas las muestras después de la ligadura de códigos de barras utilizando un fluorómetro(Tabla de materiales)según las instrucciones del fabricante. Estime el volumen de la muestra con la concentración más baja de dsDNA y luego calcule el dsDNA total encontrado en esta muestra. Utilice este número para determinar las cantidades equimolares de todas las demás muestras que se agruparán.
NOTA: Debido a que el cálculo equimolar maximizará la cantidad de dsDNA agrupado y, por lo tanto, producirá un grupo de alto volumen (>65 μL), la limpieza es necesaria para concentrar la piscina.
- Para ello, cuantifique todas las muestras después de la ligadura de códigos de barras utilizando un fluorómetro(Tabla de materiales)según las instrucciones del fabricante. Estime el volumen de la muestra con la concentración más baja de dsDNA y luego calcule el dsDNA total encontrado en esta muestra. Utilice este número para determinar las cantidades equimolares de todas las demás muestras que se agruparán.
- Limpieza y concentración de piscinas de dsDNA
- Agregue 2.5x volumen de cuentas paramagnéticas(Tabla de Materiales)a la piscina de ADN, y luego mueva suavemente el tubo para mezclar el contenido. Coloque el tubo en el rotador durante 5 minutos en RT. Gire la muestra a 2000 x g y g y gule en un imán.
- Añadir 250 μL recién preparados al 70% de etanol (en agua libre de nucleasas), teniendo cuidado de no molestar al pellet. Aspire el etanol y repita el lavado de etanol una vez.
- Después de la segunda aspiración, gire la muestra a 2000 x g y colóquela de nuevo en el imán. Pipetee cualquier etanol residual y deje que la muestra se seque durante aproximadamente 30 s.
- Retire el tubo del imán y vuelva a suspender el pellet en 60-70 μL de agua libre de nucleasa. Incubar en RT durante 2 min. Peletizar la muestra en el imán hasta que el elu esté claro, y luego retirar el elu y transferir a un tubo de microcentrífuga limpio de 1,5 ml.
- Cuantifique la piscina concentrada utilizando un fluorómetro y luego prepare una alícuota para proceder a la etapa de ligadura del adaptador: prepare 700 ng de la muestra en un volumen final de 65 μL. Mantenga el resto del grupo a 4 ° C para completar una segunda ejecución una vez que finalice la primera ejecución.
- Proceda con la ligadura del adaptador según las instrucciones del fabricante y cargue la muestra en la celda de flujo. Inicie la ejecución de secuenciación.
NOTA: Aspire aire y ~200 μL de búfer de almacenamiento desde el puerto de cebado de la celda de flujo antes de la carga de la muestra. Esto es fundamental para el cebado exitoso de la celda de flujo y la carga de muestras. Utilice una pipeta p1000 y puntas al dibujar y depositar soluciones a través del puerto de cebado de la celda de flujo.
- Secuencia la biblioteca de acuerdo con las instrucciones del fabricante.
- Abra el software operativo para la secuenciación y haga clic en Inicio. Ingrese un nombre para el experimento, una nomenclatura recomendada incluye la fecha de ejecución y el nombre del usuario. Haga clic en Continuar con la selección del kit, seleccione el kit de preparación de la biblioteca apropiado y los paquetes de expansión de código de barras utilizados, y luego haga clic en Continuar con las opciones de ejecución.
- Ajuste la duración de la ejecución a 48 h si planea preparar suficiente biblioteca para una segunda ejecución (de lo contrario, deje en el valor predeterminado 72 h). Haga clic en Continuar a Basecalling.
- Marque la opción de llamada base Config: Fast Basecalling y asegúrese de que el código de barras esté configurado en Habilitado para que los archivos FASTQ de salida se recorten de las secuencias de códigos de barras y se demultiplexen en directorios separados basados en códigos de barras. Haga clic en Continuar a la salida.
- Elija dónde guardar los datos de secuenciación de salida. Espere aproximadamente 30-50 Gb de datos si solo ahorra la salida FASTQ y >500 Gb de datos si también ahorra la salida FAST5. Desmarque la opción de filtrado Qscore: 7 | Longitud de lectura: Sin filtrar si planea continuar con el filtrado descrito en la sección 7.2, de lo contrario deje marcada y ajuste la longitud de lectura a 200.
- Haga clic en Continuar para ejecutar el programa de instalación y revise todas las configuraciones. Si la configuración es correcta, haga clic en Inicio,de lo contrario haga clic en Atrás y realice los ajustes necesarios.
- Si se desea, la celda de flujo se puede lavar según las instrucciones del fabricante y volver a cargarse con el grupo restante. Repita los pasos de la versión 6.2 para el grupo restante una vez que se haya completado la primera ejecución y se haya lavado la celda de flujo.
NOTA: Al configurar la segunda ejecución, ajuste el voltaje de polarización a -250 mV según las recomendaciones del fabricante para las celdas de flujo utilizadas anteriormente en carreras de más de 48 h.
7. Evaluación y preparación de lecturas
Nota : una estructura de directorios recomendada se representa en la figura 4. Cree los directorios que se encuentran en el escritorio,es decir, Long_Reads, Short_Reads y Trimmed_Reads, antes de continuar con los pasos de cálculo a continuación.
- Lecturas cortas (Figura 3)
NOTA: Las lecturas cortas se generan en el formato FASTQ. Los archivos contienen 4000 lecturas máximas por FASTQ. Estos a menudo se comprimen (.gz archivo) y se organizan en múltiples archivos. Dependiendo de la plataforma, los códigos de barras generalmente se recortan. Algunos programas aceptan archivos en formato comprimido, otros pueden requerir su extracción antes de importarlos. Las lecturas deben pasar los pasos de control de calidad (QC) para garantizar la precisión de los datos durante el ensamblaje del genoma. Si CLC Genomics Workbench no está disponible, se pueden usar programas alternativos para recortar y leer en corto el control de calidad, como Trimmomatic.25 o Trim Galore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) para recortar y FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) para evaluar la calidad de lectura. Se recomienda que la cobertura media de lectura corta, estimada multiplicando el número de lecturas por la longitud media de lectura y dividiendo por el tamaño del genoma, sea >100x.- Abra el software Genomics Workbench(Tabla de materiales)e importe todos los archivos FASTQ de lectura corta de extremo emparejado. Los archivos emparejados se generarán automáticamente.
- Cree una nueva carpeta en CLC_Data haciendo clic en Nuevo en la barra de herramientas superior y seleccionando Carpeta... para almacenar los archivos. Asigne a la carpeta el nombre que desee, una convención recomendada utiliza el id. Guarde todos los resultados de los pasos siguientes en esta carpeta.
- En la barra de herramientas superior, haga clic en el botón Importar y seleccione Illumina... Desplácese hasta todos los archivos de lectura corta que correspondan al ejemplo y selecciónelos. Asegúrese de que la opción lecturas emparejadas esté seleccionada y desmarque la opción Eliminar lecturas fallidas. Haga clic en Siguiente,seleccione Guardary vuelva a hacer clic en Siguiente. Elija guardar los archivos importados en la nueva carpeta creada en el paso anterior y haga clic en Finalizar.
- Cree una lista de secuencia de todos los archivos emparejados para el aislado; esto concatenará los datos de lectura en un solo archivo para simplificar el análisis.
- En la barra de herramientas superior, haga clic en el botón Nuevo y seleccione Lista de secuencias... En la lista de directorios de la izquierda, seleccione los archivos que se van a concatenar y use las flechas para moverlos a la lista de archivos seleccionados a la derecha. Haga clic en Siguiente,seleccione Guardary vuelva a hacer clic en Siguiente. Elija guardar la lista de secuencias y haga clic en Finalizar.
- Una vez generada la lista de secuencias, cámbiele el nombre inmediatamente con el ID de ejemplo.
- Ejecute la herramienta QC for Sequencing Reads en la lista de secuencias: este procedimiento evaluará los parámetros de calidad general de las lecturas generadas por NGS de lectura corta.
- Busque la herramienta QC for Sequencing Reads en el menú del cuadro de herramientas (ventana inferior izquierda). Haga doble clic en la herramienta y, a continuación, elija la lista de secuencias que desea analizar y haga clic en Siguiente.
- Asegúrese de que todas las opciones de salida estén marcadas y elija Guardar en Manejo de resultados. Haga clic en Siguiente y especifique guardar los archivos de salida, y luego haga clic en Finalizar.
- Ejecute la herramienta Recortar lecturas en la lista de secuencias: el recorte se realizará en función de la calidad, la longitud y la ambigüedad. Este proceso asume que los códigos de barras utilizados en la secuenciación se han recortado antes de este paso.
- Busque la herramienta Recortar lecturas en el cuadro de herramientas (ventana inferior izquierda). Haga doble clic en Recortar lecturas, y luego elija la lista de secuencias a analizar y haga clic en Siguiente.
- Recorte de calidad: establezca el límite de puntuación de calidad en 0,01 y deje los nucleótidos ambiguos en 2. Haga clic en Siguiente.
NOTA: Los parámetros pueden ajustarse a discreción del usuario; estos son los ajustes recomendados. - Desmarque Recorte automático del adaptador de lectura directa (solo haga esto si los adaptadores se han recortado de las lecturas antes de importar a CLC). Haga clic en Siguiente y marque Descartar lecturas por debajo de la longitud,use el valor predeterminado 15.
- Haga clic en Siguiente, marque Crear informey, a continuación, elija Guardar. Haga clic en Siguiente y especifique dónde guardar los archivos de salida. Haga clic en Finalizar.
- Exportar la lista de secuencias recortadas: el ensamblaje y análisis híbridos posteriores se completarán fuera de CLC y requieren que se exporten archivos recortados de lectura corta.
- En la navegación del directorio en la parte superior izquierda, elija el archivo recortado generado en el paso 7.1.4 y luego haga clic en Exportar en la barra de herramientas superior. Seleccione Fastq para el tipo de archivo de exportación y haga clic en Siguiente. Marque Exportar lista de secuencias emparejadas a dos archivos. Luego, haga clic en Siguiente y elija el directorio Trimmed_Reads al que exportar los archivos. Haga clic en Finalizar. Asegúrese de que los archivos recortados de lectura corta se exportaron correctamente como dos archivos (R1 y R2) con la extensión .fastq.
NOTA: La lista de secuencias recortadas debe exportarse a dos archivos, normalmente designados por CLC como R1 y R2. Esto es fundamental ya que el ensamblaje híbrido descendente requiere que la entrada de datos de lectura corta se configure como tal. - Cambie el nombre de los archivos exportados, absténgase del uso de espacios y caracteres especiales en los nombres de archivo. Para simplificar, un formato recomendado es trimmed_short_file. R1.fastq.
- En la navegación del directorio en la parte superior izquierda, elija el archivo recortado generado en el paso 7.1.4 y luego haga clic en Exportar en la barra de herramientas superior. Seleccione Fastq para el tipo de archivo de exportación y haga clic en Siguiente. Marque Exportar lista de secuencias emparejadas a dos archivos. Luego, haga clic en Siguiente y elija el directorio Trimmed_Reads al que exportar los archivos. Haga clic en Finalizar. Asegúrese de que los archivos recortados de lectura corta se exportaron correctamente como dos archivos (R1 y R2) con la extensión .fastq.
- Abra el software Genomics Workbench(Tabla de materiales)e importe todos los archivos FASTQ de lectura corta de extremo emparejado. Los archivos emparejados se generarán automáticamente.
- Lecturas largas (MinION) (Figura 3)
NOTA: La siguiente canalización para la preparación de lecturas de secuenciación Larga (MinION) para ensamblaje híbrido utiliza los programas NanoFilt y Nanostat26 ejecutados por la línea de comandos. Instale las herramientas antes de continuar y familiarícese con los conceptos básicos de UNIX para ejecutar estos comandos. Se recomiendan terminales predeterminados y Bash Shell. Una guía de lección para comandos y uso de terminales comunes se encuentra en Software Carpentry27. Las instrucciones a continuación asumen que los archivos generados se nombrarán con la nomenclatura de código de barras (NB01, NB02, etc.) y se guardarán en el directorio Long_Reads. Alternativamente, el filtrado de lectura se puede lograr utilizando MinKNOW al configurar la ejecución de secuenciación. Se recomienda que la cobertura promedio de lectura larga sea de >100x. La longitud media de lectura recomendada es de >2000 pb; por lo tanto, el número de lecturas largas necesarias es menor que el número de lecturas cortas.- Crear nuevos directorios para cada código de barras utilizado en la ejecución (barcode01, barcode02, etc.) dentro del directorio Long_Reads (Figura 4). Copie todos los archivos .fastq que corresponden a cada código de barras en la carpeta correspondiente. Combine todos los archivos .fastq para cada código de barras de cada ejecución.
- Abra Terminal y navegue a los directorios de códigos de barras dentro del directorio Long_Reads usando el comando cd: cd Desktop/Long_Reads/barcode01
- Concatenar todos los archivos .fastq por código de barras en un solo archivo .fastq ejecutando el siguiente comando: cat *.fastq > NB01.fastq
NOTA: Este comando combina todas las lecturas de cada uno de los archivos FASTQ en un solo FASTQ grande denominado NB01.fastq. - Utilice NanoStat para evaluar la calidad de lectura de la muestra ejecutando el siguiente comando: NanoStat --fastq NB01.fastq
- Registre los resultados copiando la salida en un archivo de texto o Word para futuras referencias.
- Utilice NanoFilt para filtrar lecturas MinION descartando lecturas con Q < 7 y longitud < 200 ejecutando el comando: NanoFilt -q 7 -l 200 bp NB01.fastq | gzip > NB01 _trimmed.fastq.gz
- Ejecute NanoStat en el archivo recortado generado en el paso 7.2.6 ejecutando el comando: NanoStat --fastq NB01 _trimmed.fastq.gz
- Registre los resultados copiando el resultado en un archivo de texto o Word y compárelos con los resultados del paso 7.2.4 para asegurarse de que el filtrado se realizó correctamente (Tabla 1).
- Repita los pasos 7.2.2 a 7.2.8 para cada código de barras utilizado en la ejecución de secuenciación.
Nota : el archivo NB01_trimmed.fastq.gz generado en el paso 7.2.6 se utilizará para el ensamblaje híbrido.
8. Generación de ensamblaje del genoma híbrido
NOTA: La siguiente tubería de ensamblaje utiliza Unicycler19,28,29,30 para combinar lecturas cortas y largas preparadas en las secciones 7.1 y 7.2(Figura 3). Instale Unicycler y sus dependencias y ejecute los comandos a continuación. Se supone que los archivos de lectura corta exportados en el paso 7.1.5 se denominan trimmed_short_file. R1.fastq y trimmed_short_file. R2.fastq para simplificar.
- Organice los archivos de lectura corta y los archivos de lectura larga en un único directorio denominado Trimmed_Reads. El directorio debe contener lo siguiente:
- Un archivo .fastq.gz para lecturas largas recortadas (generadas en el paso 7.2.6).
- Dos archivos .fastq (R1 y R2) para lecturas cortas recortadas (generadas en el paso 7.1.5).
- Navegue hasta el directorio Trimmed_Reads que almacena los archivos de lectura mediante el comando cd en Terminal: cd Desktop/Trimmed_Reads
- Una vez en el directorio correcto, comprima los dos archivos de lectura corta para que también estén en el formato .fastq.gz ejecutando el siguiente comando: gzip trimmed_short_file. R1.fastq
- Repita el paso 8.2 para R1 y R2. Compruebe que todos los archivos leídos estén ahora en el formato .fastq.gz y verifique que todos los archivos coincidan con el mismo aislamiento.
- Comience el conjunto híbrido mediante Unicycler ejecutando el siguiente comando:
monociclista -1 trimmed_short_file. R1.fastq.gz -2 trimmed_short_file. R2.fastq.gz -l NB01 _trimmed.fastq.gz -o unicycler_output_directory
NOTA: -o especifica el directorio en el que se guardará la salida del monociclo, el monociclo creará este directorio una vez que se ejecute el comando; no genere el directorio de antemano. El tiempo de ejecución varía según la potencia computacional de la computadora utilizada, así como el tamaño del genoma y el número de lecturas. Esto puede tomar desde 4 h hasta 1 o 2 días. Este protocolo se realizó en una máquina CentOS Linux 7 con 250 Gb de RAM, CPU Intel Xeon (R) con 2,5 GHz 12 núcleos prácticos y 48 núcleos virtuales. Alternativamente, las computadoras personales con 16 Gb de RAM y procesadores de 6 núcleos a 2,6 GHz pueden calcular estos ensamblajes en un tiempo de procesamiento más largo. - Cuando se complete la ejecución, revise el archivo monociclizador.log para asegurarse de que no haya errores: registre el número, el tamaño y el estado (completo, incompleto) de los contigs generados.
- Si se identifican contigs incompletos (denotados como incompletos en el registro de Unicycler), vuelva a ejecutar Unicycler en negrita agregando el siguiente indicador al comando en el paso 8.4: --mode bold.
NOTA: El modo en negrita reducirá el umbral de calidad aceptado para puentes de lectura largos durante el montaje; esto puede producir un ensamblaje completo, pero la calidad del ensamblaje puede verse disminuida. Se recomienda utilizar el modo negrita solo cuando sea necesario y como evidencia preliminar para que la unión contig se confirme más tarde mediante PCR.
- Si se identifican contigs incompletos (denotados como incompletos en el registro de Unicycler), vuelva a ejecutar Unicycler en negrita agregando el siguiente indicador al comando en el paso 8.4: --mode bold.
9. Evaluación de la calidad del montaje
NOTA: El siguiente protocolo utiliza Bandage31 y QUAST32,dos programas que deben configurarse antes de su uso(Figura 2 y Figura 4). El vendaje no requiere instalación una vez descargado y QUAST requiere familiaridad con el uso básico de la línea de comandos. También se recomienda evaluar la integridad del genoma utilizando Benchmarking Universal Single-Copy Orthologs (BUSCO)33.
- Vendaje: Haga clic en Archivo. A continuación, elija Load Graph y seleccione el archivo assembly.gfa que se guardó en unicycler_output_directory generado por Unicycler en el paso 8.4. Una vez cargado, haga clic en el botón Dibujar gráfico en la barra de herramientas de la izquierda y observe cómo se conectan y organizan los contigs (llamados nodos) para evaluar si el ensamblaje está completo(Figura 5).
NOTA: Los conjuntos completos están representados por contigs circulares simples enlazados en ambos extremos (Figura 5A,B). Los ensamblajes incompletos tienen múltiples contigs unidos entre sí o son lineales(Figura 5C). Los contigs lineales pequeños pueden no estar incompletos, ya que pueden indicar elementos extracromosómicos lineales. La cobertura, también llamada profundidad, se notará en el vendaje y representa la abundancia relativa de los contigs al cromosoma, normalizada en Unicycler a 1x. - QUAST
- Dentro de la Terminal, navegue a la carpeta que almacena la salida del monociclador utilizando el comando cd: cd Desktop/Trimmed_Reads/unicycler_output_directory
NOTA: Los espacios no están permitidos en la ruta hacia donde se encuentra el conjunto, es decir, ningún directorio que conduzca a la salida del monociclizador puede tener espacios en su nombre. Como alternativa, copie el archivo assembly.fasta en el escritorio para facilitar el acceso. - Ejecute QUAST ejecutando el siguiente comando: quast assembly.fasta -o quast_output_directory
- Revise los informes generados por QUAST en el directorio de salida quast_output_directory.
- Dentro de la Terminal, navegue a la carpeta que almacena la salida del monociclador utilizando el comando cd: cd Desktop/Trimmed_Reads/unicycler_output_directory
10. Anotación del genoma
NOTA: La siguiente canalización de anotaciones utiliza Prokka34,una herramienta de línea de comandos que debe instalarse antes de su uso. Alternativamente, utilice Prokka a través de la GUI automatizada K-Base(Tabla de Materiales)o anote genomas a través del servidor web RAST35. Si se depositan genomas en ncbi, se anotarán automáticamente utilizando el Prokaryotic Genome Annotation Pipeline (PGAP)36.
- Navegue dentro del Terminal hasta la carpeta que almacena la salida del monociclizador mediante el comando cd (consulte el paso 9.2.1). A continuación, ejecute Prokka ejecutando el siguiente comando: prokka --prefix sample_ID --outdir prokka_output_directory assembly.fasta
NOTA: --prefix nombrará todos los archivos de salida en función de la sample_ID especificada. --outdir creará un directorio de salida con el nombre especificado donde se guardarán todos los archivos de salida de Prokka; no cree un directorio de salida para Prokka de antemano. - Revise las anotaciones abriendo la tabla .tsv y/o cargando el archivo .gff generado en un software de análisis de secuencia para visualizar y analizar las anotaciones (Figura 6).
- Se pueden generar tipos específicos de anotaciones dependiendo de factores genéticos de interés. Se recomienda comenzar con las herramientas fáciles de usar en el servidor web del Centro de Epidemiología Genómica (www.genomicepidemiology.org/) para el análisis preliminar37,38,39,40,41. Se dispone de herramientas adicionales para la detección de sistemas CRISPR-cas y profagos (Figura 3)42,43.
11. Prácticas sugeridas para la democratización de los datos
- Cuando sea posible, deposite todos los datos de lectura en bruto, así como los genomas ensamblados en un repositorio público como NCBI Sequence Read Archive (SRA) y Genbank. Los genomas se anotan automáticamente a través de la tubería PGAP durante el proceso de deposición del NCBI.
Resultados
Este protocolo ha sido optimizado para el cultivo y secuenciación de bacterias urinarias pertenecientes a los géneros enumerados en la Figura 1. No todas las bacterias urinarias son cultivables por este método. Los medios de cultivo y las condiciones son especificados por el género en la Figura 1. Las evaluaciones ejemplares de electroforesis en gel de la integridad del gDNA se representan en la Figura 2. En la Figura 3se describe una visión general de la tubería bioinformática para el procesamiento de lectura de secuenciación, el ensamblaje del genoma y la anotación. En la Figura 4 se proporciona una guía para la estructura de directorios computacionales para simplificar la comprensión del protocolo y proporcionar un marco para una organización exitosa. Además, se incluyen genomas completos representativos de dos Klebsiella spp., K. pneumoniae y K. oxytoca,que fueron generados por este protocolo. Una representación de estos ensamblajes se proporciona en la Figura 5 y también incluye un ejemplo incompleto adicional del genoma de K. pneumoniae. Una descripción detallada de cada genoma completo totalmente anotado se muestra en la Figura 6. Finalmente, en la Tabla 1 se proporciona un resumen de las estadísticas de lectura de secuenciación para ofrecer una amplia comprensión de los datos brutos y recortados suficientes para la generación de ensamblajes de genoma cerrado de alta calidad. Además, los parámetros clave de los dos representantes completan Klebsiella spp. se enumeran los genomas. Los genomas y los datos en bruto se depositaron en Genbank bajo el BioProyecto PRJNA683049.

Figura 1: Cultivo de orina mejorado modificado de diversos géneros urinarios. Gráfico para el caldo de agar y líquido que se puede utilizar para cultivar diversos géneros urinarios. Se sugiere que todos los cultivos se realicen a 35 °C como se describe en la subsección 1.1. Los círculos representan medios apropiados para cultivar un género en particular, los colores se seleccionaron arbitrariamente para distinguir un tipo de medio de otro. CDC-AN BAP (rojo), CDC Anaerobe Sheep Blood Agar; 5% Oveja-BAP (naranja), Agar de Sangre de Oveja; BHI (verde), Infusión del corazón cerebral; TSB (amarillo), caldo de soja tríptico; Orientación CHROMagar (azul). aGardnerella vaginalis debe cultivarse en HBT Bilayer G. vaginalis Agar selectivo en atmósfera microaerofílica y bajo requisitos especiales de cultivo de caldo44. bLactobacillus iners debe cultivarse en placas rabbit-BAP al 5% y caldo NYCIII en atmósfera microaerofílica. cLactobacillus spp. puede cultivarse en MRS en condiciones microaerofílicas. Haga clic aquí para ver una versión más grande de esta figura.
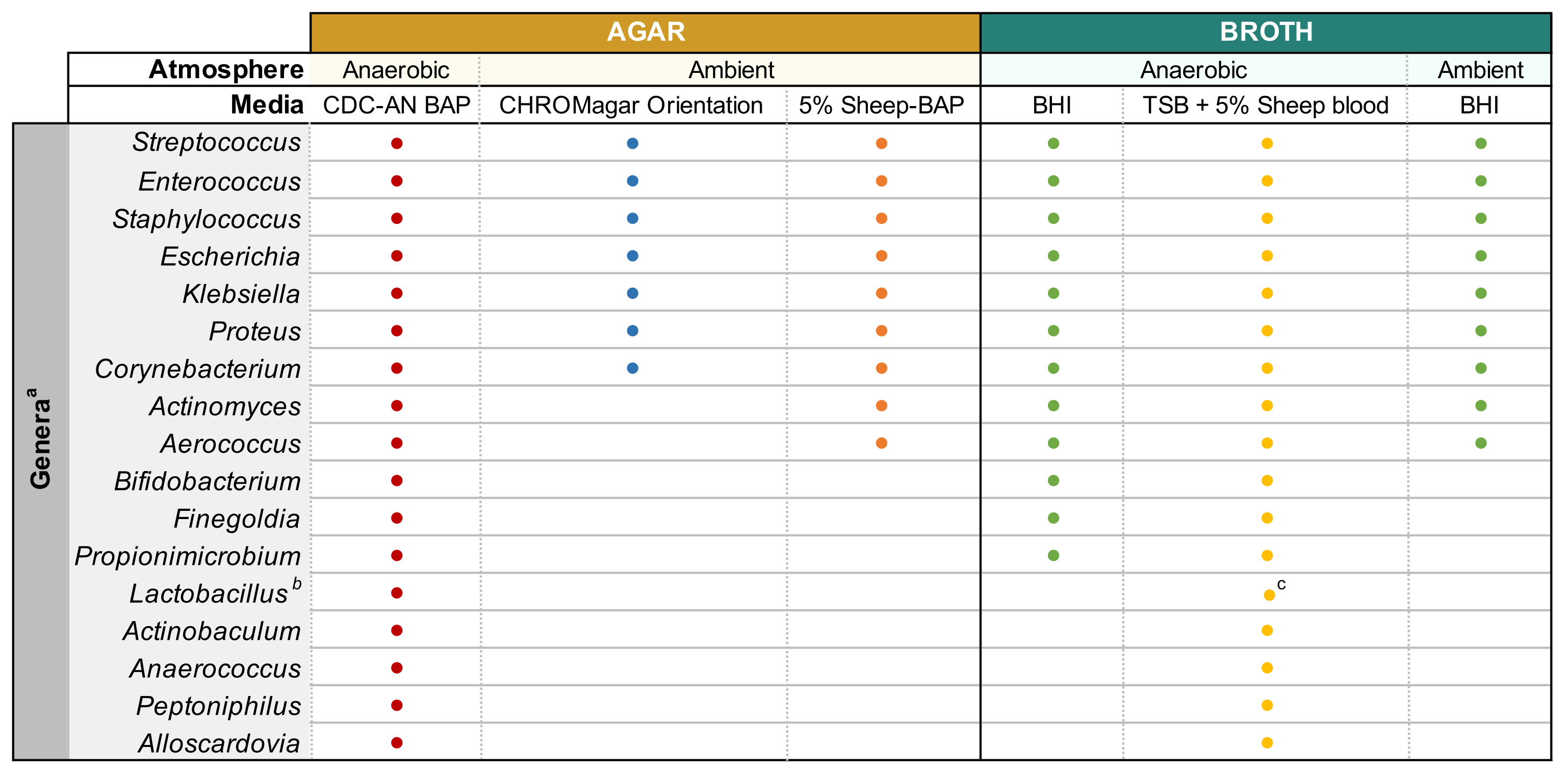{kind=link}

Figura 2: Imágenes de gel de agarosa de extracción genómica de ADN. Imágenes representativas de gel que representan los resultados de la extracción de gDNA. (A) Carril 1: escalera de 1 kb, Carril 2: gDNA intacto que representa una extracción exitosa, Carril 3: frotis que indica gDNA fragmentado. (B) Carril 1: escalera de 1 kb, carriles 2 y 3: contaminación por ARNr denotada por dos bandas entre 1,5 kb y 3 kb. Haga clic aquí para ver una versión más grande de esta figura.
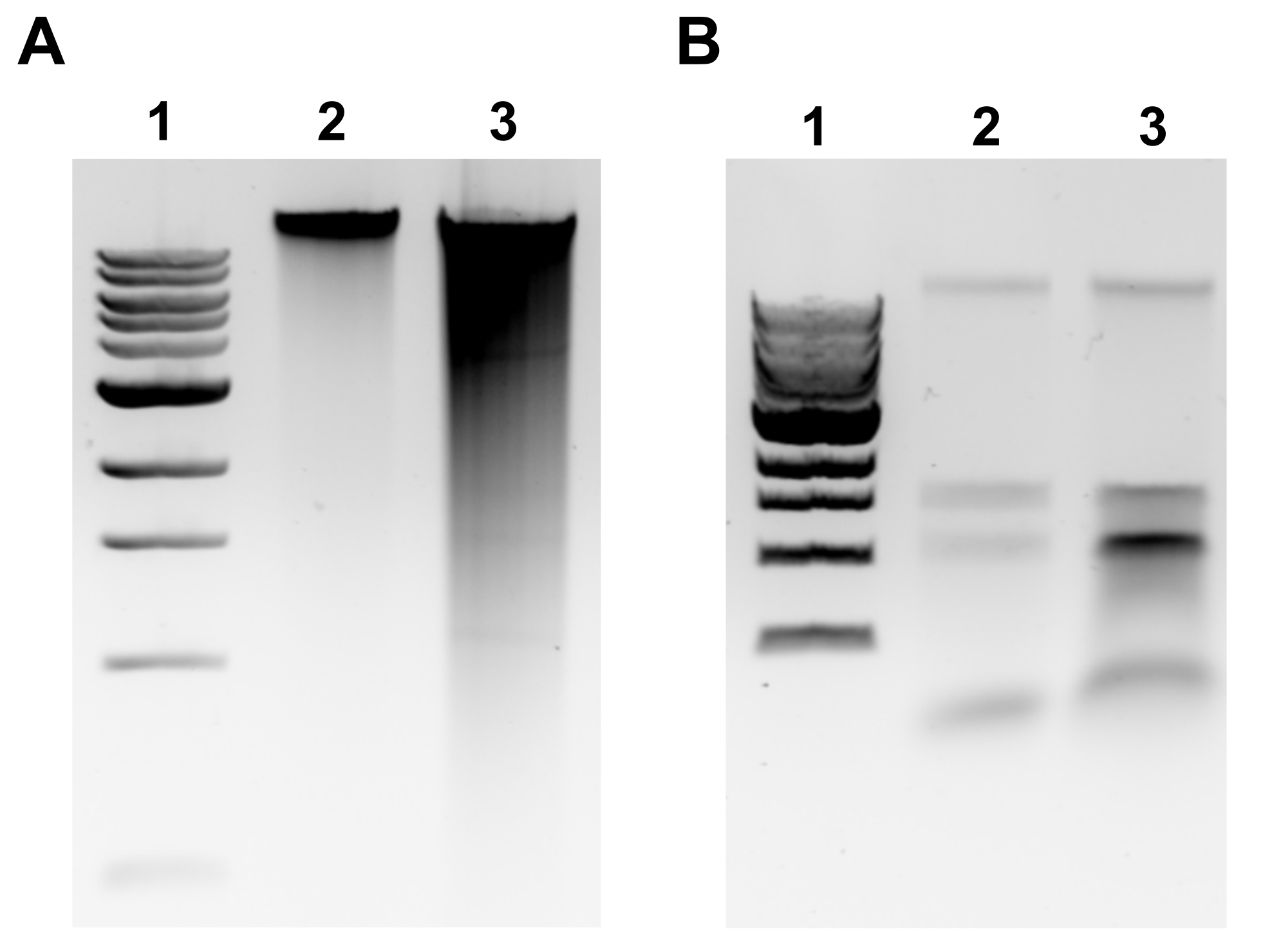{kind=link}

Figura 3:Flujo de trabajo de ensamblaje del genoma híbrido. Esquema de pasos desde el control de calidad de lectura y el preprocesamiento hasta la anotación del ensamblaje. El recorte de lectura elimina las lecturas ambiguas y de baja calidad. Los parámetros Q-score y length se indican y representan las lecturas que se conservan. El ensamblaje utiliza lecturas cortas y largas para generar un ensamblaje híbrido de novo del genoma. La calidad del ensamblaje se evalúa en función de la integridad y la corrección utilizando herramientas y parámetros especificados. El ensamblaje final del genoma se anota para todos los genes y loci específicos de interés. Haga clic aquí para ver una versión más grande de esta figura.
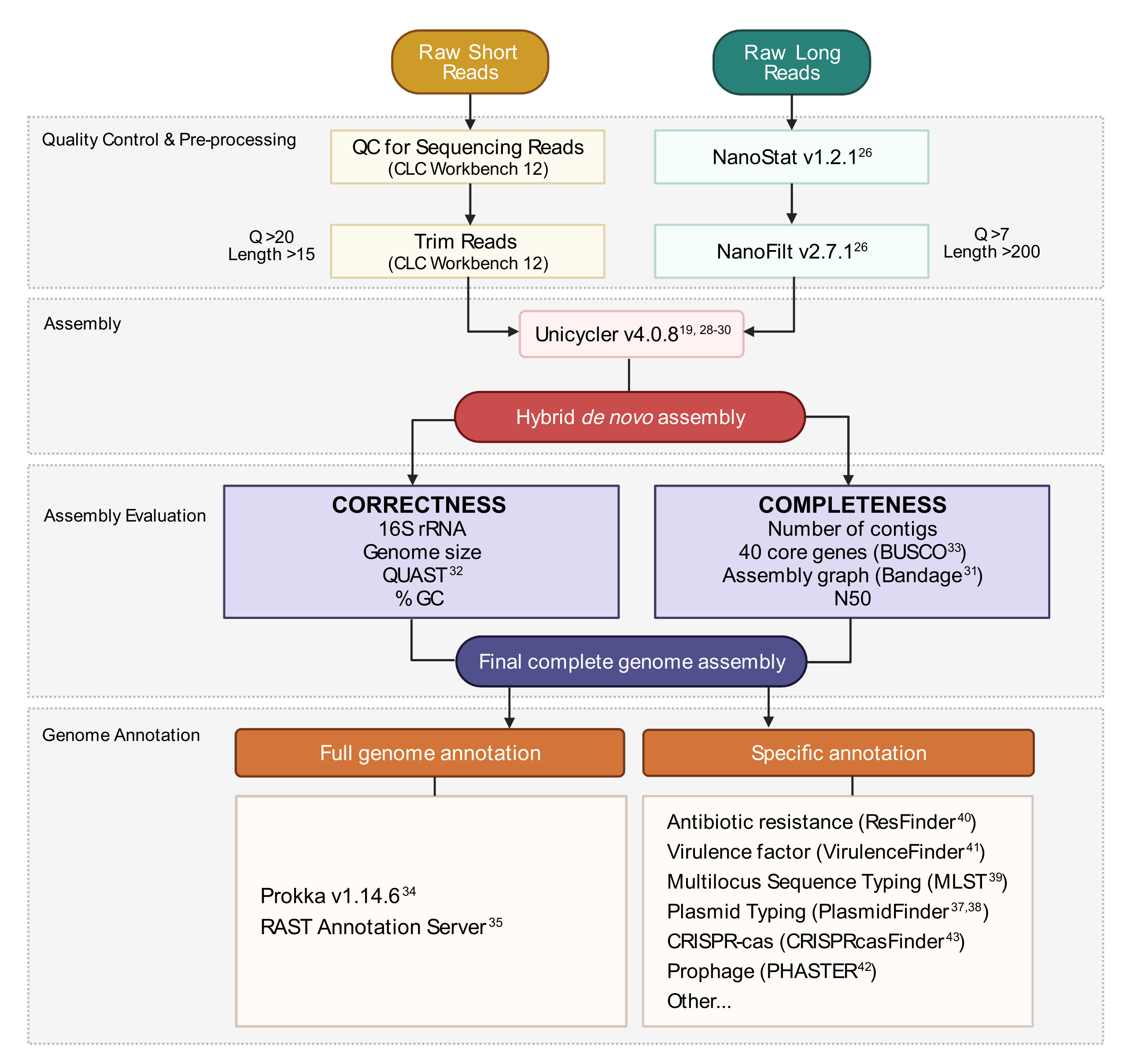{kind=link}

Figura 4:Guía de estructura de directorios de bioinformática. Un esquema de la organización recomendada de directorios y archivos para el procesamiento de lecturas cortas y largas, ensamblaje híbrido y anotación del genoma y control de calidad. Los pasos clave de procesamiento de datos de la línea de comandos se resaltan junto a los archivos y directorios correspondientes. Obtención de comandos y banderas (negrita), archivos de entrada (azul), archivos de salida o directorios (rojo), entrada de usuario como la convención de nomenclatura de archivos (magenta). Haga clic aquí para ver una versión más grande de esta figura.
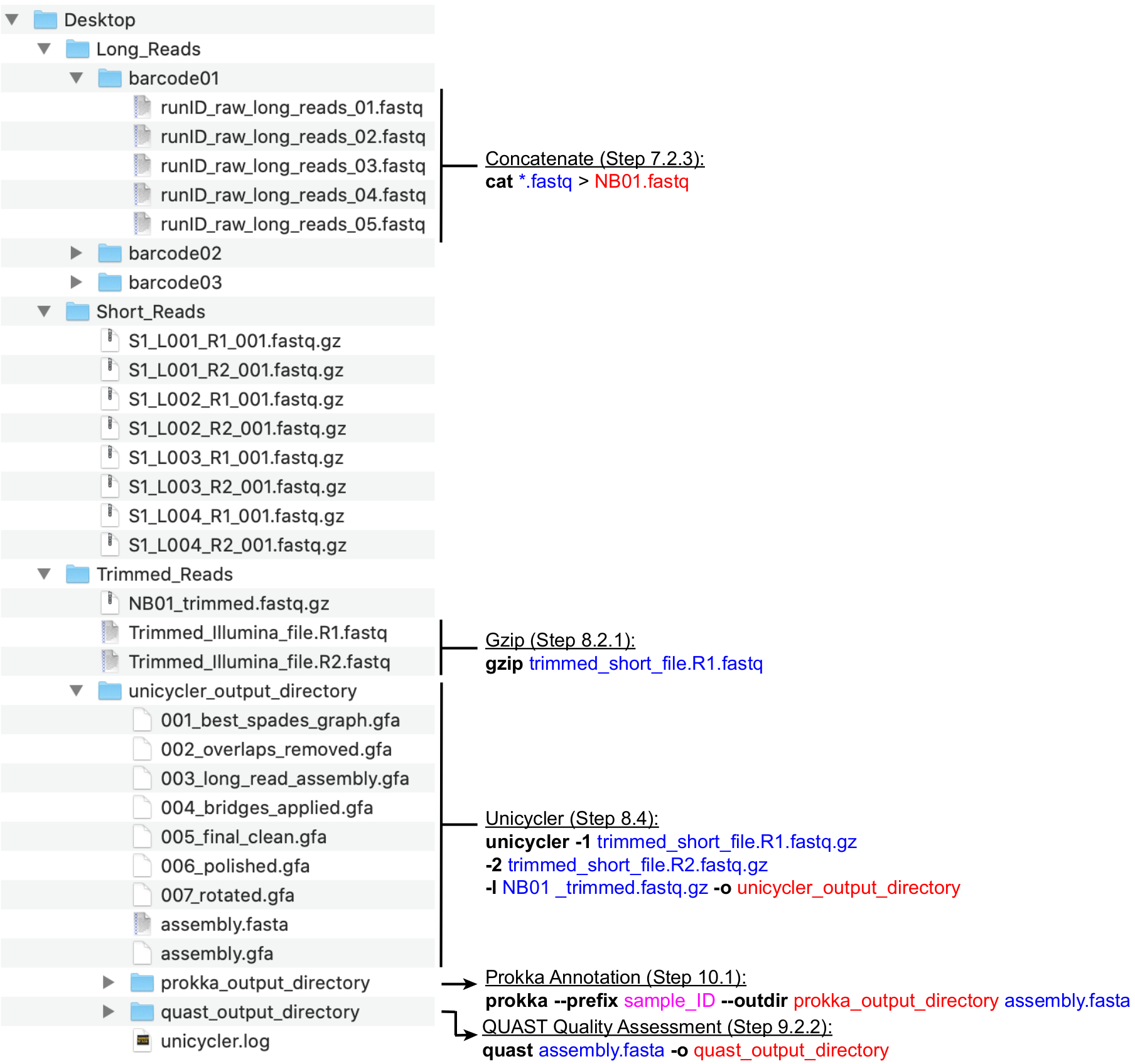{kind=link}

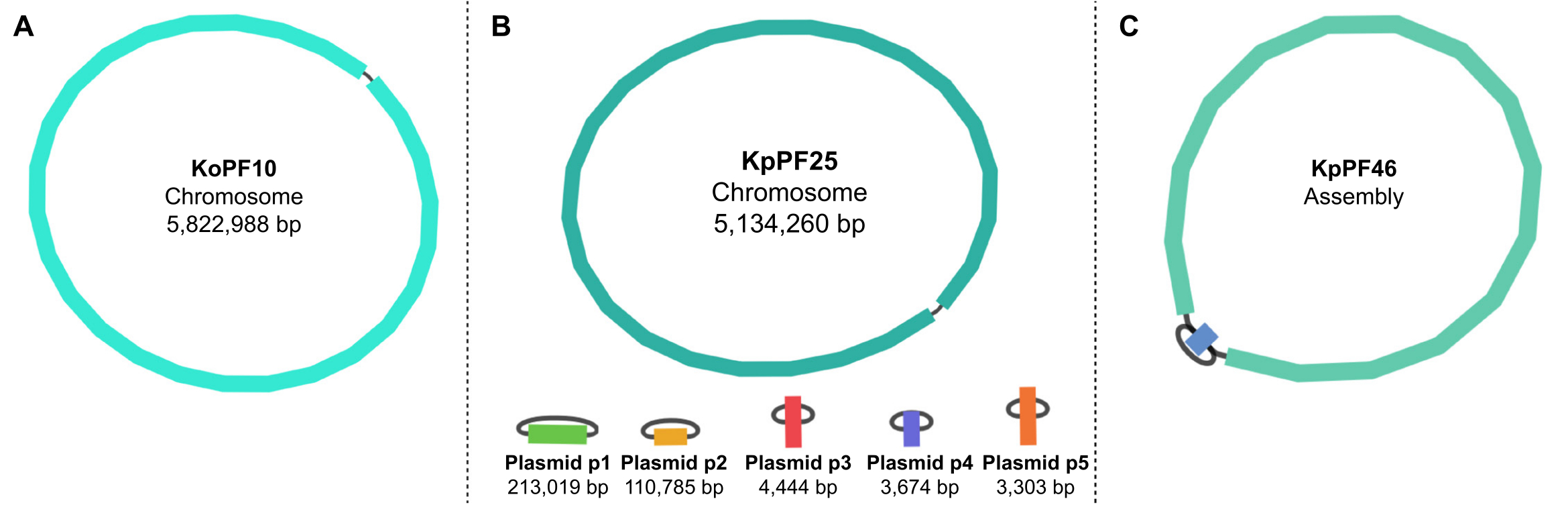
Figura 5: Gráficos de ensamblaje del genoma por vendaje. Gráficos representativos de ensamblaje completo del genoma de (A) Klebsiella oxytoca KoPF10 y (B) Klebsiella pneumoniae KpPF25 y ensamblaje incompleto del genoma de (C) Klebsiella pneumoniae KpPF46. El genoma completo de KoPF10 demuestra un solo cromosoma cerrado y el genoma completo de KpPF25 consiste en un cromosoma cerrado y cinco plásmidos cerrados. El cromosoma incompleto de KpPF46 consiste en dos contigs interconectados. El ensamblaje híbrido de novo de un monociclo genera un gráfico de ensamblaje que es visualizado por Bandage. El gráfico de ensamblaje proporciona un esquema simplista del genoma, indicando cromosomas cerrados o plásmidos por un enlazador que conecta dos extremos de un solo contig. La presencia de más de un contig interconectado indica un ensamblaje incompleto. El tamaño y la profundidad de Contig también se pueden observar en Bandage. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

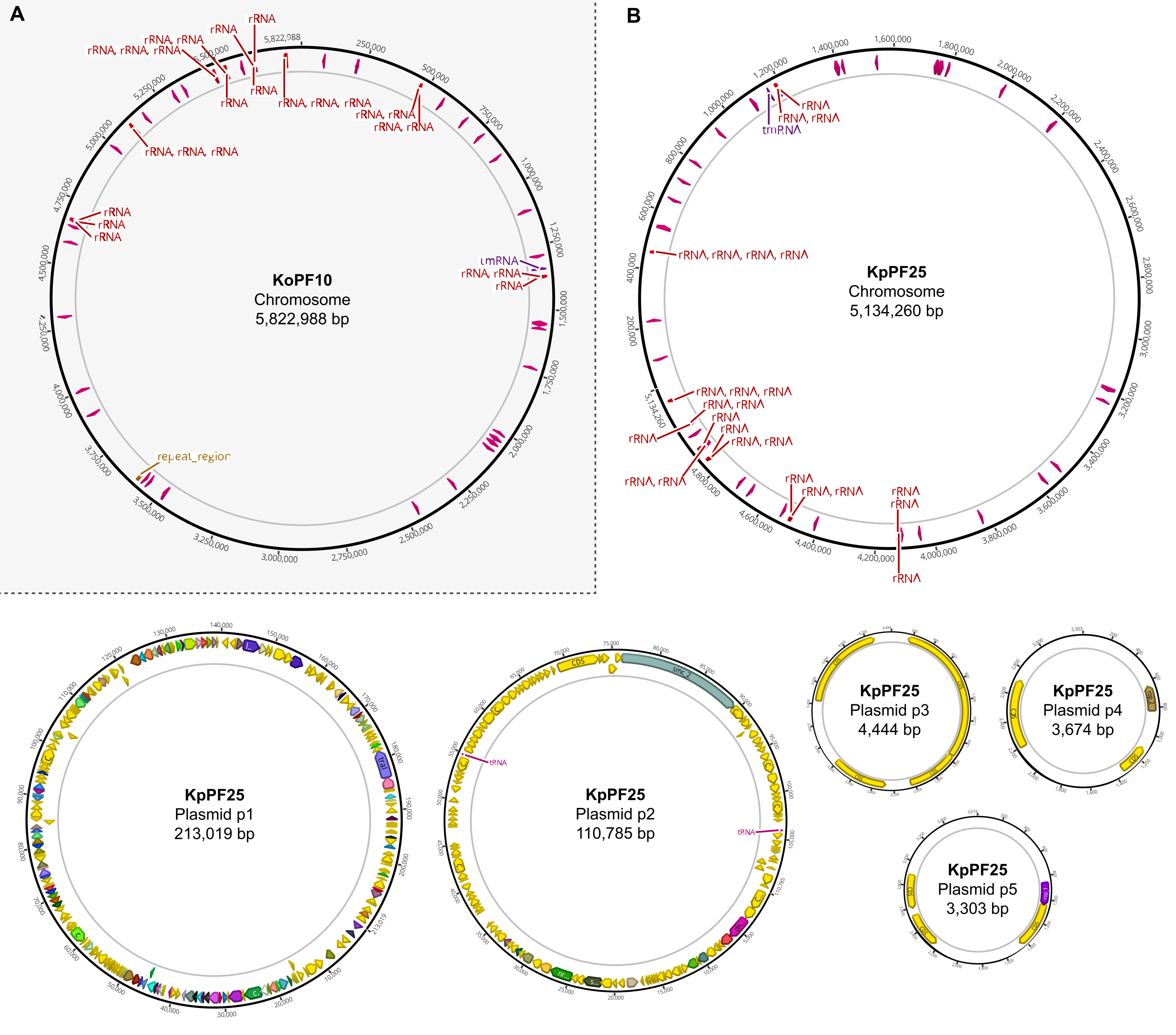
Figura 6: Mapas completos del genoma de conjuntos híbridos anotados. Mapas de ensamblaje generados por Geneious Prime para el genoma completo de (A) K. oxytoca KoPF10 y (B) K. pneumoniae KpPF25 que muestran genes anotados denotados por flechas de colores a lo largo de las columnas vertebrales de los plásmidos. Los cromosomas solo muestran genes de ARNr y ARNt para simplificar. Las anotaciones del genoma se realizaron utilizando Prokka como se indica en la sección 10 de este protocolo. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Tabla 1: Representante Klebsiella spp. características completas del montaje. Parámetros de montaje de la cepa koPF10 de K. oxitoca y de la cepa KpPF25 de K. pneumoniae. Se proporcionan los números de acceso para los datos depositados en el NCBI. Se especifica el número de lecturas antes y después del recorte para ambas tecnologías de secuenciación. N50 se proporciona solo para lecturas largas, ya que las lecturas cortas son de una longitud controlada. Plasmid replicon predijo utilizando la base de datos PlasmidFinder v2.1 Enteroebacteriaceae con parámetros establecidos en 80% de identidad y 60% de longitud. a MLST, tipo de secuencia multilocus. b CDS, Secuencias de codificación. c Plasmid replicon predicho utilizando la base de datos PlasmidFinder v2.1 Enterobacteriaceae con parámetros establecidos en 80% de identidad y 60% de longitud. d Oxford Nanopore Technologies (ONT) depositó datos leídos. e Illumina depositó datos leídos. Haga clic aquí para descargar esta tabla.
Discusión
El protocolo integral de ensamblaje del genoma híbrido descrito aquí ofrece un enfoque simplificado para el cultivo exitoso de diversa microbiota urinaria y uropatógenos, y el ensamblaje completo de sus genomas. El WGS exitoso de genomas bacterianos comienza con el aislamiento de microbios diversos y, a veces, fastidiosos para extraer su ADN genómico. Hasta la fecha, los protocolos de urocultivo existentes carecen de la sensibilidad necesaria para detectar muchas especies urinarias o implican enfoques largos y extensos que requieren tiempo y recursos prolongados11. El enfoque de cultivo de orina mejorado modificado descrito ofrece un protocolo simplificado pero completo para el aislamiento exitoso de bacterias pertenecientes a 17 géneros urinarios comunes, incluidas especies comensales potencialmente patógenas o beneficiosas, y bacterias aeróbicas o anaeróbicas facultativas y obligadas. Esto, a su vez, proporciona el material de partida necesario para la secuenciación y el ensamblaje precisos de los genomas bacterianos y para experimentos fenotípicos críticos, que contribuyen a la comprensión de la salud y la enfermedad urinaria. Además, este enfoque de cultivo modificado proporciona un diagnóstico clínico más definido de los microorganismos viables que se encuentran en las muestras de orina y permite su biobanco para futuros estudios genómicos. Sin embargo, este protocolo no está exento de limitaciones. Puede requerir largos tiempos de incubación dependiendo del organismo, así como el uso de recursos como una cámara de hipoxia o incubadoras controladas que pueden no estar fácilmente disponibles. El uso de GasPaks anaeróbicos ofrece una solución alternativa, pero estos son costosos y no siempre producen un ambiente sostenido y controlado. Finalmente, el sesgo de cultivo, así como la diversidad de muestras, pueden permitir que organismos y uropatógenos particulares superen a las bacterias fastidiosas. A pesar de estas limitaciones, este enfoque hace posible un cultivo de diversas bacterias urinarias.
La secuenciación genómica ha ganado popularidad con el avance de las tecnologías de secuenciación de próxima generación que aumentaron enormemente tanto el rendimiento como la precisión de los datos de secuenciación14,15. Junto con el desarrollo de algoritmos para el procesamiento de datos y el ensamblaje de novo, las secuencias completas del genoma están al alcance de los científicos novatos y expertos por igual15,45. El conocimiento de la organización general del genoma proporcionado por los genomas completos ofrece importantes conocimientos evolutivos y biológicos, incluida la duplicación de genes, la pérdida de genes y la transferencia horizontal degenes 14. Además, los genes importantes para la resistencia a los antimicrobianos y la virulencia a menudo se localizan en elementos móviles, que generalmente no se resuelven en borradores de ensamblajes del genoma15,16.
El protocolo aquí sigue un enfoque híbrido para la combinación de datos de secuenciación de plataformas de lectura corta y larga para generar ensamblajes completos del genoma. Si bien se centra en los genomas bacterianos urinarios, este procedimiento puede adaptarse a diversas bacterias de diversas fuentes de aislamiento. Los pasos críticos en este enfoque incluyen seguir una técnica estéril adecuada y utilizar los medios y las condiciones de cultivo apropiados para el aislamiento de bacterias urinarias puras. Además, la extracción de gDNA intacto y de alto rendimiento es esencial para generar datos de secuenciación libres de lecturas contaminantes que puedan obstaculizar el éxito del ensamblaje. Los protocolos posteriores de preparación de bibliotecas son críticos para la generación de lecturas de calidad de suficiente longitud y profundidad. Por lo tanto, es de suma importancia manejar el gDNA con cuidado durante la preparación de la biblioteca para la secuenciación de lectura larga en particular, ya que el mayor beneficio de esta tecnología es la generación de lecturas largas sin límite teórico de longitud superior. También se describen secciones para el control de calidad (QC) apropiado de las lecturas de secuenciación que elimina los datos ruidosos y mejora el resultado del ensamblaje.
A pesar del aislamiento exitoso del ADN, la preparación de la biblioteca y la secuenciación, la naturaleza de la arquitectura genómica de algunas especies aún puede proporcionar un obstáculo para la generación de un ensamblaje de genoma cerrado45,46. Las secuencias repetitivas a menudo complican el cálculo del ensamblaje y, a pesar de los datos de lectura larga, estas regiones pueden resolverse con poca confianza o no resolverse en absoluto. Por lo tanto, las lecturas largas deben ser en promedio más largas que la región de repetición más grande del genoma o la cobertura debe ser alta (>100x)19. Algunos genomas pueden permanecer incompletos y requieren enfoques manuales para su finalización. Sin embargo, los genomas incompletos ensamblados híbridos suelen estar compuestos de menos contigs que los genomas de borrador de lectura corta. Ajustar los parámetros predeterminados del algoritmo de ensamblaje o seguir cortes más estrictos para leer el control de calidad puede ayudar. Alternativamente, un enfoque sugerido es mapear lecturas largas a las regiones incompletas en busca de evidencia para la ruta de ensamblaje más probable, y luego confirmar la ruta utilizando la secuenciación de PCR y Sanger de la región amplificada. Se sugiere el mapeo de lecturas utilizando Minimap2 y Bandage ofrece una herramienta útil para la visualización de lecturas mapeadas a lo largo de contigs ensamblados que proporcionan evidencia de vinculación contig47.
Un desafío adicional para generar genomas completos radica en la familiaridad y la comodidad con las herramientas de línea de comandos. Muchas herramientas bioinformáticas se desarrollan para ofrecer oportunidades computacionales a cualquier usuario; sin embargo, su utilización se basa en una comprensión de los conceptos básicos de UNIX y la programación. Este protocolo tiene como objetivo proporcionar instrucciones suficientemente detalladas para permitir a las personas sin experiencia previa en la línea de comandos generar ensamblajes de genoma cerrado y anotarlos.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Agradecemos al Dr. Moutusee Jubaida Islam y al Dr. Luke Joyce por sus contribuciones a este protocolo. También nos gustaría reconocer al Centro del Genoma de la Universidad de Texas en Dallas por sus comentarios y apoyo. Este trabajo fue financiado por la Fundación Welch, número de premio AT-2030-20200401 a N.J.D., por los Institutos Nacionales de Salud, número de premio R01AI116610 a K.P., y por la Cátedra Felecia y John Cain en Salud de la Mujer, en poder de P.E.Z.
Materiales
| Name | Company | Catalog Number | Comments |
| Equipment: | |||
| Bioanalyzer 2100 | Agilent | G29398A | Optional but recommended |
| Centrifuge | Eppendorf | -- | Any centrifuge for spinning conicals and microcentrifuge tubes (e.g. Models 5810R/5424R) |
| Electrophoresis | BioRad Laboratories | 1645070 | |
| Gel Imaging System | BioRad Laboratories | ChemiDoc models | |
| Incubator | ThermoFisher Scientific | -- | Any CO2 Incubator (e.g. Thermo Forma model 3110) |
| Magnetic Rack | New England BioLabs | S15095 | 12-tube rack |
| MinION | Oxford Nanopore Technologies | -- | |
| Nanodrop | ThermoFisher Scientific | ND-ONE-W | |
| NextSeq 500 | Illumina | SY-415-1002 | Other Illumina models are acceptable |
| Plate Reader | BioTek | -- | Synergy H1 |
| Qubit fluorometer | ThermoFisher Scientific | Q33238 | |
| Rotator | Benchmark Scientific | H2024 | |
| Thermocycler | ThermoFisher Scientific | -- | Any thermocycler for PCR reactions (e.g. ProFlex PCR system) |
| Materials: | |||
| 10X Phosphate Buffered Saline (PBS) | Fisher Scientific | BP3991 | |
| 10X TBE buffer | -- | -- | 1M Tris,1M Boric Acid,0.2M EDTA (pH 8.0) |
| 1429R primer | Sigma Aldrich (Custom oligos) | -- | GGTTACCTTGTTACGACTT |
| 1kb Ladder | VWR | 101228-494 | |
| 1M Tris-Cl (pH 7.5) | ThermoFisher Scientific | 15567027 | |
| 6x Loading dye | Fisher Scientific | NC0783588 | |
| 8F primer | Sigma Aldrich (Custom oligos) | -- | AGAGTTTGATCCTGGCTCAG |
| Agar | Fisher Scientific | BP1423-2 | |
| Agarose | BioRad Laboratories | 63001 | |
| AMPure XP Beads | Beckman Coulter | A63880 | |
| Anaerobe Pouch System - GasPak EZ | BD Diagnostic Systems | B260683 | |
| Boric Acid | Fisher Scientific | A73-500 | |
| Brain Heart Infusion Broth | BD Diagnostic Systems | 212304 | |
| CDC Anaerobe 5% Sheep Blood Agar | BD Diagnostic Systems | L007357 | |
| CHROMagar Orientation | BD Diagnostic Systems | PA-257481.04 | |
| DNeasy Blood & Tissue | QIAGEN | 69504 | |
| DreamTaq Master Mix | ThermoFisher Scientific | K1081 | |
| Dry Anaerobic Indicator Strips | BD Diagnostic Systems | 271051 | |
| EDTA | Fisher Scientific | S311-500 | |
| Ethanol 200 Proof | Sigma Aldrich | E7023 | For molecular biology |
| Ethidium Bromide | ThermoFisher Scientific | BP130210 | |
| Flow cell priming kit | Oxford Nanopore Technologies | EXP-FLP002 | |
| Flow cell wash kit | Oxford Nanopore Technologies | EXP-WSH003 | |
| Gel Extraction Miniprep Kit | BioBasic | BS654 | |
| Ligation sequencing kit | Oxford Nanopore Technologies | SQK-LSK109 | |
| Lysozyme | Research Products International Corp | L381005.05 | |
| Mutanolysin | Sigma Aldrich | M9901-5KU | |
| Native barcoding expansion 1-12 | Oxford Nanopore Technologies | EXP-NBD104 | |
| NEB Blunt/TA Ligase Master Mix | New England BioLabs | M0367L | |
| NEBNext FFPE DNA Repair Mix | New England BioLabs | M6630L | |
| NEBNext quick ligation buffer | New England BioLabs | B6058S | |
| NEBNext Ultra II End repair / dA-tailing module | New England BioLabs | E7546L | |
| Nextera DNA CD Indexes | Illumina | 20018708 | |
| Nextera DNA Flex Library Prep - (M) Tagmentation | Illumina | 20018705 | |
| Nuclease-free water | Sigma Aldrich | W4502 | |
| Qubit 1X dsDNA HS Assay Kit | ThermoFisher Scientific | Q33230 | |
| Qubit Assay Tubes | ThermoFisher Scientific | Q32856 | |
| Quick T4 DNA Ligase | New England BioLabs | E6056L | |
| R9 Flow cell | Oxford Nanopore Technologies | FLO-MIN106D | |
| RNase A | ThermoFisher Scientific | EN0531 | |
| Sheep Blood | Hemostat Laboratories | DS13250 | |
| TE buffer | -- | -- | 10mM Tris, 1mM EDTA (pH 8.0) |
| Triton X-100 | Sigma Aldrich | T8787 | |
| Tryptic Soy Broth | BD Diagnostic Systems | 211825 | |
| Software & Bioinformatic Tools: | |||
| Bandage | -- | -- | https://rrwick.github.io/Bandage/ |
| Center for Genomic Epidemiology | -- | -- | http://www.genomicepidemiology.org/ |
| CLC Genomics Workbench 12 | QIAGEN | -- | |
| CRISPRcasFinder | -- | -- | https://crisprcas.i2bc.paris-saclay.fr/ |
| FastQC | -- | -- | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Geneious Prime | Geneious | -- | |
| gVolante (BUSCO) | -- | -- | https://gvolante.riken.jp/ |
| Kbase Prokka Wrapper | -- | -- | https://kbase.us/applist/apps/ProkkaAnnotation/annotate_contigs/release |
| Minimap2 | -- | -- | https://github.com/lh3/minimap2 |
| MinKNOW | Oxford Nanopore Technologies | -- | |
| NanoFilt | -- | -- | https://github.com/wdecoster/nanofilt |
| NanoStat | -- | -- | https://github.com/wdecoster/nanostat |
| PHASTER | -- | -- | https://phaster.ca/ |
| Prokka | -- | -- | https://github.com/tseemann/prokka |
| QUAST | -- | -- | http://quast.sourceforge.net/quast |
| Trim Galore | -- | -- | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| Trimmomatic | -- | -- | http://www.usadellab.org/cms/?page=trimmomatic |
| Unicycler | -- | -- | https://github.com/rrwick/Unicycler#necessary-read-length |
Referencias
- Brubaker, L., Wolfe, A. The urinary microbiota: a paradigm shift for bladder disorders. Current Opinion in Obstetrics & Gynecology. 28 (5), 407-412 (2016).
- Neugent, M. L., Hulyalkar, N. V., Nguyen, V. H., Zimmern, P. E., De Nisco, N. J. Advances in understanding the human urinary microbiome and its potential role in urinary tract infection. mBio. 11 (2), (2020).
- Klein, R. D., Hultgren, S. J. Urinary tract infections: microbial pathogenesis, host-pathogen interactions and new treatment strategies. Nature Reviews. Microbiology. 18 (4), 211-226 (2020).
- Horsley, H., et al. Enterococcus faecalis subverts and invades the host urothelium in patients with chronic urinary tract infection. PLoS One. 8 (12), 83637 (2013).
- Reitzer, L., Zimmern, P. Rapid growth and metabolism of uropathogenic Escherichia coli in relation to urine composition. Clinical Microbiology Reviews. 33 (1), 00101-00119 (2019).
- Snyder, J. A., et al. Transcriptome of uropathogenic Escherichia coli during urinary tract infection. Infection and Immunity. 72 (11), 6373-6381 (2004).
- Ipe, D. S., Horton, E., Ulett, G. C. The basics of bacteriuria: Strategies of microbes for persistence in urine. Frontiers in Cellular and Infection Microbiology. 6, 14 (2016).
- Babikir, I. H., et al. The impact of cathelicidin, the human antimicrobial peptide LL-37 in urinary tract infections. BMC Infectious Diseases. 18 (1), 17 (2018).
- Jancel, T., Dudas, V. Management of uncomplicated urinary tract infections. The Western Journal of Medicine. 176 (1), 51-55 (2002).
- Ventola, C. L. The antibiotic resistance crisis: part 1: causes and threats. P & T. 40 (4), 277-283 (2015).
- Price, T. K., et al. The clinical urine culture: Enhanced techniques improve detection of clinically relevant microorganisms. Journal of Clinical Microbiology. 54 (5), 1216-1222 (2016).
- Kass, E. H. Asymptomatic infections of the urinary tract. Transactions of the Association of American Physicians. 69, 56-64 (1956).
- Garcia, L. S. . Clinical microbiology procedures handbook. 3rd edn. , (2010).
- Fraser, C. M., Eisen, J. A., Nelson, K. E., Paulsen, I. T., Salzberg, S. L. The value of complete microbial genome sequencing (you get what you pay for). Journal of Bacteriology. 184 (23), 6403-6405 (2002).
- Chen, Z., Erickson, D. L., Meng, J. Benchmarking hybrid assembly approaches for genomic analyses of bacterial pathogens using Illumina and Oxford Nanopore sequencing. BMC Genomics. 21 (1), 631 (2020).
- Greig, D. R., Dallman, T. J., Hopkins, K. L., Jenkins, C. MinION nanopore sequencing identifies the position and structure of bacterial antibiotic resistance determinants in a multidrug-resistant strain of enteroaggregative Escherichia coli. Microbial Genomics. 4 (10), 000213 (2018).
- Carraro, D. M., et al. PCR-assisted contig extension: stepwise strategy for bacterial genome closure. Biotechniques. 34 (3), 626-628 (2003).
- Tettelin, H., Radune, D., Kasif, S., Khouri, H., Salzberg, S. L. Optimized multiplex PCR: efficiently closing a whole-genome shotgun sequencing project. Genomics. 62 (3), 500-507 (1999).
- Wick, R. R., Judd, L. M., Gorrie, C. L., Holt, K. E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Computational Biology. 13 (6), 1005595 (2017).
- Singhal, N., Kumar, M., Kanaujia, P. K., Virdi, J. S. MALDI-TOF mass spectrometry: an emerging technology for microbial identification and diagnosis. Frontiers in Microbiology. 6, 791 (2015).
- Turner, S., Pryer, K. M., Miao, V. P., Palmer, J. D. Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. The Journal of Eukaryotic Microbiology. 46 (4), 327-338 (1999).
- Weisburg, W. G., Barns, S. M., Pelletier, D. A., Lane, D. J. 16S ribosomal DNA amplification for phylogenetic study. Journal of Bacteriology. 173 (2), 697-703 (1991).
- Janda, J. M., Abbott, S. L. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. Journal of Clinical Microbiology. 45 (9), 2761-2764 (2007).
- Stevenson, K., McVey, A. F., Clark, I. B. N., Swain, P. S., Pilizota, T. General calibration of microbial growth in microplate readers. Science Reports. 6, 38828 (2016).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (15), 2114-2120 (2014).
- De Coster, W., D'Hert, S., Schultz, D. T., Cruts, M., Van Broeckhoven, C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 34 (15), 2666-2669 (2018).
- Wilson, G., et al. The UNIX Shell. Zenodo. , (2019).
- Bankevich, A., et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology. 19 (5), 455-477 (2012).
- Vaser, R., Sovic, I., Nagarajan, N., Sikic, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Research. 27 (5), 737-746 (2017).
- Walker, B. J., et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 9 (11), 112963 (2014).
- Wick, R. R., Schultz, M. B., Zobel, J., Holt, K. E. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics. 31 (20), 3350-3352 (2015).
- Gurevich, A., Saveliev, V., Vyahhi, N., Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 29 (8), 1072-1075 (2013).
- Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31 (19), 3210-3212 (2015).
- Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 30 (14), 2068-2069 (2014).
- Aziz, R. K., et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 9, 75 (2008).
- Tatusova, T., et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Research. 44 (14), 6614-6624 (2016).
- Carattoli, A., Hasman, H. PlasmidFinder and In Silico pMLST: Identification and Typing of Plasmid Replicons in Whole-Genome Sequencing (WGS). Methods in Molecular Biology. 2075, 285-294 (2020).
- Carattoli, A., et al. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrobial Agents and Chemotherapy. 58 (7), 3895-3903 (2014).
- Larsen, M. V., et al. Multilocus sequence typing of total-genome-sequenced bacteria. Journal of Clinical Microbiology. 50 (4), 1355-1361 (2012).
- Bortolaia, V., et al. ResFinder 4.0 for predictions of phenotypes from genotypes. The Journal of Antimicrobial Chemotherapy. 75 (12), 3491-3500 (2020).
- Joensen, K. G., et al. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. Journal of Clinical Microbiology. 52 (5), 1501-1510 (2014).
- Arndt, D., et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Research. 44 (1), 16-21 (2016).
- Couvin, D., et al. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Research. 46 (1), 246-251 (2018).
- Totten, P. A., Amsel, R., Hale, J., Piot, P., Holmes, K. K. Selective differential human blood bilayer media for isolation of Gardnerella (Haemophilus) vaginalis. Journal of Clinical Microbiology. 15 (1), 141-147 (1982).
- Nagarajan, N., Pop, M. Sequence assembly demystified. Nat Reviews. Genetics. 14 (3), 157-167 (2013).
- Phillippy, A. M., Schatz, M. C., Pop, M. Genome assembly forensics: finding the elusive mis-assembly. Genome Biology. 9 (3), 55 (2008).
- . Unicycler Wiki Available from: https://github.com/rrwick/Unicycler/wiki (2017)
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados