
Method Article
Assemblage hybride du génome de novo pour la génération de génomes complets de bactéries urinaires à l’aide de technologies de séquençage à lecture courte et longue
Dans cet article
Résumé
Ce protocole détaille une approche globale pour la culture, le séquençage et l’assemblage hybride de novo du génome des bactéries urinaires. Il fournit une procédure reproductible pour la génération de séquences génomiques complètes et circulaires utiles pour étudier les éléments génétiques chromosomiques et extrachromosomiques contribuant à la colonisation urinaire, à la pathogenèse et à la dissémination de la résistance aux antimicrobiens.
Résumé
Les séquences complètes du génome fournissent des données précieuses pour la compréhension de la diversité génétique et des facteurs de colonisation uniques des microbes urinaires. Ces données peuvent inclure des éléments génétiques mobiles, tels que les plasmides et les phages extrachromosomiques, qui contribuent à la dissémination de la résistance aux antimicrobiens et compliquent davantage le traitement des infections des voies urinaires (IVU). En plus de fournir une résolution fine de la structure du génome, les génomes complets et fermés permettent des analyses comparatives et évolutives détaillées. La génération de génomes complets de novo a longtemps été une tâche difficile en raison des limites de la technologie de séquençage disponible. Le séquençage de nouvelle génération (NGS) à extrémité appariée produit des lectures courtes de haute qualité, ce qui donne souvent des assemblages de génome précis mais fragmentés. Au contraire, le séquençage des nanopores fournit de longues lectures de qualité inférieure conduisant normalement à des assemblages complets sujets aux erreurs. De telles erreurs peuvent entraver les études d’association à l’échelle du génome ou fournir des résultats d’analyse de variantes trompeurs. Par conséquent, les approches hybrides combinant des lectures courtes et longues sont apparues comme des méthodes fiables pour obtenir des génomes bactériens fermés très précis. Il est rapporté ici une méthode complète pour la culture de diverses bactéries urinaires, l’identification des espèces par séquençage du gène de l’ARNr 16S, l’extraction de l’ADN génomique (ADNg) et la génération de lectures courtes et longues par les plateformes NGS et Nanopore, respectivement. En outre, cette méthode décrit un pipeline bioinformatique d’algorithmes de contrôle de la qualité, d’assemblage et de prédiction de gènes pour la génération de séquences complètes annotées du génome. La combinaison d’outils bioinformatiques permet de sélectionner des données de lecture de haute qualité pour l’assemblage du génome hybride et l’analyse en aval. L’approche simplifiée pour l’assemblage hybride du génome de novo décrite dans ce protocole peut être adaptée à l’utilisation dans toute bactérie cultivable.
Introduction
Le microbiome urinaire est un domaine de recherche émergent qui a brisé une idée fausse de plusieurs décennies selon laquelle les voies urinaires sont stériles chez les personnes en bonne santé. Les membres du microbiote urinaire peuvent servir à équilibrer l’environnement urinaire et à prévenir les infections des voies urinaires (IVU)1,2. Les bactéries uropathogènes envahissent les voies urinaires et emploient divers mécanismes de virulence pour déplacer le microbiote résident, coloniser l’urothélium, échapper aux réponses immunitaires et contrer les pressionsenvironnementales 3,4. L’urine est un milieu relativement limité en nutriments caractérisé par une osmolarité élevée, une disponibilité limitée en azote et en glucides, une faible oxygénation et un faible pH5,6,7. L’urine est également considérée comme antimicrobienne, composée de fortes concentrations d’urée inhibitrice et de peptides antimicrobiens tels que la cathélicidine humaine LL-378. L’étude des mécanismes utilisés par les bactéries résidentes et les uropathogènes pour coloniser les voies urinaires est essentielle pour mieux comprendre la santé des voies urinaires et développer de nouvelles stratégies de traitement des infections urinaires. De plus, à mesure que l’échec des thérapies antimicrobiennes de première ligne devient de plus en plus fréquent, il est de plus en plus important de surveiller la dissémination d’éléments génétiques mobiles porteurs de déterminants de la résistance aux antimicrobiens au sein des populations de bactéries urinaires9,10.
Pour étudier les génotypes et les phénotypes des bactéries urinaires, leur culture réussie et le séquençage ultérieur du génome entier (WGS) sont impératifs. Des méthodes dépendantes de la culture sont nécessaires pour détecter et identifier les microbes viables dans les échantillons d’urine11. La culture d’urine clinique standard consiste à plaquer l’urine sur une gélose à 5 % de sang de mouton (BAP) et une gélose MacConkey et à incuber aérobiement à 35 °C pendant 24 h12. Cependant, avec un seuil de détection de ≥105 UFC/mL13, de nombreux membres du microbiote urinaire ne sont pas rapportés par cette méthode. Les techniques de culture améliorées telles que la culture quantitative améliorée de l’urine (EQUC)11 utilisent diverses combinaisons de différents volumes d’urine, temps d’incubation, milieux de culture et conditions atmosphériques pour identifier les microbes couramment omis par la culture d’urine standard. Décrit dans ce protocole est une version modifiée de la CQE, appelée ici protocole de culture d’urine améliorée modifiée, qui permet la culture de diverses bactéries urinaires et uropathogènes en utilisant des milieux sélectifs et des conditions atmosphériques optimales, mais n’est pas intrinsèquement quantitative. L’isolement réussi des bactéries urinaires permet l’extraction de l’ADN génomique (ADNg) pour le WGS en aval et l’assemblage du génome.
Les assemblages de génomes, en particulier les assemblages complets, permettent de découvrir des facteurs génétiques susceptibles de contribuer à la colonisation, au maintien de niches et à la virulence du microbiote résident et des bactéries uropathogènes. Les ébauches d’assemblages de génomes contiennent un nombre diversifié de séquences contiguës (contigs) qui peuvent contenir des erreurs de séquençage et manquer d’informations d’orientation. Dans un assemblage complet du génome, l’orientation et la précision de chaque paire de bases ont été vérifiées14. En outre, l’obtention de séquences complètes du génome permet de mieux comprendre la structure du génome, la diversité génétique et les éléments génétiques mobiles15. Des lectures courtes à elles seules peuvent identifier la présence ou l’absence de gènes importants, mais peuvent ne pas identifier leur contextegénomique 16. Grâce aux technologies de séquençage à lecture longue telles qu’Oxford Nanopore et PacBio, la génération d’assemblages fermés de novo de génomes bactériens ne nécessite plus de méthodes fastidieuses telles que la fermeture manuelle d’assemblages de novo par PCRmultiplex 17,18. La combinaison du séquençage à lecture courte de nouvelle génération et des technologies de séquençage à lecture longue Nanopore permet la génération facile d’assemblages de génomes bactériens précis, complets et fermés à des coûts relativement faibles19. Le séquençage à lecture courte produit des assemblages de génome précis mais fragmentés composés généralement d’une moyenne de 40 à 100 contigs, tandis que le séquençage nanopore génère de longues lectures d’environ 5 à 100 kb de longueur qui sont moins précises mais peuvent servir d’échafaudages pour joindre les contigs et résoudre la synténie génomique. Les approches hybrides utilisant à la fois des technologies à lecture courte et à lecture longue peuvent produire des génomes bactériens précis et complets19.
Décrit ici est un protocole complet pour l’isolement et l’identification des bactéries de l’urine humaine, l’extraction de l’ADN génomique, le séquençage et l’assemblage complet du génome à l’aide d’une approche d’assemblage hybride. Ce protocole met particulièrement l’accent sur les étapes nécessaires pour modifier correctement les lectures générées par le séquençage à lecture courte et longue pour l’assemblage précis d’un chromosome bactérien fermé et d’éléments extrachromosomiques tels que les plasmides.
Protocole
Les bactéries ont été cultivées à partir d’urine prélevée sur des femmes consentantes dans le cadre des études 19MR0011 (UTD) et STU 032016-006 (UTSW) approuvées par le comité d’examen institutionnel.
1. Culture d’urine améliorée modifiée
REMARQUE: Toutes les étapes de culture doivent être effectuées dans des conditions stériles. Stérilisez tous les instruments, solutions et supports. Nettoyez la zone de travail avec de l’éthanol à 70%, puis installez un brûleur Bunsen et travaillez soigneusement près de la flamme pour réduire les risques de contamination. Alternativement, une armoire de biosécurité de classe II peut être utilisée pour maintenir un environnement stérile. Portez un équipement de protection individuelle (EPI) approprié pour éviter l’exposition à des microbes potentiellement pathogènes.
- Placage de l’urine stockée dans le glycérol et isolement des colonies
- Décongeler l’urine stockée de glycérol à température ambiante (RT). Une fois décongelé, vortex l’échantillon pendant 5 s pour mélanger. Dans des tubes stériles de microcentrifugation, préparer des dilutions 1:3 et 1:30 de l’urine dans une solution saline stérile 1x tamponnée au phosphate (PBS) jusqu’à un volume final de 100 μL.
REMARQUE: L’urine stockée dans le glycérol est préparée en mélangeant 500 μL d’urine non diluée et 500 μL de glycérol stérile à 50% dans des cryoviales et en stockant à -80 ° C. - Préchauffer les plaques de gélose à 37 °C pendant 15 min avant utilisation. Veuillez consulter la figure 1 pour connaître les types de milieux et les conditions de culture adaptés aux genres bactériens urinaires courants. Bien mélanger l’urine diluée par pipetage avant le placage, plaquer 100 μL de l’urine diluée sur la plaque de gélose souhaitée et étaler l’échantillon à l’aide de billes de verre stériles. Plaque 100 μL du diluant PBS 1x sur une plaque séparée comme contrôle de croissance.
REMARQUE : Si vous tentez de cultiver des espèces uropathogènes communes (p. ex., Escherichia coli, Klebsiella spp., Enterococcus faecalis, etc.), il est recommandé d’utiliser une gélose chromogène (Tableau des matériaux) car elle permet d’identifier facilement les espèces bactériennes uropathogènes (Figure 1). L’acide nalidixique à la colistine (AIIC) ou la gélose MRS sont utiles pour isoler les espèces fastidieuses à Gram positif (p. ex., Lactobacillus spp.)de l’urine connue pour contenir des uropathogènes à Gram négatif, qui peuvent surpasser les espèces fastidieuses dans les géloses non sélectives. - Incuber la plaque inversée dans les conditions atmosphériques souhaitées à 35 °C pendant une période de 24 h pour les uropathogènes et de 3 à 5 jours pour les bactéries fastidieuses (Figure 1).
- Après la période d’incubation, retirez les plaques de l’incubateur. Dans chaque plaque, choisissez les colonies qui présentent une couleur, une morphologie ou des motifs hémolytiques uniques.
- Re-streak la colonie bactérienne à l’aide d’une boucle stérile sur la gélose correspondante et incuber la plaque inversée pendant 2-5 jours dans l’atmosphère désirée pour obtenir des colonies bien isolées.
REMARQUE: Si vous utilisez le BAP pour la culture primaire, le patching des colonies sur une gélose chromogène peut fournir des informations utiles sur l’hétérogénéité de la population bactérienne dans l’échantillon.
- Décongeler l’urine stockée de glycérol à température ambiante (RT). Une fois décongelé, vortex l’échantillon pendant 5 s pour mélanger. Dans des tubes stériles de microcentrifugation, préparer des dilutions 1:3 et 1:30 de l’urine dans une solution saline stérile 1x tamponnée au phosphate (PBS) jusqu’à un volume final de 100 μL.
- Culture dans un bouillon liquide et des isolats bactériens de glycérol
- Une fois que les colonies isolées qui correspondent à la morphologie de la colonie parente sont obtenues, choisissez une seule colonie et inoculez dans 3 mL de bouillon liquide à l’aide d’une boucle d’inoculation stérile. Reportez-vous à la figure 1 pour le bouillon capable de soutenir la croissance des genres communs de microbiote urinaire. Scellez les plaques d’agar avec un parafilm et conservez-les à 4 °C pendant 2 à 4 jours. Incuber des cultures liquides dans les conditions atmosphériques souhaitées pendant 1 à 5 jours jusqu’à ce que la culture soit visiblement trouble.
- Une fois la croissance observée, vortex la culture, puis ajouter 1 mL de la culture pendant la nuit à 500 μL de glycérol stérile à 50% dans un cryovial de 2 mL; sceller et mélanger doucement par inversion. Préparez deux stocks de glycérol pour chaque colonie (l’un sert de sauvegarde) et conservez-les à -80 °C.
2. Identification des espèces bactériennes par séquençage du gène Sanger de l’ARNr 16S
REMARQUE: L’identité microbienne peut également être confirmée à l’aide de la spectrométrie de masse à temps de vol par désorption laser assistée par matrice (MALDI-TOF)20.
- Réaction en chaîne colonie-polymérase (PCR)
- Préparer 25 μL de la réaction pcR dans des tubes PCR en ajoutant 12,5 μL de 2x Taq Polymerase Master Mix, 0,5 μL de 10 μM 8F primer, 0,5 μL de 10 μM 1492R primer(Table des matériaux),et 11,5 μL d’eau sans nucléase21.
REMARQUE: Si vous effectuez une PCR pour plusieurs échantillons, faites un mélange maître de réaction de mélange de Taq Polymerase, d’amorces et d’eau stérile sans nucléase. Ensuite, aliquote 25 μL dans chaque tube PCR. - Pour effectuer la PCR de la colonie, faites glisser une colonie bien isolée de la nouvelle stries à l’aide d’un cure-dent stérile ou d’une pointe de pipette. Remettre la colonie en suspension dans le mélange réactionnel PCR préparé à l’étape 2.1.1. Mélanger doucement. Recueillir le liquide au fond du tube par un spin rapide à 2000 x g.
REMARQUE: Assurez-vous que l’échantillon est exempt de bulles d’air. Inclure un échantillon de contrôle sans gabarit (NTC) contenant uniquement le mélange de réactions PCR. - Placez les tubes d’échantillonnage dans le thermocycleur et exécutez le programme suivant: 95 °C pendant 3 min; 40 cycles de: 95 °C pendant 30 s, 51 °C pendant 30 s et 72 °C pendant 1 min 30 s; 72 °C pendant 10 min; maintenir à 10 °C.
- Préparer 25 μL de la réaction pcR dans des tubes PCR en ajoutant 12,5 μL de 2x Taq Polymerase Master Mix, 0,5 μL de 10 μM 8F primer, 0,5 μL de 10 μM 1492R primer(Table des matériaux),et 11,5 μL d’eau sans nucléase21.
- Extraction de gel et identification des espèces
- À la fin de l’exécution de la PCR, vérifiez le produit PCR sur un gel d’agarose à 1% préparé dans un tampon 0,5x Tris-Borate-EDTA (TBE). Avant de couler le gel, ajouter le bromure d’éthidium (EtBr). Ensuite, coulez le gel à l’aide de peignes pour les puits qui contiennent au moins 20 μL de volume d’échantillon.
ATTENTION : L’EtBr est un agent intercalant suspecté d’être cancérigène. Portez toujours des gants et de l’EPI lorsque vous les manipulez et éliminez les matériaux contenant de l’EtBr conformément aux directives de l’établissement. - Lorsque le gel est réglé, placez-le dans le réservoir d’électrophorèse rempli de tampon TBE 0,5x et retirez le peigne. Chargez l’échelle de 1 kb dans le premier puits et 10-20 μL de la réaction PCR dans les puits suivants. Exécuter à 100-140 V jusqu’à ce qu’il soit résolu. Visualisez le gel sous la lumière UV et confirmez la présence d’une bande clairement définie à ~ 1,5 kb qui est absente dans le puits NTC.
ATTENTION: Les rayons UV sont nocifs pour la peau et les yeux, utilisez un protecteur approprié lorsque vous visualisez le gel et portez un EPI approprié.
REMARQUE: La PCR des colonies peut échouer pour certaines bactéries; procéder à la PCR à partir d’ADNg isolé est une autre option22. - Retirez les bandes d’environ 1,5 kb à l’aide d’un rasoir et transférez les boutures de gel dans des tubes de microcentrifugation propres. Procéder au protocole d’extraction du gel conformément aux instructions du fabricant (Table des matériaux). Mesurer la concentration de l’ADN purifié par spectrophotomètre à microvolume.
REMARQUE: Une concentration >10 ng / μL est souhaitable et A260/280 entre 1,7 et 2,0 est acceptable. - Préparez deux réactions de séquençage de Sanger pour chaque échantillon, l’une en utilisant le 8F et l’autre en utilisant l’amorce 1492R dans de l’eau sans nucléase selon les directives de tout service de séquençage Sanger choisi.
- Une fois les données de séquençage reçues, téléchargez les séquences d’ADN sur le site Web BLAST (Basic Local Alignment Search Tool) du NCBI (blast.ncbi.nlm.nih.gov/Blast.cgi), choisissez Nucleotide BLAST (blastn), sélectionnez la base de données d’ARNr/ ITS 16S séquences d’ARN ribosomique (bactéries et archées) et exécutez le programme Megablast. L’isolat peut être identifié par la réponse de la plus haute qualité à une référence de la base de données.
REMARQUE: Certaines espèces bactériennes présentent une identité élevée dans leurs séquences d’ARNr 16S et peuvent être impossibles à distinguer par cette seule méthode. La spéciation nécessitera une homologie de l’ADN et des analyses biochimiques pour distinguer en toute confiance les membres du même genre23.
- À la fin de l’exécution de la PCR, vérifiez le produit PCR sur un gel d’agarose à 1% préparé dans un tampon 0,5x Tris-Borate-EDTA (TBE). Avant de couler le gel, ajouter le bromure d’éthidium (EtBr). Ensuite, coulez le gel à l’aide de peignes pour les puits qui contiennent au moins 20 μL de volume d’échantillon.
3. Extraction de l’ADN génomique (ADNg)
REMARQUE: Cette section utilise des réactifs et des colonnes de spin fournis dans le kit d’extraction d’ADNg référencé dans la Table des matériaux pour l’extraction à haut rendement d’ADN génomique de qualité de diverses espèces bactériennes. Vous trouverez ci-dessous les modifications et instructions recommandées.
- Préparez les réactifs en kit selon les instructions du fabricant.
- Préparer des cultures de 3 à 10 mL dans un bouillon stérile approprié(figure 1)en inoculant des bactéries provenant de colonies bien isolées dans le milieu et en incubant à la température et à la pression atmosphérique indiquées à la figure 1 jusqu’à ce qu’une croissance suffisante soit observée.
- Après incubation, mesurer la densité optique à 600 nm (OD600)de la culture à l’aide d’un spectrophotomètre24.
- Préparer l’échantillon pour la quantification en diluant les cultures pendant la nuit dans un rapport de 1:10. Incluez également un blanc du milieu de culture stérile pour la mesure. Calculer la densité optique en soustrayant la lecture à blanc de la lecture de l’échantillon et en multipliant par le facteur de dilution de dix.
- À l’aide de la mesure de l’OD600 et d’un rapport OD600 /ML préétabli pour l’espèce, calculez combien de millilitres de culture sont nécessaires pour obtenir 2 x 109 cellules.
- Centrifugez le volume de culture requis pendant 5 min à 5000 x g pour granuler. Aspirer le surnageant et remettre la pastille en suspension dans un tampon TE froid de 200 μL (pré-refroidir sur la glace au début de la procédure).
- Centrifuger l’échantillon pendant 2 min à 5000 x g. Retirez le surnageant, puis remettez en suspension la pastille dans 180 μL de tampon de lyse enzymatique (ELB) et ajoutez 20 μL de RNase A pré-bouillie (10 mg/mL). Pour une lyse efficace des bactéries à Gram positif, ajouter 18 μL de mutanolysine (25 kU/mL). Bien vortex, puis incuber les échantillons à 37 °C sur rotateur pendant 2 h.
REMARQUE: Il est recommandé d’utiliser l’ELB décrit dans le protocole du fabricant pour les bactéries Gram positif et Gram négatif. - Procédez conformément aux instructions du fabricant.
REMARQUE: Répétez les étapes d’élution une ou deux fois de plus pour obtenir un rendement supplémentaire en ADNg, si vous le souhaitez. - Évaluer la qualité de l’ADNg extrait comme indiqué à la rubrique 4 et stocker l’ADNg à 4 °C s’il est utilisé dans un délai de 1 semaine. Alternativement, conservez l’ADNg à -20 °C pour un stockage à long terme.
4. Évaluation de la qualité de l’ADNg extrait
- Pour évaluer la qualité par électrophorèse sur gel, préparer un gel d’agarose à 1 % tel que décrit à la sous-section 2.2. Préparez l’échantillon dans un tube propre : mélanger 1-2 μL d’ADNg extrait et 3 μL de colorant de chargement 2x sur parafilm. Exécutez le gel une fois chargé, puis visualisez-le sous la lumière UV.
REMARQUE: L’extraction réussie de l’ADNg sera évidente par une bande discrète au sommet du gel et un frottis minimal(Figure 2A). Le frottis est révélateur d’un cisaillement. Si aucune bande d’ADNg n’est évidente et/ou si le frottis est important, répétez l’extraction de l’ADNg. Envisagez de réduire les temps d’incubation dans la RNase A et la protéinase K. Si deux bandes autour de 1,5-3 kb sont observées, cela suggère une contamination par l’ARN(Figure 2B). Préparer la RNase A fraîche et répéter l’extraction. - Pour évaluer la qualité par spectrophotomètre à microvolume, mesurer la concentration d’ADNg et le rapport d’absorbance A260/280 par spectrophotomètre à microvolume. Des concentrations >50 ng/μL et A260/280 comprises entre 1,7 et 2,0 sont acceptables.
REMARQUE: Un faible rendement en ADNg peut être dû à un faible apport, un apport élevé, une contamination des nucléases, une lyse insuffisante. Les rapports d’absorbance supérieurs à la plage indiquent une contamination par l’ARN. Répétez l’extraction si la qualité de l’ADNg est médiocre. - Pour évaluer la qualité par fluoromètre, suivez les instructions du fabricant pour quantifier la concentration d’ADNg à l’aide d’un kit de dosage haute sensibilité et d’un instrument fluoromètre(Table des matériaux). Une concentration >50 ng/μL est souhaitable.
5. Séquençage à lecture courte de nouvelle génération jumelé et préparation de la bibliothèque
REMARQUE: Le séquençage à lecture courte peut être effectué sur divers instruments à des longueurs et des orientations de lecture distinctes. Le séquençage par paires de 150 pb (cycle 300 cycles) est recommandé pour les WGS bactériens. La préparation et le séquençage des bibliothèques peuvent être sous-traités à des installations de base ou à des laboratoires commerciaux.
- Préparer la bibliothèque de séquençage selon les instructions du fabricant (Table des matériaux). Suivre la concentration finale recommandée par le fabricant dans la bibliothèque de chargement; toutefois, une modification recommandée consiste à charger la bibliothèque groupée à 1,8 pM pour une génération de lecture optimale sur les instruments NextSeq.
- Bien que facultatif, utilisez un bioanalyseur(Table des matériaux)pour évaluer la distribution des fragments de bibliothèque regroupés et vous assurer que la taille du fragment est de 600 pb en moyenne.
6. Préparation de la bibliothèque de séquençage Nanopore MinION
- Préparer la bibliothèque de séquençage selon le protocole du fabricant (Table des matériaux). L’utilisation de deux kits d’extension de codes-barres permet de multiplexer jusqu’à 24 échantillons sur une seule cellule de flux. Il est recommandé d’effectuer la préparation de la bibliothèque en deux parties, 12 échantillons à la fois lors du multiplexage de 24 échantillons. Les 24 échantillons peuvent être regroupés comme décrit ci-dessous.
REMARQUE: Les échantillons peuvent être stockés à 4 ° C pendant la nuit à la fin de la ligature de code-barres natif - cela fournit un point d’arrêt dans le protocole, si nécessaire. À la fin de la section de ligature de code-barres natif du protocole de préparation de la bibliothèque, il est recommandé de regrouper les quantités équimolaires de chaque échantillon jusqu’à la masse maximale d’ADN (ng) possible.- Pour ce faire, quantifiez tous les échantillons suivant la ligature de codes-barres à l’aide d’un fluoromètre(Table des matériaux)selon les instructions du fabricant. Estimez le volume de l’échantillon avec la concentration d’ADNds la plus faible, puis calculez l’ADNds total trouvé dans cet échantillon. Utilisez ce nombre pour déterminer les quantités équimolaires de tous les autres échantillons qui seront regroupés.
REMARQUE: Étant donné que le calcul équimolaire maximisera la quantité d’ADNds mis en commun et produira ainsi un pool à volume élevé (>65 μL), un nettoyage est nécessaire pour concentrer le pool.
- Pour ce faire, quantifiez tous les échantillons suivant la ligature de codes-barres à l’aide d’un fluoromètre(Table des matériaux)selon les instructions du fabricant. Estimez le volume de l’échantillon avec la concentration d’ADNds la plus faible, puis calculez l’ADNds total trouvé dans cet échantillon. Utilisez ce nombre pour déterminer les quantités équimolaires de tous les autres échantillons qui seront regroupés.
- Nettoyage et concentration de la piscine dsDNA
- Ajouter 2,5x volume de perles paramagnétiques (Table des matériaux) au pool d’ADN, puis faire glisser doucement le tube pour mélanger le contenu. Placez le tube dans le rotateur pendant 5 min à RT. Faites tourner l’échantillon à 2000 x g et pastillez sur un aimant.
- Ajouter 250 μL d’éthanol fraîchement préparé à 70% (dans de l’eau sans nucléase), en prenant soin de ne pas déranger la pastille. Aspirer l’éthanol et répéter le lavage à l’éthanol une fois.
- Après la deuxième aspiration, faites tourner l’échantillon à 2000 x g et replacez-le sur l’aimant. Pipettez tout éthanol résiduel et laissez l’échantillon sécher pendant environ 30 s.
- Retirez le tube de l’aimant et remettez la pastille dans 60 à 70 μL d’eau sans nucléase. Incuber à RT pendant 2 min. Abreuver l’échantillon sur l’aimant jusqu’à ce que l’élute soit clair, puis retirez l’éluat et transférez-le dans un tube de microcentrifugation propre de 1,5 mL.
- Quantifiez la piscine concentrée à l’aide d’un fluoromètre, puis préparez une aliquote pour passer à l’étape de ligature de l’adaptateur : préparer 700 ng de l’échantillon dans un volume final de 65 μL. Conservez le reste de la piscine à 4 °C pour une deuxième course à terminer une fois la première course terminée.
- Procédez à la ligature de l’adaptateur selon les instructions du fabricant et chargez l’échantillon sur la cellule d’écoulement. Démarrez l’exécution du séquençage.
REMARQUE: Aspirer de l’air et environ 200 μL de tampon de stockage à partir du port d’amorçage de la cellule d’écoulement avant le chargement de l’échantillon. Ceci est essentiel pour le succès de l’amorçage de la cellule d’écoulement et du chargement de l’échantillon. Utilisez une pipette p1000 et des embouts lorsque vous dessinez et déposez des solutions à travers le port d’amorçage de la cellule d’écoulement.
- Séquencez la bibliothèque selon les instructions du fabricant.
- Ouvrez le logiciel d’exploitation pour le séquençage et cliquez sur Démarrer. Entrez un nom pour l’expérience, une nomenclature recommandée inclut la date d’exécution et le nom de l’utilisateur. Cliquez sur Continuer à la sélection du kit, sélectionnez le kit de préparation de bibliothèque approprié et le ou les packs d’extension de code-barres utilisés, puis cliquez sur Continuer à exécuter les options.
- Ajustez la durée de l’exécution à 48 h si vous prévoyez de préparer suffisamment de bibliothèque pour une deuxième exécution (sinon, laissez par défaut 72 h). Cliquez sur Continuer vers Basecalling.
- Cochez l’option d’appel de base Config: Fast Basecalling et assurez-vous que Le codage à barres est défini sur Activé afin que les fichiers FASTQ de sortie soient coupés des séquences de codes à barres et démultiplexés dans des répertoires distincts en fonction du code-barres. Cliquez sur Continuer jusqu’à la sortie.
- Choisissez où enregistrer les données de séquençage de sortie. Attendez-vous à environ 30 à 50 Go de données si vous enregistrez uniquement la sortie FASTQ et >500 Go de données si vous enregistrez également la sortie FAST5. Décochez l’option de filtrage Qscore: 7 | Readlength : Non filtré si vous prévoyez de procéder au filtrage décrit à la section 7.2, sinon laissez la case cochée et ajustez Readlength à 200.
- Cliquez sur Continuer pour exécuter le programme d’installation et passez en revue tous les paramètres. Si les paramètres sont corrects, cliquez sur Démarrer, sinon cliquez sur Retour et effectuez les ajustements nécessaires.
- Si vous le souhaitez, la cellule d’écoulement peut être lavée selon les instructions du fabricant et rechargée avec la piscine restante. Répétez les étapes de la section 6.2 pour le pool restant une fois la première exécution terminée et la cellule d’écoulement lavée.
REMARQUE: Lors de la configuration de la deuxième série, ajustez la tension de polarisation à -250 mV conformément aux recommandations du fabricant pour les cellules d’écoulement précédemment utilisées dans les séries de plus de 48 h.
7. Évaluation et préparation des lectures
Remarque : Une structure de répertoire recommandée est illustrée à la figure 4. Créez les répertoires trouvés dans le Bureau, à savoir, Long_Reads, Short_Reads et Trimmed_Reads, avant de procéder aux étapes de calcul ci-dessous.
- Lectures courtes (Graphique 3)
REMARQUE: Les lectures courtes sont générées au format FASTQ. Les fichiers contiennent 4000 lectures maximum par FASTQ. Ceux-ci sont souvent compressés (.gz archive) et organisés en plusieurs fichiers. Selon la plate-forme, les codes-barres sont généralement coupés. Certains programmes acceptent les fichiers au format compressé, d’autres peuvent nécessiter leur extraction avant l’importation. Les lectures doivent passer les étapes de contrôle de la qualité (CQ) pour assurer l’exactitude des données lors de l’assemblage du génome. Si CLC Genomics Workbench n’est pas disponible, d’autres programmes peuvent être utilisés pour couper et QC les lectures courtes telles que Trimmomatic25 ou Trim Galore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) pour le rognage et FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) pour évaluer la qualité de lecture. Il est recommandé de multiplier par >100x la couverture moyenne des lectures courtes, estimée en multipliant le nombre de lectures par la longueur moyenne des lectures et en divisant par la taille du génome.- Ouvrez le logiciel Genomics Workbench (Table of Materials) et importez tous les fichiers FASTQ à lecture courte appariés. Les fichiers jumelés seront générés automatiquement.
- Créez un nouveau dossier sous CLC_Data en cliquant sur nouveau dans la barre d’outils supérieure et en sélectionnant Dossier... pour stocker les fichiers. Nommez le dossier comme vous le souhaitez, une convention recommandée utilise l’exemple d’ID. Enregistrez toutes les sorties des étapes suivantes dans ce dossier.
- Dans la barre d’outils supérieure, cliquez sur le bouton Importer et sélectionnez Illumina... Accédez à tous les fichiers à lecture courte qui correspondent à l’exemple et sélectionnez-les. Assurez-vous que l’option lectures appariées est sélectionnée et décochez l’option Supprimer les lectures ayant échoué. Cliquez sur Suivant, sélectionnez Enregistrer, puis cliquez à nouveau sur Suivant. Choisissez d’enregistrer les fichiers importés dans le nouveau dossier créé à l’étape précédente et cliquez sur Terminer.
- Créez une liste de séquences de tous les fichiers appariés pour l’isolat ; cela concaténera les données lues dans un seul fichier pour simplifier l’analyse.
- Dans la barre d’outils supérieure, cliquez sur le bouton Nouveau et sélectionnez Liste des séquences... Dans la liste des répertoires à gauche, sélectionnez les fichiers à concaténer et utilisez les flèches pour les déplacer dans la liste des fichiers sélectionnés à droite. Cliquez sur Suivant, sélectionnez Enregistrer, puis cliquez à nouveau sur Suivant. Choisissez d’enregistrer la liste des séquences et cliquez sur Terminer.
- Une fois la liste de séquences générée, renommez-la immédiatement avec l’ID d’exemple.
- Exécutez l’outil QC pour les lectures de séquençage sur la liste des séquences : cette procédure évaluera les paramètres de qualité globaux des lectures générées par les NGS à lecture courte.
- Recherchez l’outil QC pour les lectures de séquençage dans le menu de la boîte à outils (fenêtre en bas à gauche). Double-cliquez sur l’outil, puis choisissez la liste des séquences à analyser et cliquez sur Suivant.
- Assurez-vous que toutes les options de sortie sont cochées et choisissez Enregistrer sous Gestion des résultats. Cliquez sur Suivant et spécifiez pour enregistrer les fichiers de sortie, puis cliquez sur Terminer.
- Exécutez l’outil Ajuster les lectures dans la liste des séquences : le rognage sera effectué en fonction de la qualité, de la longueur et de l’ambiguïté. Ce processus suppose que les codes-barres utilisés dans le séquençage ont été coupés avant cette étape.
- Recherchez l’outil Ajuster les lectures dans la boîte à outils (fenêtre en bas à gauche). Double-cliquez sur Trim Reads, puis choisissez la liste des séquences à analyser et cliquez sur Suivant.
- Découpage de qualité: définissez la limite de score de qualité à 0,01 et laissez les nucléotides ambigus à 2. Cliquez sur Suivant.
REMARQUE: Les paramètres peuvent être ajustés à la discrétion de l’utilisateur; ce sont les paramètres recommandés. - Décochez la case Découpage automatique de l’adaptateur en lecture (ne le faites que si les adaptateurs ont été coupés des lectures avant l’importation dans CLC). Cliquez sur Suivant et cochez Ignorer les lectures ci-dessous la longueur,utilisez la valeur par défaut 15.
- Cliquez sur Suivant, cochez Créer un rapport, puis choisissez Enregistrer. Cliquez sur Suivant et spécifiez où enregistrer les fichiers de sortie. Cliquez sur Terminer.
- Exporter la liste des séquences découpées : l’assemblage et l’analyse hybrides suivants seront effectués en dehors de CLC et nécessitent l’exportation des fichiers à lecture courte découpés.
- Dans la navigation dans le répertoire en haut à gauche, choisissez le fichier découpé généré à l’étape 7.1.4, puis cliquez sur Exporter dans la barre d’outils supérieure. Sélectionnez Fastq pour le type de fichier d’exportation et cliquez sur Suivant. Cochez exporter la liste des séquences jumelées vers deux fichiers. Ensuite, cliquez sur Suivant et choisissez le répertoire Trimmed_Reads vers lequel exporter les fichiers. Cliquez sur Terminer. Assurez-vous que les fichiers à lecture courte découpés ont été exportés avec succès en tant que deux fichiers (R1 et R2) avec l’extension .fastq.
REMARQUE : La liste des séquences découpées doit être exportée dans deux fichiers, généralement désignés par CLC comme R1 et R2. Ceci est essentiel car l’assemblage hybride en aval nécessite une entrée de données à lecture courte pour être configurée en tant que telle. - Renommez les fichiers exportés, veuillez vous abstenir d’utiliser des espaces et des caractères spéciaux dans les noms de fichiers. Pour plus de simplicité, un format recommandé est trimmed_short_file. R1.fastq.
- Dans la navigation dans le répertoire en haut à gauche, choisissez le fichier découpé généré à l’étape 7.1.4, puis cliquez sur Exporter dans la barre d’outils supérieure. Sélectionnez Fastq pour le type de fichier d’exportation et cliquez sur Suivant. Cochez exporter la liste des séquences jumelées vers deux fichiers. Ensuite, cliquez sur Suivant et choisissez le répertoire Trimmed_Reads vers lequel exporter les fichiers. Cliquez sur Terminer. Assurez-vous que les fichiers à lecture courte découpés ont été exportés avec succès en tant que deux fichiers (R1 et R2) avec l’extension .fastq.
- Ouvrez le logiciel Genomics Workbench (Table of Materials) et importez tous les fichiers FASTQ à lecture courte appariés. Les fichiers jumelés seront générés automatiquement.
- Lecture longue (MinION) (Figure 3)
REMARQUE: Le pipeline suivant pour la préparation des lectures de séquençage Long (MinION) pour l’assemblage hybride utilise les programmes NanoFilt et Nanostat26 exécutés par la ligne de commande. Installez les outils avant de continuer et familiarisez-vous avec les bases d’UNIX afin d’exécuter ces commandes. Les terminaux par défaut et Bash Shell sont recommandés. Un guide de leçon pour les commandes et l’utilisation courantes des terminaux se trouve à Software Carpentry27. Les instructions ci-dessous supposent que les fichiers générés seront nommés avec la nomenclature des codes-barres (NB01, NB02, etc.) et sont enregistrés dans le répertoire Long_Reads. Alternativement, le filtrage en lecture peut être effectué à l’aide de MinKNOW lors de la configuration de l’exécution de séquençage. Il est recommandé que la couverture moyenne en lecture longue soit de >100x. La longueur moyenne de lecture recommandée est de >2000 pb; par conséquent, le nombre de lectures longues nécessaires est inférieur au nombre de lectures courtes.- Créez de nouveaux répertoires pour chaque code-barres utilisé dans l’exécution (code-barres01, code-barres02, etc.) dans le répertoire Long_Reads(Figure 4). Copiez tous les fichiers .fastq qui correspondent à chaque code-barres dans le dossier approprié. Combinez tous les fichiers .fastq pour chaque code-barres de chaque exécution.
- Ouvrez Terminal et accédez aux répertoires de codes à barres dans le répertoire Long_Reads à l’aide de la commande cd : cd Desktop/Long_Reads/barcode01
- Concaténez tous les fichiers .fastq par code-barres en un seul fichier .fastq en exécutant la commande suivante : cat *.fastq > NB01.fastq
REMARQUE : Cette commande combine toutes les lectures de chacun des fichiers FASTQ en un seul FASTQ volumineux nommé NB01.fastq. - Utilisez NanoStat pour évaluer la qualité de lecture de l’échantillon en exécutant la commande suivante : NanoStat --fastq NB01.fastq
- Enregistrez les résultats en copiant la sortie dans un fichier texte ou Word pour référence ultérieure.
- Utilisez NanoFilt pour filtrer les lectures MinION en rejetant les lectures avec Q < 7 et la longueur < 200 en exécutant la commande: NanoFilt -q 7 -l 200 bp NB01.fastq | gzip > NB01 _trimmed.fastq.gz
- Exécutez NanoStat sur le fichier découpé généré à l’étape 7.2.6 en exécutant la commande: NanoStat --fastq NB01 _trimmed.fastq.gz
- Enregistrez les résultats en copiant la sortie dans un fichier texte ou Word et comparez-les aux résultats de l’étape 7.2.4 pour vous assurer que le filtrage a réussi (Tableau 1).
- Répétez les étapes 7.2.2 à 7.2.8 pour chaque code-barres utilisé dans l’exécution du séquençage.
Remarque : Le fichier NB01_trimmed.fastq.gz généré à l’étape 7.2.6 sera utilisé pour l’assemblage hybride.
8. Génération d’un assemblage de génome hybride
REMARQUE : Le pipeline d’assemblage suivant utilise Unicycler19,28,29,30 pour combiner des lectures courtes et longues préparées dans les sections 7.1 et 7.2 ( Figure3). Installez Unicycler et ses dépendances et exécutez les commandes ci-dessous. Les fichiers à lecture courte exportés à l’étape 7.1.5 sont supposés être nommés trimmed_short_file. R1.fastq et trimmed_short_file. R2.fastq pour plus de simplicité.
- Organisez les fichiers à lecture courte et les fichiers à lecture longue dans un répertoire unique nommé Trimmed_Reads. Le répertoire doit contenir les éléments suivants :
- Un fichier .fastq.gz pour les lectures longues découpées (généré à l’étape 7.2.6).
- Deux fichiers .fastq (R1 et R2) pour les lectures courtes découpées (générés à l’étape 7.1.5).
- Accédez au répertoire Trimmed_Reads qui stocke les fichiers lus à l’aide de la commande cd dans Terminal : cd Desktop/Trimmed_Reads
- Une fois dans le bon répertoire, compressez les deux fichiers à lecture courte afin qu’ils soient également au format .fastq.gz en exécutant la commande suivante: gzip trimmed_short_file. R1.fastq
- Répétez l’étape 8.2 pour R1 et R2. Vérifiez que tous les fichiers lus sont maintenant au format .fastq.gz et vérifiez que tous les fichiers correspondent au même isolat.
- Commencez l’assemblage hybride à l’aide d’Unicycler en exécutant la commande suivante :
monocycle -1 trimmed_short_file. R1.fastq.gz -2 trimmed_short_file. R2.fastq.gz -l NB01 _trimmed.fastq.gz -o unicycler_output_directory
REMARQUE: -o spécifie le répertoire dans lequel la sortie Unicycler sera enregistrée, Unicycler créera ce répertoire une fois la commande exécutée; ne générez pas le répertoire au préalable. Le temps d’exécution varie en fonction de la puissance de calcul de l’ordinateur utilisé ainsi que de la taille du génome et du nombre de lectures. Cela peut prendre de 4 h à 1 ou 2 jours. Ce protocole a été réalisé sur une machine CentOS Linux 7 avec 250 Gb de RAM, processeur Intel Xeon (R) avec 2,5 GHz 12 cœurs pratiques et 48 cœurs virtuels. Alternativement, les ordinateurs personnels avec 16 Gb de RAM et 2,6 GHz processeurs 6 cœurs peuvent calculer ces assemblages à un temps de traitement plus long. - Une fois l’exécution terminée, examinez le fichier unicycler.log pour vous assurer qu’il n’y a pas d’erreurs - enregistrez le nombre, la taille et l’état (complet, incomplet) des contigs générés.
- Si des contigs incomplets sont identifiés (notés comme incomplets dans le journal Unicycler), réexécutez Unicycler en mode gras en ajoutant l’indicateur suivant à la commande à l’étape 8.4 : --mode gras.
REMARQUE: Le mode gras abaissera le seuil de qualité accepté pour les ponts à lecture longue pendant l’assemblage; cela peut donner un assemblage complet, mais la qualité de l’assemblage peut être diminuée. Il est recommandé d’utiliser le mode gras uniquement lorsque cela est nécessaire et comme preuve préliminaire de l’assemblage contig à confirmer ultérieurement par PCR.
- Si des contigs incomplets sont identifiés (notés comme incomplets dans le journal Unicycler), réexécutez Unicycler en mode gras en ajoutant l’indicateur suivant à la commande à l’étape 8.4 : --mode gras.
9. Évaluation de la qualité de l’assemblage
REMARQUE : Le protocole suivant utilise Bandage31 et QUAST32, deux programmes qui doivent être configurés avant utilisation (Figure 2 et Figure 4). Bandage ne nécessite pas d’installation une fois téléchargé et QUAST nécessite une connaissance de l’utilisation de base de la ligne de commande. Il est également recommandé d’évaluer l’exhaustivité du génome à l’aide de l’analyse comparative des orthologues universels à copie unique (BUSCO)33.
- Bandage : Cliquez sur Fichier. Ensuite, choisissez Load Graph et sélectionnez le fichier assembly.gfa qui a été enregistré dans unicycler_output_directory généré par Unicycler à l’étape 8.4. Une fois chargé, cliquez sur le bouton Dessiner le graphique dans la barre d’outils de gauche et regardez comment les contigs (appelés nœuds) sont connectés et organisés pour évaluer si l’assemblage est complet (Figure 5).
REMARQUE: Les assemblages complets sont représentés par des contigs circulaires simples liés aux deux extrémités (Figure 5A, B). Les assemblages incomplets ont plusieurs contigs liés entre eux ou sont linéaires(Figure 5C). Les petits contigs linéaires peuvent ne pas être incomplets car ils peuvent indiquer des éléments extrachromosomiques linéaires. La couverture, également appelée profondeur, sera notée en bandage et représente l’abondance relative des contigs au chromosome, normalisés dans Unicycler à 1x. - QUAST
- Dans le terminal, accédez au dossier qui stocke la sortie Unicycler à l’aide de la commande cd : cd Desktop/Trimmed_Reads/unicycler_output_directory
REMARQUE : Les espaces ne sont pas autorisés dans le chemin d’accès à l’emplacement de l’assemblage, c’est-à-dire qu’aucun répertoire menant à la sortie Unicycler ne peut avoir d’espaces dans son nom. Vous pouvez également copier le fichier assembly.fasta sur le Bureau pour y accéder facilement. - Exécutez QUAST en exécutant la commande suivante : quast assembly.fasta -o quast_output_directory
- Passez en revue les rapports générés par QUAST dans le répertoire de sortie quast_output_directory.
- Dans le terminal, accédez au dossier qui stocke la sortie Unicycler à l’aide de la commande cd : cd Desktop/Trimmed_Reads/unicycler_output_directory
10. Annotation du génome
REMARQUE: Le pipeline d’annotation ci-dessous utilise Prokka34, un outil de ligne de commande qui doit être installé avant utilisation. Alternativement, utilisez Prokka via l’interface graphique automatisée K-Base(Table des matériaux)ou annotez les génomes via le serveur Web RAST35. Si vous déposez des génomes dans ncBI, ils seront automatiquement annotés à l’aide du pipeline d’annotation du génome procaryote (PGAP)36.
- Naviguez dans le terminal jusqu’au dossier qui stocke la sortie Unicycler à l’aide de la commande cd (voir étape 9.2.1). Ensuite, exécutez Prokka en exécutant la commande suivante : prokka --prefix sample_ID --outdir prokka_output_directory assembly.fasta
REMARQUE: --prefix nommera tous les fichiers de sortie en fonction de la sample_ID spécifiée. --outdir créera un répertoire de sortie avec le nom spécifié où tous les fichiers de sortie Prokka seront enregistrés; ne créez pas de répertoire de sortie pour Prokka au préalable. - Passez en revue les annotations en ouvrant la table .tsv et/ou en téléchargeant le fichier .gff généré dans un logiciel d’analyse de séquence pour visualiser et analyser les annotations (Figure 6).
- Des types spécifiques d’annotations peuvent être générés en fonction de facteurs génétiques d’intérêt. Il est recommandé de commencer par les outils conviviaux sur le serveur Web du Center for Genomic Epidemiology (www.genomicepidemiology.org/) pourl’analysepréliminaire37,38,39,40,41. Des outils supplémentaires pour la détection des systèmes CRISPR-cas et des prophages sont disponibles (Figure 3)42,43.
11. Pratiques suggérées pour la démocratisation des données
- Dans la mesure du possible, déposez toutes les données de lecture brutes ainsi que les génomes assemblés dans un référentiel public tel que NCBI Sequence Read Archive (SRA) et Genbank. Les génomes sont automatiquement annotés via le pipeline PGAP pendant le processus de dépôt NCBI.
Résultats
Ce protocole a été optimisé pour la culture et le séquençage de bactéries urinaires appartenant aux genres énumérés à la figure 1. Toutes les bactéries urinaires ne sont pas cultivables par cette méthode. Les milieux de culture et les conditions sont spécifiés par le genre à la figure 1. Des évaluations exemplaires de l’intégrité de l’ADNg par électrophorèse sur gel sont illustrées à la figure 2. Une vue d’ensemble du pipeline de bioinformatique pour le séquençage du traitement de la lecture, de l’assemblage du génome et de l’annotation est décrite à la figure 3. Un guide pour la structure de répertoire de calcul est fourni à la figure 4 pour simplifier la compréhension du protocole et fournir un cadre pour une organisation réussie. En outre, sont inclus les génomes complets représentatifs de deux Klebsiella spp., K. pneumoniae et K. oxytoca, qui ont été générés par ce protocole. Une représentation de ces assemblages est fournie à la figure 5 et comprend également un exemple incomplet supplémentaire du génome de K. pneumoniae. Un aperçu détaillé de chaque génome complet entièrement annoté est illustré à la figure 6. Enfin, un résumé des statistiques de lecture de séquençage est fourni dans le tableau 1 afin d’offrir une compréhension générale des données brutes et découpées suffisantes pour la génération d’assemblages de génomes fermés de haute qualité. En outre, les paramètres clés des deux représentants complètent Klebsiella spp. les génomes sont répertoriés. Les génomes et les données brutes ont été déposés à Genbank dans le cadre du BioProject PRJNA683049.

Figure 1: Culture urinaire améliorée modifiée de divers genres urinaires. Tableau de la gélose et du bouillon liquide qui peuvent être utilisés pour cultiver divers genres urinaires. Il est suggéré d’effectuer toute culture à 35 °C comme décrit à la sous-section 1.1. Les cercles représentent des milieux appropriés pour la culture d’un genre particulier, les couleurs ont été choisies arbitrairement pour distinguer un type de média d’un autre. CDC-AN BAP (rouge), CDC Anaerobe Sheep Blood Agar; 5% BAP de mouton (orange), gélose de sang de mouton; BHI (vert), Infusion cœur cérébral; BST (jaune), bouillon de soja tryptique; Orientation CHROMagar (bleu). aGardnerella vaginalis doit être cultivée sur HBT Bilayer G. vaginalis Gélose sélective en atmosphère microaérophile et selon les exigences spéciales de culture de bouillon44. bLes iners de lactobacilles doivent être cultivés sur des plaques de BAP de lapin à 5 % et du bouillon NYCIII dans une atmosphère microaérophile. cLactobacillus spp. peut être cultivé sur MRS dans des conditions microaérophiles. Veuillez cliquer ici pour voir une version agrandie de cette figure.
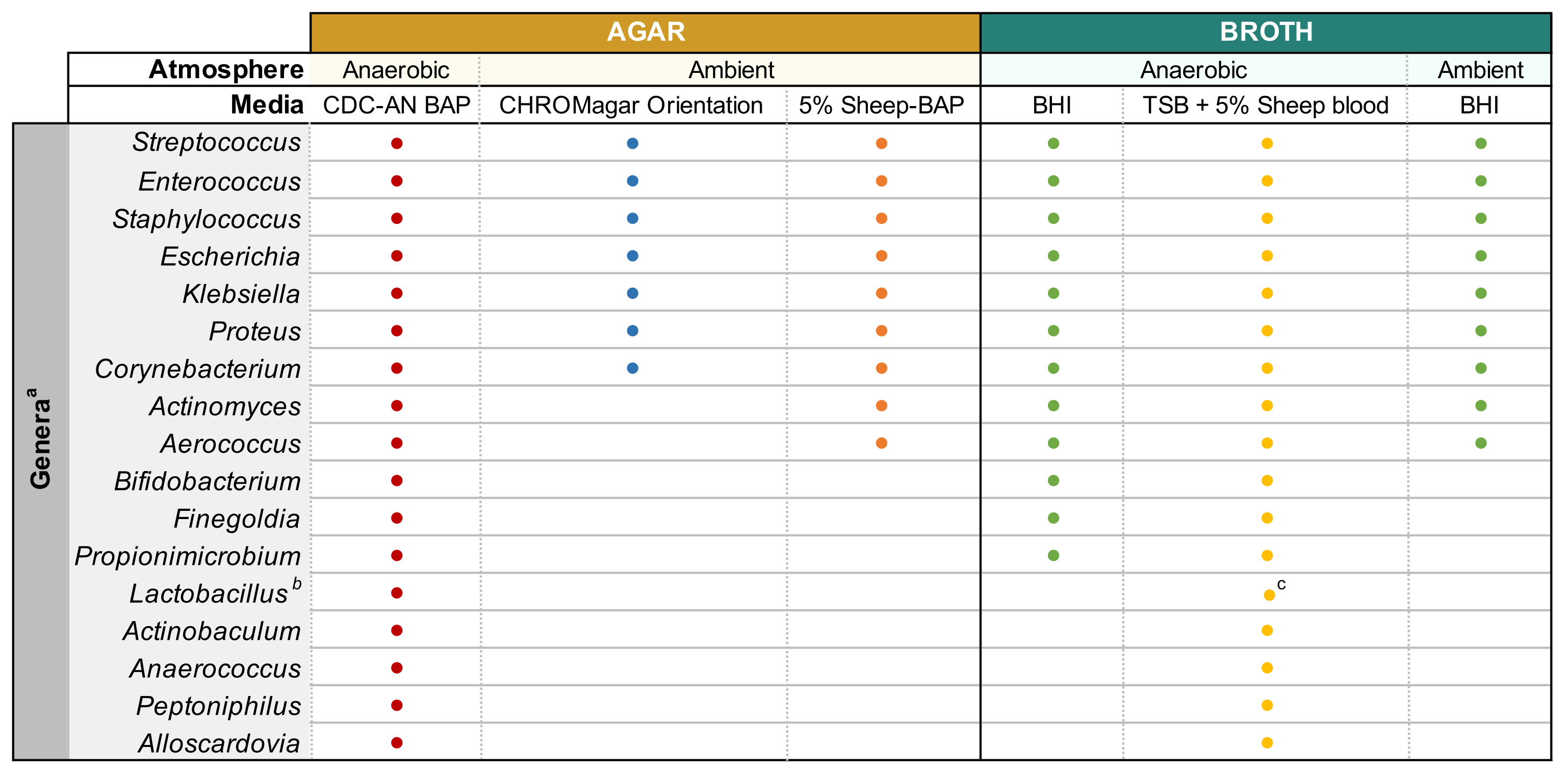{kind=link}

Figure 2: Imagesde gel d’agarose d’extraction génomique de l’ADN. Images de gel représentatives représentant les résultats de l’extraction de l’ADNg. (A) Voie 1: échelle de 1 kb, Voie 2: ADNg intact représentant une extraction réussie, Voie 3: frottis indiquant un ADNg fragmenté. (B) Voie 1: échelle de 1 kb, voies 2 et 3: contamination par l’ARNr indiquée par deux bandes comprises entre 1,5 kb et 3 kb. Veuillez cliquer ici pour voir une version agrandie de cette figure.
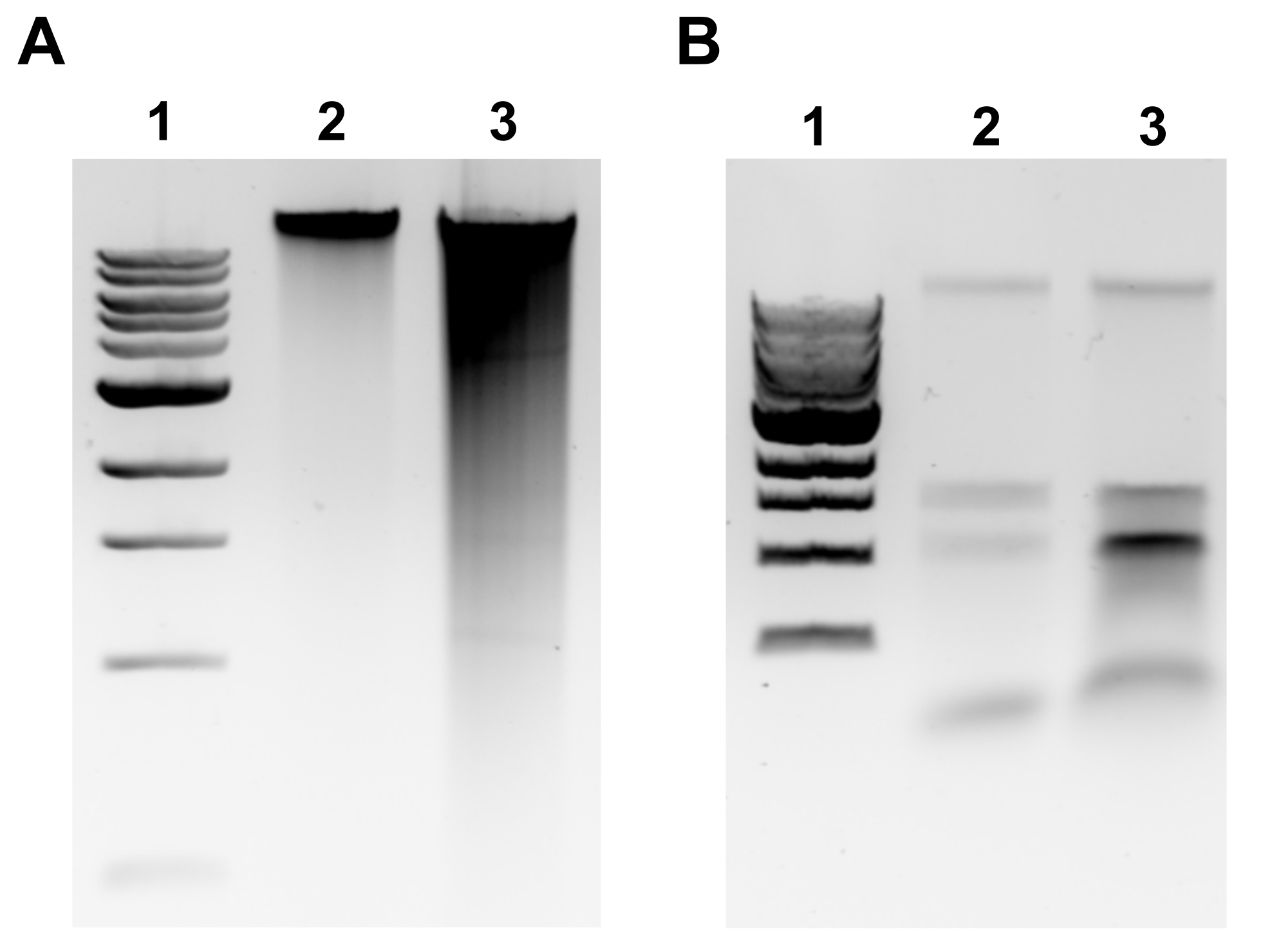{kind=link}

Figure 3: Flux de travail d’assemblage du génome hybride. Schéma des étapes du contrôle qualité de lecture et du prétraitement à l’annotation d’assemblage. Le découpage de lecture supprime les lectures ambiguës et de mauvaise qualité. Les paramètres Q-score et length sont indiqués et représentent les lectures qui sont conservées. L’assemblage utilise à la fois des lectures courtes et longues pour générer un assemblage hybride de génome de novo. La qualité de l’assemblage est évaluée en fonction de l’exhaustivité et de l’exactitude à l’aide d’outils et de paramètres spécifiés. L’assemblage final du génome est annoté pour tous les gènes et loci spécifiques d’intérêt. Veuillez cliquer ici pour voir une version agrandie de cette figure.
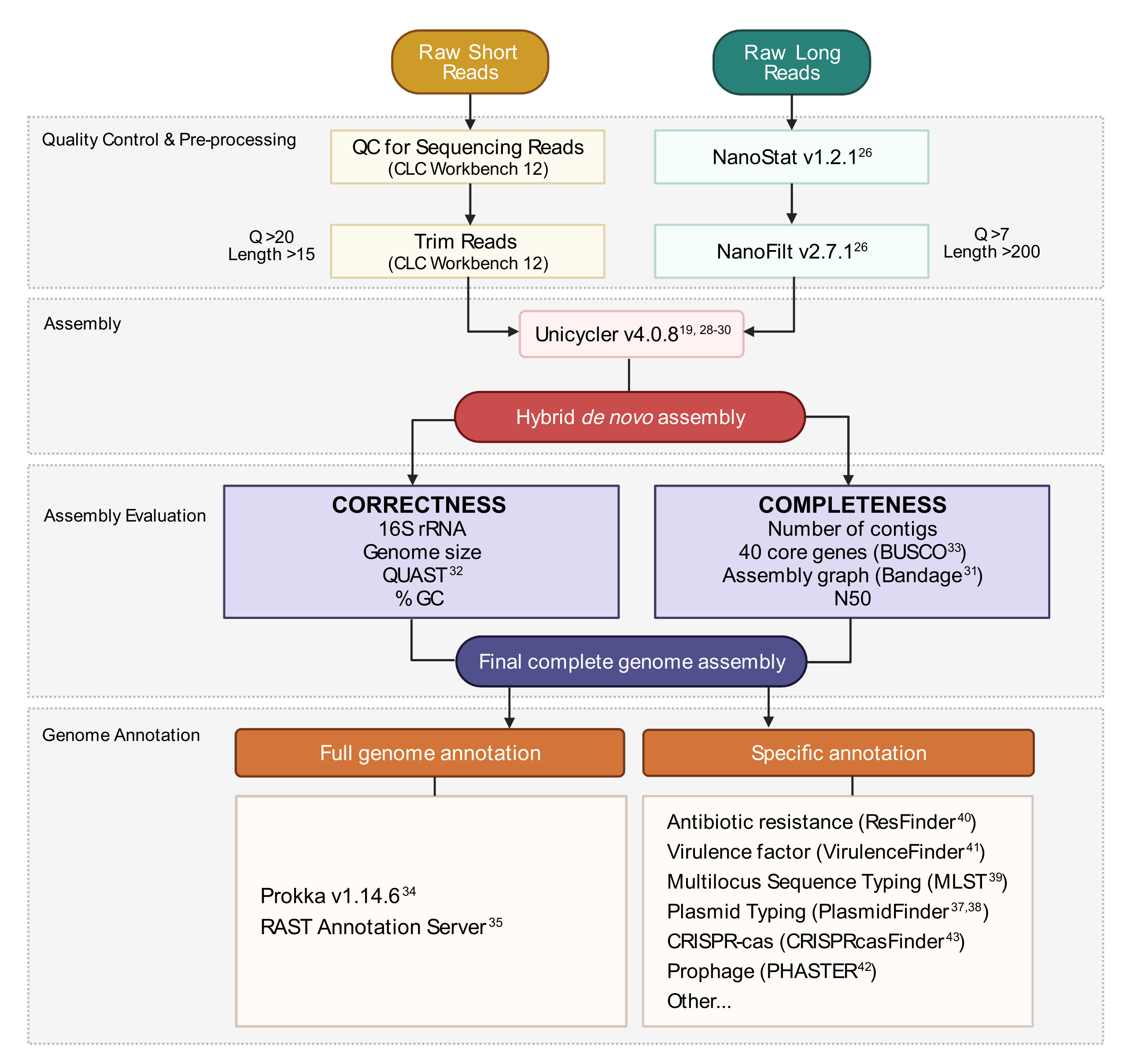{kind=link}

Figure 4: Guide de la structure des répertoires de bioinformatique. Schéma de l’organisation recommandée des répertoires et des fichiers pour le traitement des lectures courtes et longues, l’assemblage hybride, l’annotation du génome et le contrôle qualité. Les principales étapes de traitement des données en ligne de commande sont mises en surbrillance à côté des fichiers et répertoires correspondants. Déclenchement de commandes et d’indicateurs (gras), fichiers d’entrée (bleu), fichiers ou répertoires de sortie (rouge), entrée utilisateur telle que la convention de dénomination de fichier (magenta). Veuillez cliquer ici pour voir une version agrandie de cette figure.
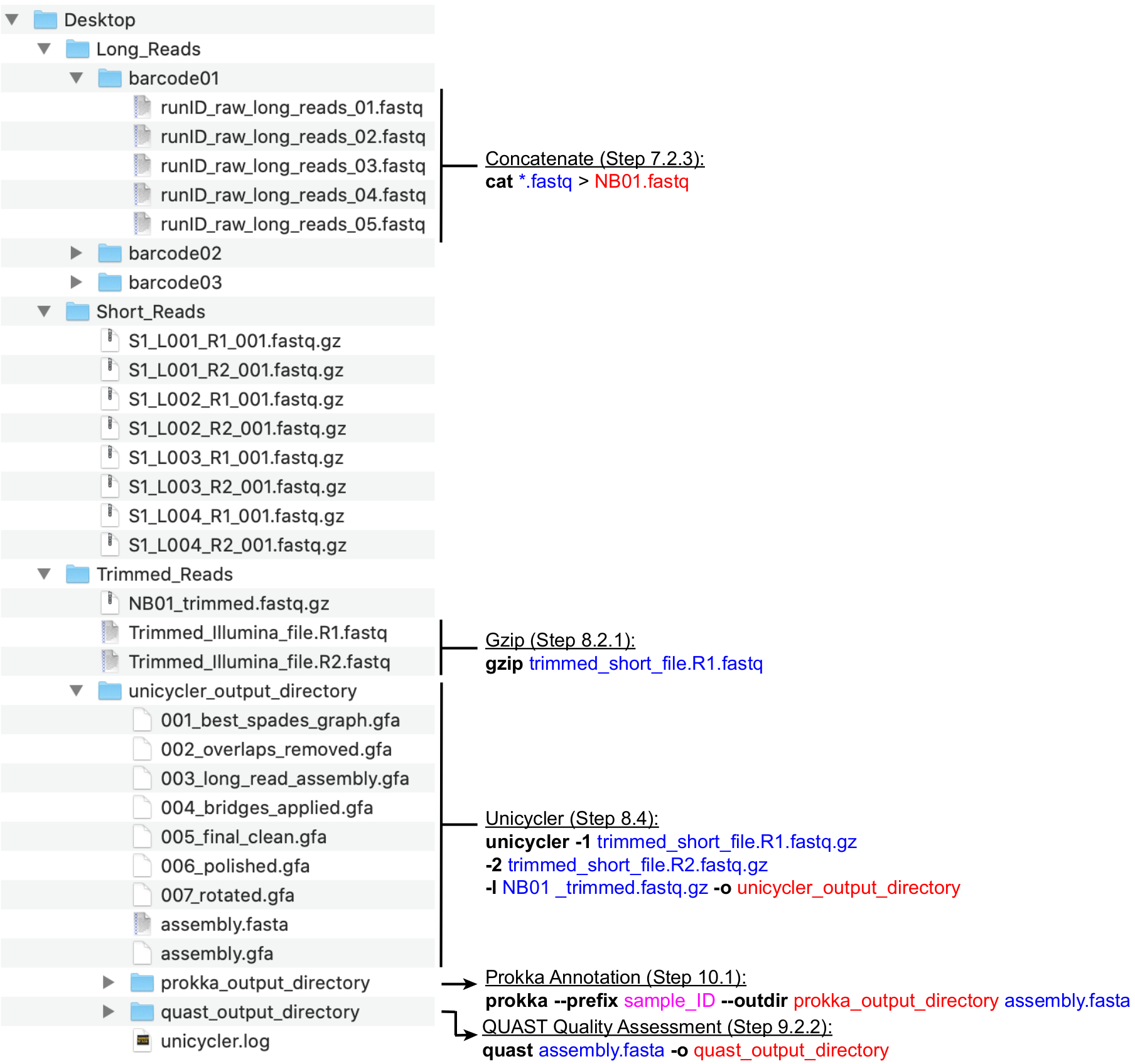{kind=link}

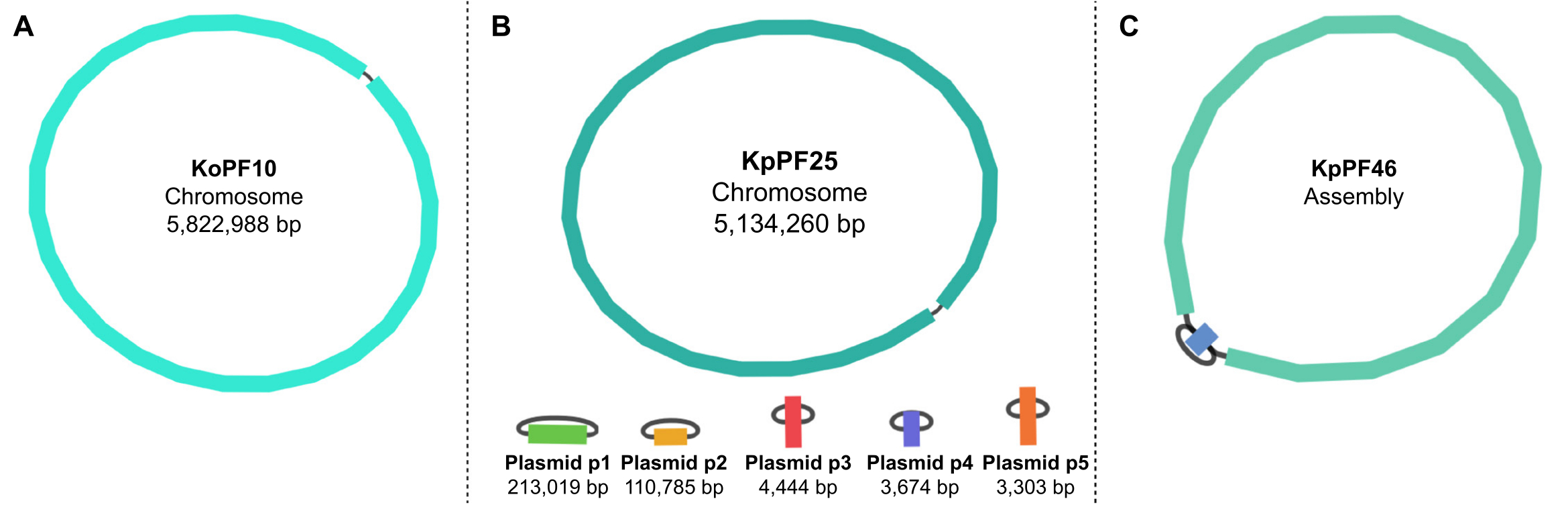
Figure 5: Graphiques d’assemblage du génome par bandage. Graphes représentatifs complets de l’assemblage du génome de (A) Klebsiella oxytoca KoPF10 et (B) Klebsiella pneumoniae KpPF25 et assemblage incomplet du génome de (C) Klebsiella pneumoniae KpPF46. Le génome complet de KoPF10 démontre un seul chromosome fermé et le génome complet de KpPF25 se compose d’un chromosome fermé et de cinq plasmides fermés. Le chromosome incomplet de KpPF46 est constitué de deux contigs interconnectés. L’assemblage hybride de novo Unicycler génère un graphique d’assemblage qui est visualisé par Bandage. Le graphique d’assemblage fournit un schéma simpliste du génome, indiquant un chromosome fermé ou des plasmides par un liant reliant deux extrémités d’un même contig. La présence de plus d’un contig interconnecté indique un assemblage incomplet. La taille et la profondeur de Contig peuvent également être notées dans Bandage. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

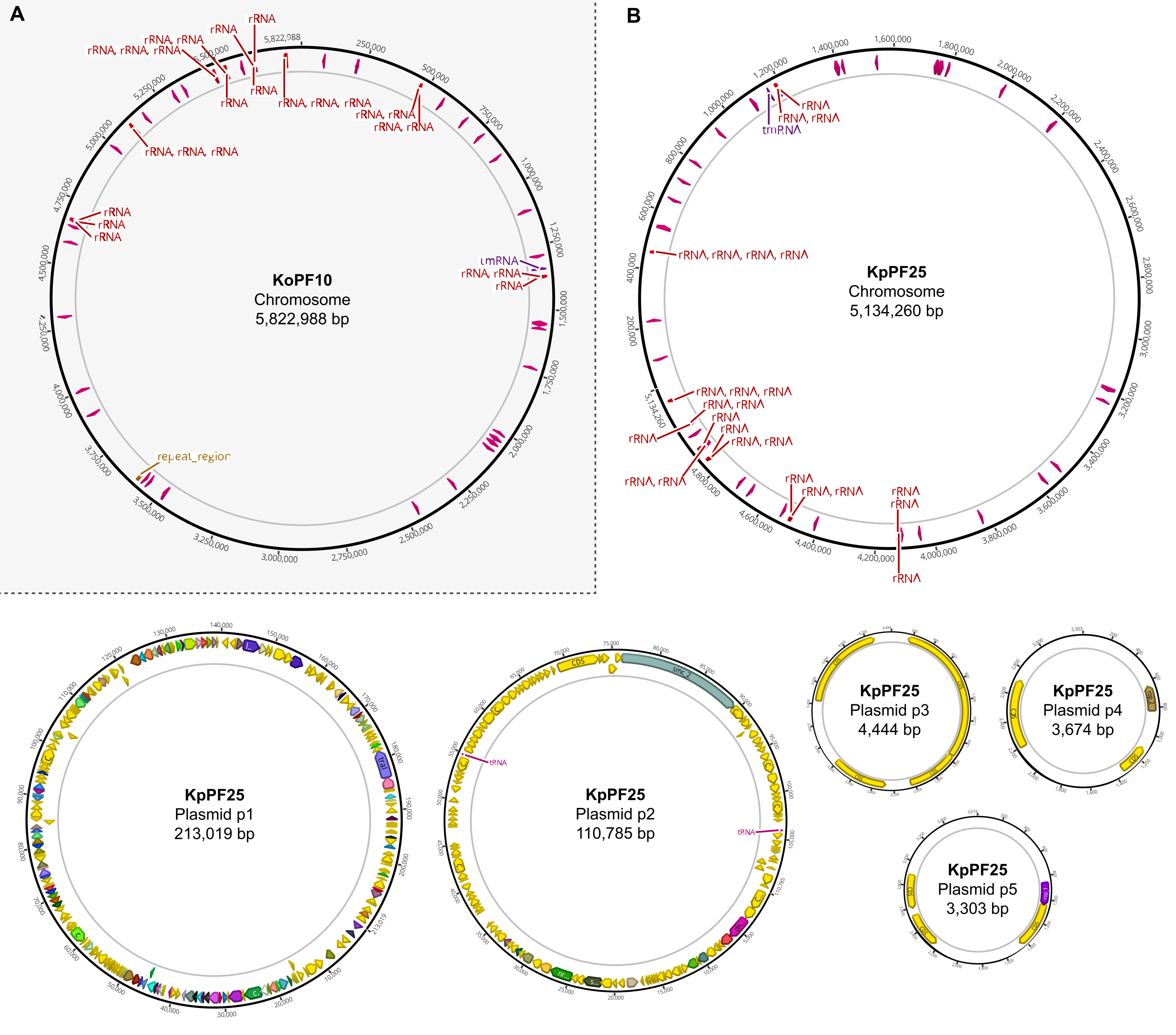
Figure 6: Cartes complètes du génome d’assemblages hybrides annotés. Cartes d’assemblage générées par Geneious Prime pour le génome complet de (A) K. oxytoca KoPF10 et (B) K. pneumoniae KpPF25 montrant des gènes annotés désignés par des flèches colorées le long des épines dorsales plasmidiques. Les chromosomes ne montrent que des gènes d’ARNr et d’ARNt pour plus de simplicité. Les annotations du génome ont été effectuées à l’aide de Prokka comme indiqué à la section 10 de ce protocole. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Tableau 1: Caractéristiques représentatives de Klebsiella spp. Paramètres d’assemblage de la souche K. oxytoca KoPF10 et de la souche K. pneumoniae KpPF25. Les numéros d’acquisition pour les données déposées sur ncBI sont fournis. Le nombre de lectures avant et après le rognage est spécifié pour les deux technologies de séquençage. N50 est fourni pour les lectures longues uniquement car les lectures courtes sont d’une longueur contrôlée. Réplication plasmidique prédite à l’aide de la base de données PlasmidFinder v2.1 Enteroebacteriaceae avec des paramètres définis sur 80% d’identité et 60% de longueur. un MLST, type de séquence multilocus. b CDS, séquences de codage. c Réplication plasmidique prédite à l’aide de la base de données PlasmidFinder v2.1 Enterobacteriaceae avec des paramètres définis sur 80% d’identité et 60% de longueur. d Oxford Nanopore Technologies (ONT) a déposé des données de lecture. e Illumina a déposé des données de lecture. Veuillez cliquer ici pour télécharger ce tableau.
Discussion
Le protocole complet d’assemblage du génome hybride décrit ici offre une approche simplifiée pour la culture réussie de divers microbiotes urinaires et uropathogènes, et l’assemblage complet de leurs génomes. Le succès des WGS des génomes bactériens commence par l’isolement de microbes divers et parfois fastidieux afin d’extraire leur ADN génomique. À ce jour, les protocoles de culture d’urine existants n’ont pas la sensibilité nécessaire pour détecter de nombreuses espèces urinaires ou impliquent des approches longues et étendues qui nécessitent un temps et des ressourcesprolongés 11. L’approche de culture d’urine améliorée modifiée décrite offre un protocole simplifié mais complet pour l’isolement réussi des bactéries appartenant à 17 genres urinaires courants, y compris les espèces commensales potentiellement pathogènes ou bénéfiques, et les bactéries aérobies ou anaérobies facultatives et obligatoires. Cela fournit à son tour le matériel de départ nécessaire pour le séquençage et l’assemblage précis des génomes bactériens et pour les expériences phénotypiques critiques, qui contribuent à la compréhension de la santé et de la maladie urinaires. De plus, cette approche de culture modifiée permet un diagnostic clinique plus précis des micro-organismes viables présents dans les échantillons d’urine et permet leur biobanque pour de futures études génomiques. Cependant, ce protocole n’est pas sans limites. Il peut nécessiter de longs temps d’incubation selon l’organisme ainsi que l’utilisation de ressources telles qu’une chambre d’hypoxie ou des incubateurs contrôlés qui peuvent ne pas être facilement disponibles. L’utilisation de GasPaks anaérobies offre une solution alternative, mais ceux-ci sont coûteux et ne produisent pas toujours un environnement durable et contrôlé. Enfin, le biais de culture ainsi que la diversité des échantillons peuvent permettre à des organismes et des uropathogènes particuliers de surpasser les bactéries fastidieuses. Malgré ces limites, une culture de diverses bactéries urinaires est rendue possible par cette approche.
Le séquençage génomique a gagné en popularité avec l’avancement des technologies de séquençage de nouvelle génération qui ont considérablement augmenté à la fois le rendement et la précision des données de séquençage14,15. Couplées au développement d’algorithmes pour le traitement des données et l’assemblage de novo, les séquences complètes du génome sont à portée de main de scientifiques novices et experts15,45. La connaissance de l’organisation globale du génome fournie par les génomes complets offre des informations évolutives et biologiques importantes, y compris la duplication des gènes, la perte de gènes et le transfert horizontal de gènes14. De plus, les gènes importants pour la résistance aux antimicrobiens et la virulence sont souvent localisés sur des éléments mobiles, qui ne sont généralement pas résolus dans les assemblages de génomesprovisoires 15,16.
Le protocole présenté dans le présent document suit une approche hybride pour la combinaison de données de séquençage provenant de plates-formes à lecture courte et à lecture longue afin de générer des assemblages complets du génome. Bien que axée sur les génomes bactériens urinaires, cette procédure peut être adaptée à diverses bactéries provenant de diverses sources d’isolement. Les étapes critiques de cette approche comprennent le suivi d’une technique stérile adéquate et l’utilisation de milieux et de conditions de culture appropriés pour l’isolement des bactéries urinaires pures. En outre, l’extraction d’ADNg intact à haut rendement est essentielle pour générer des données de séquençage exemptes de lectures contaminantes susceptibles d’entraver le succès de l’assemblage. Les protocoles de préparation de bibliothèque ultérieurs sont essentiels pour la génération de lectures de qualité d’une longueur et d’une profondeur suffisantes. Par conséquent, il est extrêmement important de manipuler l’ADNg avec soin lors de la préparation de la bibliothèque pour le séquençage à lecture longue en particulier, car le plus grand avantage de cette technologie est la génération de lectures longues sans limite théorique de longueur supérieure. Des sections sont également décrites pour le contrôle de la qualité (CQ) approprié des lectures de séquençage qui élimine les données bruyantes et améliore les résultats d’assemblage.
Malgré le succès de l’isolement de l’ADN, de la préparation de la bibliothèque et du séquençage, la nature de l’architecture génomique de certaines espèces peut encore constituer un obstacle à la génération d’un assemblage de génome fermé45,46. Les séquences répétitives compliquent souvent le calcul de l’assemblage et malgré de longues données de lecture, ces régions peuvent être résolues avec une faible confiance, voire pas du tout. Les lectures longues doivent donc être en moyenne plus longues que la plus grande région de répétition du génome ou la couverture doit être élevée (>100x)19. Certains génomes peuvent rester incomplets et nécessiter des approches manuelles pour être complétés. Néanmoins, les génomes incomplets assemblés hybrides sont généralement composés de moins de contigs que les génomes brouillons à lecture courte. Il peut être utile d’ajuster les paramètres par défaut de l’algorithme d’assemblage ou de suivre des seuils plus stricts pour le contrôle qualité en lecture. Alternativement, une approche suggérée consiste à mapper les lectures longues aux régions incomplètes à la recherche de preuves du chemin d’assemblage le plus probable, puis à confirmer le chemin en utilisant la PCR et le séquençage de Sanger de la région amplifiée. La cartographie des lectures à l’aide de Minimap2 est suggérée et Bandage offre un outil utile pour la visualisation des lectures cartographiées le long des contigs assemblés fournissant des preuves de la liaison contig47.
Un défi supplémentaire pour générer des génomes complets réside dans la familiarité et le confort avec les outils de ligne de commande. De nombreux outils bioinformatiques sont développés pour offrir des opportunités de calcul à tout utilisateur; cependant, leur utilisation repose sur une compréhension des bases d’UNIX et de la programmation. Ce protocole vise à fournir des instructions suffisamment détaillées pour permettre aux personnes sans expérience préalable de la ligne de commande de générer des assemblages de génomes fermés et de les annoter.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Nous remercions le Dr Moutusee Jubaida Islam et le Dr Luke Joyce pour leurs contributions à ce protocole. Nous tenons également à remercier l’Université du Texas au Dallas Genome Center pour ses commentaires et son soutien. Ce travail a été financé par la Welch Foundation, numéro de prix AT-2030-20200401 à N.J.D., par les National Institutes of Health, numéro de prix R01AI116610 à K.P., et par la chaire Felecia et John Cain en santé des femmes, détenue par P.E.Z.
matériels
| Name | Company | Catalog Number | Comments |
| Equipment: | |||
| Bioanalyzer 2100 | Agilent | G29398A | Optional but recommended |
| Centrifuge | Eppendorf | -- | Any centrifuge for spinning conicals and microcentrifuge tubes (e.g. Models 5810R/5424R) |
| Electrophoresis | BioRad Laboratories | 1645070 | |
| Gel Imaging System | BioRad Laboratories | ChemiDoc models | |
| Incubator | ThermoFisher Scientific | -- | Any CO2 Incubator (e.g. Thermo Forma model 3110) |
| Magnetic Rack | New England BioLabs | S15095 | 12-tube rack |
| MinION | Oxford Nanopore Technologies | -- | |
| Nanodrop | ThermoFisher Scientific | ND-ONE-W | |
| NextSeq 500 | Illumina | SY-415-1002 | Other Illumina models are acceptable |
| Plate Reader | BioTek | -- | Synergy H1 |
| Qubit fluorometer | ThermoFisher Scientific | Q33238 | |
| Rotator | Benchmark Scientific | H2024 | |
| Thermocycler | ThermoFisher Scientific | -- | Any thermocycler for PCR reactions (e.g. ProFlex PCR system) |
| Materials: | |||
| 10X Phosphate Buffered Saline (PBS) | Fisher Scientific | BP3991 | |
| 10X TBE buffer | -- | -- | 1M Tris,1M Boric Acid,0.2M EDTA (pH 8.0) |
| 1429R primer | Sigma Aldrich (Custom oligos) | -- | GGTTACCTTGTTACGACTT |
| 1kb Ladder | VWR | 101228-494 | |
| 1M Tris-Cl (pH 7.5) | ThermoFisher Scientific | 15567027 | |
| 6x Loading dye | Fisher Scientific | NC0783588 | |
| 8F primer | Sigma Aldrich (Custom oligos) | -- | AGAGTTTGATCCTGGCTCAG |
| Agar | Fisher Scientific | BP1423-2 | |
| Agarose | BioRad Laboratories | 63001 | |
| AMPure XP Beads | Beckman Coulter | A63880 | |
| Anaerobe Pouch System - GasPak EZ | BD Diagnostic Systems | B260683 | |
| Boric Acid | Fisher Scientific | A73-500 | |
| Brain Heart Infusion Broth | BD Diagnostic Systems | 212304 | |
| CDC Anaerobe 5% Sheep Blood Agar | BD Diagnostic Systems | L007357 | |
| CHROMagar Orientation | BD Diagnostic Systems | PA-257481.04 | |
| DNeasy Blood & Tissue | QIAGEN | 69504 | |
| DreamTaq Master Mix | ThermoFisher Scientific | K1081 | |
| Dry Anaerobic Indicator Strips | BD Diagnostic Systems | 271051 | |
| EDTA | Fisher Scientific | S311-500 | |
| Ethanol 200 Proof | Sigma Aldrich | E7023 | For molecular biology |
| Ethidium Bromide | ThermoFisher Scientific | BP130210 | |
| Flow cell priming kit | Oxford Nanopore Technologies | EXP-FLP002 | |
| Flow cell wash kit | Oxford Nanopore Technologies | EXP-WSH003 | |
| Gel Extraction Miniprep Kit | BioBasic | BS654 | |
| Ligation sequencing kit | Oxford Nanopore Technologies | SQK-LSK109 | |
| Lysozyme | Research Products International Corp | L381005.05 | |
| Mutanolysin | Sigma Aldrich | M9901-5KU | |
| Native barcoding expansion 1-12 | Oxford Nanopore Technologies | EXP-NBD104 | |
| NEB Blunt/TA Ligase Master Mix | New England BioLabs | M0367L | |
| NEBNext FFPE DNA Repair Mix | New England BioLabs | M6630L | |
| NEBNext quick ligation buffer | New England BioLabs | B6058S | |
| NEBNext Ultra II End repair / dA-tailing module | New England BioLabs | E7546L | |
| Nextera DNA CD Indexes | Illumina | 20018708 | |
| Nextera DNA Flex Library Prep - (M) Tagmentation | Illumina | 20018705 | |
| Nuclease-free water | Sigma Aldrich | W4502 | |
| Qubit 1X dsDNA HS Assay Kit | ThermoFisher Scientific | Q33230 | |
| Qubit Assay Tubes | ThermoFisher Scientific | Q32856 | |
| Quick T4 DNA Ligase | New England BioLabs | E6056L | |
| R9 Flow cell | Oxford Nanopore Technologies | FLO-MIN106D | |
| RNase A | ThermoFisher Scientific | EN0531 | |
| Sheep Blood | Hemostat Laboratories | DS13250 | |
| TE buffer | -- | -- | 10mM Tris, 1mM EDTA (pH 8.0) |
| Triton X-100 | Sigma Aldrich | T8787 | |
| Tryptic Soy Broth | BD Diagnostic Systems | 211825 | |
| Software & Bioinformatic Tools: | |||
| Bandage | -- | -- | https://rrwick.github.io/Bandage/ |
| Center for Genomic Epidemiology | -- | -- | http://www.genomicepidemiology.org/ |
| CLC Genomics Workbench 12 | QIAGEN | -- | |
| CRISPRcasFinder | -- | -- | https://crisprcas.i2bc.paris-saclay.fr/ |
| FastQC | -- | -- | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Geneious Prime | Geneious | -- | |
| gVolante (BUSCO) | -- | -- | https://gvolante.riken.jp/ |
| Kbase Prokka Wrapper | -- | -- | https://kbase.us/applist/apps/ProkkaAnnotation/annotate_contigs/release |
| Minimap2 | -- | -- | https://github.com/lh3/minimap2 |
| MinKNOW | Oxford Nanopore Technologies | -- | |
| NanoFilt | -- | -- | https://github.com/wdecoster/nanofilt |
| NanoStat | -- | -- | https://github.com/wdecoster/nanostat |
| PHASTER | -- | -- | https://phaster.ca/ |
| Prokka | -- | -- | https://github.com/tseemann/prokka |
| QUAST | -- | -- | http://quast.sourceforge.net/quast |
| Trim Galore | -- | -- | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| Trimmomatic | -- | -- | http://www.usadellab.org/cms/?page=trimmomatic |
| Unicycler | -- | -- | https://github.com/rrwick/Unicycler#necessary-read-length |
Références
- Brubaker, L., Wolfe, A. The urinary microbiota: a paradigm shift for bladder disorders. Current Opinion in Obstetrics & Gynecology. 28 (5), 407-412 (2016).
- Neugent, M. L., Hulyalkar, N. V., Nguyen, V. H., Zimmern, P. E., De Nisco, N. J. Advances in understanding the human urinary microbiome and its potential role in urinary tract infection. mBio. 11 (2), (2020).
- Klein, R. D., Hultgren, S. J. Urinary tract infections: microbial pathogenesis, host-pathogen interactions and new treatment strategies. Nature Reviews. Microbiology. 18 (4), 211-226 (2020).
- Horsley, H., et al. Enterococcus faecalis subverts and invades the host urothelium in patients with chronic urinary tract infection. PLoS One. 8 (12), 83637 (2013).
- Reitzer, L., Zimmern, P. Rapid growth and metabolism of uropathogenic Escherichia coli in relation to urine composition. Clinical Microbiology Reviews. 33 (1), 00101-00119 (2019).
- Snyder, J. A., et al. Transcriptome of uropathogenic Escherichia coli during urinary tract infection. Infection and Immunity. 72 (11), 6373-6381 (2004).
- Ipe, D. S., Horton, E., Ulett, G. C. The basics of bacteriuria: Strategies of microbes for persistence in urine. Frontiers in Cellular and Infection Microbiology. 6, 14 (2016).
- Babikir, I. H., et al. The impact of cathelicidin, the human antimicrobial peptide LL-37 in urinary tract infections. BMC Infectious Diseases. 18 (1), 17 (2018).
- Jancel, T., Dudas, V. Management of uncomplicated urinary tract infections. The Western Journal of Medicine. 176 (1), 51-55 (2002).
- Ventola, C. L. The antibiotic resistance crisis: part 1: causes and threats. P & T. 40 (4), 277-283 (2015).
- Price, T. K., et al. The clinical urine culture: Enhanced techniques improve detection of clinically relevant microorganisms. Journal of Clinical Microbiology. 54 (5), 1216-1222 (2016).
- Kass, E. H. Asymptomatic infections of the urinary tract. Transactions of the Association of American Physicians. 69, 56-64 (1956).
- Garcia, L. S. . Clinical microbiology procedures handbook. 3rd edn. , (2010).
- Fraser, C. M., Eisen, J. A., Nelson, K. E., Paulsen, I. T., Salzberg, S. L. The value of complete microbial genome sequencing (you get what you pay for). Journal of Bacteriology. 184 (23), 6403-6405 (2002).
- Chen, Z., Erickson, D. L., Meng, J. Benchmarking hybrid assembly approaches for genomic analyses of bacterial pathogens using Illumina and Oxford Nanopore sequencing. BMC Genomics. 21 (1), 631 (2020).
- Greig, D. R., Dallman, T. J., Hopkins, K. L., Jenkins, C. MinION nanopore sequencing identifies the position and structure of bacterial antibiotic resistance determinants in a multidrug-resistant strain of enteroaggregative Escherichia coli. Microbial Genomics. 4 (10), 000213 (2018).
- Carraro, D. M., et al. PCR-assisted contig extension: stepwise strategy for bacterial genome closure. Biotechniques. 34 (3), 626-628 (2003).
- Tettelin, H., Radune, D., Kasif, S., Khouri, H., Salzberg, S. L. Optimized multiplex PCR: efficiently closing a whole-genome shotgun sequencing project. Genomics. 62 (3), 500-507 (1999).
- Wick, R. R., Judd, L. M., Gorrie, C. L., Holt, K. E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Computational Biology. 13 (6), 1005595 (2017).
- Singhal, N., Kumar, M., Kanaujia, P. K., Virdi, J. S. MALDI-TOF mass spectrometry: an emerging technology for microbial identification and diagnosis. Frontiers in Microbiology. 6, 791 (2015).
- Turner, S., Pryer, K. M., Miao, V. P., Palmer, J. D. Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. The Journal of Eukaryotic Microbiology. 46 (4), 327-338 (1999).
- Weisburg, W. G., Barns, S. M., Pelletier, D. A., Lane, D. J. 16S ribosomal DNA amplification for phylogenetic study. Journal of Bacteriology. 173 (2), 697-703 (1991).
- Janda, J. M., Abbott, S. L. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. Journal of Clinical Microbiology. 45 (9), 2761-2764 (2007).
- Stevenson, K., McVey, A. F., Clark, I. B. N., Swain, P. S., Pilizota, T. General calibration of microbial growth in microplate readers. Science Reports. 6, 38828 (2016).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (15), 2114-2120 (2014).
- De Coster, W., D'Hert, S., Schultz, D. T., Cruts, M., Van Broeckhoven, C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 34 (15), 2666-2669 (2018).
- Wilson, G., et al. The UNIX Shell. Zenodo. , (2019).
- Bankevich, A., et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology. 19 (5), 455-477 (2012).
- Vaser, R., Sovic, I., Nagarajan, N., Sikic, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Research. 27 (5), 737-746 (2017).
- Walker, B. J., et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 9 (11), 112963 (2014).
- Wick, R. R., Schultz, M. B., Zobel, J., Holt, K. E. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics. 31 (20), 3350-3352 (2015).
- Gurevich, A., Saveliev, V., Vyahhi, N., Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 29 (8), 1072-1075 (2013).
- Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31 (19), 3210-3212 (2015).
- Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 30 (14), 2068-2069 (2014).
- Aziz, R. K., et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 9, 75 (2008).
- Tatusova, T., et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Research. 44 (14), 6614-6624 (2016).
- Carattoli, A., Hasman, H. PlasmidFinder and In Silico pMLST: Identification and Typing of Plasmid Replicons in Whole-Genome Sequencing (WGS). Methods in Molecular Biology. 2075, 285-294 (2020).
- Carattoli, A., et al. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrobial Agents and Chemotherapy. 58 (7), 3895-3903 (2014).
- Larsen, M. V., et al. Multilocus sequence typing of total-genome-sequenced bacteria. Journal of Clinical Microbiology. 50 (4), 1355-1361 (2012).
- Bortolaia, V., et al. ResFinder 4.0 for predictions of phenotypes from genotypes. The Journal of Antimicrobial Chemotherapy. 75 (12), 3491-3500 (2020).
- Joensen, K. G., et al. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. Journal of Clinical Microbiology. 52 (5), 1501-1510 (2014).
- Arndt, D., et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Research. 44 (1), 16-21 (2016).
- Couvin, D., et al. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Research. 46 (1), 246-251 (2018).
- Totten, P. A., Amsel, R., Hale, J., Piot, P., Holmes, K. K. Selective differential human blood bilayer media for isolation of Gardnerella (Haemophilus) vaginalis. Journal of Clinical Microbiology. 15 (1), 141-147 (1982).
- Nagarajan, N., Pop, M. Sequence assembly demystified. Nat Reviews. Genetics. 14 (3), 157-167 (2013).
- Phillippy, A. M., Schatz, M. C., Pop, M. Genome assembly forensics: finding the elusive mis-assembly. Genome Biology. 9 (3), 55 (2008).
- . Unicycler Wiki Available from: https://github.com/rrwick/Unicycler/wiki (2017)
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.