
Method Article
Гибридная сборка генома De Novo для генерации полных геномов мочевых бактерий с использованием технологий короткого и длинного считывания секвенирования
В этой статье
Резюме
Этот протокол детализирует комплексный подход к культивированию, секвенированию и сборке гибридного генома мочевых бактерий de novo. Он обеспечивает воспроизводимую процедуру для генерации полных, круговых последовательностей генома, полезных для изучения как хромосомных, так и внехромосомных генетических элементов, способствующих колонизации мочи, патогенезу и распространению устойчивости к противомикробным препаратам.
Аннотация
Полные последовательности генома предоставляют ценные данные для понимания генетического разнообразия и уникальных факторов колонизации мочевых микробов. Эти данные могут включать подвижные генетические элементы, такие как плазмиды и внехромосомный фаг, которые способствуют распространению устойчивости к противомикробным препаратам и еще больше усложняют лечение инфекции мочевыводящих путей (ИМП). В дополнение к обеспечению точного разрешения структуры генома, полные, закрытые геномы позволяют проводить детальную сравнительную геномику и эволюционный анализ. Генерация полных геномов de novo уже давно является сложной задачей из-за ограничений доступной технологии секвенирования. Парное секвенирование следующего поколения (NGS) обеспечивает высококачественное короткое чтение, часто приводящее к точным, но фрагментированным сборкам генома. Напротив, секвенирование nanopore обеспечивает длительное чтение более низкого качества, что обычно приводит к подверженным ошибкам полным сборкам. Такие ошибки могут препятствовать общегеномным исследованиям ассоциаций или давать вводящие в заблуждение результаты анализа вариантов. Поэтому гибридные подходы, сочетающие как короткие, так и длинные чтения, стали надежными методами достижения высокоточных закрытых бактериальных геномов. В настоящем документе представлен комплексный способ культивирования разнообразных мочевых бактерий, идентификации видов путем секвенирования гена 16S рРНК, экстракции геномной ДНК (гДНК) и генерации коротких и длинных считываний платформами NGS и Nanopore соответственно. Кроме того, этот метод описывает биоинформационный конвейер алгоритмов контроля качества, сборки и прогнозирования генов для генерации аннотированных полных последовательностей генома. Сочетание биоинформационных инструментов позволяет выбирать высококачественные считываемые данные для сборки гибридного генома и последующего анализа. Упрощенный подход к гибридной сборке генома de novo, описанный в этом протоколе, может быть адаптирован для использования в любых культивируемых бактериях.
Введение
Мочевой микробиом является новой областью исследований, которая разрушила многолетнее заблуждение о том, что мочевыводящие пути стерильны у здоровых людей. Члены мочевой микробиоты могут служить для балансировки мочевой среды и предотвращения инфекции мочевыводящих путей (ИМП)1,2. Уропатогенные бактерии вторгаются в мочевыводящие пути и используют различные механизмы вирулентности для вытеснения резидентной микробиоты, колонизации уротелия, уклонения от иммунных реакций и противодействия давлению окружающей среды3,4. Моча представляет собой относительно питательную среду, характеризующуюся высокой осмолярностью, ограниченной доступностью азота и углеводов, низкой оксигенацией и низким рН5,6,7. Моча также считается противомикробной, состоящей из высоких концентраций ингибирующей мочевины и антимикробных пептидов, таких как человеческий кателицидин LL-378. Изучение механизмов, используемых как резидентными бактериями, так и уропатогенами для колонизации мочевыводящих путей, имеет решающее значение для дальнейшего понимания здоровья мочевыводящих путей и разработки новых стратегий лечения ИМП. Кроме того, по мере того, как неудача передовой антимикробной терапии становится все более распространенной, все более важно контролировать распространение мобильных генетических элементов, несущих детерминанты устойчивости к противомикробным препаратам, в популяциях мочевых бактерий9,10.
Для исследования генотипов и фенотипов мочевых бактерий необходима их успешная культивирование и последующее секвенирование всего генома (WGS). Культурозависимые методы необходимы для обнаружения и идентификации жизнеспособных микробов в образцах мочи11. Стандартный клинический посев мочи включает нанесение мочи на 5% агар овечьей крови (BAP) и агар Макконки и аэробную инкубацию при 35 °C в течение 24 ч12. Однако при пороге обнаружения ≥105 КОЕ/мл13многие члены мочевой микробиоты не сообщаются этим методом. Улучшенные методы культивирования, такие как Enhanced Quantitative Urine Culture (EQUC)11, используют различные комбинации различных объемов мочи, времени инкубации, питательных сред и атмосферных условий для идентификации микробов, обычно пропускаемых стандартным посевом мочи. В этом протоколе описана модифицированная версия EQUC, называемая здесь модифицированным протоколом расширенной культуры мочи, которая позволяет культивировать различные мочевые бактерии и уропатогены с использованием селективных сред и оптимальных атмосферных условий, но по своей сути не является количественной. Успешная изоляция мочевых бактерий позволяет экстрагировать геномную ДНК (гДНК) для последующей WGS и сборки генома.
Сборки генома, в частности, полные сборки, позволяют обнаружить генетические факторы, которые могут способствовать колонизации, поддержанию ниши и вирулентности как среди резидентной микробиоты, так и среди уропатогенных бактерий. Черновые сборки генома содержат разнообразное количество смежных последовательностей (contigs), которые могут содержать ошибки секвенирования и отсутствие информации об ориентации. В полной сборке генома как ориентация, так и точность каждой пары оснований были проверены14. Кроме того, получение полных последовательностей генома дает представление о структуре генома, генетическом разнообразии и мобильных генетических элементах15. Короткие чтения сами по себе могут идентифицировать наличие или отсутствие важных генов, но не могут точно определить их геномный контекст16. Благодаря технологиям секвенирования с длительным считыванием, таким как Oxford Nanopore и PacBio, создание закрытых de novo сборок бактериальных геномов больше не требует напряженных методов, таких как ручное закрытие сборок de novo мультиплекснойПЦР 17,18. Сочетание технологий секвенирования с коротким считыванием Next Generation и технологий секвенирования с длинным считыванием Nanopore позволяет легко генерировать точные, полные и закрытые сборки бактериального генома при относительно низкихзатратах19. Короткочитаемое секвенирование производит точные, но фрагментированные сборки генома, обычно состоящие в среднем из 40-100 контигов, в то время как секвенирование нанопор генерирует длинные чтения длиной около 5-100 кб, которые менее точны, но могут служить каркасами для соединения контигов и разрешения геномной синтении. Гибридные подходы, использующие как технологии короткого, так и длинного чтения, могут производить точные и полные бактериальные геномы19.
Здесь описан комплексный протокол для выделения и идентификации бактерий из мочи человека, геномной экстракции ДНК, секвенирования и полной сборки генома с использованием гибридного подхода к сборке. Этот протокол обеспечивает особый акцент на шагах, необходимых для правильной модификации считываний, генерируемых коротким и длинным секвенированием для точной сборки закрытой бактериальной хромосомы и внехромосомных элементов, таких как плазмиды.
протокол
Бактерии культивировались из мочи, собранной у женщин по обоюдному согласию, в рамках одобренных институциональным наблюдательным советом исследований 19MR0011 (UTD) и STU 032016-006 (UTSW).
1. Модифицированный усиленный посев мочи
ПРИМЕЧАНИЕ: Все этапы культивирования должны проводиться в стерильных условиях. Стерилизуйте все инструменты, решения и носители. Очистите рабочую зону 70% этанолом, затем установите горелку Бунзена и тщательно работайте рядом с пламенем, чтобы уменьшить вероятность загрязнения. В качестве альтернативы, шкаф биобезопасности класса II может использоваться для поддержания стерильной среды. Носите соответствующие средства индивидуальной защиты (СИЗ), чтобы избежать воздействия потенциально патогенных микробов.
- Покрытие глицериновой мочой и выделение колоний
- Разморозить глицериновую мочу при комнатной температуре (РТ). После размораживания вихрь образца в течение 5 с для смешивания. В стерильных микроцентрифужных пробирках готовят 1:3 и 1:30 разведения мочи в стерильном 1x фосфатно-буферном физиологическом растворе (PBS) до конечного объема 100 мкл.
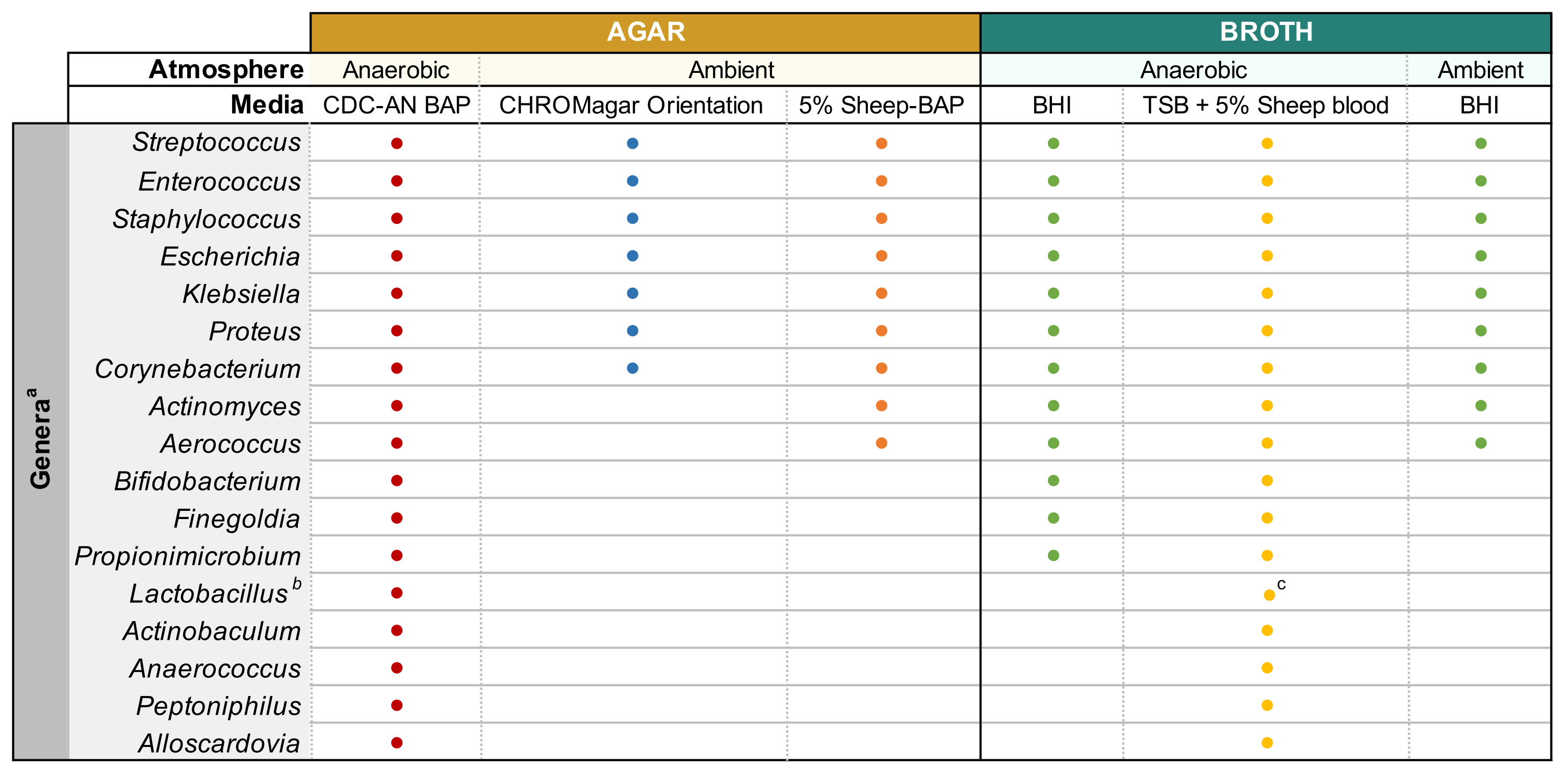
ПРИМЕЧАНИЕ: Глицериновую мочу получают путем смешивания 500 мкл неразбавленной мочи и 500 мкл 50% стерильного глицерина в криовиалах и хранения при -80 °C. - Предварительно нагревайте тарелки с агаром при 37 °C в течение 15 мин перед использованием. См. Рисунок 1 для типов сред и условий культивирования, подходящих для распространенных бактериальных родов мочи. Хорошо перемешайте разбавленную мочу путем пипетки перед нанесением, пластину 100 мкл разбавленной мочи на нужную агаровую пластину и распределите образец с помощью стерильных стеклянных шариков. Пластина 100 мкл 1x разбавителя PBS на отдельной пластине в качестве контроля роста.
ПРИМЕЧАНИЕ: При попытке культивирования общих уропатогенных видов (например, Escherichia coli, Klebsiella spp., Enterococcus faecalis и т. Д.), Рекомендуется использовать хромогенный агар(Таблица материалов),поскольку это позволяет легко идентифицировать уропатогенные виды бактерий(рисунок 1). Колистина налидиксовая кислота (CNA) или агар MRS полезны для выделения привередливых грамположительных видов (например, Lactobacillus spp.)из мочи, которая, как известно, содержит грамотрицательные уропатогены, которые могут превзойти привередливые виды в неселективных агарах. - Инкубируют перевернутую в нужном атмосферном состоянии пластину при 35 °С в течение 24 ч для уропатогенов и 3-5 дней для привередливых бактерий(рисунок 1).
- После инкубационного периода снимите пластины с инкубатора. Из каждой тарелки выберите колонии, которые демонстрируют уникальный цвет, морфологию или гемолитические узоры.
- Повторно проведите бактериальную колонию с помощью стерильной петли на соответствующий агар и инкубируйте перевернутую пластину в течение 2-5 дней в желаемой атмосфере для получения хорошо изолированных колоний.
ПРИМЕЧАНИЕ: При использовании BAP для первичной культуры, нанесение колоний на хромогенный агар может дать полезную информацию о неоднородности бактериальной популяции в образце.
- Разморозить глицериновую мочу при комнатной температуре (РТ). После размораживания вихрь образца в течение 5 с для смешивания. В стерильных микроцентрифужных пробирках готовят 1:3 и 1:30 разведения мочи в стерильном 1x фосфатно-буферном физиологическом растворе (PBS) до конечного объема 100 мкл.
- Культивирование в жидком бульоне и бактериальных изолятах с глицериновым чулками
- Как только будут получены изолированные колонии, которые соответствуют морфологии родительской колонии, выберите одну колонию и привите в 3 мл жидкого бульона, используя стерильную петлю прививки. Обратитесь к рисунку 1 для отвара, способного поддерживать рост общих родов микробиоты мочи. Запечатайте агаровые пластины парапленкой и храните при 4 °C в течение 2-4 дней. Инкубировать жидкие культуры в нужных атмосферных условиях в течение 1-5 дней до тех пор, пока культура не станет заметно мутной.
- После того, как наблюдается рост, вихрь культуры, а затем добавляют 1 мл ночной культуры к 500 мкл стерильного 50% глицерина в криовиальном 2 мл; запечатывают и аккуратно перемешивают путем инверсии. Подготовьте два запаса глицерина для каждой колонии (один служит резервным) и храните при -80 °C.
2. Идентификация видов бактерий по секвенированию гена Сэнгера 16S рРНК
ПРИМЕЧАНИЕ: Микробная идентичность может быть альтернативно подтверждена с помощью матричной лазерной десорбционной ионизационной масс-спектрометрии времени полета (MALDI-TOF)20.
- Колониеполимеразная цепная реакция (ПЦР)
- Готовят 25 мкл реакции ПЦР в пробирках ПЦР, добавляя 12,5 мкл 2x Taq Polymerase Master Mix, 0,5 мкл праймера 10 мкМ 8F, 0,5 мкл праймера 10 мкМ 1492R(Таблица материалов)и 11,5 мкл безнуклеазнойводы 21.
ПРИМЕЧАНИЕ: Если вы выполняете ПЦР для нескольких образцов, сделайте реакционную мастер-смесь из смеси Taq Polymerase, праймеров и стерильной воды без нуклеазы. Затем аликвота по 25 мкл в каждую трубку ПЦР. - Чтобы выполнить колоние-ПЦР, проведите пальцем по хорошо изолированной колонии от повторной полосы с помощью стерильной зубочистки или наконечника пипетки. Повторное суспендирование колонии в реакционной смеси ПЦР, полученной на стадии 2.1.1. Аккуратно перемешать. Соберите жидкость на дне трубки быстрым отжимом при 2000 х г.
ПРИМЕЧАНИЕ: Убедитесь, что образец не содержит пузырьков воздуха. Включите контрольный образец без шаблона (NTC), содержащий только реакционную смесь ПЦР. - Поместите пробирки в термоциклер и выполните следующую программу: 95 °C в течение 3 мин; 40 циклов: 95 °C в течение 30 с, 51 °C в течение 30 с и 72 °C в течение 1 мин 30 с; 72 °C в течение 10 мин; удерживать при 10 °C.
- Готовят 25 мкл реакции ПЦР в пробирках ПЦР, добавляя 12,5 мкл 2x Taq Polymerase Master Mix, 0,5 мкл праймера 10 мкМ 8F, 0,5 мкл праймера 10 мкМ 1492R(Таблица материалов)и 11,5 мкл безнуклеазнойводы 21.
- Экстракция геля и идентификация видов
- По завершении ПЦР проверьте продукт ПЦР на 1% агарозном геле, приготовленном в буфере 0,5x Tris-Borate-EDTA (TBE). Перед отливкой геля добавьте бромид этидия (EtBr). Затем отлить гель с помощью гребней для скважин, которые вмещают не менее 20 мкл объема образца.
ВНИМАНИЕ: EtBr является интеркалирующим агентом, подозреваемым в канцерогене. Всегда надевайте перчатки и СИЗ при обращении с ним и утилизируйте материалы, содержащие EtBr, в соответствии с руководящими принципами учреждения. - Когда гель установлен, поместите гель в резервуар для электрофореза, заполненный буфером 0,5x TBE, и удалите гребень. Загрузите лестницу 1 кб в первую скважину и 10-20 мкл реакции ПЦР в последующие скважины. Работайте при напряжении 100-140 В до тех пор, пока не будет решено. Визуализируйте гель под ультрафиолетовым светом и подтвердите наличие четко определенной полосы при ~1,5 кб, которая отсутствует в NTC хорошо.
ВНИМАНИЕ: УФ-лучи вредны для кожи и глаз, используйте соответствующую защиту при визуализации геля и носите соответствующие СИЗ.
ПРИМЕЧАНИЕ: Колониальная ПЦР может быть неудачной для некоторых бактерий; продолжение ПЦР из изолированной гДНК является альтернативным вариантом22. - Рассейте полосы размером ~1,5 кб с помощью бритвы и переложите гелевые черенки в чистые микроцентрифужные трубки. Приступают к протоколу экстракции геля в соответствии с инструкциями производителя(Таблица материалов). Измерьте концентрацию очищенной ДНК микрообъемным спектрофотометром.
ПРИМЕЧАНИЕ: Концентрация >10 нг/мкл является желательной, а A260/280 в пределах 1,7-2,0 является приемлемой. - Подготовьте две реакции секвенирования Сэнгера для каждого образца, одну с использованием 8F, а другую с использованием праймера 1492R в воде без нуклеазы в соответствии с руководящими принципами любой выбранной службы секвенирования Сэнгера.
- Как только данные секвенирования получены, загрузите последовательности ДНК на веб-сайт NCBI Basic Local Alignment Search Tool (BLAST) (blast.ncbi.nlm.nih.gov/Blast.cgi), выберите Nucleotide BLAST (blastn), выберите базу данных рРНК / ITS 16S рибосомных последовательностей РНК (бактерии и археи) и запустите программу Megablast. Изолят может быть идентифицирован по наиболее качественному обращению к ссылке из базы данных.
ПРИМЕЧАНИЕ: Некоторые виды бактерий демонстрируют высокую идентичность в своих 16S рРНК-последовательностях и могут быть неразличимы только этим методом. Видообразование потребует гомологии ДНК и биохимического анализа, чтобы уверенно различать представителей одного рода23.
- По завершении ПЦР проверьте продукт ПЦР на 1% агарозном геле, приготовленном в буфере 0,5x Tris-Borate-EDTA (TBE). Перед отливкой геля добавьте бромид этидия (EtBr). Затем отлить гель с помощью гребней для скважин, которые вмещают не менее 20 мкл объема образца.
3. Экстракция геномной ДНК (гДНК)
ПРИМЕЧАНИЕ: В этом разделе используются реагенты и спин-колонки, представленные в наборе для экстракции гДНК, упомянутом в Таблице материалов, для извлечения высокого урожая качественной геномной ДНК из различных видов бактерий. Ниже приведены рекомендуемые изменения и инструкции.
- Подготовьте набор реагентов в соответствии с инструкциями производителя.
- Готовят 3-10 мл культур в соответствующем стерильном бульоне(Рисунок 1)путем инокуляции бактерий из хорошо изолированных колоний в среду и инкубации при температуре и атмосферном давлении, отмеченных на Фиг.1, до тех пор, пока не будет наблюдаться достаточный рост.
- После инкубации измеряют оптическую плотность при 600 нм (OD600)культуры с помощью спектрофотометра24.
- Подготовьте образец к количественной оценке, разбавляя культуры на ночь в соотношении 1:10. Включите заготовку стерильной питательной среды для измерения. Рассчитайте оптическую плотность, вычитая пустое считывание из показаний образца и умножая на коэффициент разбавления, равный десяти.
- Используя измерение OD600 и заранее установленное соотношение OD600 к КОЕ/мл для вида, рассчитайте, сколько миллилитров культуры необходимо для получения 2 х 109 клеток.
- Центрифугировать необходимый объем культуры в течение 5 мин при 5000 х г на гранулу. Аспирировать супернатант и повторно суспендировать гранулу в 200 мкл холодного TE-буфера (предварительно охладить на льду в начале процедуры).
- Центрифугирование образца в течение 2 мин при 5000 х г. Удалите супернатант, а затем повторно суспендируйте гранулу в 180 мкл ферментативного лизисного буфера (ELB) и добавьте 20 мкл предварительно кипяченой РНКазы А (10 мг/мл). Для эффективного лизиса грамположительных бактерий добавляют 18 мкл мутанолизина (25 кЕд/мл). Вихрь хорошо, а затем инкубируют образцы при 37 °C на ротаторе в течение 2 ч.
ПРИМЕЧАНИЕ: Рекомендуется использовать ELB, описанный в протоколе производителя, как для грамположительных, так и для грамотрицательных бактерий. - Действуйте в соответствии с инструкциями производителя.
ПРИМЕЧАНИЕ: Повторите этапы элюирования еще один или два раза, чтобы получить дополнительный выход гДНК, если это необходимо. - Оцените качество экстрагированной гДНК в соответствии с инструкциями в разделе 4 и храните гДНК при 4 °C, если она будет использоваться в течение 1 недели. В качестве альтернативы, держите гДНК при -20 °C для длительного хранения.
4. Оценка качества экстрагированной гДНК
- Для оценки качества геля электрофорезом готовят 1% агарозный гель, как описано в подразделе 2.2. Подготовьте образец в чистой пробирке: смешайте 1-2 мкл экстрагированной гДНК и 3 мкл 2-кратного нагрузочного красителя на парапленке. Запустите гель после загрузки, а затем визуализируйте его под ультрафиолетовым светом.
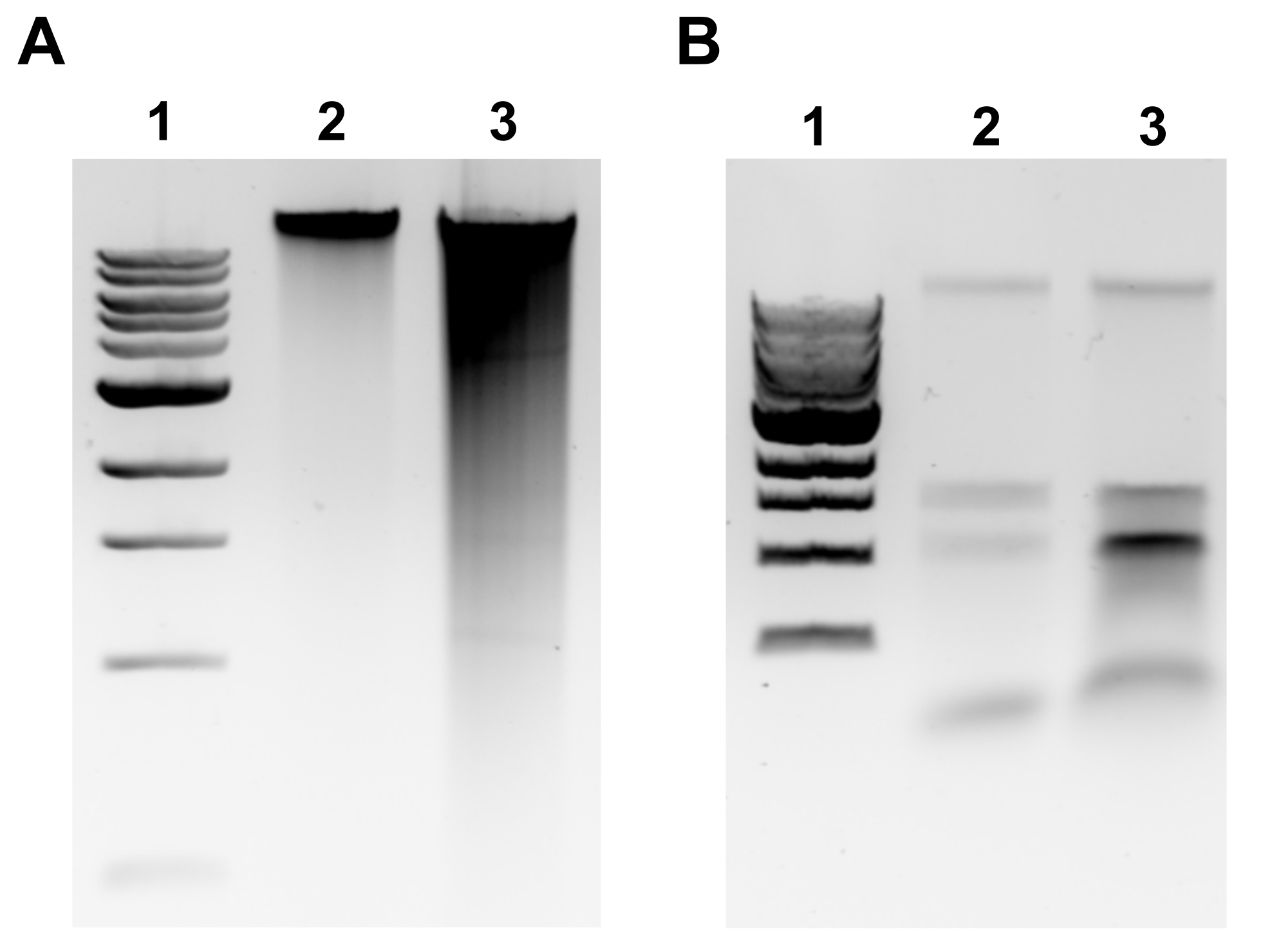
ПРИМЕЧАНИЕ: Успешная экстракция гДНК будет очевидна дискретной полосой в верхней части геля и минимальным размазыванием(рисунок 2A). Смазывание свидетельствует о стрижке. Если полоса гДНК не очевидна и/или размазывание является существенным, повторите экстракцию гДНК. Рассмотрите возможность сокращения времени инкубации в РНКазе А и протеиназе К. Если наблюдаются две полосы около 1,5-3 кб, это говорит о загрязнении РНК(рисунок 2B). Приготовьте свежую РНКазу А и повторите экстракцию. - Для оценки качества микрообъемным спектрофотометром измеряют концентрацию гДНК и коэффициент поглощения А260/280 микрообъемным спектрофотометром. Допустимыми являются концентрации >50 нг/мкл и A260/280 в пределах 1,7-2,0.
ПРИМЕЧАНИЕ: Низкий выход гДНК может быть обусловлен низким входом, высоким входом, загрязнением нуклеазами, недостаточным лизисом. Коэффициенты поглощения выше диапазона указывают на загрязнение РНК. Повторите экстракцию, если качество гДНК плохое. - Для оценки качества с помощью флуорометра следуйте инструкциям производителя по количественной оценке концентрации гДНК с использованием высокочувствительного набора для анализа и прибора флуорометра(Таблица материалов). Желательна концентрация >50 нг/мкл.
5. Сопряженное секвенирование короткого чтения следующего поколения и подготовка библиотеки
ПРИМЕЧАНИЕ: Краткосчитываемое секвенирование может выполняться на различных приборах с различной длиной считывания и ориентацией. Парное секвенирование 150 bp (300 циклов) рекомендуется для бактериальных WGS. Как подготовка библиотек, так и последовательность могут быть переданы на аутсорсинг основным объектам или коммерческим лабораториям.
- Подготовьте библиотеку секвенирования в соответствии с инструкциями производителя(Таблица материалов). Следуйте рекомендованной производителем концентрации библиотеки конечной загрузки; однако рекомендуется загрузить библиотеку пула на уровне 1,8 пМ для оптимальной генерации чтения на инструментах NextSeq.
- Хотя это необязательно, используйте биоанализатор(Таблица материалов)для оценки распределения фрагментов объединенной библиотеки и обеспечения того, чтобы размер фрагмента составлял в среднем 600 бит в секунду.
6. Подготовка библиотеки секвенирования Nanopore MinION
- Подготовьте библиотеку секвенирования по протоколу производителя(Таблица материалов). Использование двух комплектов расширения штрихкодов позволяет мультиплексировать до 24 образцов на одной проточной ячейке. Рекомендуется выполнять подготовку библиотеки в двух частях, по 12 образцов одновременно при мультиплексировании 24 образцов. Все 24 образца могут быть объединены в пул, как описано ниже.
ПРИМЕЧАНИЕ: Образцы могут храниться при температуре 4 °C в течение ночи после завершения лигирования нативного штрих-кода - это обеспечивает остановку в протоколе, если это необходимо. В конце раздела «Лигирование нативного штрих-кода» протокола подготовки библиотеки рекомендуется объединить эквимолярные количества каждого образца до максимально возможной массы ДНК (нг).- Для этого необходимо количественно оценить все образцы после лигирования штрих-кода с помощью флуорометра(Таблица материалов)в соответствии с инструкциями производителя. Оцените объем образца с самой низкой концентрацией дцДНК, а затем рассчитайте общую дцДНК, обнаруженную в этом образце. Используйте это число для определения эквимолярных количеств всех других образцов, которые будут объединены вместе.
ПРИМЕЧАНИЕ: Поскольку эквимолярное вычисление максимизирует количество объединенной dsDNA и, таким образом, даст пул большого объема (>65 мкл), очистка необходима для концентрации пула.
- Для этого необходимо количественно оценить все образцы после лигирования штрих-кода с помощью флуорометра(Таблица материалов)в соответствии с инструкциями производителя. Оцените объем образца с самой низкой концентрацией дцДНК, а затем рассчитайте общую дцДНК, обнаруженную в этом образце. Используйте это число для определения эквимолярных количеств всех других образцов, которые будут объединены вместе.
- Очистка и концентрация бассейна dsDNA
- Добавьте 2,5-кратный объем парамагнитных шариков(Таблица материалов)в пул ДНК, а затем осторожно проведите по трубке, чтобы перемешать содержимое. Поместите трубку в ротатор на 5 мин при RT. Открутите образец при 2000 х г и гранулу на магните.
- Добавьте 250 мкл свежеприготовленного 70% этанола (в воде, свободной от нуклеаз), заботясь о том, чтобы не потревожить гранулу. Аспирируйте этанол и повторите промывку этанола один раз.
- После второй аспирации открутите образец на 2000 х г и поместите его обратно на магнит. Пипетка отключите любой остаточный этанол и дайте образцу высохнуть в течение примерно 30 с.
- Извлеките трубку из магнита и повторно суспендируйте гранулу в 60-70 мкл воды, свободной от нуклеазы. Инкубировать на RT в течение 2 мин. Нанесите образец на магнит до тех пор, пока элют не станет прозрачным, а затем удалите элют и переложите в чистую микроцентрифужную трубку объемом 1,5 мл.
- Количественно оценить концентрированный бассейн с помощью флуорометра, а затем подготовить аликвоту, чтобы перейти к этапу перевязки адаптера: подготовить 700 нг образца в конечном объеме 65 мкл. Сохраните оставшуюся часть бассейна при 4 °C для второго запуска, который будет завершен после завершения первого запуска.
- Продолжайте перевязку адаптером в соответствии с инструкциями производителя и загрузите образец в проточную ячейку. Запустите запуск виртуализации.
ПРИМЕЧАНИЕ: Аспиратный воздух и ~200 мкл буфера хранения из праймирующего отверстия проточной ячейки перед загрузкой образца. Это имеет решающее значение для успешной подкачки проточной ячейки и загрузки образца. Используйте пипетку p1000 и наконечники при рисовании и нанесении растворов через заправочный порт проточной ячейки.
- Упорядочивайте библиотеку в соответствии с инструкциями производителя.
- Откройте операционное программное обеспечение для последовательности и нажмите кнопку Пуск. Введите имя эксперимента, рекомендуемая номенклатура включает дату запуска и имя пользователя. Нажмите «Продолжить выбор комплекта»,выберите соответствующий комплект для подготовки библиотеки и пакет расширения штрих-кода, а затем нажмите «Продолжить запуск параметров».
- Отрегулируйте продолжительность выполнения до 48 ч, если планируете подготовить достаточную библиотеку для второго запуска (в противном случае оставьте значение по умолчанию 72 ч). Нажмите «Продолжить к базовому вызову».
- Проверьте опцию basecalling Config: Fast Basecalling и убедитесь, что для параметра Штрихкодирование установлено значение Включено, чтобы выходные файлы FASTQ были обрезаны из последовательностей штрих-кодов и демультиплексированы в отдельные каталоги на основе штрих-кода. Нажмите «Продолжить вывод».
- Выберите, где сохранять выходные данные секвенирования. Ожидайте приблизительно 30-50 Гб данных, если только сохранить выход FASTQ, и >500 Гб данных, если также сохранить выход FAST5. Снимите флажок Фильтровать параметр Qscore: 7 | Длина чтения: Не фильтруется, если планируется продолжить фильтрацию, описанную в разделе 7.2, в противном случае оставьте флажок и установите readlength на 200.
- Нажмите «Продолжить, чтобы запустить программу установки» и просмотрите все настройки. Если настройки верны, нажмите «Пуск»,в противном случае нажмите «Назад» и внесите необходимые коррективы.
- При желании проточная ячейка может быть промыта в соответствии с инструкциями завода-изготовителя и перегружена оставшимся бассейном. Повторите шаги, описанные в версии 6.2, для оставшегося пула после завершения первого запуска и промывки проточной ячейки.
ПРИМЕЧАНИЕ: При настройке второго запуска отрегулируйте напряжение смещения до -250 мВ в соответствии с рекомендациями производителя для проточных ячеек, ранее использовавшихся в пробегах более 48 ч.
7. Оценка и подготовка чтений
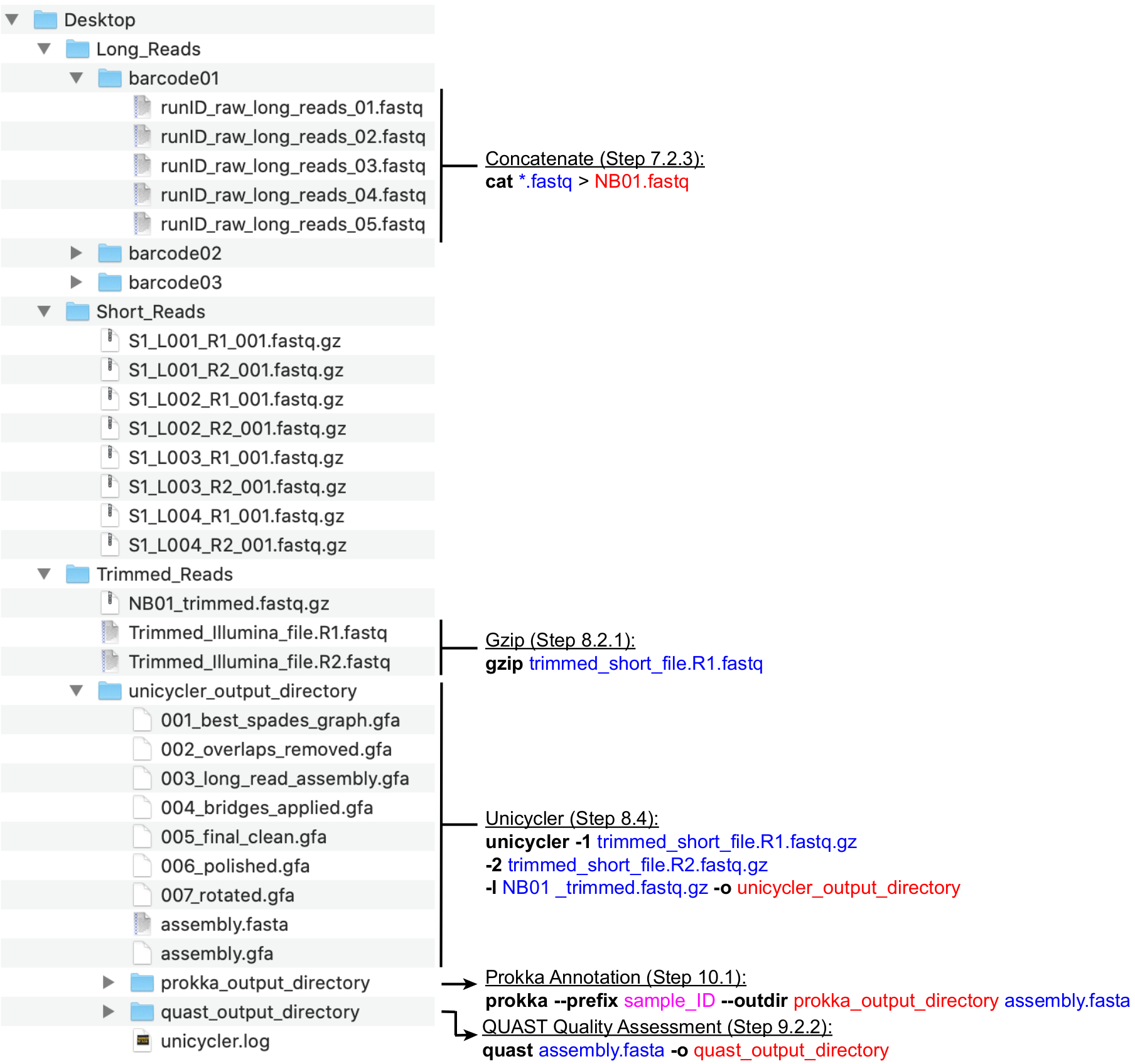
ПРИМЕЧАНИЕ: Рекомендуемая структура каталогов показана на рисунке 4. Создайте каталоги, найденные на рабочем столе,а именно: Long_Reads, Short_Reads и Trimmed_Reads, прежде чем приступить к выполнению описанных ниже шагов вычисления.
- Короткие чтения (Рисунок 3)
ПРИМЕЧАНИЕ: Короткие чтения генерируются в формате FASTQ. Файлы содержат максимум 4000 операций чтения на FASTQ. Они часто сжимаются (.gz архиве) и организуются в несколько файлов. В зависимости от платформы штрих-коды обычно обрезаются. Некоторые программы принимают файлы в сжатом формате, другие могут потребовать их извлечения перед импортом. Считыватели должны пройти этапы контроля качества (QC) для обеспечения точности данных во время сборки генома. Если CLC Genomics Workbench недоступен, для обрезки и короткого чтения QC могут использоваться альтернативные программы, такие как Trimmomatic25 или Trim Galore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) для обрезки и FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) для оценки качества чтения. Средний охват коротким чтением, оцениваемый путем умножения числа считываний на среднюю длину чтения и деления на размер генома, рекомендуется >100x.- Откройте программное обеспечение Genomics Workbench(Таблица материалов)и импортируйте все сопряженные файлы быстрого чтения fastQ. Сопряженные файлы будут сгенерированы автоматически.
- Создайте новую папку в разделе CLC_Data, нажав на кнопку Создать на верхней панели инструментов и выбрав Папка... , чтобы сохранить файлы. Присвойте папке желаемое имя, рекомендуется использовать идентификатор примера. Сохраните все выходные данные из следующих шагов в эту папку.
- На верхней панели инструментов нажмите на кнопку Импорт и выберите Illumina... Перейдите к образцу и выберите все файлы краткого чтения, соответствующие образцу. Убедитесь, что выбран параметр сопряженного чтения, и снимите флажок Удалить неудачные чтения. Нажмите «Далее»,выберите «Сохранить»и снова нажмите «Далее». Выберите сохранение импортированных файлов в новой папке, созданной на предыдущем шаге, и нажмите «Готово».
- Создайте список последовательностей всех сопряженных файлов для изолята; это объединит считываемые данные в один файл для простоты анализа.
- На верхней панели инструментов нажмите на кнопку Создать и выберите Список последовательностей... В списке каталогов слева выберите файлы для объединения и с помощью стрелок переместите их в список выбранных файлов справа. Нажмите «Далее»,выберите «Сохранить»и снова нажмите «Далее». Выберите сохранение списка последовательностей и нажмите готово.
- После создания списка последовательностей немедленно переименуйте его с идентификатором образца.
- Запустите средство контроля качества для секвенирования чтения в списке последовательностей: эта процедура оценит общие параметры качества считываний, генерируемых короткочитаемым NGS.
- Найдите инструмент контроля качества для секвенирования чтения в меню панели инструментов (левое нижнее окно). Дважды щелкните инструмент, а затем выберите список последовательностей для анализа и нажмите далее.
- Убедитесь, что отмечены все параметры вывода, и выберите Сохранить в разделе Обработка результатов. Нажмите кнопку Далее и укажите, чтобы сохранить выходные файлы, а затем нажмите кнопку Готово.
- Запустите инструмент «Обрезать чтение» в списке последовательностей: обрезка будет выполняться на основе качества, длины и неоднозначности. Этот процесс предполагает, что штрих-коды, используемые в секвенировании, были обрезаны до этого шага.
- Найдите инструмент «Обрезать чтение» на панели инструментов (левое нижнее окно). Дважды щелкните На Trim Reads, а затем выберите список последовательностей для анализа и нажмите Далее.
- Обрезка качества: установите предел показателя качества на 0,01 и оставьте неоднозначные нуклеотиды на уровне 2. Нажмите далее.
ПРИМЕЧАНИЕ: Параметры могут быть скорректированы по усмотрению пользователя; это рекомендуемые параметры. - Снимите флажок Автоматическая сквозная обрезка адаптеров (делайте это только в том случае, если адаптеры были обрезаны из чтения перед импортом в CLC). Нажмите «Далее» и установите флажок «Отменить чтение ниже длины»,используйте значение по умолчанию 15.
- Нажмите кнопку Далее,установите флажок Создать отчет,а затем выберите Сохранить. Нажмите далее и укажите, где сохранить выходные файлы. Нажмите кнопку Готово.
- Экспорт списка обрезанных последовательностей: последующая гибридная сборка и анализ будут завершены вне CLC и потребуют экспорта обрезанных файлов для короткого чтения.
- В навигации по каталогу в левом верхнем углу выберите обрезанный файл, созданный на шаге 7.1.4, а затем нажмите «Экспорт» на верхней панели инструментов. Выберите Fastq для типа файла экспорта и нажмите далее. Установите флажок Экспортировать список сопряженных последовательностей в два файла. Затем нажмите «Далее» и выберите каталог Trimmed_Reads для экспорта файлов. Нажмите кнопку Готово. Убедитесь, что обрезанные файлы краткого чтения были успешно экспортированы как два файла (R1 и R2) с расширением .fastq.
ПРИМЕЧАНИЕ: Обрезанный список последовательностей должен быть экспортирован в два файла, обычно обозначаемых CLC как R1 и R2. Это очень важно, так как последующая гибридная сборка требует ввода данных с коротким чтением, который должен быть настроен как таковой. - Переименуйте экспортированные файлы, пожалуйста, воздержитесь от использования пробелов и специальных символов в именах файлов. Для простоты рекомендуется формат trimmed_short_file. R1.fastq.
- В навигации по каталогу в левом верхнем углу выберите обрезанный файл, созданный на шаге 7.1.4, а затем нажмите «Экспорт» на верхней панели инструментов. Выберите Fastq для типа файла экспорта и нажмите далее. Установите флажок Экспортировать список сопряженных последовательностей в два файла. Затем нажмите «Далее» и выберите каталог Trimmed_Reads для экспорта файлов. Нажмите кнопку Готово. Убедитесь, что обрезанные файлы краткого чтения были успешно экспортированы как два файла (R1 и R2) с расширением .fastq.
- Откройте программное обеспечение Genomics Workbench(Таблица материалов)и импортируйте все сопряженные файлы быстрого чтения fastQ. Сопряженные файлы будут сгенерированы автоматически.
- Длинные (MinION) чтения (Рисунок 3)
ПРИМЕЧАНИЕ: Следующий конвейер для подготовки чтения секвенирования Long (MinION) для гибридной сборки использует программы26 NanoFilt и Nanostat, выполняемые командной строкой. Установите инструменты перед продолжением и ознакомьтесь с основами UNIX, чтобы выполнить эти команды. Рекомендуется использовать терминалы по умолчанию и Bash Shell. Руководство по общим командам терминала и их использованию можно найти в Software Carpentry27. Приведенные ниже инструкции предполагают, что сгенерированные файлы будут названы с номенклатурой штрих-кода (NB01, NB02 и т. Д.) И сохранены в каталоге Long_Reads. Кроме того, фильтрация чтения может быть выполнена с помощью MinKNOW при настройке выполнения виртуализации. Среднее покрытие длинного чтения рекомендуется >100x. Рекомендуемая средняя длина чтения составляет >2000 bp; поэтому необходимое число длинных считываний меньше, чем число коротких считываний.- Создайте новые каталоги для каждого штрих-кода, используемого в прогоне (штрих-код01, штрих-код02 и т.д.) в каталоге Long_Reads(рисунок 4). Скопируйте все файлы .fastq, соответствующие каждому штрих-коду, в соответствующую папку. Объедините все файлы .fastq для каждого штрих-кода с каждого запуска.
- Откройте терминал и перейдите к каталогам штрих-кодов в каталоге Long_Reads с помощью команды cd: cd Desktop/Long_Reads/barcode01
- Объедините все файлы .fastq на штрих-код в один файл .fastq, выполнив следующую команду: cat *.fastq > NB01.fastq
ПРИМЕЧАНИЕ: Эта команда объединяет все операции чтения из каждого файла FASTQ в один большой FASTQ с именем NB01.fastq. - Используйте NanoStat для оценки качества чтения образца, выполнив следующую команду: NanoStat --fastq NB01.fastq
- Запишите результаты, скопировав выходные данные в текстовый файл или файл Word для дальнейшего использования.
- Использование NanoFilt для фильтрации MinION считывает отбрасывание считываний с Q < 7 и длиной < 200 путем выполнения команды: NanoFilt -q 7 -l 200 bp NB01.fastq | gzip > NB01 _trimmed.fastq.gz
- Запустите NanoStat для обрезанного файла, созданного на шаге 7.2.6, выполнив команду: NanoStat --fastq NB01 _trimmed.fastq.gz
- Запишите результаты, скопировав выходные данные в текстовый файл или файл Word, и сравните их с результатами шага 7.2.4, чтобы убедиться, что фильтрация прошла успешно(таблица 1).
- Повторите шаги с 7.2.2 по 7.2.8 для каждого штрих-кода, используемого в цикле виртуализации.
ПРИМЕЧАНИЕ: Файл NB01_trimmed.fastq.gz, созданный на шаге 7.2.6, будет использоваться для гибридной сборки.
8. Создание гибридной сборки генома
ПРИМЕЧАНИЕ: Следующий сборочный конвейер использует Unicycler19,28,29,30 для объединения коротких и длинных считываний, подготовленных в разделах 7.1 и 7.2(рисунок 3). Установите Unicycler и его зависимости и выполните команды ниже. Предполагается, что файлы краткого чтения, экспортированные на шаге 7.1.5, называются trimmed_short_file. R1.fastq и trimmed_short_file. R2.fastq для простоты.
- Организуйте файлы краткого и длинного чтения в одном каталоге с именем Trimmed_Reads. Каталог должен содержать следующее:
- Файл .fastq.gz для обрезанного длительного чтения (созданный на шаге 7.2.6).
- Два файла .fastq (R1 и R2) для обрезанного короткого чтения (созданы на шаге 7.1.5).
- Перейдите в Trimmed_Reads каталога, в котором хранятся прочитанные файлы, с помощью команды cd в Terminal: cd Desktop/Trimmed_Reads
- Оказавшись в правильном каталоге, заархивируйте два коротких файла чтения, чтобы они также были в формате .fastq.gz выполнив следующую команду: gzip trimmed_short_file. R1.fastq
- Повторите шаг 8.2 для R1 и R2. Убедитесь, что все прочитанные файлы теперь находятся в формате .fastq.gz и убедитесь, что все файлы соответствуют одному и тому же изолированию.
- Начните гибридную сборку с помощью Unicycler, выполнив следующую команду:
одноколесный велосипед -1 trimmed_short_file. R1.fastq.gz -2 trimmed_short_file. R2.fastq.gz -l NB01 _trimmed.fastq.gz -o unicycler_output_directory
ПРИМЕЧАНИЕ: -o указывает каталог, в котором будут сохранены выходные данные Unicycler, Unicycler создаст этот каталог после выполнения команды; не создавайте каталог заранее. Время выполнения зависит от вычислительной мощности используемого компьютера, а также размера генома и количества считываний. Это может занять от 4 часов до 1 или 2 дней. Этот протокол выполнялся на машине CentOS Linux 7 с 250 Гб оперативной памяти, процессором Intel Xeon (R) с 2,5 ГГц, 12 практическими ядрами и 48 виртуальными ядрами. Кроме того, персональные компьютеры с 16 ГБ ОЗУ и 6-ядерными процессорами с тактовой частотой 2,6 ГГц могут вычислять эти сборки с более длительным временем обработки. - Когда запуск будет завершен, просмотрите файл unicycler.log чтобы убедиться в отсутствии ошибок - запишите количество, размер и состояние (завершено, неполно) сгенерированных контигов.
- Если неполные контиги идентифицированы (обозначены как неполные в журнале Unicycler), повторно запустите Unicycler жирным шрифтом, добавив следующий флаг к команде на шаге 8.4: --mode bold.
ПРИМЕЧАНИЕ: Полужирный режим снизит порог качества, принятый для мостов длительного считывания во время сборки; это может привести к полной сборке, но качество сборки может быть снижено. Рекомендуется использовать жирный шрифт только тогда, когда это необходимо, и в качестве предварительного доказательства для присоединения contig, которое впоследствии будет подтверждено ПЦР.
- Если неполные контиги идентифицированы (обозначены как неполные в журнале Unicycler), повторно запустите Unicycler жирным шрифтом, добавив следующий флаг к команде на шаге 8.4: --mode bold.
9. Оценка качества сборки
ПРИМЕЧАНИЕ: Следующий протокол использует Bandage31 и QUAST32,две программы, которые должны быть настроены перед использованием(рисунок 2 и рисунок 4). Bandage не требует установки после загрузки, а QUAST требует знакомства с базовым использованием командной строки. Также рекомендуется оценивать полноту генома с помощью Benchmarking Universal Single-Copy Orthologs (BUSCO)33.
- Повязка: Нажмите на Файл. Затем выберите Загрузить граф и выберите файл assembly.gfa, который был сохранен в unicycler_output_directory, созданном Unicycler на шаге 8.4. После загрузки нажмите кнопку Draw Graph на левой панели инструментов и посмотрите, как соединяются и организуются контиги (называемые узлами), чтобы оценить, завершена ли сборка(рисунок 5).
ПРИМЕЧАНИЕ: Полные сборки представлены одиночными круговыми контигами, связанными на обоих концах(рисунок 5A,B). Неполные сборки имеют несколько контигов, связанных друг с другом или линейных(рисунок 5C). Малые линейные контиги не могут быть неполными, поскольку они могут указывать на линейные внехромосомные элементы. Покрытие, также называемое глубиной, будет отмечено в повязке и представляет собой относительное обилие присоединений к хромосоме, нормализованное у Унициклера до 1х. - КВАСТ
- В терминале перейдите в папку, в которой хранятся выходные данные Unicycler, с помощью команды cd: cd Desktop/Trimmed_Reads/unicycler_output_directory
ПРИМЕЧАНИЕ: Пробелы не допускаются в пути к месту расположения сборки, т.е. никакие каталоги, ведущие к выходным данным Unicycler, не могут содержать пробелы в своем имени. Кроме того, можно скопировать файл assembly.fasta на рабочий стол для удобного доступа. - Запустите QUAST, выполнив следующую команду: quast assembly.fasta -o quast_output_directory
- Просмотрите отчеты, созданные QUAST, в выходном каталоге quast_output_directory.
- В терминале перейдите в папку, в которой хранятся выходные данные Unicycler, с помощью команды cd: cd Desktop/Trimmed_Reads/unicycler_output_directory
10. Аннотация генома
ПРИМЕЧАНИЕ: Приведенный ниже конвейер аннотаций использует Prokka34,средство командной строки, которое должно быть установлено перед использованием. В качестве альтернативы можно использовать Prokka через автоматизированный графический интерфейс K-Base(Таблица материалов)или аннотировать геномы через веб-сервер RAST35. При депонировании геномов в NCBI они будут автоматически аннотированы с помощью конвейера аннотирования генома прокариоты (PGAP)36.
- Перейдите в терминале в папку, в которой хранятся выходные данные Unicycler с помощью команды cd (см. шаг 9.2.1). Затем запустите Prokka, выполнив следующую команду: prokka --префикс sample_ID --outdir prokka_output_directory assembly.fasta
ПРИМЕЧАНИЕ: --prefix будет называть все выходные файлы на основе указанного sample_ID. --outdir создаст выходной каталог с указанным именем, где будут сохранены все выходные файлы Prokka; не создавайте выходной каталог для Prokka заранее. - Просмотрите аннотации, открыв таблицу .tsv и/или загрузив файл .gff, сгенерированный в программное обеспечение для анализа последовательностей для визуализации и анализа аннотаций(рисунок 6).
- Конкретные типы аннотаций могут быть сгенерированы в зависимости от генетических факторов, представляющих интерес. Рекомендуется начать с удобных инструментов на веб-сервере Центра геномной эпидемиологии (www.genomicepidemiology.org/) для предварительного анализа37,38,39,40,41. Доступны дополнительные инструменты для обнаружения систем CRISPR-cas и профагов(Рисунок 3)42,43.
11. Предлагаемые методы демократизации данных
- Когда это возможно, депонируйте все необработанные данные о считывании, а также собранные геномы в общедоступном хранилище, таком как NCBI Sequence Read Archive (SRA) и Genbank. Геномы автоматически аннотируются через конвейер PGAP в процессе осаждения NCBI.
Результаты
Этот протокол был оптимизирован для культивирования и секвенирования мочевых бактерий, принадлежащих к родам, перечисленным на рисунке 1. Не все мочевые бактерии культивируются этим методом. Питательные среды и условия указаны по роду на рисунке 1. Примерные оценки целостности гДНК гелевым электрофорезом показаны на рисунке 2. Обзор конвейера биоинформатики для секвенирования обработки считывания, сборки генома и аннотации описан на рисунке 3. Руководство по вычислительной структуре каталогов приведено на рисунке 4, чтобы упростить понимание протокола и обеспечить основу для успешной организации. Кроме того, включены репрезентативные полные геномы двух Klebsiella spp., K. pneumoniae и K. oxytoca,которые были сгенерированы этим протоколом. Представление этих сборок приведено на рисунке 5, а также включает дополнительный неполный пример генома K. pneumoniae. Подробный обзор каждого полностью аннотированного полного генома показан на рисунке 6. Наконец, резюме статистики чтения секвенирования приведено в таблице 1, чтобы предложить широкое понимание необработанных и усеченных данных, достаточных для генерации высококачественных закрытых сборок генома. Дополнительно ключевые параметры двух представителей комплектуют Klebsiella spp. геномы перечислены. Геномы и необработанные данные были депонированы в Генбанке в рамках БиоПроекта PRJNA683049.

Рисунок 1:Модифицированный усиленный посев мочи различных мочевых родов. Диаграмма для агара и жидкого бульона, который может быть использован для культивирования различных мочевых родов. Все культивирование предлагается проводить при температуре 35°С, как описано в подразделе 1.1. Круги представляют собой носители, подходящие для культивирования определенного рода, цвета были произвольно выбраны, чтобы отличить один тип медиа от другого. CDC-AN BAP (красный), CDC Анаэроб Овечий Кровавый Агар; 5% Sheep-BAP (оранжевый), Sheep Blood Agar; BHI (зеленый), инфузия сердца мозга; TSB (желтый), Триптический соевый бульон; Ориентация CHROMagar (синий). aGardnerella vaginalis следует культивировать на HBT Bilayer G. vaginalis Селективный агар в микроаэрофильной атмосфере и под специальными требованиями к культуре бульона44. bLactobacillus iners следует культивировать на 5% пластинах Rabbit-BAP и бульоне NYCIII в микроаэрофильной атмосфере. cLactobacillus spp. может культивироваться на МРС в микроаэрофильных условиях. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

Рисунок 2:Геномная экстракция ДНК изображения агарозного геля. Репрезентативные изображения геля, изображающие результаты экстракции гДНК. (A) Полоса 1: лестница размером 1 кб, полоса 2: неповрежденная гДНК, представляющая успешную экстракцию, полоса 3: смазывание, указывающее на фрагментированную гДНК. (B) Полоса 1: лестница 1 кб, полосы 2 и 3: загрязнение рРНК, обозначаемое двумя полосами между 1,5 кб и 3 кб. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

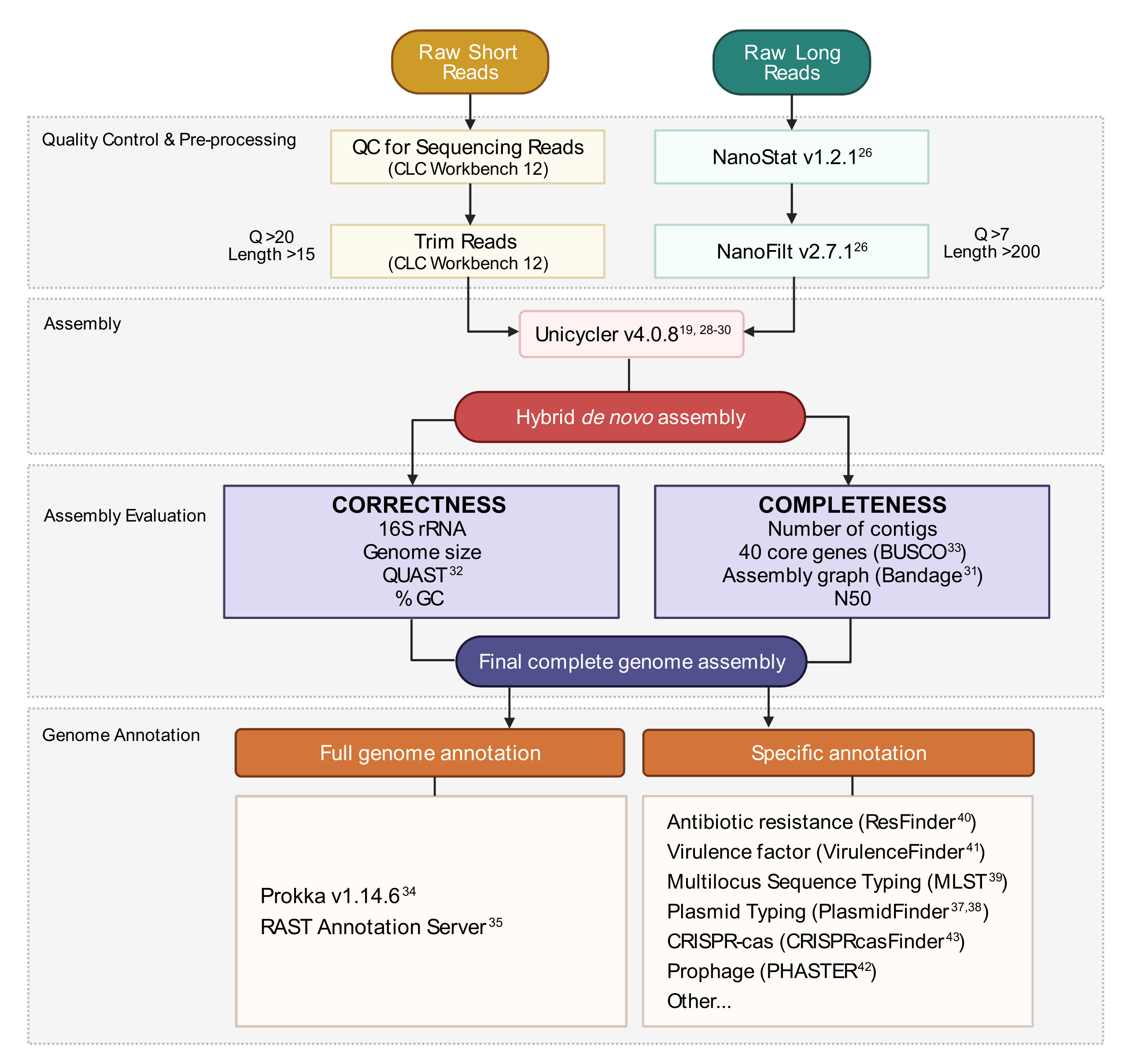
Рисунок 3:Рабочий процесс сборки гибридного генома. Схема этапов от контроля качества чтения и предварительной обработки до аннотации сборки. Обрезка чтения удаляет неоднозначные и некачественные чтения. Параметры Q-score и length указываются и представляют собой сохраненные показания. Сборка использует как короткие, так и длинные чтения для создания гибридной сборки генома de novo. Качество сборки оценивается на основе полноты и корректности с использованием заданных инструментов и параметров. Окончательная сборка генома аннотируется для всех генов и конкретных локусов, представляющих интерес. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

Рисунок 4:Руководство по структуре каталогов биоинформатики. Схема рекомендуемой организации каталогов и файлов для обработки коротких и длинных считываний, гибридной сборки, аннотации генома и контроля качества. Основные этапы обработки данных командной строки выделяются рядом с соответствующими файлами и каталогами. Получение команд и флагов (полужирный шрифт), входных файлов (синий), выходных файлов или каталогов (красный), пользовательского ввода, такого как соглашение об именовании файлов (magenta). Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

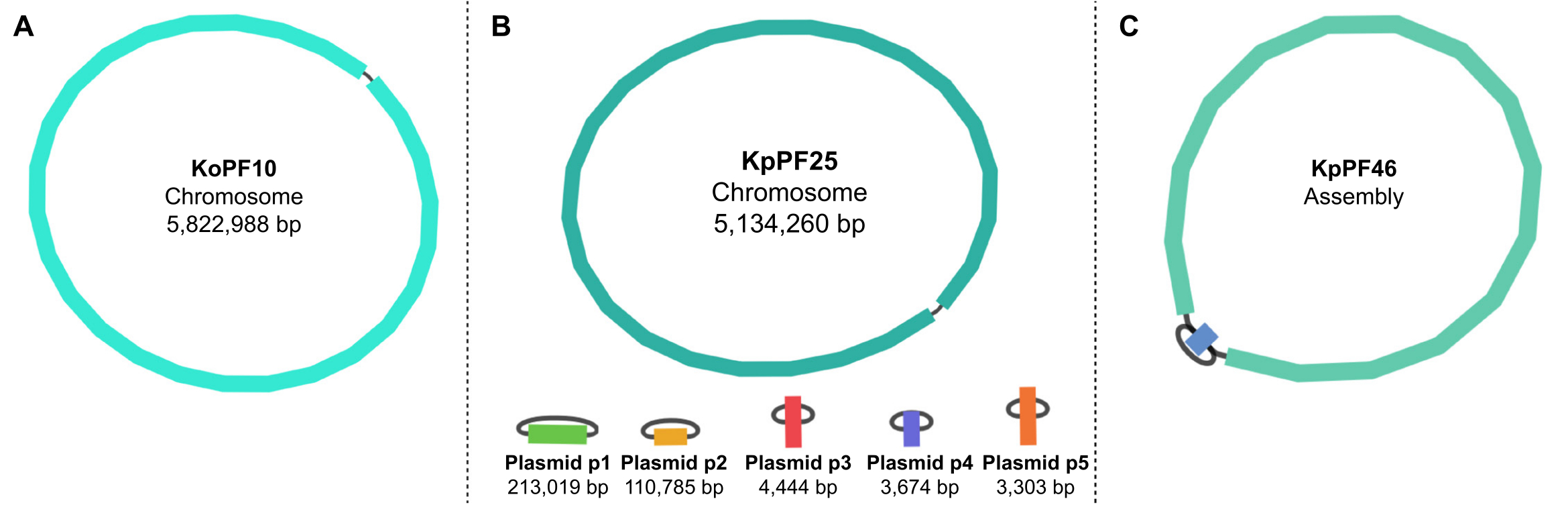
Рисунок 5:Графики сборки генома с помощью bandage. Репрезентативные полные графики сборки генома(A) Klebsiella oxytoca KoPF10 и(B) Klebsiella pneumoniae KpPF25 и неполная сборка генома(C) Klebsiella pneumoniae KpPF46. Полный геном KoPF10 демонстрирует одну закрытую хромосому, а полный геном KpPF25 состоит из закрытой хромосомы и пяти закрытых плазмид. Неполная хромосома KpPF46 состоит из двух взаимосвязанных контигов. Unicycler hybrid de novo assembly генерирует график сборки, который визуализируется Bandage. Сборочный граф обеспечивает упрощенную схему генома, указывающую на замкнутую хромосому или плазмиды с помощью линкера, соединяющего два конца одного контига. Наличие более одного взаимосвязанного контига указывает на неполную сборку. Размер и глубина Contig также можно отметить в Bandage. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

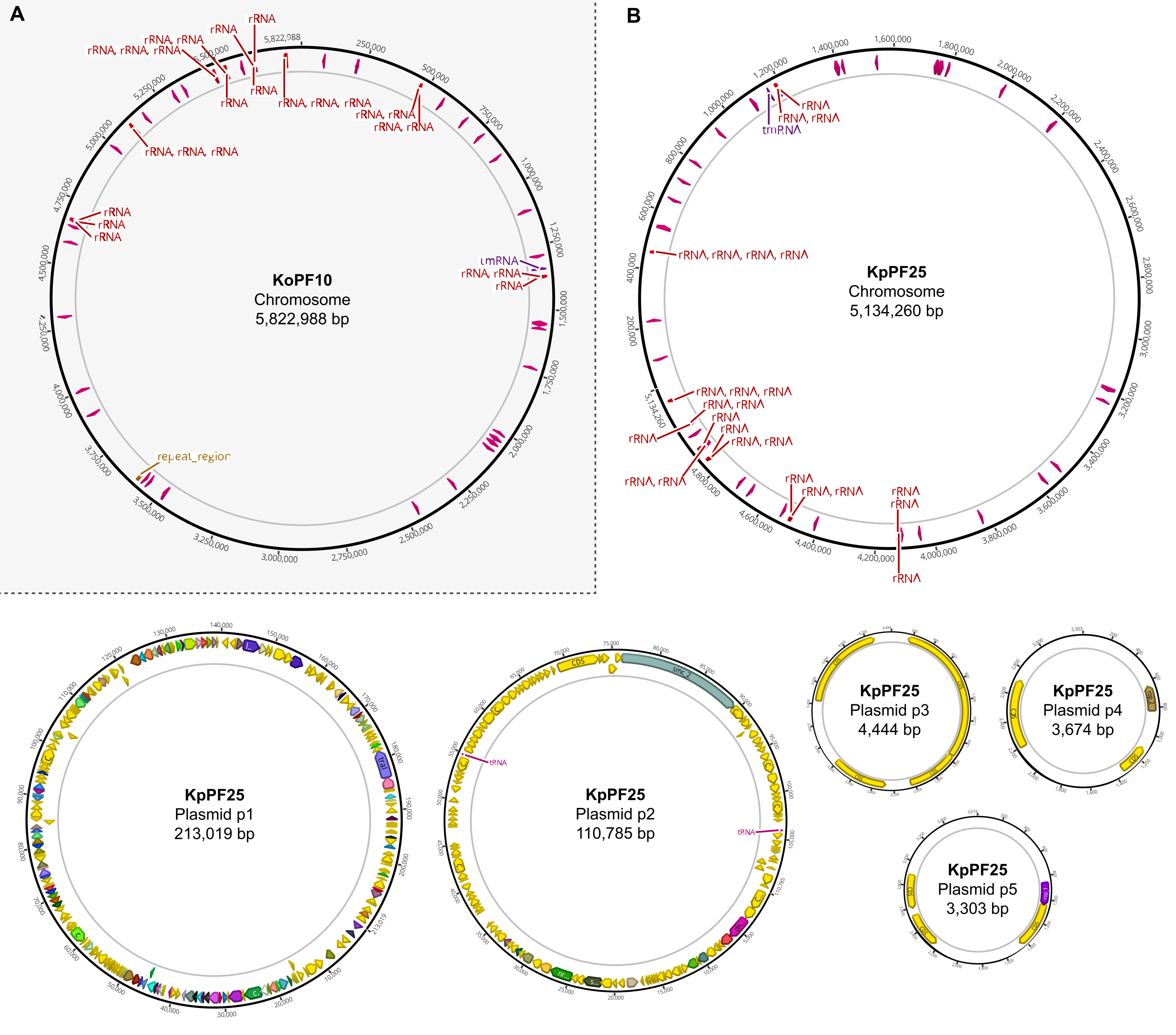
Рисунок 6:Полные карты генома аннотированных гибридных сборок. Сборочные карты, сгенерированные Geneious Prime для полного генома(A) K. oxytoca KoPF10 и(B) K. pneumoniae KpPF25, показывающие аннотированные гены, обозначаемые цветными стрелками вдоль плазмидных магистралей. Хромосомы показывают только гены рРНК и тРНК для простоты. Аннотации генома выполнялись с использованием Prokka, как указано в разделе 10 этого протокола. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

Таблица 1: Представитель Klebsiella spp.полные сборочные характеристики. Приведены параметры сборки штамма K. oxytoca KoPF10 и K. pneumoniae штамма KpPF25. Приведены номера присоединения для депонированных данных по NCBI. Количество считываний как до, так и после обрезки определяется для обеих технологий виртуализации. N50 предоставляется только для длинных считываний, поскольку короткие чтения имеют контролируемую длину. Плазмидный репликон предсказан с использованием базы данных PlasmidFinder v2.1 Enteroebacteriaceae с параметрами, установленными на 80% идентичности и 60% длины. mlST, тип последовательности multilocus. b CDS, Кодирование последовательностей. c Плазмидный репликон предсказан с использованием базы данных PlasmidFinder v2.1 Enterobacteriaceae с параметрами, установленными на 80% идентичности и 60% длины. d Оксфордские нанопоровые технологии (ONT) депонировали данные о считывании. e Illumina депонировала данные о считывании. Пожалуйста, нажмите здесь, чтобы загрузить эту таблицу.
Обсуждение
Комплексный протокол сборки гибридного генома, описанный здесь, предлагает упрощенный подход для успешного культивирования разнообразной мочевой микробиоты и уропатогенов, а также полной сборки их геномов. Успешная WGS бактериальных геномов начинается с выделения разнообразных и иногда привередливых микробов с целью извлечения их геномной ДНК. На сегодняшний день существующие протоколы посева мочи либо не обладают необходимой чувствительностью для обнаружения многих видов мочеиспускания, либо включают длительные и обширные подходы, требующие длительного времени и ресурсов11. Описанный модифицированный улучшенный подход к посеву мочи предлагает упрощенный, но всеобъемлющий протокол для успешной изоляции бактерий, принадлежащих к 17 общим мочевым родам, включая потенциально патогенные или полезные комменсальные виды, а также факультативные и облигатные аэробные или анаэробные бактерии. Это, в свою очередь, обеспечивает необходимый исходный материал для точного секвенирования и сборки бактериальных геномов и для критических фенотипических экспериментов, которые способствуют пониманию здоровья и заболеваний мочеиспускания. Кроме того, этот модифицированный культуральный подход обеспечивает более определенную клиническую диагностику жизнеспособных микроорганизмов, обнаруженных в образцах мочи, и позволяет проводить их биобанк для будущих геномных исследований. Однако этот протокол не лишен ограничений. Это может потребовать длительного времени инкубации в зависимости от организма, а также использования ресурсов, таких как камера гипоксии или контролируемые инкубаторы, которые могут быть недоступны. Использование анаэробных газовых пакетов предлагает альтернативное решение, но они являются дорогостоящими и не всегда создают устойчивую и контролируемую среду. Наконец, смещение культуры, а также разнообразие образцов могут позволить определенным организмам и уропатогенам превзойти привередливые бактерии. Несмотря на эти ограничения, культура разнообразных мочевых бактерий становится возможной благодаря такому подходу.
Геномное секвенирование приобрело популярность с развитием технологий секвенирования следующего поколения, которые значительно увеличили как выход, так и точность данных секвенирования14,15. В сочетании с разработкой алгоритмов обработки данных и сборки de novo, полные последовательности генома находятся на кончиках пальцев как начинающих, так и опытных ученых15,45. Знания об общей организации генома, обеспечиваемые полными геномами, предлагают важные эволюционные и биологические идеи, включая дупликацию генов, потерю генов и горизонтальный перенос генов14. Кроме того, гены, важные для устойчивости к противомикробным препаратам и вирулентности, часто локализуются на подвижных элементах, которые обычно не разрешаются в черновых сборках генома15,16.
Протокол в настоящем документе следует гибридному подходу для комбинации данных секвенирования с платформ короткого и длинного чтения для создания полных сборок генома. Хотя эта процедура сосредоточена на геномах мочевых бактерий, она может быть адаптирована к различным бактериям из различных источников изоляции. Критические шаги в этом подходе включают следование адекватной стерильной технике и использование соответствующих сред и условий культивирования для выделения чистых мочевых бактерий. Кроме того, извлечение неповрежденной высокопроизводительной гДНК имеет важное значение для генерации данных секвенирования, свободных от загрязняющих считываний, которые могут препятствовать успеху сборки. Последующие протоколы подготовки библиотек имеют решающее значение для создания качественных чтений достаточной длины и глубины. Поэтому крайне важно обращаться с гДНК с осторожностью во время подготовки библиотеки к секвенированию длительного чтения, в частности, поскольку самым большим преимуществом этой технологии является генерация длинных считываний без теоретического верхнего предела длины. Также выделены разделы для надлежащего контроля качества (QC) последовательности считывания, которая устраняет шумные данные и улучшает результаты сборки.
Несмотря на успешную изоляцию ДНК, подготовку библиотек и секвенирование, природа геномной архитектуры некоторых видов все еще может служить препятствием для генерации закрытой сборки генома45,46. Повторяющиеся последовательности часто усложняют вычисления сборки, и, несмотря на данные длительного чтения, эти области могут быть разрешены с низкой степенью достоверности или вообще не разрешаться. Таким образом, длительное чтение должно быть в среднем длиннее, чем самая большая повторяющаяся область в геноме, или покрытие должно быть высоким (>100x)19. Некоторые геномы могут оставаться неполными и требуют ручных подходов для завершения. Тем не менее, гибридные собранные неполные геномы, как правило, состоят из меньшего количества контигов, чем короткочитаемые черновые геномы. Настройка параметров алгоритма сборки по умолчанию или следование более строгим отсечкам для контроля качества чтения может помочь. В качестве альтернативы, один из предлагаемых подходов заключается в отображении длинных считываний в неполные области в поисках доказательств наиболее вероятного пути сборки, а затем в подтверждении пути с использованием СЕКВР и секвенирования Сэнгера амплифицированной области. Предлагается картографирование считываний с использованием Minimap2, и Bandage предлагает полезный инструмент для визуализации сопоставленных считываний вдоль собранных контигов, предоставляя доказательства связи contig47.
Дополнительная проблема при создании полных геномов заключается в знакомстве и комфорте с инструментами командной строки. Многие биоинформационные инструменты разработаны, чтобы предложить вычислительные возможности любому пользователю; однако их использование зависит от понимания основ UNIX и программирования. Этот протокол направлен на предоставление достаточно подробных инструкций, позволяющих людям без предварительного опыта работы в командной строке генерировать закрытые сборки генома и аннотировать их.
Раскрытие информации
Авторам нечего раскрывать.
Благодарности
Мы благодарим д-ра Мутузи Джубайду Ислама и д-ра Люка Джойса за их вклад в этот протокол. Мы также хотели бы поблагодарить Техасский университет в Далласском геномном центре за их отзывы и поддержку. Эта работа финансировалась Фондом Уэлча, номером награды AT-2030-20200401 для N.J.D., Национальными институтами здравоохранения, номером награды R01AI116610 для K.P., и кафедрой Фелесии и Джона Кейна в области женского здоровья, проводимой P.E.Z.
Материалы
| Name | Company | Catalog Number | Comments |
| Equipment: | |||
| Bioanalyzer 2100 | Agilent | G29398A | Optional but recommended |
| Centrifuge | Eppendorf | -- | Any centrifuge for spinning conicals and microcentrifuge tubes (e.g. Models 5810R/5424R) |
| Electrophoresis | BioRad Laboratories | 1645070 | |
| Gel Imaging System | BioRad Laboratories | ChemiDoc models | |
| Incubator | ThermoFisher Scientific | -- | Any CO2 Incubator (e.g. Thermo Forma model 3110) |
| Magnetic Rack | New England BioLabs | S15095 | 12-tube rack |
| MinION | Oxford Nanopore Technologies | -- | |
| Nanodrop | ThermoFisher Scientific | ND-ONE-W | |
| NextSeq 500 | Illumina | SY-415-1002 | Other Illumina models are acceptable |
| Plate Reader | BioTek | -- | Synergy H1 |
| Qubit fluorometer | ThermoFisher Scientific | Q33238 | |
| Rotator | Benchmark Scientific | H2024 | |
| Thermocycler | ThermoFisher Scientific | -- | Any thermocycler for PCR reactions (e.g. ProFlex PCR system) |
| Materials: | |||
| 10X Phosphate Buffered Saline (PBS) | Fisher Scientific | BP3991 | |
| 10X TBE buffer | -- | -- | 1M Tris,1M Boric Acid,0.2M EDTA (pH 8.0) |
| 1429R primer | Sigma Aldrich (Custom oligos) | -- | GGTTACCTTGTTACGACTT |
| 1kb Ladder | VWR | 101228-494 | |
| 1M Tris-Cl (pH 7.5) | ThermoFisher Scientific | 15567027 | |
| 6x Loading dye | Fisher Scientific | NC0783588 | |
| 8F primer | Sigma Aldrich (Custom oligos) | -- | AGAGTTTGATCCTGGCTCAG |
| Agar | Fisher Scientific | BP1423-2 | |
| Agarose | BioRad Laboratories | 63001 | |
| AMPure XP Beads | Beckman Coulter | A63880 | |
| Anaerobe Pouch System - GasPak EZ | BD Diagnostic Systems | B260683 | |
| Boric Acid | Fisher Scientific | A73-500 | |
| Brain Heart Infusion Broth | BD Diagnostic Systems | 212304 | |
| CDC Anaerobe 5% Sheep Blood Agar | BD Diagnostic Systems | L007357 | |
| CHROMagar Orientation | BD Diagnostic Systems | PA-257481.04 | |
| DNeasy Blood & Tissue | QIAGEN | 69504 | |
| DreamTaq Master Mix | ThermoFisher Scientific | K1081 | |
| Dry Anaerobic Indicator Strips | BD Diagnostic Systems | 271051 | |
| EDTA | Fisher Scientific | S311-500 | |
| Ethanol 200 Proof | Sigma Aldrich | E7023 | For molecular biology |
| Ethidium Bromide | ThermoFisher Scientific | BP130210 | |
| Flow cell priming kit | Oxford Nanopore Technologies | EXP-FLP002 | |
| Flow cell wash kit | Oxford Nanopore Technologies | EXP-WSH003 | |
| Gel Extraction Miniprep Kit | BioBasic | BS654 | |
| Ligation sequencing kit | Oxford Nanopore Technologies | SQK-LSK109 | |
| Lysozyme | Research Products International Corp | L381005.05 | |
| Mutanolysin | Sigma Aldrich | M9901-5KU | |
| Native barcoding expansion 1-12 | Oxford Nanopore Technologies | EXP-NBD104 | |
| NEB Blunt/TA Ligase Master Mix | New England BioLabs | M0367L | |
| NEBNext FFPE DNA Repair Mix | New England BioLabs | M6630L | |
| NEBNext quick ligation buffer | New England BioLabs | B6058S | |
| NEBNext Ultra II End repair / dA-tailing module | New England BioLabs | E7546L | |
| Nextera DNA CD Indexes | Illumina | 20018708 | |
| Nextera DNA Flex Library Prep - (M) Tagmentation | Illumina | 20018705 | |
| Nuclease-free water | Sigma Aldrich | W4502 | |
| Qubit 1X dsDNA HS Assay Kit | ThermoFisher Scientific | Q33230 | |
| Qubit Assay Tubes | ThermoFisher Scientific | Q32856 | |
| Quick T4 DNA Ligase | New England BioLabs | E6056L | |
| R9 Flow cell | Oxford Nanopore Technologies | FLO-MIN106D | |
| RNase A | ThermoFisher Scientific | EN0531 | |
| Sheep Blood | Hemostat Laboratories | DS13250 | |
| TE buffer | -- | -- | 10mM Tris, 1mM EDTA (pH 8.0) |
| Triton X-100 | Sigma Aldrich | T8787 | |
| Tryptic Soy Broth | BD Diagnostic Systems | 211825 | |
| Software & Bioinformatic Tools: | |||
| Bandage | -- | -- | https://rrwick.github.io/Bandage/ |
| Center for Genomic Epidemiology | -- | -- | http://www.genomicepidemiology.org/ |
| CLC Genomics Workbench 12 | QIAGEN | -- | |
| CRISPRcasFinder | -- | -- | https://crisprcas.i2bc.paris-saclay.fr/ |
| FastQC | -- | -- | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Geneious Prime | Geneious | -- | |
| gVolante (BUSCO) | -- | -- | https://gvolante.riken.jp/ |
| Kbase Prokka Wrapper | -- | -- | https://kbase.us/applist/apps/ProkkaAnnotation/annotate_contigs/release |
| Minimap2 | -- | -- | https://github.com/lh3/minimap2 |
| MinKNOW | Oxford Nanopore Technologies | -- | |
| NanoFilt | -- | -- | https://github.com/wdecoster/nanofilt |
| NanoStat | -- | -- | https://github.com/wdecoster/nanostat |
| PHASTER | -- | -- | https://phaster.ca/ |
| Prokka | -- | -- | https://github.com/tseemann/prokka |
| QUAST | -- | -- | http://quast.sourceforge.net/quast |
| Trim Galore | -- | -- | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| Trimmomatic | -- | -- | http://www.usadellab.org/cms/?page=trimmomatic |
| Unicycler | -- | -- | https://github.com/rrwick/Unicycler#necessary-read-length |
Ссылки
- Brubaker, L., Wolfe, A. The urinary microbiota: a paradigm shift for bladder disorders. Current Opinion in Obstetrics & Gynecology. 28 (5), 407-412 (2016).
- Neugent, M. L., Hulyalkar, N. V., Nguyen, V. H., Zimmern, P. E., De Nisco, N. J. Advances in understanding the human urinary microbiome and its potential role in urinary tract infection. mBio. 11 (2), (2020).
- Klein, R. D., Hultgren, S. J. Urinary tract infections: microbial pathogenesis, host-pathogen interactions and new treatment strategies. Nature Reviews. Microbiology. 18 (4), 211-226 (2020).
- Horsley, H., et al. Enterococcus faecalis subverts and invades the host urothelium in patients with chronic urinary tract infection. PLoS One. 8 (12), 83637 (2013).
- Reitzer, L., Zimmern, P. Rapid growth and metabolism of uropathogenic Escherichia coli in relation to urine composition. Clinical Microbiology Reviews. 33 (1), 00101-00119 (2019).
- Snyder, J. A., et al. Transcriptome of uropathogenic Escherichia coli during urinary tract infection. Infection and Immunity. 72 (11), 6373-6381 (2004).
- Ipe, D. S., Horton, E., Ulett, G. C. The basics of bacteriuria: Strategies of microbes for persistence in urine. Frontiers in Cellular and Infection Microbiology. 6, 14 (2016).
- Babikir, I. H., et al. The impact of cathelicidin, the human antimicrobial peptide LL-37 in urinary tract infections. BMC Infectious Diseases. 18 (1), 17 (2018).
- Jancel, T., Dudas, V. Management of uncomplicated urinary tract infections. The Western Journal of Medicine. 176 (1), 51-55 (2002).
- Ventola, C. L. The antibiotic resistance crisis: part 1: causes and threats. P & T. 40 (4), 277-283 (2015).
- Price, T. K., et al. The clinical urine culture: Enhanced techniques improve detection of clinically relevant microorganisms. Journal of Clinical Microbiology. 54 (5), 1216-1222 (2016).
- Kass, E. H. Asymptomatic infections of the urinary tract. Transactions of the Association of American Physicians. 69, 56-64 (1956).
- Garcia, L. S. . Clinical microbiology procedures handbook. 3rd edn. , (2010).
- Fraser, C. M., Eisen, J. A., Nelson, K. E., Paulsen, I. T., Salzberg, S. L. The value of complete microbial genome sequencing (you get what you pay for). Journal of Bacteriology. 184 (23), 6403-6405 (2002).
- Chen, Z., Erickson, D. L., Meng, J. Benchmarking hybrid assembly approaches for genomic analyses of bacterial pathogens using Illumina and Oxford Nanopore sequencing. BMC Genomics. 21 (1), 631 (2020).
- Greig, D. R., Dallman, T. J., Hopkins, K. L., Jenkins, C. MinION nanopore sequencing identifies the position and structure of bacterial antibiotic resistance determinants in a multidrug-resistant strain of enteroaggregative Escherichia coli. Microbial Genomics. 4 (10), 000213 (2018).
- Carraro, D. M., et al. PCR-assisted contig extension: stepwise strategy for bacterial genome closure. Biotechniques. 34 (3), 626-628 (2003).
- Tettelin, H., Radune, D., Kasif, S., Khouri, H., Salzberg, S. L. Optimized multiplex PCR: efficiently closing a whole-genome shotgun sequencing project. Genomics. 62 (3), 500-507 (1999).
- Wick, R. R., Judd, L. M., Gorrie, C. L., Holt, K. E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Computational Biology. 13 (6), 1005595 (2017).
- Singhal, N., Kumar, M., Kanaujia, P. K., Virdi, J. S. MALDI-TOF mass spectrometry: an emerging technology for microbial identification and diagnosis. Frontiers in Microbiology. 6, 791 (2015).
- Turner, S., Pryer, K. M., Miao, V. P., Palmer, J. D. Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. The Journal of Eukaryotic Microbiology. 46 (4), 327-338 (1999).
- Weisburg, W. G., Barns, S. M., Pelletier, D. A., Lane, D. J. 16S ribosomal DNA amplification for phylogenetic study. Journal of Bacteriology. 173 (2), 697-703 (1991).
- Janda, J. M., Abbott, S. L. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. Journal of Clinical Microbiology. 45 (9), 2761-2764 (2007).
- Stevenson, K., McVey, A. F., Clark, I. B. N., Swain, P. S., Pilizota, T. General calibration of microbial growth in microplate readers. Science Reports. 6, 38828 (2016).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (15), 2114-2120 (2014).
- De Coster, W., D'Hert, S., Schultz, D. T., Cruts, M., Van Broeckhoven, C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 34 (15), 2666-2669 (2018).
- Wilson, G., et al. The UNIX Shell. Zenodo. , (2019).
- Bankevich, A., et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology. 19 (5), 455-477 (2012).
- Vaser, R., Sovic, I., Nagarajan, N., Sikic, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Research. 27 (5), 737-746 (2017).
- Walker, B. J., et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 9 (11), 112963 (2014).
- Wick, R. R., Schultz, M. B., Zobel, J., Holt, K. E. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics. 31 (20), 3350-3352 (2015).
- Gurevich, A., Saveliev, V., Vyahhi, N., Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 29 (8), 1072-1075 (2013).
- Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31 (19), 3210-3212 (2015).
- Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 30 (14), 2068-2069 (2014).
- Aziz, R. K., et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 9, 75 (2008).
- Tatusova, T., et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Research. 44 (14), 6614-6624 (2016).
- Carattoli, A., Hasman, H. PlasmidFinder and In Silico pMLST: Identification and Typing of Plasmid Replicons in Whole-Genome Sequencing (WGS). Methods in Molecular Biology. 2075, 285-294 (2020).
- Carattoli, A., et al. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrobial Agents and Chemotherapy. 58 (7), 3895-3903 (2014).
- Larsen, M. V., et al. Multilocus sequence typing of total-genome-sequenced bacteria. Journal of Clinical Microbiology. 50 (4), 1355-1361 (2012).
- Bortolaia, V., et al. ResFinder 4.0 for predictions of phenotypes from genotypes. The Journal of Antimicrobial Chemotherapy. 75 (12), 3491-3500 (2020).
- Joensen, K. G., et al. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. Journal of Clinical Microbiology. 52 (5), 1501-1510 (2014).
- Arndt, D., et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Research. 44 (1), 16-21 (2016).
- Couvin, D., et al. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Research. 46 (1), 246-251 (2018).
- Totten, P. A., Amsel, R., Hale, J., Piot, P., Holmes, K. K. Selective differential human blood bilayer media for isolation of Gardnerella (Haemophilus) vaginalis. Journal of Clinical Microbiology. 15 (1), 141-147 (1982).
- Nagarajan, N., Pop, M. Sequence assembly demystified. Nat Reviews. Genetics. 14 (3), 157-167 (2013).
- Phillippy, A. M., Schatz, M. C., Pop, M. Genome assembly forensics: finding the elusive mis-assembly. Genome Biology. 9 (3), 55 (2008).
- . Unicycler Wiki Available from: https://github.com/rrwick/Unicycler/wiki (2017)
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеThis article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены