
Method Article
Montagem híbrida de genoma de Novo para a geração de genomas completos de bactérias urinárias usando tecnologias de sequenciamento de leitura curta e longa
Neste Artigo
Resumo
Este protocolo detalha uma abordagem abrangente para a montagem de um genoma híbrido de bactérias urinárias. Fornece um procedimento reprodutível para a geração de sequências completas de genomas circulares úteis no estudo de elementos genéticos cromossômicos e extracromosômicos que contribuem para a colonização urinária, patogênese e disseminação de resistência antimicrobiana.
Resumo
Sequências completas de genomas fornecem dados valiosos para a compreensão da diversidade genética e fatores de colonização únicos de micróbios urinários. Esses dados podem incluir elementos genéticos móveis, como plasmídeos e fágio extracromosomal, que contribuem para a disseminação da resistência antimicrobiana e complicam ainda mais o tratamento da infecção do trato urinário (ITU). Além de fornecer uma boa resolução da estrutura do genoma, genomas completos e fechados permitem a genômica comparativa detalhada e análises evolutivas. A geração de genomas completos de novo tem sido uma tarefa desafiadora devido às limitações da tecnologia de sequenciamento disponível. O Sequenciamento de Próxima Geração (NGS) de ponta emparelhada produz leituras curtas de alta qualidade, muitas vezes resultando em conjuntos precisos, mas fragmentados de genomas. Pelo contrário, o sequenciamento de Nanopore fornece leituras longas de menor qualidade normalmente levando a conjuntos completos propensos a erros. Tais erros podem dificultar estudos de associação em todo o genoma ou fornecer resultados enganosos de análise de variantes. Portanto, abordagens híbridas que combinam leituras curtas e longas surgiram como métodos confiáveis para alcançar genomas bacterianos fechados altamente precisos. Relatado aqui é um método abrangente para a cultura de diversas bactérias urinárias, identificação de espécies por sequenciamento genético 16S rRNA, extração de DNA genômico (gDNA), e geração de leituras curtas e longas por plataformas NGS e Nanopore, respectivamente. Além disso, este método descreve um pipeline bioinforático de algoritmos de controle de qualidade, montagem e previsão genética para a geração de sequências completas de genoma anotadas. A combinação de ferramentas bioinformáticas permite a seleção de dados de leitura de alta qualidade para montagem de genoma híbrido e análise a jusante. A abordagem simplificada para o conjunto híbrido de novo genoma descrito neste protocolo pode ser adaptada para uso em qualquer bactéria cultural.
Introdução
O microbioma urinário é uma área emergente de pesquisa que destruiu um equívoco de décadas de que o trato urinário é estéril em indivíduos saudáveis. Os membros da microbiota urinária podem servir para equilibrar o ambiente urinário e prevenir a infecção do trato urinário (ITU)1,2. Bactérias uropatômicas invadem o trato urinário e empregam diversos mecanismos de virulência para deslocar a microbiota residente, colonizar o urotélio, evitar respostas imunológicas e neutralizar pressões ambientais3,4. A urina é um meio relativamente limitado por nutrientes caracterizado por alta osmolaridade, disponibilidade limitada de nitrogênio e carboidratos, baixa oxigenação e baixo pH5,6,7. A urina também é considerada antimicrobiana, composta de altas concentrações de ureia inibitória e peptídeos antimicrobianos, como o cathelicidina humano LL-378. Investigar mecanismos empregados tanto por bactérias residentes quanto por uropatógenos para colonizar o trato urinário é fundamental para entender melhor a saúde do trato urinário e desenvolver novas estratégias para o tratamento da ITU. Além disso, à medida que a falha das terapias antimicrobianas de linha de frente se torna mais comum, é cada vez mais importante monitorar a disseminação de elementos genéticos móveis que carregam determinantes de resistência antimicrobiana dentro de populações de bactérias urinárias9,10.
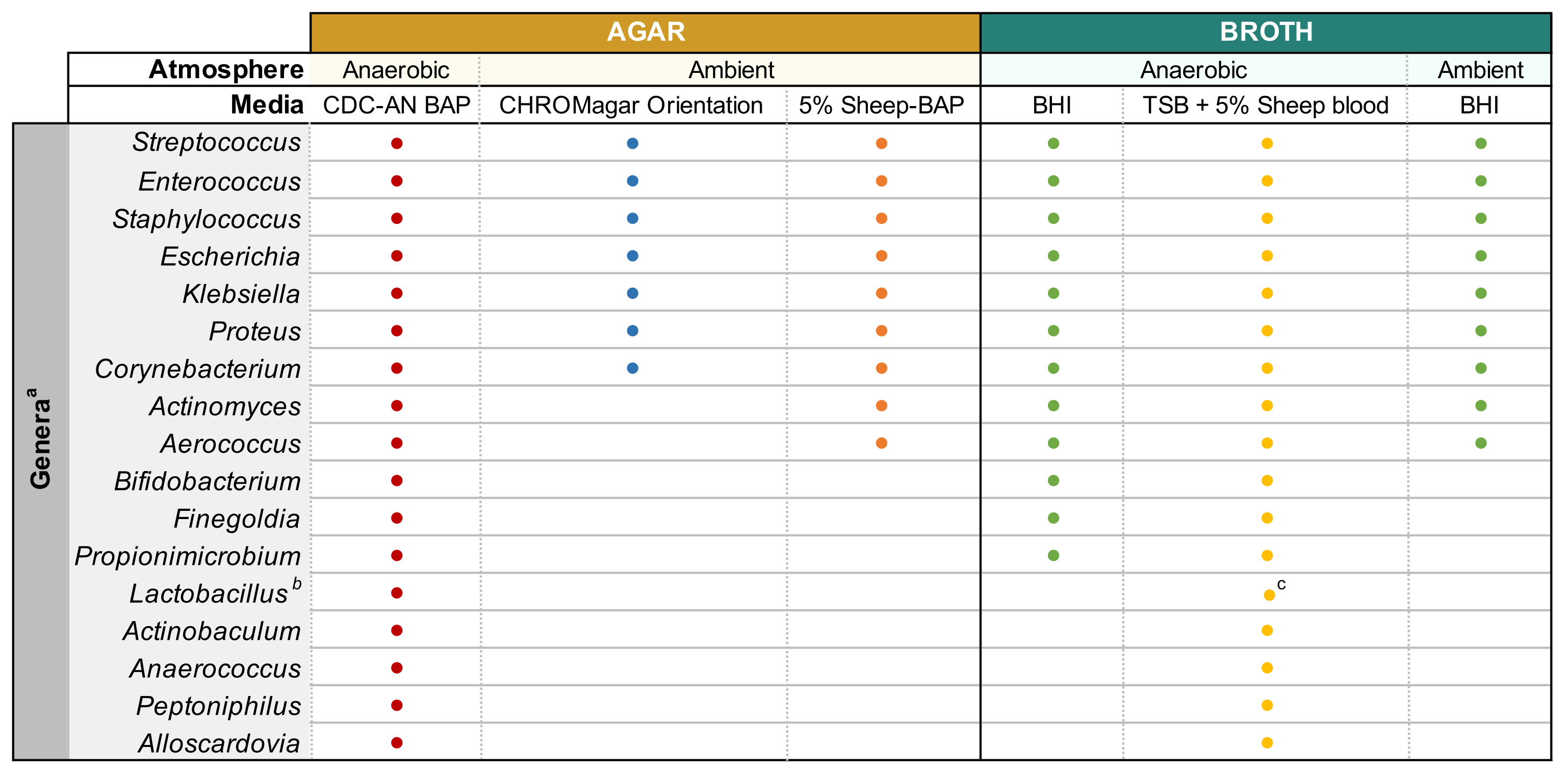
Para investigar genótipos e fenótipos de bactérias urinárias, sua cultura bem sucedida e sequenciamento de genomas inteiros subsequentes (WGS) é imperativo. Métodos dependentes da cultura são necessários para detectar e identificar micróbios viáveis em amostras de urina11. A cultura clínica de urina padrão envolve a criação de urina em 5% de ágar de sangue de ovelha (BAP) e ágar MacConkey e incubação aerobicamente a 35 °C por 24 h12. No entanto, com um limiar de detecção de≥10 5 CFU/mL13, muitos membros da microbiota urinária não são relatados por este método. Técnicas aprimoradas de cultivo, como a Cultura Quantitativa Aprimorada da Urina (EQUC)11 empregam várias combinações de diferentes volumes de urina, tempos de incubação, mídia cultural e condições atmosféricas para identificar micróbios comumente perdidos pela cultura de urina padrão. Descrito neste protocolo é uma versão modificada do EQUC, denominada aqui protocolo Modified Enhanced Urine Culture, que permite a cultivo de diversas bactérias urinárias e uropatógenos usando mídia seletiva e condições atmosféricas ideais, mas não é inerentemente quantitativo. O isolamento bem sucedido das bactérias urinárias permite a extração de DNA genômico (gDNA) para WGS a jusante e montagem de genoma.
Montagens de genomas, conjuntos completos em particular, permitem a descoberta de fatores genéticos que podem contribuir para a colonização, manutenção de nicho e virulência entre as microbiotas residentes e as bactérias uropatogênicas. Os conjuntos de genomas de rascunho contêm um número diversificado de sequências conguosas (contigs) que podem conter erros de sequenciamento e falta de informações de orientação. Em um conjunto completo do genoma, tanto a orientação quanto a precisão de cada par de base foram verificadas14. Além disso, a obtenção de sequências completas de genomas fornece uma visão da estrutura do genoma, da diversidade genética e dos elementos genéticosmóveis 15. Leituras curtas por si só podem identificar a presença ou ausência de genes importantes, mas podem não identificar seu contexto genômico16. Com a habilitação de tecnologias de sequenciamento de leitura longa, como Oxford Nanopore e PacBio, gerar conjuntos fechados de novos genomas bacterianos não requer mais métodos extenuantes, como o fechamento manual de novos conjuntos pelo multiplex PCR17,18. A combinação de tecnologias de sequenciamento de leitura curta da Próxima Geração e nanopore de sequenciamento de longa leitura permite a geração fácil de conjuntos precisos, completos e fechados de genoma bacteriano a custos relativamente baixos19. Sequenciamento de leitura curta produz conjuntos precisos e fragmentados de genoma geralmente consistindo de uma média de 40-100 contigs, enquanto o sequenciamento de nanoporo gera leituras longas de cerca de 5-100 kb de comprimento que são menos precisas, mas podem servir como andaimes para se juntar a contigs e resolver a sintensia genômica. Abordagens híbridas utilizando tecnologias de leitura curta e longa podem produzir genomas bacterianos precisos e completos19.
Descrito aqui é um protocolo abrangente para o isolamento e identificação de bactérias da urina humana, extração genômica de DNA, sequenciamento e montagem completa do genoma usando uma abordagem híbrida de montagem. Este protocolo fornece uma ênfase especial nas etapas necessárias para modificar adequadamente as leituras geradas pelo sequenciamento de leitura curta e longa leitura para a montagem precisa de um cromossomo bacteriano fechado e elementos extracromosomais, como plasmídeos.
Protocolo
As bactérias foram cultivadas a partir da urina coletada de mulheres consentidas como parte de estudos aprovados pelo conselho de revisão institucional 19MR0011 (UTD) e STU 032016-006 (UTSW).
1. Cultura de urina aprimorada modificada
NOTA: Todas as etapas culturais devem ser realizadas em condições estéreis. Esterilize todos os instrumentos, soluções e mídia. Limpe a área de trabalho com 70% de etanol, depois configure um queimador Bunsen e trabalhe cuidadosamente perto da chama para reduzir as chances de contaminação. Alternativamente, um armário de biossegurança classe II pode ser usado para manter um ambiente estéril. Use equipamentos de proteção individual (EPI) adequados para evitar exposição a micróbios potencialmente patogênicos.
- Emplacando urina abastecida com glicerol e isolamento de colônia
- Descongele urina abastecida com glicerol à temperatura ambiente (RT). Uma vez descongelado, vórtice a amostra para 5 s para misturar. Em tubos de microcentrifuuge estéreis, prepare 1:3 e 1:30 diluições da urina em salina estéril 1x Tamponado de Fosfato (PBS) a um volume final de 100 μL.
NOTA: A urina estocada de glicerol é preparada misturando 500 μL de urina não diluída e 500 μL de glicerol estéril de 50% em criovials e armazenando a -80 °C. - Placas de ágar pré-aquecidas a 37 °C por 15 minutos antes de usar. Consulte a Figura 1 para tipos de mídia e condições de cultura adequadas aos gêneros bacterianos urinários comuns. Misture bem a urina diluída por pipetação antes de chapear, placa de 100 μL da urina diluída na placa de ágar desejada e espalhe a amostra usando contas de vidro estéreis. Placa 100 μL do 1x PBS diluído em uma placa separada como um controle de crescimento sem crescimento.
NOTA: Se tentar cultivar espécies uropatômicas comuns (por exemplo, Escherichia coli, Klebsiella spp., Enterococcus faecalis, etc.), recomenda-se o uso de ágar cromogênico(Tabela de Materiais),pois permite fácil identificação de espécies bacterianas uropatômicas(Figura 1). O ácido nalidúlico colistina (CNA) ou ágar MRS são úteis para isolar espécies gram-positivas relanceiras (por exemplo, Lactobacillus spp.) da urina conhecida por conter uropatógenos Gram-negativos, que podem superar as espécies exigentes em ágares não-seletivos. - Incubar a placa invertida na condição atmosférica desejada a 35 °C por um período de 24h para uropatógenos e 3-5 dias para bactérias exigentes(Figura 1).
- Após o período de incubação, remova as placas da incubadora. De cada prato, escolha as colônias que exibem uma cor única, morfologia ou padrões hemoléticos.
- Re-listrar a colônia bacteriana usando um laço estéril no ágar correspondente e incubar a placa invertida por 2-5 dias na atmosfera desejada para obter colônias bem isoladas.
NOTA: Se utilizar BAP para cultura primária, as colônias de remendação em ágar cromogênico podem fornecer informações úteis sobre a heterogeneidade da população bacteriana na amostra.
- Descongele urina abastecida com glicerol à temperatura ambiente (RT). Uma vez descongelado, vórtice a amostra para 5 s para misturar. Em tubos de microcentrifuuge estéreis, prepare 1:3 e 1:30 diluições da urina em salina estéril 1x Tamponado de Fosfato (PBS) a um volume final de 100 μL.
- Cultivo em caldo líquido e isolados bacterianos de estocagem de glicerol
- Uma vez que as colônias isoladas que correspondem à morfologia da colônia-mãe são obtidas, escolha uma única colônia e inocula em 3 mL de caldo líquido usando um laço de inoculação estéril. Consulte a Figura 1 para caldo capaz de suportar o crescimento de gêneros de microbiota urinária comum. Sele as placas de ágar com parafilm e armazene-as a 4 °C durante 2-4 dias. Incubar culturas líquidas nas condições atmosféricas desejadas por 1-5 dias até que a cultura esteja visivelmente turva.
- Após o crescimento ser observado, o vórtice da cultura e, em seguida, adicione 1 mL da cultura da noite para o dia a 500 μL de glicerol estéril em um criovial de 2 mL; selar e misturar suavemente por inversão. Prepare dois estoques de glicerol para cada colônia (um serve como backup) e armazene a -80 °C.
2. Identificação de espécies bacterianas pelo sequenciamento do gene Sanger de 16S rRNA
NOTA: A identidade microbiana pode ser confirmada alternativamente usando o Tempo de Ionização de Desorção a Laser Assistido por Matriz (MALDI-TOF)20.
- Reação da cadeia Colônia-Polimerase (PCR)
- Prepare um 25 μL da reação PCR em tubos PCR adicionando 12,5 μL de 2x Taq Polymerase Master Mix, 0,5 μL de 10 μM 8F primer, 0,5 μL de 10 μM 1492R primer(Tabela de Materiais), e 11,5 μL de água sem nuclease21.
NOTA: Se realizar PCR para várias amostras, faça uma mistura mestre de reação de mistura de Polimerase Taq, primers e água livre de nuclease estéril. Em seguida, alíquota de 25 μL em cada tubo PCR. - Para realizar a colônia-PCR, deslize uma colônia bem isolada da re-raia usando um palito de dente estéril ou ponta de pipeta. Resuspense a colônia no mix de reação PCR preparado na etapa 2.1.1. Misture suavemente. Colete o líquido na parte inferior do tubo por um giro rápido a 2000 x g.
NOTA: Certifique-se de que a amostra está livre de bolhas de ar. Inclua uma amostra de controle sem modelo (NTC) contendo apenas o mix de reação PCR. - Coloque os tubos de amostra no termociclador e execute o seguinte programa: 95 °C por 3 min; 40 ciclos de: 95 °C para 30 s, 51 °C para 30 s e 72 °C para 1 min 30 s; 72 °C por 10 min; mantenha a 10 °C.
- Prepare um 25 μL da reação PCR em tubos PCR adicionando 12,5 μL de 2x Taq Polymerase Master Mix, 0,5 μL de 10 μM 8F primer, 0,5 μL de 10 μM 1492R primer(Tabela de Materiais), e 11,5 μL de água sem nuclease21.
- Extração de gel e identificação de espécies
- Após a conclusão da execução do PCR, verifique o produto PCR em um gel de 1% de agarose preparado em 0,5x Tris-Borate-EDTA (TBE). Antes de lançar o gel, adicione brometo de ethidium (EtBr). Em seguida, lance o gel usando pentes para poços que possuem pelo menos 20 μL de volume amostral.
ATENÇÃO: O EtBr é um agente intercalante suspeito de ser cancerígeno. Use sempre luvas e EPI ao manuseá-lo e descarte de materiais contendo EtBr de acordo com as diretrizes da instituição. - Quando o gel estiver pronto, coloque o gel no tanque de eletroforese preenchido com tampão TBE de 0,5x e remova o pente. Carregue a escada de 1 kb no primeiro poço e 10-20 μL da reação pcr em poços subsequentes. Corra em 100-140 V até resolver. Visualize o gel sob luz UV e confirme a presença de uma banda claramente definida em ~1,5 kb que está ausente no poço NTC.
ATENÇÃO: Os raios UV são prejudiciais à pele e aos olhos, usam um protetor apropriado ao visualizar o gel e usam EPI apropriados.
NOTA: Colônia PCR pode não ter sucesso para algumas bactérias; procedido com PCR de gDNA isolado é uma opção alternativa22. - Extite as faixas de ~1,5 kb usando uma navalha e transfira os cortes de gel em tubos de microcentrifusagem limpos. Proceder com o protocolo de extração de gel de acordo com as instruções do fabricante(Tabela de Materiais). Meça a concentração do DNA purificado pelo espectrofotômetro de microvolume.
NOTA: É desejável uma concentração >10 ng/μL, e a A260/280 entre 1,7-2.0 é aceitável. - Prepare duas reações de sequenciamento de Sanger para cada amostra, uma usando o 8F e outra usando a cartilha 1492R em água sem nuclease de acordo com as diretrizes de qualquer serviço de sequenciamento Sanger escolhido.
- Uma vez recebidos os dados de sequenciamento, carregue as sequências de DNA no site da NCBI Basic Local Alignment Search Tool (BLAST) (blast.ncbi.nlm.nih.gov/Blast.cgi), escolha o Nucleotide BLAST (blastn), selecione o banco de dados rRNA/ITS 16S sequências de RNA ribossômico (Bacteria e Archaea) e execute o programa Megablast. O isolado pode ser identificado pela maior qualidade atingida por uma referência do banco de dados.
NOTA: Algumas espécies bacterianas apresentam alta identidade em suas sequências de rRNA 16S e podem ser indistinguíveis apenas por este método. A especiação exigirá a disselogia de DNA e análises bioquímicas para distinguir com confiança membros do mesmo gênero23.
- Após a conclusão da execução do PCR, verifique o produto PCR em um gel de 1% de agarose preparado em 0,5x Tris-Borate-EDTA (TBE). Antes de lançar o gel, adicione brometo de ethidium (EtBr). Em seguida, lance o gel usando pentes para poços que possuem pelo menos 20 μL de volume amostral.
3. Extração de DNA genômico (gDNA)
NOTA: Esta seção utiliza reagentes e colunas spin fornecidas no kit de extração gDNA referenciado na Tabela de Materiais para a extração de alto rendimento de DNA genômico de qualidade de diversas espécies bacterianas. Fornecido abaixo são recomendados modificações e instruções.
- Prepare os reagentes do kit por instruções do fabricante.
- Prepare culturas de 3-10 mL em caldo estéril apropriado(Figura 1) inoculando bactérias de colônias bem isoladas na mídia e incubando à temperatura e pressão atmosférica observada na Figura 1 até que o crescimento suficiente seja observado.
- Após a incubação, meça a densidade óptica em 600 nm (OD600) da cultura usando um espectrofotômetro24.
- Prepare a amostra para quantificação diluindo culturas durante a noite na proporção 1:10. Inclua um branco dos meios de cultura estéreis para medição também. Calcule a densidade óptica subtraindo a leitura em branco da leitura da amostra e multiplicando-se pelo fator de diluição de dez.
- Usando a medição OD600 e uma relação OD600 a CFU/mL pré-estabelecida para a espécie, calcule quantos mililitros de cultura são necessários para obter 2 x 109 células.
- Centrifugar o volume de cultura necessário por 5 min a 5000 x g para pelota. Aspire o supernasciente e resuspense a pelota em 200 μL tampão te frio (pré-frio no gelo no início do procedimento).
- Centrifugar a amostra por 2 min a 5000 x g. Remova o supernasciente e, em seguida, resuspenque a pelota em 180 μL de Tampão de Lise Enzimática (ELB) e adicione 20 μL de RNase A pré-fervido (10 mg/mL). Para a lise eficiente das bactérias Gram-positivas, adicione 18 μL de mutanolise (25 kU/mL). Vórtice bem, e, em seguida, incubar as amostras a 37 °C no rotador por 2 h.
NOTA: Recomenda-se utilizar o ELB descrito no protocolo do fabricante para bactérias Gram-positivas e Gram-negativas. - Proceda de acordo com as instruções do fabricante.
NOTA: Repita as etapas de elução por mais uma ou duas vezes para obter rendimento adicional de gDNA, se desejar. - Avalie a qualidade do gDNA extraído conforme instruído na seção 4 e armazene gDNA a 4 °C se ele será usado dentro de 1 semana. Alternativamente, mantenha o gDNA a -20 °C para armazenamento a longo prazo.
4. Avaliar a qualidade do gDNA extraído
- Para avaliar a qualidade por eletroforese gel, prepare 1% de gel de agarose conforme descrito na subseção 2.2. Prepare a amostra em um tubo limpo: misture 1-2 μL de gDNA extraído e 3 μL de corante de carregamento de 2x no parafilme. Execute o gel uma vez carregado e, em seguida, visualize-o sob a luz UV.
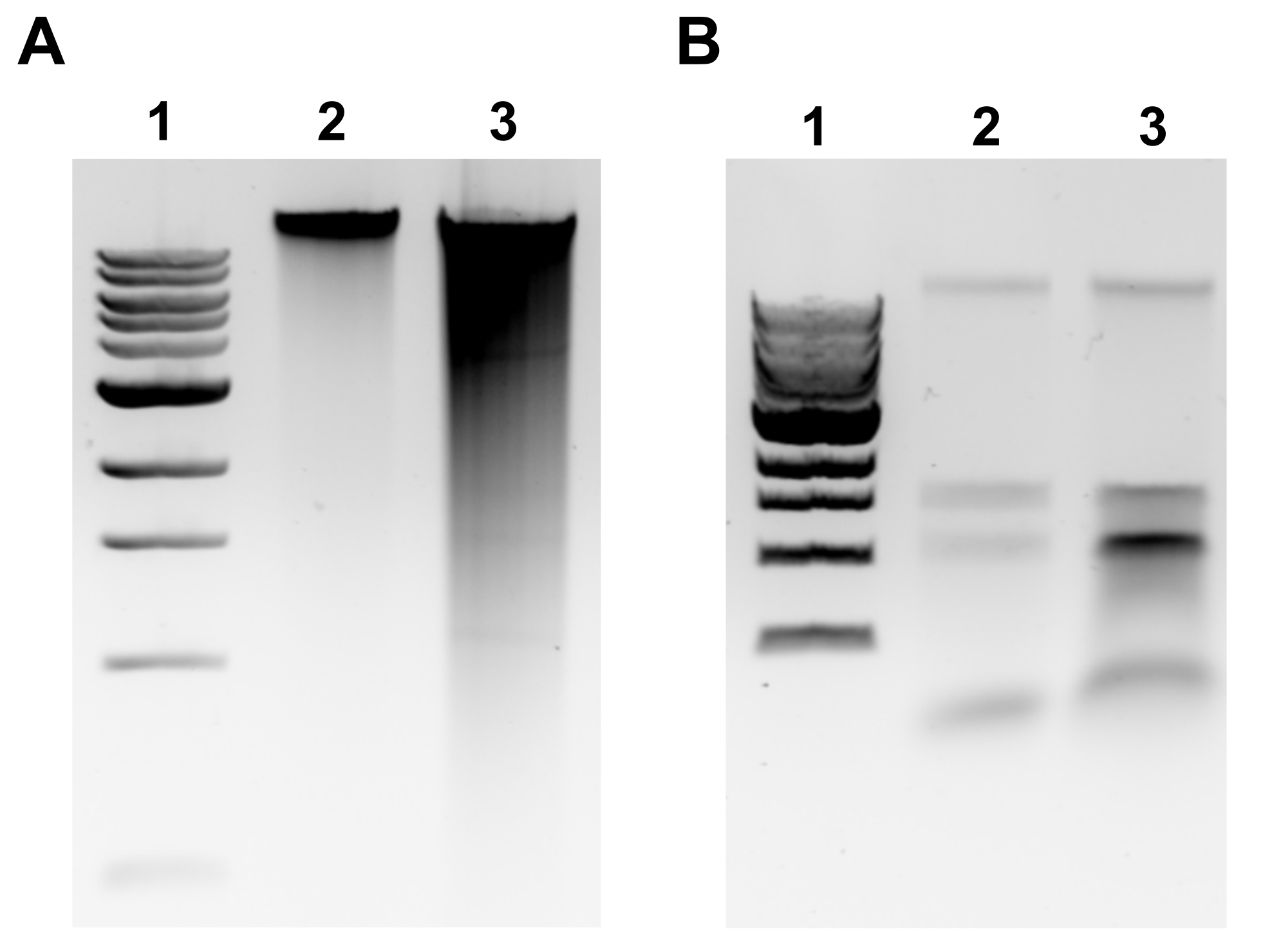
NOTA: A extração de GDNA bem sucedida será evidente por uma banda discreta na parte superior do gel e no mínimo de manchas(Figura 2A). A mancha é um indicativo de tesoura. Se nenhuma banda gDNA for evidente e/ou manchar for substancial, repita a extração gDNA. Considere reduzir os tempos de incubação em RNase A e Proteinase K. Se duas bandas em torno de 1,5-3 kb forem observadas, isso sugere contaminação de RNA(Figura 2B). Prepare RNase A fresco e repita a extração. - Para avaliar a qualidade pelo espectrofotômetro microvolume, meça a concentração gDNA e a razão de absorvância A260/280 por espectrofotômetro de microvolume. Concentrações >50 ng/μL e A260/280 entre 1,7-2.0 são aceitáveis.
NOTA: O baixo rendimento do GDNA pode ser devido à baixa entrada, alta entrada, contaminação de nucleases, lise insuficiente. As razões de absorvência acima da faixa indicam contaminação por RNA. Repita a extração se a qualidade gDNA estiver ruim. - Para avaliar a qualidade por fluorômetro, siga as instruções do fabricante para quantificar a concentração de GDNA utilizando kit de ensaio de alta sensibilidade e instrumento fluorômetro(Tabela de Materiais). A concentração >50 ng/μL é desejável.
5. Sequenciamento de leitura curta de próxima geração emparelhado e preparação da biblioteca
NOTA: O sequenciamento de leitura curta pode ser executado em vários instrumentos em comprimentos e orientações de leitura distintas. Recomenda-se sequenciamento de extremidade pareada de 150 bp (300 ciclos) para WGS bacteriana. Tanto a preparação da biblioteca quanto o sequenciamento podem ser terceirizados para instalações centrais ou laboratórios comerciais.
- Prepare a biblioteca de sequenciamento de acordo com as instruções do fabricante(Tabela de Materiais). Siga a concentração final da biblioteca de carregamento recomendada pelo fabricante; no entanto, uma modificação recomendada é carregar a biblioteca em pooled em 1,8 pM para uma geração de leitura ideal nos instrumentos NextSeq.
- Embora opcional, utilize um Bioanalyzer (Tabela de Materiais) para avaliar a distribuição do fragmento da biblioteca agrupada e garantir que o tamanho do fragmento seja de 600 bp em média.
6. Preparação da biblioteca de sequenciamento nanopore MinION
- Prepare a biblioteca de sequenciamento de acordo com o protocolo do fabricante (Tabela de Materiais). O uso de dois kits de expansão de código de barras permite multiplexagem de até 24 amostras em uma única célula de fluxo. Recomenda-se realizar a preparação da biblioteca em duas partes, 12 amostras por vez quando multiplexar 24 amostras. Todas as 24 amostras podem ser agrupadas conforme descrito abaixo.
NOTA: As amostras podem ser armazenadas a 4 °C durante a noite ao terminar a Ligadura de Código de Barras Nativo - isso fornece um ponto de parada no protocolo, se necessário. No final da seção de ligadura de código de barras nativo do protocolo de preparação da biblioteca, recomenda-se reunir quantidades equimolares de cada amostra até o máximo de massa de DNA (ng) possível.- Para isso, quantifique todas as amostras após a ligadura de código de barras usando um fluorômetro(Tabela de Materiais) por instruções do fabricante. Estime o volume da amostra com a menor concentração de DSDNA e, em seguida, calcule o total de dsDNA encontrado nesta amostra. Use este número para determinar as quantidades equimolares de todas as outras amostras que serão agrupadas.
NOTA: Como o cálculo equimolar maximizará a quantidade de dsDNA agrupado e, assim, produzirá uma piscina de alto volume (>65 μL), a limpeza é necessária para concentrar a piscina.
- Para isso, quantifique todas as amostras após a ligadura de código de barras usando um fluorômetro(Tabela de Materiais) por instruções do fabricante. Estime o volume da amostra com a menor concentração de DSDNA e, em seguida, calcule o total de dsDNA encontrado nesta amostra. Use este número para determinar as quantidades equimolares de todas as outras amostras que serão agrupadas.
- limpeza e concentração da piscina dsDNA
- Adicione 2,5x de volume de contas paramagnéticas(Tabela de Materiais) à piscina de DNA e, em seguida, gire suavemente o tubo para misturar o conteúdo. Coloque o tubo no rotador por 5 minutos na RT. Gire a amostra a 2000 x g e pelota em um ímã.
- Adicione 250 μL recém-preparado 70% de etanol (em água livre nuclease), tomando cuidado para não perturbar a pelota. Aspire o etanol e repita a lavagem do etanol uma vez.
- Após a segunda aspiração, gire a amostra a 2000 x g e coloque-a de volta no ímã. Pipeta de qualquer etanol residual e permite que a amostra seque por aproximadamente 30 s.
- Remova o tubo do ímã e resuspenda a pelota em 60-70 μL de água livre de nuclease. Incubar na RT por 2 min. Pelota a amostra no ímã até que o elto esteja limpo e, em seguida, remova o elto e transfira para um tubo de microcentrifuge limpo de 1,5 mL.
- Quantifique a piscina concentrada usando um fluorômetro e, em seguida, prepare uma alíquota para prosseguir para a etapa de ligadura do adaptador: prepare 700 ng da amostra em volume final de 65 μL. Mantenha o restante da piscina a 4 °C para que uma segunda corrida seja concluída assim que a primeira corrida estiver concluída.
- Proceda com a ligadura adaptador conforme instruído pelo fabricante e carregue a amostra na célula de fluxo. Inicie a corrida de sequenciamento.
NOTA: Ar aspirado e ~200 μL de tampão de armazenamento da porta de escoramento da célula de fluxo antes do carregamento da amostra. Isso é fundamental para o sucesso do escoramento de células de fluxo e carregamento de amostras. Use uma pipeta p1000 e dicas ao desenhar e depositar soluções através da porta de escoramento da célula de fluxo.
- Sequencie a biblioteca de acordo com as instruções do fabricante.
- Abra o software operacional para sequenciar e clique em Iniciar. Insira um nome para o experimento, uma nomenclatura recomendada inclui a data de execução e o nome do usuário. Clique em Continuar a Seleção de Kits,selecione o kit de preparação da biblioteca apropriado e pacotes de expansão de código de barras usados e clique em Continuar a executar opções.
- Ajuste o comprimento de execução para 48 h se planejar preparar biblioteca suficiente para uma segunda corrida (caso contrário, deixe no padrão 72 h). Clique em Continuar para Basear .
- Verifique a opção de chamada base Config: Base rápida e certifique-se de que a codificação de barras esteja definida como Ativada para que os arquivos FASTQ de saída sejam cortados das sequências de código de barras e desmultiplexados em diretórios separados com base em código de barras. Clique em Continuar para Saída.
- Escolha onde salvar dados de sequenciamento de saída. Espere aproximadamente 30-50 Gb de dados se apenas salvar a saída FASTQ e >500 Gb de dados se também salvar a saída FAST5. Desmarque a opção de filtragem Qscore: 7 | Relength: Sem filtro se planeja continuar com a filtragem descrita na seção 7.2, de outra forma deixe verificado e ajuste Readlength para 200.
- Clique em Continuar a configurar e revise todas as configurações. Se as configurações estiverem corretas, clique em Iniciar,clique em Voltar e faça os ajustes necessários.
- Se desejar, a célula de fluxo pode ser lavada de acordo com as instruções do fabricante e recarregada com o restante da piscina. Repita as etapas em 6.2 para o pool restante uma vez que a primeira corrida esteja completa e a célula de fluxo tenha sido lavada.
NOTA: Ao configurar a segunda corrida, ajuste a tensão de viés para -250 mV de acordo com as recomendações do fabricante para células de fluxo anteriormente utilizadas em corridas superiores a 48 h.
7. Avaliar e preparar leituras
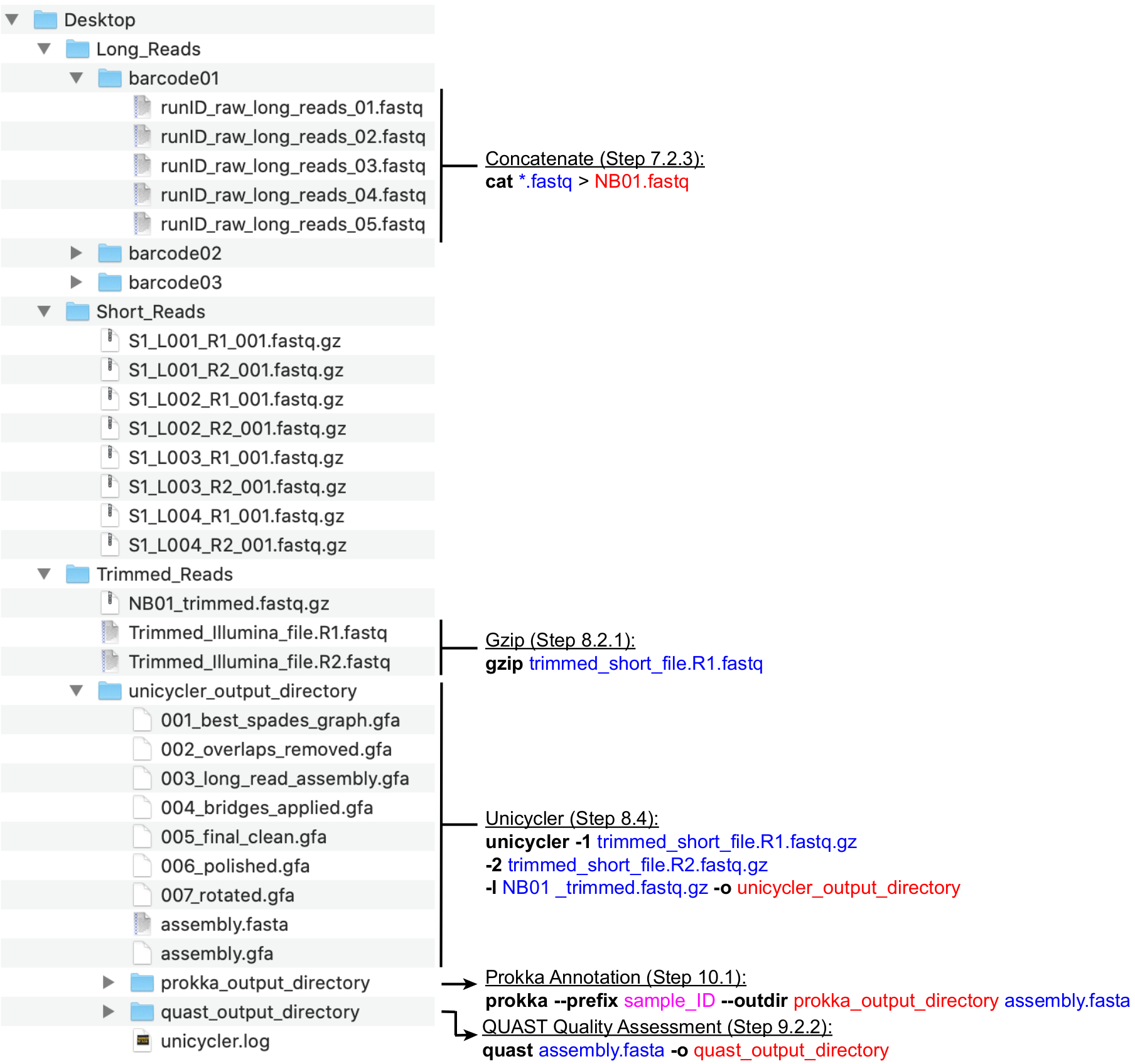
NOTA: Uma estrutura de diretório recomendada é retratada na Figura 4. Crie os diretórios encontrados na Área de Trabalho, ou seja, Long_Reads, Short_Reads e Trimmed_Reads, antes de prosseguir com as etapas de computação abaixo.
- Leituras curtas (Figura 3)
NOTA: Leituras curtas são geradas no formato FASTQ. Os arquivos contêm 4000 leituras máximas por FASTQ. Estes são frequentemente fechados (.gz arquivo) e organizados em vários arquivos. Dependendo da plataforma, os códigos de barras são tipicamente aparados. Alguns programas aceitam arquivos no formato com zíper, outros podem exigir sua extração antes da importação. As leituras devem passar por etapas de controle de qualidade (QC) para garantir a precisão dos dados durante o montagem do genoma. Se a bancada de trabalho CLC Genomics não estiver disponível, programas alternativos podem ser usados para aparar e leituras curtas de QC, como Trimmomatic25 ou Trim Galore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) para aparamento e FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) para avaliar a qualidade da leitura. Recomenda-se que a cobertura média de leitura curta, estimada pela multiplicação do número de leituras por comprimento médio de leitura e divisão pelo tamanho do genoma, seja >100x.- Abra o software de bancada de trabalho genômica(Tabela de Materiais) e importe todos os arquivos FASTQ de leitura curta emparelhados. Os arquivos emparelhados serão gerados automaticamente.
- Crie uma nova pasta sob CLC_Data clicando no Novo na barra de ferramentas superior e selecionando Pasta... para armazenar os arquivos. Nomeie a pasta conforme desejado, uma convenção recomendada está usando o ID da amostra. Salve toda a saída das etapas a seguir para esta pasta.
- Na barra de ferramentas superior, clique no botão Importar e selecione Illumina... Navegue e selecione todos os arquivos de leitura curta que correspondem à amostra. Certifique-se de que a opção de leituras emparelhadas está selecionada e desmarque a opção Remover leituras com falha. Clique em Next, selecione Salvare clique em Next novamente. Escolha salvar os arquivos importados na nova pasta criada na etapa anterior e clique em Concluir.
- Criar uma sequência de todos os arquivos emparelhados para o isolado; isso irá concatenar dados de leitura em um único arquivo para simplicidade de análise.
- Na barra de ferramentas superior, clique no botão Novo e selecione Lista de sequências... Na lista de diretórios à esquerda, selecione os arquivos a serem concatenados e use as setas para movê-los para a lista de arquivos selecionados à direita. Clique em Next, selecione Salvare clique em Next novamente. Escolha salvar a lista de sequências e clique em Concluir.
- Uma vez que a sequência é gerada, renomeie-a imediatamente com o ID da amostra.
- Execute a ferramenta QC for Sequencing Reads na lista de sequências: Este procedimento avaliará os parâmetros gerais de qualidade das leituras geradas pelo NGS de leitura curta.
- Procure a ferramenta QC for Sequencing Reads no menu caixa de ferramentas (janela inferior esquerda). Clique duas vezes na ferramenta e escolha a lista de sequências a serem analisadas e clique em Next.
- Certifique-se de que todas as opções de saída sejam verificadas e escolha Salvar sob manipulação de resultados. Clique em 'Próximo' e especifique para salvar os arquivos de saída e clique em Concluir.
- Execute a ferramenta Trim Reads na lista de sequências: O corte será feito com base na qualidade, comprimento e ambiguidade. Este processo pressupõe que os códigos de barras usados no sequenciamento tenham sido aparados antes desta etapa.
- Procure a ferramenta Trim Reads na caixa de ferramentas (janela inferior-esquerda). Clique duas vezes em Trim Readse, em seguida, escolha a lista de sequência a ser analisada e clique em Next.
- Aparação de qualidade: defina o limite de pontuação de qualidade para 0,01 e deixe nucleotídeos ambíguos em 2. Clique em Next.
NOTA: Os parâmetros podem ser ajustados a critério do usuário; estas são as configurações recomendadas. - Desmarcar o ajuste automático do adaptador de leitura automática (só faça isso se os adaptadores tiverem sido cortados das leituras antes de importar para clc). Clique em Next e verifique Descarte Leituras Abaixo do Comprimento, use o padrão 15.
- Clique em Next, verifique Criar relatórioe, em seguida, escolha Salvar. Clique em 'Próximo' e especifique onde salvar os arquivos de saída. Clique em Concluir.
- Exporte a sequência aparada: o conjunto e a análise híbrido subsequentes serão concluídos fora do CLC e requer que os arquivos de leitura curta sejam exportados.
- A partir da navegação do diretório no canto superior esquerdo, escolha o arquivo aparado gerado na etapa 7.1.4 e clique em Exportar na barra de ferramentas superior. Selecione Fastq para o tipo de arquivo de exportação e clique em Next. Verifique a lista de sequência emparelhada de exportação para dois arquivos. Em seguida, clique em Next e escolha o diretório de Trimmed_Reads para exportar os arquivos. Clique em Concluir. Certifique-se de que os arquivos de leitura curta aparados foram exportados com sucesso como dois arquivos (R1 e R2) com a extensão .fastq.
NOTA: A sequência aparada deve ser exportada em dois arquivos, tipicamente designados por CLC como R1 e R2. Isso é fundamental, pois o conjunto híbrido a jusante requer entrada de dados de leitura curta para ser configurada como tal. - Renomeie os arquivos exportados, por favor, abstenha-se do uso de espaços e caracteres especiais em nomes de arquivos. Para simplificar, um formato recomendado é trimmed_short_file. R1.fastq.
- A partir da navegação do diretório no canto superior esquerdo, escolha o arquivo aparado gerado na etapa 7.1.4 e clique em Exportar na barra de ferramentas superior. Selecione Fastq para o tipo de arquivo de exportação e clique em Next. Verifique a lista de sequência emparelhada de exportação para dois arquivos. Em seguida, clique em Next e escolha o diretório de Trimmed_Reads para exportar os arquivos. Clique em Concluir. Certifique-se de que os arquivos de leitura curta aparados foram exportados com sucesso como dois arquivos (R1 e R2) com a extensão .fastq.
- Abra o software de bancada de trabalho genômica(Tabela de Materiais) e importe todos os arquivos FASTQ de leitura curta emparelhados. Os arquivos emparelhados serão gerados automaticamente.
- Leituras longas (MinION)(Figura 3)
NOTA: O seguinte pipeline para a preparação de leituras de sequenciamento Long (MinION) para montagem híbrida utiliza os programas NanoFilt e Nanostat26 executados pela linha de comando. Instale as ferramentas antes de prosseguir e esteja familiarizado com o básico do UNIX para executar esses comandos. Os terminais padrão e o Bash Shell são recomendados. Um guia de aula para comandos e uso de terminais comuns é encontrado no Software Carpentry27. As instruções abaixo supõem que os arquivos gerados serão nomeados com a nomenclatura de código de barras (NB01, NB02, etc.) e são salvos no diretório Long_Reads. Alternativamente, a filtragem de leitura pode ser realizada usando o MinKNOW ao configurar a execução de sequenciamento. Recomenda-se que a cobertura média de leitura longa seja >100x. O comprimento médio de leitura recomendado é de >2000 bp; portanto, o número de leituras longas necessárias é menor do que o número de leituras curtas.- Criar novos diretórios para cada código de barras usado na execução (código de barras001, código de barras02, etc.) dentro do diretório Long_Reads(Figura 4). Copie todos os arquivos .fastq que correspondem a cada código de barras na pasta apropriada. Combine todos os arquivos .fastq para cada código de barras de cada execução.
- Abra terminal e navegue até os diretórios de código de barras dentro do diretório Long_Reads usando o comando cd: cd Desktop/Long_Reads/código de barras001
- Concatenar todos os arquivos .fastq por código de barras em um único arquivo .fastq executando o seguinte comando: cat *.fastq > NB01.fastq
NOTA: Este comando combina todas as leituras de cada um dos arquivos FASTQ em um GRANDE e único FASTQ chamado NB01.fastq. - Use o NanoStat para avaliar a qualidade de leitura da amostra executando o seguinte comando: NanoStat --fastq NB01.fastq
- Registo os resultados copiando a saída em um texto ou arquivo do Word para referência futura.
- Use NanoFilt para filtrar leituras de leituras de descarte com Q < 7 e comprimento < 200 executando o comando: NanoFilt -q 7 -l 200 bp NB01.fastq | gzip > NB01 _trimmed.fastq.gz
- Execute o NanoStat no arquivo aparado gerado na etapa 7.2.6 executando o comando: NanoStat --fastq NB01 _trimmed.fastq.gz
- Registre os resultados copiando a saída em um texto ou arquivo do Word e compare com os resultados da etapa 7.2.4 para garantir que a filtragem tenha sido bem sucedida(Tabela 1).
- Repetir as etapas 7.2.2 a 7.2.8 para cada código de barras usado na execução do sequenciamento.
NOTA: O arquivo NB01_trimmed.fastq.gz gerado na etapa 7.2.6 será usado para montagem híbrida.
8. Geração de montagem de genoma híbrido
NOTA: O seguinte pipeline de montagem utiliza o Unicycler19,28,29,30 para combinar leituras curtas e longas preparadas nas seções 7.1 e 7.2(Figura 3). Instale o Unicycler e suas dependências e execute os comandos abaixo. Os arquivos de leitura curta exportados na etapa 7.1.5 são considerados trimmed_short_file. R1.fastq e trimmed_short_file. R2.fastq para simplicidade.
- Organize os arquivos de leitura curta e arquivos de leitura longa em um único diretório chamado Trimmed_Reads. O diretório deve conter o seguinte:
- Um arquivo .fastq.gz para leituras longas aparadas (gerada na etapa 7.2.6).
- Dois arquivos .fastq (R1 e R2) para leituras curtas aparadas (geradas na etapa 7.1.5).
- Navegue até o diretório Trimmed_Reads que armazena os arquivos de leitura usando o comando cd em Terminal: cd Desktop/Trimmed_Reads
- Uma vez no diretório correto, feche os dois arquivos de leitura curta para que eles também estejam no formato .fastq.gz executando o seguinte comando: gzip trimmed_short_file. R1.fastq
- Repita o passo 8.2 tanto para R1 quanto para R2. Verifique se todos os arquivos de leitura estão agora no formato .fastq.gz e verifique se todos os arquivos correspondem ao mesmo isolado.
- Inicie o conjunto híbrido usando o Unicycler executando o seguinte comando:
uniciclador -1 trimmed_short_file. R1.fastq.gz -2 trimmed_short_file. R2.fastq.gz -l NB01 _trimmed.fastq.gz -o unicycler_output_directory
NOTA: -o especifica o diretório no qual a saída do Unicycler será salva, o Unicycler criará este diretório assim que o comando for executado; não gerar o diretório de antemão. O tempo de execução varia de acordo com o poder computacional do computador usado, bem como o tamanho do genoma e o número de leituras. Isso pode levar de 4h a 1 ou 2 dias. Este protocolo foi realizado em uma máquina CentOS Linux 7 com 250 Gb de RAM, CPU Intel Xeon (R) com 2,5 GHz 12 núcleos práticos e 48 núcleos virtuais. Alternativamente, computadores pessoais com 16 Gb de RAM e processadores de 6 núcleos de 2,6 GHz podem calcular esses conjuntos em um tempo de processamento mais longo. - Quando a execução estiver concluída, revise o arquivo uniciclista.log para garantir que não haja erros - registe o número, o tamanho e o status (completos, incompletos) das conchas geradas.
- Se forem identificados contigs incompletos (denotados como incompletos no registro Unicycler), recarnuda o Unicycler no modo negrito adicionando o seguinte sinalizador ao comando na etapa 8.4: -modo em negrito.
NOTA: O modo arrojado reduzirá o limite de qualidade aceito para pontes de leitura longa durante a montagem; isso pode render uma montagem completa, mas a qualidade da montagem pode ser diminuída. Recomenda-se utilizar o modo arrojado somente quando necessário e como evidência preliminar para a adesão da Contig a ser posteriormente confirmada pelo PCR.
- Se forem identificados contigs incompletos (denotados como incompletos no registro Unicycler), recarnuda o Unicycler no modo negrito adicionando o seguinte sinalizador ao comando na etapa 8.4: -modo em negrito.
9. Avaliar a qualidade da montagem
NOTA: O protocolo a seguir utiliza a Bandagem31 e o QUAST32, dois programas que devem ser configurados antes do uso(Figura 2 e Figura 4). O curativo não requer instalação uma vez baixado e o QUAST requer familiaridade com o uso básico da linha de comando. Também é recomendado avaliar a completude do genoma usando Ortologs Universais de Cópia Única (BUSCO)33.
- Bandagem: Clique no arquivo. Em seguida, escolha o Gráfico de carga e selecione o arquivo assembly.gfa que foi salvo para unicycler_output_directory gerado pelo Unicycler na etapa 8.4. Uma vez carregado, clique no botão Desenhar gráfico na barra de ferramentas do lado esquerdo e veja como os contigs (chamados nós) são conectados e organizados para avaliar se o conjunto está completo(Figura 5).
NOTA: Os conjuntos completos são representados por contigas circulares únicas ligadas em ambas as extremidades(Figura 5A,B). Conjuntos incompletos têm múltiplas contigas ligadas ou são lineares(Figura 5C). Pequenas contigas lineares podem não estar incompletas, pois podem indicar elementos extracromosomais lineares. A cobertura, também chamada de profundidade, será notada em curativo e representa a abundância relativa dos tigs ao cromossomo, normalizado em Unicycler para 1x. - QUAST
- Dentro do Terminal, navegue até a pasta que armazena a saída do Unicycler usando o comando cd: cd Desktop/Trimmed_Reads/unicycler_output_directory
NOTA: Espaços não são permitidos no caminho para onde o conjunto está localizado, ou seja, nenhum diretório que leve à saída do Unicycler pode ter espaços em seu nome. Alternativamente, copie o arquivo assembly.fasta para a Área de Trabalho para fácil acesso. - Execute o QUAST executando o seguinte comando: quast assembly.fasta -o quast_output_directory
- Revise os relatórios gerados pelo QUAST no diretório de saída quast_output_directory.
- Dentro do Terminal, navegue até a pasta que armazena a saída do Unicycler usando o comando cd: cd Desktop/Trimmed_Reads/unicycler_output_directory
10. Anotação do genoma
NOTA: O pipeline de anotação abaixo utiliza o Prokka34, uma ferramenta de linha de comando que deve ser instalada antes do uso. Alternativamente, use o Prokka através da base automática de GUI(Tabela de Materiais) ou anote genomas através do servidor web RAST35. Se depositar genomas no NCBI, eles serão automaticamente anotados usando o Pipeline de Anotação do Genoma Procariótico (PGAP)36.
- Navegue dentro do Terminal até a pasta que armazena a saída do Unicycler usando o comando cd (ver passo 9.2.1). Em seguida, execute Prokka executando o seguinte comando: prokka -prefixo sample_ID -outdir prokka_output_directory assembly.fasta
NOTA: --prefixo nomeará todos os arquivos de saída com base no sample_ID especificado. --Outdir criará um diretório de saída com o nome especificado onde todos os arquivos de saída Prokka serão salvos; não crie um diretório de saída para Prokka de antemão. - Revise as anotações abrindo a tabela .tsv e/ou carregando o arquivo .gff gerado em um software de análise de sequência para visualizar e analisar as anotações(Figura 6).
- Tipos específicos de anotações podem ser gerados dependendo de fatores genéticos de interesse. Recomenda-se começar com as ferramentas fáceis de usar no servidor web do Centro de Epidemiologia Genômica (www.genomicepidemiology.org/) para análise preliminar37,38,39,40,41. Ferramentas adicionais para detecção de sistemas CRISPR-cas e prophage estão disponíveis(Figura 3)42,43.
11. Práticas sugeridas para democratização de dados
- Quando possível, deposite todos os dados de leitura bruta, bem como genomas montados em um repositório público, como NCBI Sequence Read Archive (SRA) e Genbank. Os genomas são automaticamente anotados através do pipeline PGAP durante o processo de deposição do NCBI.
Resultados
Este protocolo foi otimizado para a cultura e sequenciamento de bactérias urinárias pertencentes aos gêneros listados na Figura 1. Nem todas as bactérias urinárias são culturais por este método. Os meios de cultura e as condições são especificados pelo gênero na Figura 1. Avaliações exemplares de eletroforese de gel da integridade gDNA são retratadas na Figura 2. Uma visão geral do pipeline bioinforático para sequenciamento de leitura, montagem de genoma e anotação é descrita na Figura 3. Um guia para a estrutura do diretório computacional é fornecido na Figura 4 para simplificar a compreensão do protocolo e fornecer estrutura para uma organização bem-sucedida. Além disso, estão incluídos genomas completos representativos de dois Klebsiella spp., K. pneumoniae e K. oxytoca,que foram gerados por este protocolo. Uma representação dessas assembleias é fornecida na Figura 5 e também inclui um exemplo adicional incompleto do genoma K. pneumoniae. Uma visão geral detalhada de cada genoma completo totalmente anotado é mostrada na Figura 6. Finalmente, um resumo das estatísticas de leitura de sequenciamento é fornecido na Tabela 1 para oferecer uma ampla compreensão dos dados brutos e aparados suficientes para a geração de conjuntos de genomas fechados de alta qualidade. Além disso, os principais parâmetros dos dois representantes completam Klebsiella spp. genomas estão listados. Genomas e dados brutos foram depositados no Genbank sob o BioProject PRJNA683049.

Figura 1: Cultura de urina modificada de diversos gêneros urinários. Gráfico para o ágar e caldo líquido que podem ser usados para cultivar diversos gêneros urinários. Toda a cultura é sugerida para ser realizada a 35 °C como descrito na subseção 1.1. Os círculos representam mídias apropriadas para a cultura de um determinado gênero, as cores foram arbitrariamente selecionadas para distinguir um tipo de mídia do outro. CDC-AN BAP (vermelho), Ágar de Sangue de Ovelhas CDC Anaerobe; 5% Ovino-BAP (laranja), Ágar de Sangue de Ovelha; BHI (verde), Infusão do Coração Cerebral; TSB (amarelo), Caldo de Soja Tptic; Orientação CHROMagar (azul). aGardnerella vaginalis deve ser cultivada em HBT Bilayer G. vaginalis Ágar seletivo em atmosfera microaerofílica e sob requisitos especiais de cultura de caldo44. bLactobacillus iners deve ser cultivado em placas de 5% Rabbit-BAP e caldo NYCIII em atmosfera microaerofílica. cLactobacillus spp. pode ser cultivada em MRS em condições microaerófilas. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: Extração genômica de DNA agarose imagens em gel. Imagens de gel representativas que retratam os resultados da extração do GDNA. (A) Pista 1: 1 kb escada, Faixa 2: gDNA intacta representando extração bem sucedida, Faixa 3: mancha indicando gDNA fragmentada. (B) Pista 1: 1 kb escada, Lanes 2 & 3: contaminação rRNA denotada por duas bandas entre 1,5 kb e 3 kb. Clique aqui para ver uma versão maior desta figura.
{kind=link}

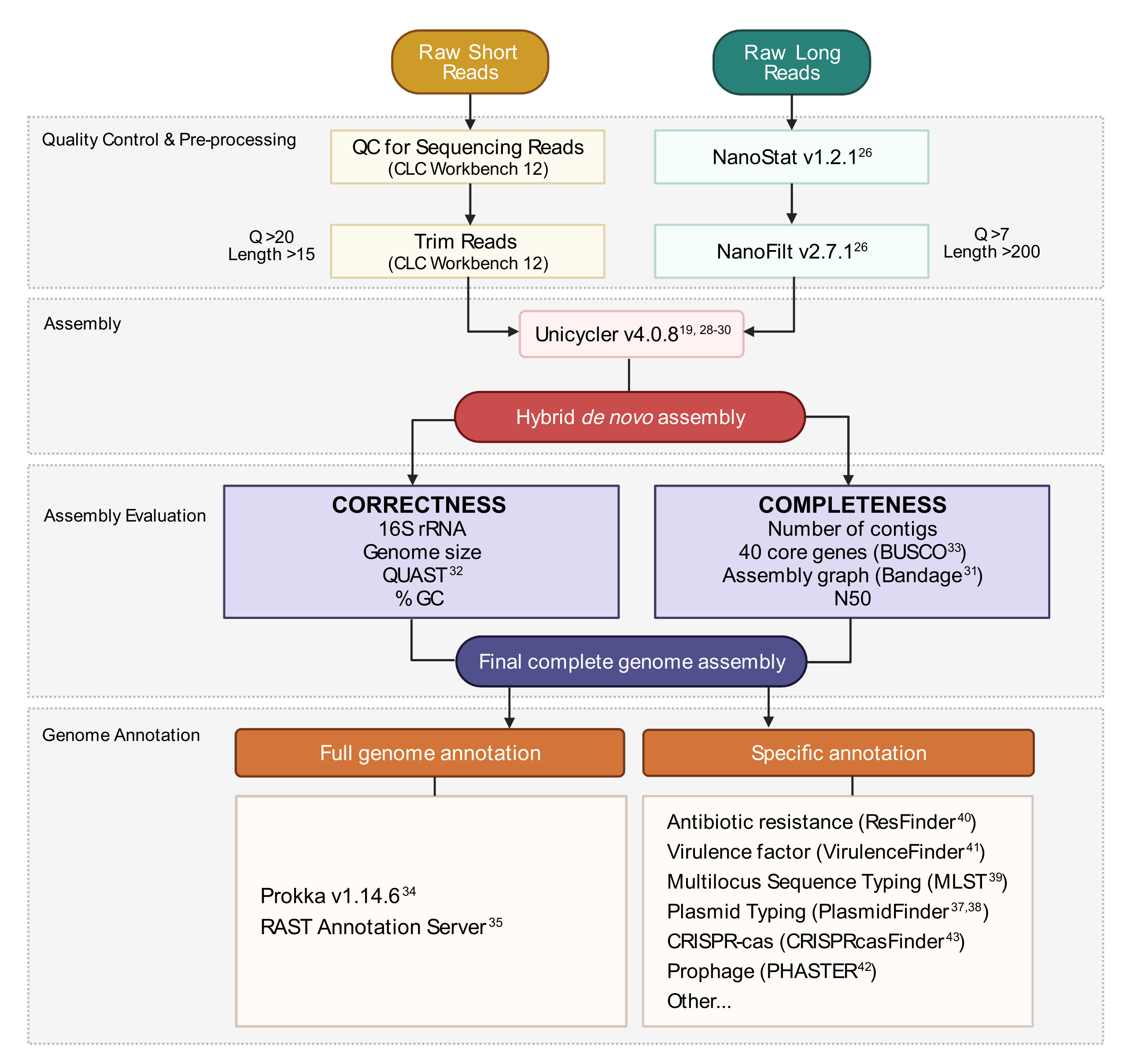
Figura 3: Fluxo de trabalho de montagem de genoma híbrido. Esquema de passos desde o controle de qualidade de leitura e pré-processamento até a anotação de montagem. A leitura de aparamento remove leituras ambíguas e de baixa qualidade. Os parâmetros de escore q e comprimento são indicados e representam as leituras que são retidas. A montagem utiliza leituras curtas e longas para gerar um conjunto híbrido de genoma de novo. A qualidade da montagem é avaliada com base na completude e correção utilizando ferramentas e parâmetros especificados. O conjunto final do genoma é anotado para todos os genes e loci específico de interesse. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: Guia de estrutura dediretório de bioinformática. Um esquema de organização recomendada de diretórios e arquivos para o processamento de leituras curtas e longas, montagem híbrida e anotação de genoma e QC. As principais etapas de processamento de dados da linha de comando são destacadas ao lado de arquivos e diretórios correspondentes. Obtenção de comandos e bandeiras (em negrito), arquivos de entrada (azul), arquivos de saída ou diretórios (vermelho), entrada do usuário, como convenção de nomeação de arquivos (magenta). Clique aqui para ver uma versão maior desta figura.
{kind=link}

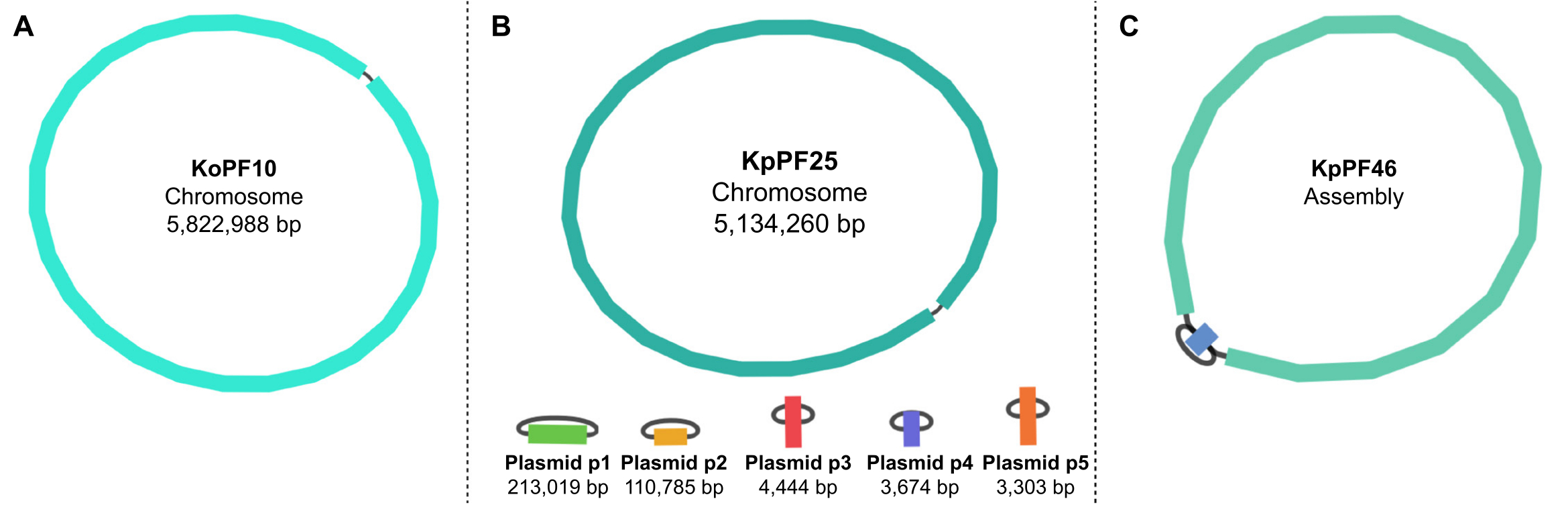
Figura 5: Gráficos de montagem do genoma por Bandge. Gráficos de montagem completa do genoma representativo de (A) Klebsiella oxytoca KoPF10 e (B) Klebsiella pneumoniae KpPF25 e conjunto de genoma incompleto de (C) Klebsiella pneumoniae KpPF46. O genoma completo de KoPF10 demonstra um único cromossomo fechado e o genoma completo de KpPF25 consiste em um cromossomo fechado e cinco plasmídeos fechados. O cromossomo incompleto de KpPF46 consiste em dois contigs interligados. O conjunto uniciclista híbrido de novo gera um gráfico de montagem que é visualizado pela Bandage. O gráfico de montagem fornece um esquema simplista do genoma, indicando cromossomo fechado ou plasmídeos por um linker conectando duas extremidades de um único contig. A presença de mais de um contig interconectado indica montagem incompleta. O tamanho e a profundidade do Contig também podem ser notados no Curativo. Clique aqui para ver uma versão maior desta figura.
{kind=link}

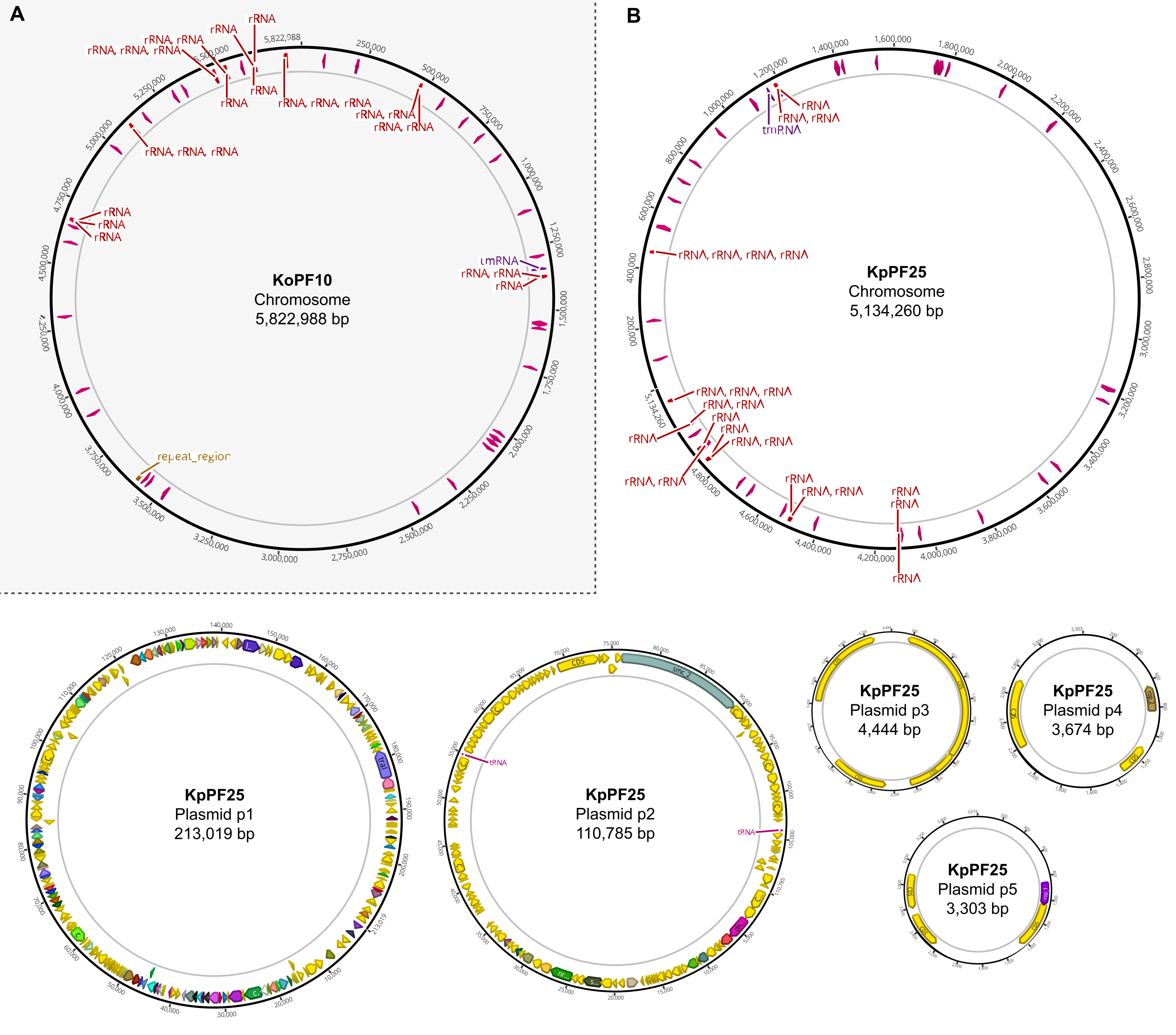
Figura 6: Mapas completos do genoma de conjuntos híbridos anotados. Mapas de montagem gerados por Geneious Prime para o genoma completo de (A) K. oxytoca KoPF10 e (B) K. pneumoniae KpPF25 mostrando genes anotados denotados por setas coloridas ao longo de espinhas plasmidas. Cromossomos só mostram genes de rRNA e tRNA para simplicidade. As anotações do genoma foram realizadas utilizando Prokka, conforme indicado na seção 10 deste protocolo. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Tabela 1: Representante Klebsiella spp. características completas de montagem. Parâmetros de montagem da cepa de K. oxytoca KoPF10 e K. pneumoniae strain KpPF25. Os números de adesão para os dados depositados no NCBI são fornecidos. O número de leituras antes e depois do corte são especificados para ambas as tecnologias de sequenciamento. N50 é fornecido para leituras longas apenas uma vez que as leituras curtas são de um comprimento controlado. Plasmid replicon previu usando o banco de dados PlasmidFinder v2.1 Enteroebacteriaceae com parâmetros definidos para 80% de identidade e 60% de comprimento. um tipo de sequência MLST, Multilocus. b CDS, Sequências de codificação. c Plasmid replicon previu usando o banco de dados PlasmidFinder v2.1 Enterobacteriaceae com parâmetros definidos para 80% de identidade e 60% de comprimento. d Oxford Nanopore Technologies (ONT) depositou dados de leitura. e Illumina depositou dados de leitura. Clique aqui para baixar esta Tabela.
Discussão
O protocolo abrangente de montagem de genomas híbridos descrito aqui oferece uma abordagem simplificada para a cultura bem sucedida de diversas microbiota urinária e uropatógenos, e a montagem completa de seus genomas. O sucesso do WGS de genomas bacterianos começa com o isolamento de micróbios diversos e às vezes exigentes, a fim de extrair seu DNA genômico. Até o momento, os protocolos de cultura urinária existentes ou não têm a sensibilidade necessária para detectar muitas espécies urinárias ou envolvem abordagens longas e extensas que requerem tempo e recursos prolongados11. A abordagem modificativa da cultura da urina aprimorada descrita oferece um protocolo simplificado, mas abrangente, para o isolamento bem-sucedido de bactérias pertencentes a 17 gêneros urinários comuns, incluindo espécies commensais potencialmente patogênicas ou benéficas, e bactérias aeróbicas ou aeróbicas facultativas e obrigatórias. Isso, por sua vez, fornece o material inicial necessário para sequenciamento preciso e montagem de genomas bacterianos e para experimentos fenotípicos críticos, que contribuem para a compreensão da saúde urinária e da doença. Além disso, essa abordagem cultural modificada prevê um diagnóstico clínico mais definido de microrganismos viáveis encontrados em amostras de urina e permite seu biobanco para futuros estudos genômicos. No entanto, este protocolo não é sem limitações. Pode exigir longos tempos de incubação, dependendo do organismo, bem como do uso de recursos como uma câmara de hipóxia ou incubadoras controladas que podem não estar prontamente disponíveis. O uso de GasPaks anaeróbicos oferece uma solução alternativa, mas estes são caros e nem sempre produzem um ambiente sustentado e controlado. Finalmente, o viés cultural, bem como a diversidade de amostras podem permitir que organismos particulares e uropatógenos superem as bactérias exigentes. Apesar dessas limitações, uma cultura de diversas bactérias urinárias é possível por essa abordagem.
O sequenciamento genômico ganhou popularidade com o avanço das tecnologias de Sequenciamento de Próxima Geração, que aumentaram tremendamente o rendimento e a precisão dos dados de sequenciamento14,15. Juntamente com o desenvolvimento de algoritmos para processamento de dados e montagem de novo, sequências completas de genomas estão na ponta dos dedos de cientistas novatos e especialistasapenas 15,45. O conhecimento da organização geral do genoma fornecida por genomas completos oferece importantes insights evolutivos e biológicos, incluindo duplicação genética, perda genética e transferência horizontal de genes14. Além disso, genes importantes para a resistência antimicrobiana e virulência são frequentemente localizados em elementos móveis, que normalmente não são resolvidos nos conjuntos degenomas 15,16.
O protocolo aqui segue uma abordagem híbrida para a combinação de dados de sequenciamento de plataformas de leitura curta e longa leitura para gerar conjuntos completos de genomas. Embora focado em genomas bacterianos urinários, este procedimento pode ser adaptado a diversas bactérias de várias fontes de isolamento. As etapas críticas nesta abordagem incluem seguir a técnica estéril adequada e utilizar condições adequadas de mídia e cultura para o isolamento de bactérias urinárias puras. Além disso, a extração de gDNA intacto e de alto rendimento é essencial para gerar dados de sequenciamento livres de leituras contaminantes que podem dificultar o sucesso da montagem. Os protocolos subsequentes de preparação da biblioteca são fundamentais para a geração de leituras de qualidade de comprimento e profundidade suficientes. Portanto, é de total importância lidar com gDNA com cuidado durante a preparação da biblioteca para sequenciamento de leitura longa em particular, já que o maior benefício desta tecnologia é a geração de leituras longas sem limite teórico de comprimento superior. Também estão delineadas seções para o controle de qualidade apropriado (QC) de leituras de sequenciamento que eliminam dados barulhentos e melhoram o resultado da montagem.
Apesar do sucesso do isolamento do DNA, da preparação da biblioteca e do sequenciamento, a natureza da arquitetura genômica de algumas espécies ainda pode fornecer um obstáculo para a geração de um conjunto de genomasfechados 45,46. Sequências repetitivas muitas vezes complicam a computação de montagem e, apesar dos dados de leitura longa, essas regiões podem ser resolvidas com baixa confiança, ou não. As leituras longas, portanto, devem ser, em média, mais longas do que a maior região de repetição do genoma ou a cobertura deve ser alta (>100x)19. Alguns genomas podem permanecer incompletos e requerem abordagens manuais para conclusão. No entanto, genomas incompletos montados em híbridos são tipicamente compostos de menos contigs do que genomas de rascunho de leitura curta. Ajustar os parâmetros padrão do algoritmo de montagem ou seguir cortes mais rigorosos para ler QC pode ajudar. Alternativamente, uma abordagem sugerida é mapear leituras longas para as regiões incompletas em busca de evidências para o caminho de montagem mais provável e, em seguida, confirmar o caminho utilizando pcr e sequenciamento Sanger da região amplificada. O mapeamento de leituras usando o Minimap2 é sugerido e o Bandge oferece uma ferramenta útil para a visualização de leituras mapeadas ao longo de contigas montadas fornecendo evidências para a ligação de contig47.
Um desafio adicional para gerar genomas completos está na familiaridade e conforto com ferramentas de linha de comando. Muitas ferramentas bioinformáticas são desenvolvidas para oferecer oportunidades computacionais a qualquer usuário; no entanto, sua utilização depende de um entendimento com o básico da UNIX e programação. Este protocolo visa fornecer instruções suficientemente detalhadas para permitir que indivíduos sem experiência prévia de linha de comando gerem conjuntos de genomas fechados e as anotem.
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
Agradecemos ao Dr. Moutusee Jubaida Islam e ao Dr. Luke Joyce por suas contribuições para este protocolo. Também gostaríamos de reconhecer a Universidade do Texas no Centro de Genoma de Dallas por seu feedback e apoio. Este trabalho foi financiado pela Fundação Welch, número do prêmio AT-2030-2020401 para n.J.D., pelos Institutos Nacionais de Saúde, prêmio número R01AI11610 para K.P., e pela Felecia e John Cain Chair in Women's Health, realizada pela P.E.Z.
Materiais
| Name | Company | Catalog Number | Comments |
| Equipment: | |||
| Bioanalyzer 2100 | Agilent | G29398A | Optional but recommended |
| Centrifuge | Eppendorf | -- | Any centrifuge for spinning conicals and microcentrifuge tubes (e.g. Models 5810R/5424R) |
| Electrophoresis | BioRad Laboratories | 1645070 | |
| Gel Imaging System | BioRad Laboratories | ChemiDoc models | |
| Incubator | ThermoFisher Scientific | -- | Any CO2 Incubator (e.g. Thermo Forma model 3110) |
| Magnetic Rack | New England BioLabs | S15095 | 12-tube rack |
| MinION | Oxford Nanopore Technologies | -- | |
| Nanodrop | ThermoFisher Scientific | ND-ONE-W | |
| NextSeq 500 | Illumina | SY-415-1002 | Other Illumina models are acceptable |
| Plate Reader | BioTek | -- | Synergy H1 |
| Qubit fluorometer | ThermoFisher Scientific | Q33238 | |
| Rotator | Benchmark Scientific | H2024 | |
| Thermocycler | ThermoFisher Scientific | -- | Any thermocycler for PCR reactions (e.g. ProFlex PCR system) |
| Materials: | |||
| 10X Phosphate Buffered Saline (PBS) | Fisher Scientific | BP3991 | |
| 10X TBE buffer | -- | -- | 1M Tris,1M Boric Acid,0.2M EDTA (pH 8.0) |
| 1429R primer | Sigma Aldrich (Custom oligos) | -- | GGTTACCTTGTTACGACTT |
| 1kb Ladder | VWR | 101228-494 | |
| 1M Tris-Cl (pH 7.5) | ThermoFisher Scientific | 15567027 | |
| 6x Loading dye | Fisher Scientific | NC0783588 | |
| 8F primer | Sigma Aldrich (Custom oligos) | -- | AGAGTTTGATCCTGGCTCAG |
| Agar | Fisher Scientific | BP1423-2 | |
| Agarose | BioRad Laboratories | 63001 | |
| AMPure XP Beads | Beckman Coulter | A63880 | |
| Anaerobe Pouch System - GasPak EZ | BD Diagnostic Systems | B260683 | |
| Boric Acid | Fisher Scientific | A73-500 | |
| Brain Heart Infusion Broth | BD Diagnostic Systems | 212304 | |
| CDC Anaerobe 5% Sheep Blood Agar | BD Diagnostic Systems | L007357 | |
| CHROMagar Orientation | BD Diagnostic Systems | PA-257481.04 | |
| DNeasy Blood & Tissue | QIAGEN | 69504 | |
| DreamTaq Master Mix | ThermoFisher Scientific | K1081 | |
| Dry Anaerobic Indicator Strips | BD Diagnostic Systems | 271051 | |
| EDTA | Fisher Scientific | S311-500 | |
| Ethanol 200 Proof | Sigma Aldrich | E7023 | For molecular biology |
| Ethidium Bromide | ThermoFisher Scientific | BP130210 | |
| Flow cell priming kit | Oxford Nanopore Technologies | EXP-FLP002 | |
| Flow cell wash kit | Oxford Nanopore Technologies | EXP-WSH003 | |
| Gel Extraction Miniprep Kit | BioBasic | BS654 | |
| Ligation sequencing kit | Oxford Nanopore Technologies | SQK-LSK109 | |
| Lysozyme | Research Products International Corp | L381005.05 | |
| Mutanolysin | Sigma Aldrich | M9901-5KU | |
| Native barcoding expansion 1-12 | Oxford Nanopore Technologies | EXP-NBD104 | |
| NEB Blunt/TA Ligase Master Mix | New England BioLabs | M0367L | |
| NEBNext FFPE DNA Repair Mix | New England BioLabs | M6630L | |
| NEBNext quick ligation buffer | New England BioLabs | B6058S | |
| NEBNext Ultra II End repair / dA-tailing module | New England BioLabs | E7546L | |
| Nextera DNA CD Indexes | Illumina | 20018708 | |
| Nextera DNA Flex Library Prep - (M) Tagmentation | Illumina | 20018705 | |
| Nuclease-free water | Sigma Aldrich | W4502 | |
| Qubit 1X dsDNA HS Assay Kit | ThermoFisher Scientific | Q33230 | |
| Qubit Assay Tubes | ThermoFisher Scientific | Q32856 | |
| Quick T4 DNA Ligase | New England BioLabs | E6056L | |
| R9 Flow cell | Oxford Nanopore Technologies | FLO-MIN106D | |
| RNase A | ThermoFisher Scientific | EN0531 | |
| Sheep Blood | Hemostat Laboratories | DS13250 | |
| TE buffer | -- | -- | 10mM Tris, 1mM EDTA (pH 8.0) |
| Triton X-100 | Sigma Aldrich | T8787 | |
| Tryptic Soy Broth | BD Diagnostic Systems | 211825 | |
| Software & Bioinformatic Tools: | |||
| Bandage | -- | -- | https://rrwick.github.io/Bandage/ |
| Center for Genomic Epidemiology | -- | -- | http://www.genomicepidemiology.org/ |
| CLC Genomics Workbench 12 | QIAGEN | -- | |
| CRISPRcasFinder | -- | -- | https://crisprcas.i2bc.paris-saclay.fr/ |
| FastQC | -- | -- | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Geneious Prime | Geneious | -- | |
| gVolante (BUSCO) | -- | -- | https://gvolante.riken.jp/ |
| Kbase Prokka Wrapper | -- | -- | https://kbase.us/applist/apps/ProkkaAnnotation/annotate_contigs/release |
| Minimap2 | -- | -- | https://github.com/lh3/minimap2 |
| MinKNOW | Oxford Nanopore Technologies | -- | |
| NanoFilt | -- | -- | https://github.com/wdecoster/nanofilt |
| NanoStat | -- | -- | https://github.com/wdecoster/nanostat |
| PHASTER | -- | -- | https://phaster.ca/ |
| Prokka | -- | -- | https://github.com/tseemann/prokka |
| QUAST | -- | -- | http://quast.sourceforge.net/quast |
| Trim Galore | -- | -- | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| Trimmomatic | -- | -- | http://www.usadellab.org/cms/?page=trimmomatic |
| Unicycler | -- | -- | https://github.com/rrwick/Unicycler#necessary-read-length |
Referências
- Brubaker, L., Wolfe, A. The urinary microbiota: a paradigm shift for bladder disorders. Current Opinion in Obstetrics & Gynecology. 28 (5), 407-412 (2016).
- Neugent, M. L., Hulyalkar, N. V., Nguyen, V. H., Zimmern, P. E., De Nisco, N. J. Advances in understanding the human urinary microbiome and its potential role in urinary tract infection. mBio. 11 (2), (2020).
- Klein, R. D., Hultgren, S. J. Urinary tract infections: microbial pathogenesis, host-pathogen interactions and new treatment strategies. Nature Reviews. Microbiology. 18 (4), 211-226 (2020).
- Horsley, H., et al. Enterococcus faecalis subverts and invades the host urothelium in patients with chronic urinary tract infection. PLoS One. 8 (12), 83637 (2013).
- Reitzer, L., Zimmern, P. Rapid growth and metabolism of uropathogenic Escherichia coli in relation to urine composition. Clinical Microbiology Reviews. 33 (1), 00101-00119 (2019).
- Snyder, J. A., et al. Transcriptome of uropathogenic Escherichia coli during urinary tract infection. Infection and Immunity. 72 (11), 6373-6381 (2004).
- Ipe, D. S., Horton, E., Ulett, G. C. The basics of bacteriuria: Strategies of microbes for persistence in urine. Frontiers in Cellular and Infection Microbiology. 6, 14 (2016).
- Babikir, I. H., et al. The impact of cathelicidin, the human antimicrobial peptide LL-37 in urinary tract infections. BMC Infectious Diseases. 18 (1), 17 (2018).
- Jancel, T., Dudas, V. Management of uncomplicated urinary tract infections. The Western Journal of Medicine. 176 (1), 51-55 (2002).
- Ventola, C. L. The antibiotic resistance crisis: part 1: causes and threats. P & T. 40 (4), 277-283 (2015).
- Price, T. K., et al. The clinical urine culture: Enhanced techniques improve detection of clinically relevant microorganisms. Journal of Clinical Microbiology. 54 (5), 1216-1222 (2016).
- Kass, E. H. Asymptomatic infections of the urinary tract. Transactions of the Association of American Physicians. 69, 56-64 (1956).
- Garcia, L. S. . Clinical microbiology procedures handbook. 3rd edn. , (2010).
- Fraser, C. M., Eisen, J. A., Nelson, K. E., Paulsen, I. T., Salzberg, S. L. The value of complete microbial genome sequencing (you get what you pay for). Journal of Bacteriology. 184 (23), 6403-6405 (2002).
- Chen, Z., Erickson, D. L., Meng, J. Benchmarking hybrid assembly approaches for genomic analyses of bacterial pathogens using Illumina and Oxford Nanopore sequencing. BMC Genomics. 21 (1), 631 (2020).
- Greig, D. R., Dallman, T. J., Hopkins, K. L., Jenkins, C. MinION nanopore sequencing identifies the position and structure of bacterial antibiotic resistance determinants in a multidrug-resistant strain of enteroaggregative Escherichia coli. Microbial Genomics. 4 (10), 000213 (2018).
- Carraro, D. M., et al. PCR-assisted contig extension: stepwise strategy for bacterial genome closure. Biotechniques. 34 (3), 626-628 (2003).
- Tettelin, H., Radune, D., Kasif, S., Khouri, H., Salzberg, S. L. Optimized multiplex PCR: efficiently closing a whole-genome shotgun sequencing project. Genomics. 62 (3), 500-507 (1999).
- Wick, R. R., Judd, L. M., Gorrie, C. L., Holt, K. E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Computational Biology. 13 (6), 1005595 (2017).
- Singhal, N., Kumar, M., Kanaujia, P. K., Virdi, J. S. MALDI-TOF mass spectrometry: an emerging technology for microbial identification and diagnosis. Frontiers in Microbiology. 6, 791 (2015).
- Turner, S., Pryer, K. M., Miao, V. P., Palmer, J. D. Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. The Journal of Eukaryotic Microbiology. 46 (4), 327-338 (1999).
- Weisburg, W. G., Barns, S. M., Pelletier, D. A., Lane, D. J. 16S ribosomal DNA amplification for phylogenetic study. Journal of Bacteriology. 173 (2), 697-703 (1991).
- Janda, J. M., Abbott, S. L. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. Journal of Clinical Microbiology. 45 (9), 2761-2764 (2007).
- Stevenson, K., McVey, A. F., Clark, I. B. N., Swain, P. S., Pilizota, T. General calibration of microbial growth in microplate readers. Science Reports. 6, 38828 (2016).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (15), 2114-2120 (2014).
- De Coster, W., D'Hert, S., Schultz, D. T., Cruts, M., Van Broeckhoven, C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 34 (15), 2666-2669 (2018).
- Wilson, G., et al. The UNIX Shell. Zenodo. , (2019).
- Bankevich, A., et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology. 19 (5), 455-477 (2012).
- Vaser, R., Sovic, I., Nagarajan, N., Sikic, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Research. 27 (5), 737-746 (2017).
- Walker, B. J., et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 9 (11), 112963 (2014).
- Wick, R. R., Schultz, M. B., Zobel, J., Holt, K. E. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics. 31 (20), 3350-3352 (2015).
- Gurevich, A., Saveliev, V., Vyahhi, N., Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 29 (8), 1072-1075 (2013).
- Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31 (19), 3210-3212 (2015).
- Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 30 (14), 2068-2069 (2014).
- Aziz, R. K., et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 9, 75 (2008).
- Tatusova, T., et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Research. 44 (14), 6614-6624 (2016).
- Carattoli, A., Hasman, H. PlasmidFinder and In Silico pMLST: Identification and Typing of Plasmid Replicons in Whole-Genome Sequencing (WGS). Methods in Molecular Biology. 2075, 285-294 (2020).
- Carattoli, A., et al. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrobial Agents and Chemotherapy. 58 (7), 3895-3903 (2014).
- Larsen, M. V., et al. Multilocus sequence typing of total-genome-sequenced bacteria. Journal of Clinical Microbiology. 50 (4), 1355-1361 (2012).
- Bortolaia, V., et al. ResFinder 4.0 for predictions of phenotypes from genotypes. The Journal of Antimicrobial Chemotherapy. 75 (12), 3491-3500 (2020).
- Joensen, K. G., et al. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. Journal of Clinical Microbiology. 52 (5), 1501-1510 (2014).
- Arndt, D., et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Research. 44 (1), 16-21 (2016).
- Couvin, D., et al. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Research. 46 (1), 246-251 (2018).
- Totten, P. A., Amsel, R., Hale, J., Piot, P., Holmes, K. K. Selective differential human blood bilayer media for isolation of Gardnerella (Haemophilus) vaginalis. Journal of Clinical Microbiology. 15 (1), 141-147 (1982).
- Nagarajan, N., Pop, M. Sequence assembly demystified. Nat Reviews. Genetics. 14 (3), 157-167 (2013).
- Phillippy, A. M., Schatz, M. C., Pop, M. Genome assembly forensics: finding the elusive mis-assembly. Genome Biology. 9 (3), 55 (2008).
- . Unicycler Wiki Available from: https://github.com/rrwick/Unicycler/wiki (2017)
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados