
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
DetectSyn: un método fluorescente rápido e imparcial para detectar cambios en la densidad de la sinapsis
En este artículo
Resumen
DetectSyn es un ensayo fluorescente rápido e imparcial que mide los cambios en el número relativo de sinapsis (compromiso pre y postsináptico) en todos los tratamientos o estados de enfermedad. Esta técnica utiliza una técnica de ligadura de proximidad que se puede utilizar tanto en neuronas cultivadas como en tejido fijo.
Resumen
Las sinapsis son el sitio de comunicación entre las neuronas. La fuerza del circuito neuronal está relacionada con la densidad sináptica, y la descomposición de las sinapsis es característica de estados de enfermedad como el trastorno depresivo mayor (TDM) y la enfermedad de Alzheimer. Las técnicas tradicionales para investigar los números de sinapsis incluyen la expresión genética de marcadores fluorescentes (por ejemplo, proteína fluorescente verde (GFP)), colorantes que llenan una neurona (por ejemplo, colorante de carbocianina, DiI) y detección inmunofluorescente de marcadores de columna vertebral (por ejemplo, densidad postsináptica 95 (PSD95)). Una advertencia importante a estas técnicas de proxy es que solo identifican cambios postsinápticos. Sin embargo, una sinapsis es una conexión entre un terminal presináptico y una columna vertebral postsináptica. El estándar de oro para medir la formación / eliminación de sinapsis requiere técnicas de microscopía electrónica o tomografía de matriz que consumen mucho tiempo. Estas técnicas requieren capacitación especializada y equipo costoso. Además, solo se puede evaluar un número limitado de neuronas y se utilizan para representar cambios en toda una región del cerebro. DetectSyn es una técnica fluorescente rápida que identifica cambios en la formación o eliminación de sinapsis debido a un estado de enfermedad o actividad farmacológica. DetectSyn utiliza un ensayo de ligadura de proximidad rápida para detectar proteínas pre y postsinápticas yuxtapuestas y microscopía fluorescente estándar, una técnica fácilmente disponible para la mayoría de los laboratorios. La detección fluorescente de la puntería resultante permite un análisis rápido e imparcial de los experimentos. DetectSyn proporciona resultados más representativos que la microscopía electrónica porque se pueden analizar áreas más grandes que un número limitado de neuronas fluorescentes. Además, DetectSyn funciona para neuronas cultivadas in vitro y cortes de tejido fijo. Finalmente, se proporciona un método para analizar los datos recopilados de esta técnica. En general, DetectSyn ofrece un procedimiento para detectar cambios relativos en la densidad de sinapsis a través de tratamientos o estados de enfermedad y es más accesible que las técnicas tradicionales.
Introducción
Las sinapsis son la unidad fundamental de comunicación entre neuronas1. Muchas sinapsis entre neuronas dentro de las mismas regiones dan lugar a circuitos que median el comportamiento2. Las sinapsis consisten en un terminal presináptico de una neurona que libera neurotransmisores o neuropéptidos que transmiten información a los receptores postsinápticos de otra neurona. La suma de señales presinápticas determina si la neurona postsináptica disparará un potencial de acción y propagará el mensaje a otras neuronas.
La sinaptopatología, la descomposición de las sinapsis, surge en enfermedades y trastornos marcados por la disminución del volumen neural, como la enfermedad de Alzheimer y el trastorno depresivo mayor, lo que resulta en circuitos que ya no funcionan de manera óptima 3,4,5. La restauración de la densidad de la sinapsis probablemente subyace a la eficacia de los posibles tratamientos para estos trastornos. Por ejemplo, recientemente se demostró que el aumento de las sinapsis subyace a la eficacia conductual de los antidepresivos rápidos6. Para detectar rápidamente posibles tratamientos de sinaptopatología, los investigadores requieren técnicas que identifiquen rápidamente los cambios en los números de sinapsis.
Las metodologías actuales consumen mucho tiempo y son costosas (microscopía electrónica, tomografía de matriz), o solo examinan los cambios postsinápticos sin incorporar el compromiso presináptico (análisis de la columna vertebral, inmunofluorescencia / colocalización). Los colorantes como DiI o las proteínas fluorescentes como GFP ayudan a visualizar las neuronas y caracterizar las espinas postsinápticas. Sin embargo, el análisis de la columna vertebral utiliza proporciones definidas por el investigador para determinar la morfología, lo que puede disminuir la reproducibilidad7. Además, todavía se está descubriendo cómo las diferentes clases de columna vertebral se relacionan con las sinapsis funcionales8. La formación de la columna vertebral puede ser transitoria y puede reflejar plasticidad postsináptica, pero estas espinas podrían eliminarse antes de estabilizarse en una sinapsis con una neurona presináptica9.
La colocalización proporciona un mejor indicador de las sinapsis que el análisis de la columna vertebral porque se puede inmunotintar las proteínas presinápticas y postsinápticas. Sin embargo, las proteínas sinápticas pueden producir valores bajos de colocalización porque las proteínas se yuxtaponen y pueden no superponerse consistentemente. Por lo tanto, debido a que las proteínas no están totalmente superpuestas, las técnicas de colocalización pueden no medir con precisión los cambios en la formación de sinapsis debido a esta información faltante. Finalmente, aunque tanto la microscopía electrónica (EM) como la tomografía de matriz proporcionan imágenes de alta resolución de sinapsis, consumen mucho tiempo. La EM requiere además equipo especializado, y los investigadores están limitados a pequeños volúmenes de tejido para cualquier experimento dado. Si bien la tomografía de matriz proporciona elegantemente la capacidad de detectar muchas proteínas en secciones ultrafinas y se puede combinar con EM10, esta técnica puede ser demasiado laboriosa y estar más allá del alcance de los experimentos que necesitan escanear rápidamente en busca de cambios en la formación de sinapsis.
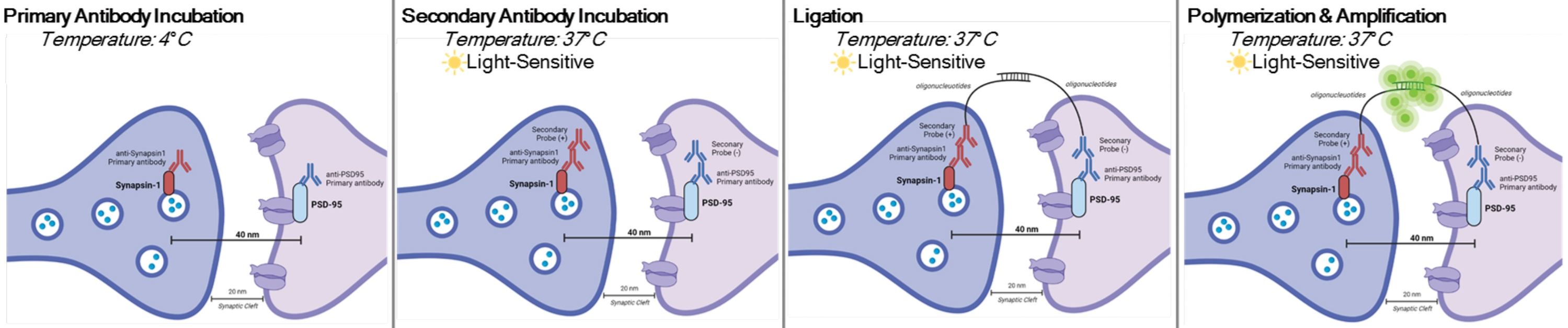
DetectSyn es una aplicación específica del ensayo de ligadura de proximidad Duolink. El ensayo de PLA permite la detección general de interacciones proteína-proteína. DetectSyn une las medidas postsinápticas proxy amplificando una señal fluorescente emitida por proteínas pre y postsinápticas etiquetadas dentro de los 40 nm entre sí. Si las proteínas sinápticas están dentro de los 40 nm, como dentro de una hendidura sináptica, entonces los anticuerpos secundarios, que contienen sondas de ADN, se hibridarán en ADN circular. Este ADN circular hibridado expresa una sonda fluorescente, que luego se amplifica y detecta con técnicas estándar de microscopía fluorescente (ver Figura 1). Crucialmente, a diferencia de la EM y la tomografía de matriz, esta técnica no requiere equipo especializado y toma aproximadamente la misma cantidad de tiempo que la inmunohistoquímica estándar. La accesibilidad de esta técnica, por lo tanto, permite a los investigadores fuera de las instituciones intensivas en investigación participar en la investigación sinaptopatología. Además, esta técnica puede examinar los cambios en la densidad sináptica en múltiples regiones del cerebro dentro de un solo experimento, ofreciendo una representación más holística de los cambios sinápticos debido a una enfermedad o tratamiento.
Protocolo
El aislamiento de células y tejidos de animales se realizó de acuerdo con la Guía para el Cuidado y Uso de Animales de Laboratorio de los Institutos Nacionales de Salud y fue aprobado por el Comité Institucional de Cuidado y Uso de Animales de Wake Forest
NOTA: Este protocolo se utiliza en muestras ya tratadas y fijadas según paradigmas y requisitos experimentales específicos. Para fines de demostración, la formación de sinapsis debido al tratamiento antidepresivo rápido se utiliza para resaltar esta técnica de detección de sinapsis6. Las neuronas previamente cultivadas en cápsulas, tratadas, fijadas en paraformaldehído al 4% (PFA) y almacenadas en solución salina tamponada con 1x fosfato (PBS) se utilizarán para resaltar los procedimientos in vitro. Se utilizará tejido del hipocampo previamente cortado (25 μm de espesor) de ratones tratados, perfundido transcárdicamente con PBS helado y PFA al 4%, y luego almacenado en crioprotector para resaltar los procedimientos de rebanada. Consulte11,12 para obtener más información sobre cómo cultivar neuronas o perfundir roedores transcárdicos. Consulte la Figura 1 para obtener una representación gráfica de este procedimiento.

Figura 1: Representación gráfica del ensayo DetectSyn. Después de permeabilizar las membranas celulares, los anticuerpos primarios para Synapsin1 y PSD95 se unen a estas proteínas sinápticas. Los secundarios con etiquetas de oligonucleótidos se unen a los anticuerpos primarios. Si Synapsin1 y PSD95 están dentro de los 40 nm, como en una sinapsis, entonces los oligonucleótidos interactúan y se amplifica una etiqueta fluorescente. Esta señal fluorescente puede ser fotografiada a través de microscopía estándar y analizada. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
1. Enjuague las muestras
- Enjuague las muestras con 500 μL de 1x PBS + 0.75% de glicina durante 5 min 3 veces con agitación suave en un agitador orbitario para eliminar pfa residual o crioprotector.
2. Bloquear y permeabilizar muestras
- Preparar solución de bloqueo y permeabilización (10% de suero de burro normal, 0,25% Tween 20) en 1x PBS. Prepárese lo suficiente para usar para incubaciones de bloqueo, primarias y secundarias.
- A las muestras (por ejemplo, hojas de cubierta o rodajas flotantes) en placas de 24 pocillos, agregue 500 μL de solución de bloqueo y permeabilización. Asegúrese de que cada pozo contenga una muestra diferente y esté debidamente etiquetado para evitar que se cambien las muestras.
- Incubar las muestras a temperatura ambiente (RT) durante 60 min para las células cultivadas o 2 h para el tejido en rodajas. Use un agitador orbital para una agitación suave.
3. Incubar muestras en anticuerpos primarios
- Preparar anticuerpos primarios en tampón de bloqueo:
- Preparar densidad postsináptica 95 (PSD95; 1:500, policlonal de conejo), Synapsin1 (1:500, monoclonal de ratón), MAP2 (1:400, policlonal de pollo)
- Preparar una alícuota de control negativo que omita uno de los pares sinápticos (por ejemplo, sin PSD95)
- Retire con cuidado la solución de bloqueo con una pipeta Pasteur de plástico. Trate de eliminar tanto como sea posible sin perturbar las células o desgarrar el tejido.
- Para células cultivadas:
- Forra una gran placa de Petri de plástico con parafilm. Transfiera cuidadosamente las fundas a la parapelícula con fórceps.
- Agregue cuidadosamente 60 μL de la solución de anticuerpos primarios a la parte superior de los cobertores. Asegúrese de no derramar la solución de anticuerpos primarios sobre el lado de la cubierta.
- Para proporcionar humedad y evitar que las muestras se sequen durante el período de incubación, agregue agua ultrapura a una placa de Petri más pequeña y coloque cuidadosamente la pequeña placa de Petri alrededor de los cobertores.
- Cubra la placa de Petri grande e incube las células cultivadas durante 1 h en RT.
- Para el tejido en rodajas:
- Agregue cuidadosamente 250 μL de la solución de anticuerpos primarios a las rodajas que flotan libremente en una placa de 24 pocillos.
- Cubra la placa e incube el tejido durante la noche a 4 °C con una agitación suave en un agitador orbitario.
4. Lavar las muestras, luego incubar en anticuerpos secundarios
- Prepare anticuerpos secundarios en el tampón de bloqueo:
- Prepare Burro anti-ratón (1:5), burro anti-conejo (1:5), burro anti-pollo (1:400).
- En este paso, se pueden obtener controles técnicos adicionales preparando una alícuota secundaria que omite la secundaria anti-ratón o anti-conejo.
- Para células cultivadas:
- Usando fórceps, golpee cuidadosamente la solución primaria de las hojas de cubierta en una toalla de papel
- Usando fórceps, transfiera cuidadosamente las cubiertas a su placa original de 24 pocillos llena de 500 μL de 1x PBS.
- Para el tejido en rodajas:
- Retire con cuidado la solución primaria de anticuerpos con una pipeta Pasteur de plástico. Trate de eliminar tanto como sea posible sin desgarrar el tejido.
- Añadir 500 μL de 1x PBS
- Lave las muestras durante 10 min 3 veces en 1x PBS con agitación suave en un agitador orbital. Durante este tiempo, lleve todos los tampones de lavado a RT.
- Durante este tiempo, cambie la parapelícula en la gran placa de Petri
- Para células cultivadas:
- Usando fórceps, transfiera cuidadosamente las fundas de regreso a la placa de Petri grande parafilmada
- Agregue cuidadosamente 40 μL de la solución de anticuerpos secundarios a la parte superior de los cobertores. Asegúrese de no derramar la solución secundaria de anticuerpos sobre el lado de la cubierta.
- Si es necesario, agregue más agua ultrapura a una placa de Petri más pequeña y coloque cuidadosamente la pequeña placa de Petri alrededor de los cobertores.
- Cubra la gran placa de Petri
- Para el tejido en rodajas:
- Agregue cuidadosamente 250 μL de la solución secundaria de anticuerpos a las rodajas que flotan libremente en una placa de 24 pocillos.
- Cubra la placa
NOTA: De aquí en adelante, proteja las muestras de la luz envolviendo la parte superior de las placas con papel de aluminio.
- Incubar las muestras a 37 °C durante 1 h
5. Ligadura
- Mezcle el material de ligadura 1:5 en agua de grado molecular.
- Al igual que en la sección 4, transfiera cuidadosamente las fundas y retire la mezcla secundaria del tejido en rodajas.
- Lavar las muestras en 500 μL de tampón de lavado A
- Para las células cultivadas, lavar 2 veces durante 5 min. Para el tejido en rodajas, lavar 2 veces durante 10 min. Use una agitación suave en un agitador orbital para ambos.
- Durante este tiempo, cambie la parapelícula en la gran placa de Petri
- Mientras mantiene la ligasa en un bloque frío, diluya la ligasa 1:40 en el stock de ligadura del paso 5.1. Realice esta dilución inmediatamente antes de agregar la ligasa a las muestras.
- Al igual que en la sección 4, retire la mayor cantidad posible del tampón de lavado A de las muestras antes de agregar la ligasa.
- Para células cultivadas: Transfiera los recubiertos a la placa de Petri parafilmada. Agregue 40 μL de la mezcla de ligadura a los cubrehojas, coloque pequeñas placas de Petri llenas de agua alrededor de las hojas de cubierta y cubra la placa de Petri grande.
- Para el tejido en rodajas: Agregue 250 μL de la mezcla de ligadura del paso 5.4 a cada pocillo y cubra la placa.
- Incubar las muestras durante 30 min a 37 °C.
6. Amplificación
- Mezcle el material de amplificación 1:5 en agua de grado molecular.
- Al igual que en la sección 4, transfiera cuidadosamente las fundas y retire la mezcla de ligadura del tejido cortado.
- Lavar muestras en 500 μL de tampón de lavado A
- Para las células cultivadas, lavar 2 veces durante 2 min. Para el tejido en rodajas, lavar 2 veces durante 10 min. Use una agitación suave en un agitador orbital para ambos.
- Durante este tiempo, cambie la parapelícula en la gran placa de Petri
- Realice esta dilución inmediatamente antes de agregar la polimerasa a las muestras. Mientras mantiene la polimerasa en un bloque frío, diluya la polimerasa
- Para las células cultivadas, diluir la polimerasa 1:80 en el material de amplificación del paso 6.1.
- Para el tejido en rodajas, diluya la polimerasa 1:40 en el material de amplificación del paso 6.1.
- Al igual que en el paso 4, retire la mayor cantidad posible del tampón de lavado A de las muestras antes de agregar la polimerasa.
- Para células cultivadas: Transfiera los recubiertos a la placa de Petri parafilmada. Agregue 40 μL de la mezcla de amplificación del paso 6.4.1 a los cubrecos, coloque pequeñas placas de Petri llenas de agua alrededor de los cubrecos y cubra la placa de Petri grande. Incubar las muestras durante 100 min a 37 °C.
- Para el tejido en rodajas: Agregue 250 μL de la mezcla de amplificación del paso 6.4.2 a cada pocillo y cubra la placa. Incubar las muestras durante 2 h a 37 °C.
NOTA: Durante este tiempo, prepare y etiquete las diapositivas.
7. Montaje
- Al igual que en el Paso 4, transfiera cuidadosamente las hojas de cubierta y retire la mezcla de amplificación del tejido en rodajas.
- Lave las muestras en 500 μL de tampón de lavado B 2 veces durante 10 minutos con agitación suave en un agitador orbital.
- Lave las muestras en 500 μL de tampón B de lavado al 1% durante 1 min con agitación suave en un agitador orbital.
- Para células cultivadas:
- Coloque 3 μL de soporte de montaje en una diapositiva
- Retire el exceso de tampón de lavado de la cubierta y luego coloque la cubierta (con las celdas hacia abajo) en el medio de montaje. Selle los lados con una pequeña cantidad de esmalte de uñas transparente para sellar la cubierta en su lugar.
- Para el tejido en rodajas:
- Transfiera cuidadosamente una rebanada de tejido al portaobjetos preparado y organícela, de modo que la rebanada quede plana. Deje caer entre 5-10 μL de medio de montaje (la cantidad dependerá del tamaño de la rebanada) sobre la rebanada de tejido
- Coloque cuidadosamente una cubierta de vidrio sobre una rodaja de tejido y selle con una pequeña cantidad de esmalte de uñas transparente a lo largo del borde para sellar la cubierta en su lugar.
- Espere al menos 15 minutos antes de analizar bajo el microscopio o guárdelo a -20 °C.
8. Obtener imágenes digitales con un microscopio confocal
- Optimice la configuración de adquisición (por ejemplo, potencia del láser, ganancia, desplazamiento) en todas las muestras de todos los tratamientos. Asegúrese de que la optimización incluya disminuir el ruido de fondo y mejorar la señal sin sobresaturar la intensidad de las señales fluorescentes. Una vez que se determina la configuración, aplique la misma configuración de adquisición en todas las imágenes obtenidas.
NOTA: Los siguientes detalles de adquisición se pueden utilizar con un microscopio confocal Nikon A1 y el software Nikon NIS AR Elements. - Coloque la diapositiva con la muestra en el escenario y busque el plano focal para la muestra utilizando DAPI a través del ocular.
- Apague el puerto ocular haciendo clic en Puerto ocular y elija un botón de configuración óptica para ajustar la configuración.
- Ajuste la ganancia, el desplazamiento y la potencia del láser para cada canal fluorescente para disminuir el ruido de fondo y mejorar la señal fluorescente. Asegúrese de que la señal fluorescente no se sobresature como se menciona en los pasos 8.5-8.6.
- Monitoree la sobresaturación utilizando un pseudocolor para la señal fluorescente. En la parte inferior de la imagen en vivo, haga clic con el botón derecho en la pestaña etiquetada con el canal fluorescente que se está utilizando actualmente.
- A continuación, elija Coloración de canal y elija un pseudocolor como Rainbow Dark para visualizar la intensidad de la fluorescencia en un pseudocolor similar a un mapa de calor. En Rainbow Dark, los colores más fríos indican menos intensidad fluorescente, y los colores más calientes indican más intensidad fluorescente.
- Una vez que todos los canales fluorescentes estén optimizados, haga clic con el botón derecho en el botón de configuración óptica previamente elegido y elija Asignar configuración de cámara actual para este botón.
- Verifique que los ajustes elegidos sean suficientes para una muestra aleatoria de cada grupo de tratamiento. Si los ajustes elegidos sobresaturan alguna de estas muestras, repita el paso 8.4 para eliminar la sobresaturación.
- Para las neuronas cultivadas, siga los pasos 8.10-8.16.
- Usando el puerto ocular, busque una neurona con dendritas que tengan una superposición mínima con otras dendritas.
- Apague el puerto ocular y use el canal DAPI para visualizar el cuerpo celular de la neurona elegida. Haga doble clic en el centro del soma para centrar la neurona en el centro del campo de visión.
- Usando el canal MAP2, encuentre el mejor plano de enfoque para la señal MAP2 con escaneo en vivo.
- En la pestaña Adquisición de ND , haga clic en Guardar en archivo y elija un archivo para guardar la imagen en Examinar. A continuación, introduzca el nombre de archivo.
- En la pestaña Z , seleccione la opción Modo simétrico definido por rango . Establezca el enfoque en el mejor plano MAP2 y haga clic en el botón Relativo para establecer este plano focal como el centro de la pila z.
- Ajuste el rango a 5 μm con pasos de 1 μm y asegúrese de marcar Cerrar obturador activo durante el movimiento Z. En la pestaña Longitud de onda , seleccione el nombre del botón óptico con los ajustes de adquisición configurados previamente en Optical Conf. Luego, haga clic en Ejecutar ahora.
- Repita los pasos 8.1.10-8.1.15 para aproximadamente 10 neuronas por recubierto/tratamiento.
- Para el tejido en rodajas, siga los pasos 8.18-8.22.
- Usando el eye-port, busque la región de interés. Por ejemplo, localice CA1 del hipocampo.
- Apague el puerto ocular y utilice el canal MAP2 para encontrar el mejor plano de enfoque para la señal MAP2 con el escaneo en vivo.
- En el menú Adquirir , elija Escanear imagen grande. A continuación, seleccione el nombre del botón óptico con los ajustes de adquisición configurados previamente en el panel Captura del panel que se abre. Además, asegúrese de seleccionar el objetivo correcto en este panel.
- En el panel Área y en el ocular, utilice las teclas de flecha para establecer los límites de la región de interés. A continuación, haga clic en Guardar imagen grande en archivo y cree un nombre de archivo de ruta de guardado para la imagen.
- En el panel Configuración , asegúrese de que la opción Captura multicanal esté activada y, a continuación, elija el nombre del botón óptico con los ajustes de adquisición configurados previamente en Optical Conf.
NOTA: Una pila z para una imagen grande es posible, pero aumentará el tiempo de escaneo.
9. Análisis
- De forma similar a la configuración de adquisición, utilice muestras de todos los tratamientos para optimizar la configuración del umbral. Asegúrese de que la optimización del umbral se centre en disminuir el ruido de fondo y mejorar la señal sin sobresaturar la intensidad de las señales fluorescentes. Una vez que se determinen estos ajustes, aplique los mismos ajustes de umbral en todas las imágenes utilizadas para el análisis, tal como se describe en los pasos 9.2 a 9.3.
- En ImageJ, la opción de umbral se encuentra en el menú Imagen > Ajustar > Umbral. Elija la opción Fondo oscuro si la imagen tiene un fondo oscuro.
- A continuación, ajuste los límites superior e inferior del umbral según la configuración de umbral optimizada previamente determinada, luego haga clic en Aplicar.
- Para las células cultivadas, utilice el canal MAP2 y una herramienta de región de interés (ROI) a mano alzada para dibujar un ROI para cada neurona, incluidas las dendritas y el soma. Para el tejido en rodajas, dibuje un ROI a mano alzada dentro de la imagen de la rebanada que encapsule el área de interés (por ejemplo, el estrato radiatum del CA1 dentro del hipocampo).
- Obtener el área del ROI. En ImageJ, mida el área en el menú Analizar > medir.
- Detecte el número de puntos dentro de cada ROI utilizando una herramienta de detección automática como Análisis de partículas en ImageJ siguiendo los pasos 9.7-9.9.
- Busque la opción Análisis de partículas en el menú Analizar > Analizar partículas. Primero, defina el diámetro del tamaño de la punta, típicamente 0.1-3 μm2.
- A continuación, elija la opción Máscaras superpuestas en el menú desplegable Mostrar y marque la opción Mostrar resultados. Luego, haga clic en Aceptar.
- Si no se detectan puntos con el rango de diámetro elegido, ajuste el rango hasta que se detecten todos los puntos con este análisis. Asegúrese de utilizar la misma configuración de análisis de partículas para todas las imágenes.
- Divida el número de puntos por el área de una región individual de interés siguiendo los pasos 9.11-9.13.
- Copie y pegue los resultados de cada imagen de la ventana emergente Resultados de ImageJ en una hoja de cálculo.
- Primero, identifique de qué archivo y muestra se obtuvieron los datos. Luego, divida el área del ROI por el número de puntos.
- A continuación, borre los datos de la ventana emergente Resultados y repita los pasos 9.2-9.12.
- Normalizar los resultados a las muestras de control: Promediar los resultados (número de áreas punteadas/ROI) para las muestras de control. Luego, divida los resultados obtenidos de todas las muestras por el promedio del control para obtener los resultados normalizados. El nuevo promedio de las muestras de control debe ser igual a 1.
Resultados
Los datos modificados de Heaney et al.6 se presentan para demostrar un experimento en el que se espera una mayor formación de sinapsis (consulte6 para obtener más información y una discusión más profunda del mecanismo). Anteriormente, se demostró que los antidepresivos rápidos requieren la activación del receptor metabotrópico inhibidor, GABAB (gamma-aminobutyric acid subtype B), para serefectivos 13. Además, los datos anteriores indicaron ...
Discusión
DetectSyn es un ensayo rápido que utiliza un ensayo de ligadura de proximidad para detectar proteínas dentro de los 40 nm entre sí, lo que permite la detección de la formación de sinapsis. Esta técnica mejora los ensayos fluorescentes actuales, que sirven solo como mediciones proxy para la formación de sinapsis. DetectSyn detecta cambios cuantificables en las proteínas sinápticas localizadas dentro de los 40 nm, es decir, dentro de la hendidura sináptica, entre sí. Además, DetectSyn es más rentable y lleva m...
Divulgaciones
Los autores no reportan ningún conflicto de intereses.
Agradecimientos
Este trabajo fue apoyado por los Institutos Nacionales de Salud NINDS R01 NS105005 (KRG) y NS105005-03S1 (KRG), el Departamento de Defensa USAMRMC W81XWH-14-1-0061 (KRG), NIAAA R01AA016852, NIAAA T32AA007565 (CFH), y una subvención de FRAXA Research (CFH) y la Asociación de Alzheimer, AARG-NTF-21-852843 (KRG), AARF-19-614794-RAPID (KRG).
Materiales
| Name | Company | Catalog Number | Comments |
| 10x PBS | Fisher Scientific | BP39920 | PBS made in house works, as well. |
| 24 well plates | Fisher Scientific | FB012929 | For tissue slices, pre-sterilized plates may be unnecessary. |
| 50 mL conical tubes | Fisher Scientific | 14-432-22 | |
| Aluminium foil | Fisher Scientific | 15-078-290 | |
| Chicken anti-MAP2 antibody | Abcam | ab5392 | |
| Clear nail polish | Fisher Scientific | NC1849418 | Other clear nail polish works, as well. |
| Cold block | Fisher Scientific | 13131012 | |
| Computer workstation | HP | ||
| Confocal or fluorescent microscope | Nikon | A1R HD25 | |
| Donkey anti-chicken FITC | Fisher Scientific | SA1-72000 | |
| Duolink donkey anti-Mouse PLUS | Sigma | DUO92001 | |
| Duolink donkey anti-Rabbit MINUS | Sigma | DUO92005 | |
| Duolink In Situ Detection Reagents Far Red | Sigma | DUO92013 | Contains ligation stock, amplification stock, ligase, and polymerase. |
| Duolink In Situ Mounting Medium with DAPI | Sigma | DUO82040 | |
| Duolink In Situ Wash Buffers, Fluorescence | Sigma | DUO82049 | Contains Wash Buffer A and Wash Buffer B; dilute Wash Buffer B to 1% in diH20 for 1% Wash Buffer B. |
| Fine-tipped paintbrush | Fisher Scientific | NC9691026 | Sable hair, size 00 or 000, can also find at craft stores |
| Fisherbrand Cover Glasses: Rectangles | Fisher Scientific | 12545MP | Cover glass is unnecessary for cultured neurons already on glass coverslips. |
| Fisherbrand Superfrost Plus Microscope Slides | Fisher Scientific | 1255015 | For cultured neurons already on glass coverslips, Superfrost slides may be unnecessary. |
| Freezer, -20°C | VWR | 76449-108 | |
| Glass coverslips | Fisher Scientific | 125480 | |
| Glycine | Fisher Scientific | BP381-1 | |
| Image processing software | e.g. NIS Elements, ImageJ | ||
| Incubator | Fisher Scientific | 15-015-2633 | |
| Large petri dish, 100mm | Fisher Scientific | FB0875712 | |
| Molecular grade water | Fisher Scientific | BP24701 | |
| Mouse anti-Synapsin1 antibody | Synaptic Systems | 106-011 | |
| Normal donkey serum | Jackson ImmunoResearch | 017-000-121 | |
| Orbital shaker | Fisher Scientific | 02-106-1013 | |
| Parafilm | Fisher Scientific | 13-374-10 | |
| Pipette tips | Fisher Scientific | 02-707-025 | |
| Pipettes | Fisher Scientific | 14-388-100 | Working volumes range from 3 µL to 500 µL |
| Plastic pasteur pipette | Fisher Scientific | 02-708-006 | |
| Precision tweezers/foreceps | Fisher Scientific | 12-000-122 | |
| Rabbit anti-PSD95 antibody | Abcam | ab18258 | Other antibody pairs may work, as well, with optimization. |
| Refrigerator | VWR | 76470-402 | |
| Small petri dish, 60 mm | Fisher Scientific | FB0875713A | |
| Timer | Fisher Scientific | 14-649-17 | |
| Tween 20 | Fisher Scientific | BP337-100 |
Referencias
- Südhof, T. C. Towards an understanding of synapse formation. Neuron. 100 (2), 276-293 (2018).
- Bliss, T. V., Collingridge, G. L. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 361 (6407), 31-39 (1993).
- Heaney, C. F., Raab-Graham, K. F. Dysregulated protein synthesis in major depressive disorder. The Oxford Handbook of Neuronal Protein Synthesis. , 510-532 (2018).
- Masliah, E., Crews, L., Hansen, L. Synaptic remodeling during aging and in Alzheimer's disease. Journal of Alzheimer's Disease. 9, 91-99 (2006).
- van Spronsen, M., Hoogenraad, C. C. Synapse pathology in psychiatric and neurologic disease. Current Neurology and Neuroscience Reports. 10 (3), 207-214 (2010).
- Heaney, C. F., Namjoshi, S. V., Uneri, A., Bach, E. C., Weiner, J. L., Raab-Graham, K. F. Role of FMRP in rapid antidepressant effects and synapse regulation. Molecular Psychiatry. 26 (6), 2350-2362 (2021).
- Pchitskaya, E., Bezprozvanny, I. Dendritic spines shape analysis-Classification or clusterization? Perspective. Frontiers in Synaptic Neuroscience. 12, 31 (2020).
- Alvarez, V. A., Sabatini, B. L. Anatomical and physiological plasticity of dendritic spines. Annual Review of Neuroscience. 30 (1), 79-97 (2007).
- Berry, K. P., Nedivi, E. Spine Dynamics: Are they all the same. Neuron. 96 (1), 43-55 (2017).
- Micheva, K. D., Smith, S. J. Array tomography: A new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron. 55 (1), 25-36 (2007).
- Kaech, S., Banker, G. Culturing hippocampal neurons. Nature Protocols. 1 (5), 2406-2415 (2006).
- Gage, G. J., Kipke, D. R., Shain, W. Whole animal perfusion fixation for rodents. Journal of Visualized Experiments: JoVE. (65), e3564 (2012).
- Workman, E. R., Niere, F., Raab-Graham, K. F. mTORC1-dependent protein synthesis underlying rapid antidepressant effect requires GABABR signaling. Neuropharmacology. 73, 192-203 (2013).
- Li, N., et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 329 (5994), 959-964 (2010).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados