
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
DetectSyn:シナプス密度の変化を検出するための迅速で偏りのない蛍光法
要約
DetectSynは、治療または疾患状態にわたる相対シナプス(シナプス前およびシナプス後関与)数の変化を測定する、偏りのない迅速な蛍光アッセイです。この技術は、培養ニューロンおよび固定組織の両方において使用することができる近接ライゲーション技術を利用する。
要約
シナプスはニューロン間のコミュニケーションの部位です。神経回路の強さはシナプス密度に関係しており、シナプスの破壊は大うつ病性障害(MDD)やアルツハイマー病などの疾患状態の特徴です。シナプス数を調べるための伝統的な技術には、蛍光マーカー(例えば、緑色蛍光タンパク質(GFP))の遺伝子発現、ニューロンを満たす色素(例えば、カルボシアニン色素、DiI)、および脊椎マーカーの免疫蛍光検出(例えば、シナプス後密度95(PSD95))が含まれる。これらのプロキシ技術の主な注意点は、シナプス後の変化のみを識別することです。しかし、シナプスはシナプス前終末とシナプス後脊椎との間の接続である。シナプスの形成/除去を測定するためのゴールドスタンダードには、時間のかかる電子顕微鏡またはアレイ断層撮影技術が必要です。これらの技術には、専門的なトレーニングと高価な機器が必要です。さらに、限られた数のニューロンしか評価できず、脳領域全体に対する変化を表すために使用される。DetectSynは、疾患状態または薬物活性に起因するシナプス形成または消失の変化を識別する迅速な蛍光技術である。DetectSynは、迅速な近接ライゲーションアッセイを利用して、並置されたシナプス前部およびシナプス後部タンパク質と標準的な蛍光顕微鏡法を検出します。得られた穿刺の蛍光検出により、実験の迅速かつ公平な分析が可能になります。DetectSynは、限られた数の蛍光ニューロンよりも広い領域を分析できるため、電子顕微鏡よりも代表的な結果を提供します。さらに、DetectSynは 、インビトロ 培養ニューロンおよび固定組織スライスに対して機能します。最後に、この技術から収集されたデータを分析するための方法が提供される。全体として、DetectSynは、治療または疾患状態にわたるシナプス密度の相対的な変化を検出するための手順を提供し、従来の技術よりもアクセスしやすい。
概要
シナプスは、ニューロン間のコミュニケーションの基本単位である1。同じ領域内のニューロン間の多くのシナプスは、行動を媒介する回路を生じる2。シナプスは、神経伝達物質または神経ペプチドを放出する1つのニューロンからのシナプス前末端からなり、別のニューロンのシナプス後受容体に情報を中継する。シナプス前部シグナルの総和は、シナプス後ニューロンが活動電位を発火させ、メッセージを他のニューロンに伝播するかどうかを決定する。
シナプス病理学、シナプスの分解は、アルツハイマー病および大うつ病性障害のような神経体積の減少によって特徴付けられる疾患および障害において生じ、もはや最適に実行しない回路をもたらす3,4,5。シナプス密度の回復は、これらの障害に対する潜在的な治療法の有効性の根底にある可能性が高い。例えば、シナプスの増加が急速な抗うつ薬の行動効果の根底にあることが最近実証されました6。可能なシナプス病理学的治療を迅速にスクリーニングするために、研究者はシナプス数の変化を迅速に特定する技術を必要とする。
現在の方法論は、時間と費用がかかるか(電子顕微鏡、アレイ断層撮影)、またはシナプス前部の関与(脊椎分析、免疫蛍光/共局在化)を組み込まずにシナプス後の変化のみを調べる。DiIのような色素やGFPのような蛍光タンパク質は、ニューロンを視覚化し、シナプス後棘を特徴付けるのに役立ちます。しかし、脊椎解析では、研究者が定義した比率を使用して形態を決定し、再現性を低下させる可能性があります7。さらに、異なる脊椎クラスが機能的シナプスとどのように関連しているかは、まだ明らかにされていない8。脊椎形成は一過性であり得、シナプス後可塑性を反映する可能性があるが、これらの脊椎はシナプス前ニューロン9を有するシナプスに安定する前に排除され得る。
共局在化は、シナプス前およびシナプス後タンパク質を免疫染色することができるため、脊椎分析よりもシナプスのより良いプロキシを提供する。しかしながら、シナプスタンパク質は、タンパク質が並置され、一貫して重なり合わない可能性があるため、低い共局在値をもたらす可能性がある。したがって、タンパク質は完全に重ね合わされていないため、共局在化技術は、この欠落した情報のためにシナプス形成の変化を正確に測定できない可能性がある。最後に、電子顕微鏡(EM)とアレイ断層撮影法の両方がシナプスの高解像度画像を提供しますが、時間がかかります。EMはさらに特殊な装置を必要とし、研究者は任意の実験のために少量の組織に制限されている。アレイ断層撮影法は、超薄切片上の多くのタンパク質を優雅にスクリーニングする能力を提供し、EM10と組み合わせることができるが、この技術はあまりにも労働集約的であり、シナプス形成の変化を迅速にスキャンする必要がある実験の範囲を超えている可能性がある。
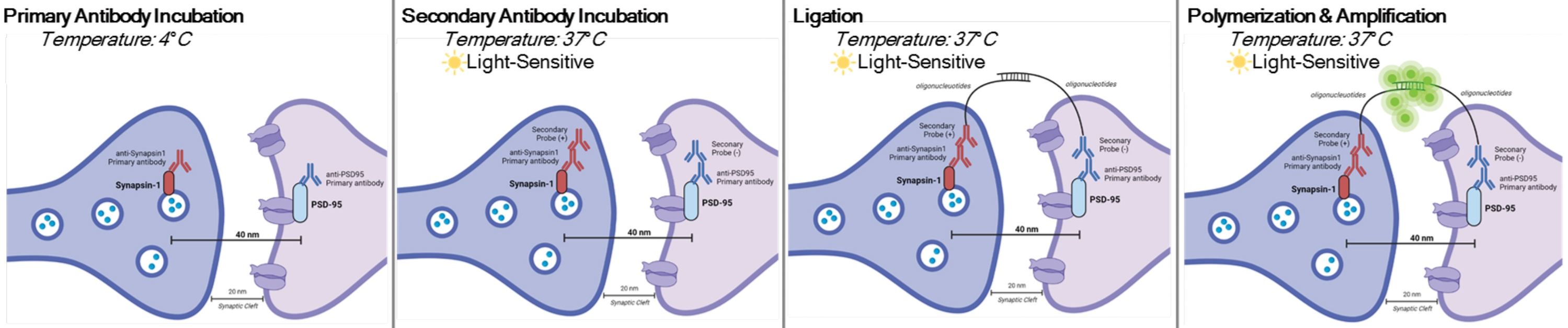
DetectSynは、デュオリンク近接ライゲーションアッセイの特定のアプリケーションです。PLAアッセイは、タンパク質間相互作用の一般的な検出を可能にします。DetectSynは、タグ付きのシナプス前およびシナプス後タンパク質によって放出される蛍光シグナルを互いに40nm以内に増幅することによって、シナプス後部のプロキシブリッジ測定を行います。シナプスタンパク質がシナプス間隙内と同様に40nm以内にある場合、DNAプローブを含む二次抗体は環状DNAにハイブリダイズする。このハイブリダイズした環状DNAは蛍光プローブを発現し、次いでこれを増幅し、標準的な蛍光顕微鏡技術で検出する( 図1参照)。重要なことに、EMやアレイ断層撮影法とは異なり、この技術は特別な装置を必要とせず、標準的な免疫組織化学とほぼ同じ時間がかかります。したがって、この技術のアクセシビリティにより、研究集約型機関以外の研究者はシナプト病理学研究に参加することができます。さらに、この技術は、単一の実験内で複数の脳領域におけるシナプス密度の変化を調べることができ、疾患または治療によるシナプス変化のより全体的な表現を提供する。
プロトコル
動物からの細胞および組織の単離は、国立衛生研究所の実験動物の世話と使用のためのガイドに従い、ウェイクフォレストの施設動物ケアおよび使用委員会によって承認されました
注:このプロトコルは、特定の実験パラダイムおよび要件に従って既に処理および固定されたサンプルで使用されます。実証目的のために、迅速な抗うつ薬治療によるシナプス形成が、このシナプス検出技術6を強調するために使用される。以前にカバースリップで培養し、処理し、4%パラホルムアルデヒド(PFA)で固定し、1xリン酸緩衝生理食塩水(PBS)に保存したニューロンは、インビトロ手順を強調するために使用される。処置したマウスから以前にスライスされた海馬組織(厚さ25μm)を、氷冷PBSおよび4%PFAで経心的に灌流し、次いで凍結保護剤に保存して、スライス手順を強調するために使用される。ニューロンを培養する方法、またはげっ歯類を経心的に灌流する方法の詳細については、11,12を参照してください。この手順をグラフィカルに表すには、図 1 を参照してください。

図1:DetectSynアッセイのグラフ表示。 細胞膜を透過処理した後、シナプシン1およびPSD95に対する一次抗体は、これらのシナプスタンパク質に結合する。次いで、オリゴヌクレオチドタグを有する二次抗体が一次抗体に結合する。シナプス1とPSD95がシナプスのように40nm以内にある場合、オリゴヌクレオチドは相互作用し、蛍光タグが増幅される。この蛍光シグナルは、標準的な顕微鏡検査によって画像化され、分析され得る。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
1. サンプルをすすぐ
- サンプルを 500 μL の 1x PBS + 0.75% グリシンで 5 分間 3 回すすぎ、オービタル シェーカーで穏やかに攪拌し、残留 PFA または凍結保護剤を除去します。
2. サンプルのブロックと透過
- ブロッキングおよび透過処理溶液(10%正常ロバ血清、0.25%Tween 20)を1x PBS中に調製する。ブロッキング、一次、および二次インキュベーションに使用するのに十分な準備をしてください。
- 24ウェルプレートのサンプル(カバースリップやフリーフローティングスライスなど)に、500 μLのブロッキングおよび透過処理溶液を加えます。各ウェルに異なるサンプルが含まれており、サンプルが切り替えられないように適切にラベル付けされていることを確認します。
- サンプルを室温(RT)で培養細胞の場合は60分間、スライスした組織の場合は2時間インキュベートします。穏やかな攪拌のためにオービタルシェーカーを使用してください。
3. 一次抗体でサンプルをインキュベートする
- ブロッキングバッファー中に一次抗体を調製する:
- シナプス後密度95(PSD95;1:500、ウサギポリクローナル)、シナプシン1(1:500、マウスモノクローナル)、MAP2(1:400、ニワトリポリクローナル)を準備する
- シナプス対の1つを省略した陰性対照アリコートを準備する(例えば、PSD95なし)
- プラスチック製のパスツールピペットでブロッキング溶液を慎重に取り除きます。細胞を乱したり、組織を引き裂いたりすることなく、できるだけ多く取り除くようにしてください。
- 培養細胞の場合:
- 大きなプラスチック製のシャーレにパラフィルムを敷きます。鉗子を使用してカバースリップをパラフィルムに慎重に移します。
- カバースリップの上部に60 μLの一次抗体溶液を慎重に加えます。一次抗体溶液をカバースリップの側面にこぼさないようにしてください。
- 湿度を確保し、インキュベーション期間中にサンプルが乾燥するのを防ぐために、小さなペトリ皿に超純水を加え、カバースリップの周りに小さなペトリ皿を慎重に配置します。
- 大きなシャーレを覆い、RTで培養細胞を1時間インキュベートする。
- スライスされた組織の場合:
- 250 μL の一次抗体溶液を 24 ウェルプレート内のフリーフローティングスライスに慎重に加えます。
- プレートを覆い、組織を軌道シェーカー上で穏やかな攪拌で4°Cで一晩インキュベートする。
4. サンプルを洗浄し、二次抗体でインキュベートする
- ブロッキングバッファー中に二次抗体を調製する:
- ロバのアンチマウス(1:5)、ロバのアンチウサギ(1:5)、ロバのアンチチキン(1:400)を準備します。
- このステップでは、追加の技術的対照は、二次的な抗マウスまたは抗ウサギのいずれかを省略した二次アリコートを調製することによって得ることができる。
- 培養細胞の場合:
- 鉗子を使用して、カバースリップからペーパータオルに一次溶液を慎重にタップします
- 鉗子を使用して、カバースリップを500 μLの1x PBSで満たされた元の24ウェルプレートに慎重に戻します。
- スライスされた組織の場合:
- 一次抗体溶液をプラスチック製のパスツールピペットで慎重に取り出します。組織を引き裂かずにできるだけ取り除くようにしてください。
- 500 μL の 1x PBS を追加
- オービタルシェーカー上で穏やかな攪拌で1x PBSでサンプルを10分間3回洗浄する。この間、すべての洗浄バッファーをRTに持参してください。
- この間、大きなペトリ皿のパラフィルムを交換してください
- 培養細胞の場合:
- 鉗子を使用して、カバースリップをパラフィルムされた大きなペトリ皿に慎重に戻します
- カバースリップの上部に40μLの二次抗体溶液を慎重に加える。二次抗体溶液をカバースリップの側面にこぼさないようにしてください。
- 必要に応じて、小さなシャーレに超純水を加え、カバースリップの周りに小さなシャーレを慎重に配置します。
- 大きなペトリ皿を覆う
- スライスされた組織の場合:
- 250 μL の二次抗体溶液を 24 ウェルプレート内のフリーフローティングスライスに慎重に加えます。
- プレートを覆う
注:ここからは、プレートの上部をホイルで包んでサンプルを光から保護します。
- サンプルを37°Cで1時間インキュベートする
5. ライゲーション
- ライゲーションストックを分子グレードの水に1:5で混合する。
- セクション4と同様に、カバースリップを慎重に移し、スライスした組織から二次ミックスを除去します。
- サンプルを 500 μL の洗浄バッファー A で洗浄する
- 培養細胞については、5分間2回洗浄する。薄切した組織については、10分間2回洗浄する。両方のために軌道シェーカーで穏やかな攪拌を使用してください。
- この間、大きなペトリ皿のパラフィルムを交換してください
- リガーゼをコールドブロック上に保持しながら、ステップ5.1からライゲーションストックでリガーゼを1:40に希釈する。この希釈は、リガーゼを試料に添加する直前に行う。
- セクション4と同様に、リガーゼを添加する前に、サンプルからできるだけ多くの洗浄バッファーAを除去してください。
- 培養細胞の場合:カバースリップをパラフィルムしたペトリ皿に戻す。40μLのライゲーションミックスをカバースリップに加え、カバースリップの周りに小さな水で満たされたペトリ皿を配置し、大きなペトリ皿を覆う。
- 薄切組織の場合:ステップ5.4のライゲーションミックス250 μLを各ウェルに追加し、プレートを覆います。
- サンプルを37°Cで30分間インキュベートする。
6. 増幅
- 増幅ストックを分子グレードの水に1:5で混合する。
- セクション4と同様に、カバースリップを慎重に移し、スライスした組織からライゲーションミックスを除去します。
- サンプルを 500 μL の洗浄バッファー A で洗浄する
- 培養細胞については、2分間2回洗浄する。薄切した組織については、10分間2回洗浄する。両方のために軌道シェーカーで穏やかな攪拌を使用してください。
- この間、大きなペトリ皿のパラフィルムを交換してください
- ポリメラーゼをサンプルに添加する直前にこの希釈を行う。ポリメラーゼをコールドブロックに保ちながら、ポリメラーゼを希釈する
- 培養細胞の場合、ステップ6.1から増幅ストック中のポリメラーゼを1:80に希釈する。
- 薄切された組織の場合、ステップ6.1から増幅ストック中のポリメラーゼを1:40に希釈する。
- ステップ4と同様に、ポリメラーゼを添加する前に、サンプルからできるだけ多くの洗浄バッファーAを除去します。
- 培養細胞の場合:カバースリップをパラフィルムしたペトリ皿に戻します。ステップ6.4.1の増幅ミックスをカバースリップに40μL加え、カバースリップの周りに小さな水で満たされたペトリ皿を配置し、大きなペトリ皿を覆う。サンプルを37°Cで100分間インキュベートする。
- 薄切組織の場合:ステップ6.4.2の増幅ミックス250 μLを各ウェルに追加し、プレートを覆います。サンプルを37°Cで2時間インキュベートする。
メモ: この間、スライドを準備してラベルを付けます。
7. 取り付け
- ステップ4と同様に、カバースリップを慎重に移し、スライスした組織から増幅ミックスを除去する。
- サンプルを500 μLの洗浄バッファーBで2回、オービタルシェーカーで穏やかに攪拌しながら10分間洗浄します。
- サンプルを500 μLの1%洗浄バッファーBで1分間洗浄し、オービタルシェーカーで穏やかに攪拌します。
- 培養細胞の場合:
- 3 μL のマウントメディアをスライドにドロップする
- カバースリップから余分な洗浄バッファーを取り出し、カバースリップを(細胞を下に向けて)マウントメディアに入れます。カバースリップを所定の位置に密封するために、少量の透明なマニキュアで側面をシールします。
- スライスされた組織の場合:
- 組織スライスを準備されたスライドに慎重に移して配置し、スライスが平らに横たわるようにします。5~10 μLのマウント培地(量はスライスのサイズによって異なります)を組織スライスに滴下します。
- ガラスカバースリップをティッシュスライスの上に置き、端に沿って少量の透明なマニキュアで密封して、カバースリップを所定の位置に密封します。
- 顕微鏡下で分析する前に少なくとも15分間待つか、-20°Cで保存してください。
8. 共焦点顕微鏡でデジタル画像を取得する
- すべての治療からのサンプルにわたって集録設定(レーザーパワー、ゲイン、オフセットなど)を最適化します。最適化に、バックグラウンドノイズの減少と、蛍光シグナルの強度を過飽和にすることなくシグナルの強化が含まれることを確認します。設定が決まったら、取得したすべての画像に同じ取得設定を適用します。
メモ:以下の取得の詳細は、ニコンA1共焦点顕微鏡およびニコンNISのARエレメントソフトウェアで使用できます。 - サンプル付きのスライドをステージ上にセットし、アイピースを通してDAPIを使用してサンプルの焦点面を見つけます。
- アイポートをクリックして アイ ポートをオフにし、光学構成ボタンを選択して設定を調整します。
- 各蛍光チャンネルのゲイン、オフセット、レーザー出力を調整して、バックグラウンドノイズを低減し、蛍光シグナルを強化します。ステップ8.5-8.6で述べたように蛍光シグナルが過飽和にならないようにしてください。
- 蛍光シグナルの疑似色を使用して過飽和を監視します。ライブ画像の下部にある、現在使用されている蛍光チャンネルのラベルが付いたタブを右クリックします。
- 次に、「 チャンネルカラーリング」 を選択し、「 レインボーダーク」 などの疑似カラーを選択して、蛍光強度をヒートマップのような疑似カラーで視覚化します。レインボーダークでは、涼しい色は蛍光強度が低いことを示し、熱い色は蛍光強度が高いことを示します。
- すべての蛍光チャンネルを最適化したら、以前に選択した光学構成ボタンを右クリックし、このボタンに [現在のカメラ設定を割り当てる ]を選択します。
- 選択した設定が、各処理群のランダムサンプルに対して十分であることを確認します。選択した設定でこれらのサンプルのいずれかが過飽和になる場合は、手順8.4を繰り返して過飽和を除去します。
- 培養ニューロンの場合は、手順 8.10 ~ 8.16 に従います。
- 眼口を使用して、他の樹状突起との重複が最小限に抑えられた樹状突起を有するニューロンを検索する。
- アイポートをオフにし、DAPIチャネルを使用して、選択したニューロンの細胞体を視覚化します。ソーマの中心をダブルクリックして、ニューロンを視野の中央に配置します。
- MAP2チャンネルを使用して、ライブスキャンでMAP2信号に最適な焦点面を見つけます。
- [ ND取得] タブで、[ ファイルに保存 ]をクリックし、[ 参照]で画像を保存するファイルを選択します。次に、ファイル名を入力します。
- 「 Z 」タブで、「 範囲で定義される対称モード」 オプションを選択します。フォーカスを最適な MAP2 平面に設定し、[ 相対] ボタンをクリックして 、このフォーカル プレーンを Z スタックの中央として設定します。
- 1μmステップでレンジを5μmに設定し、 Z移動中にアクティブシャッターを閉じるにチェックを入れてください。「 波長」 タブで、「Optical Conf」で以前に構成した集録設定で オプティカル・ボタンの名前を選択します。次に、[ 今すぐ実行]をクリックします。
- カバースリップ/治療あたり約10個のニューロンについて、ステップ8.1.10-8.1.15を繰り返します。
- 薄切組織の場合は、手順8.18~8.22に従ってください。
- アイポートを使用して、関心領域を検索します。たとえば、海馬のCA1を見つけます。
- アイポートをオフにし、MAP2チャンネルを使用して、ライブスキャンでMAP2信号に最適な焦点面を見つけます。
- [ 取得] メニューで、[ 大きな画像をスキャン] を選択します。次に、 開いたパネルから キャプチャパネルで、以前に構成した取得設定で光学式ボタンの名前を選択します。また、このパネルで正しい目的を選択してください。
- 領域パネルと接眼レンズの下で、矢印キーを使用して対象 領域 の境界を設定します。次に、[ 大画像をファイルに 保存]をクリックし、画像の保存パスファイル名を作成します。
- セットアップパネルで、マルチチャンネルキャプチャがチェックされていることを確認し、Optical Conf で以前に構成した集録設定でオプティカルボタンの名前を選択します。
メモ: 大きな画像用の Z スタックを使用することは可能ですが、スキャン時間が長くなります。
9. 分析
- 集録設定と同様に、すべての処理のサンプルを使用してしきい値設定を最適化します。スレッショルドの最適化が、蛍光シグナルの強度を過飽和にすることなく、バックグラウンドノイズを低減し、シグナルを強化することに焦点を当てていることを確認してください。これらの設定が決定されたら、手順 9.2 ~ 9.3 で説明したように、解析に使用するすべての画像に同じしきい値設定を適用します。
- ImageJ では、しきい値オプションはメニューの [画像] > [しきい値の調整] の下>あります。画像の背景が暗い場合は、[ 暗い背景] オプションを選択します。
- 次に、以前に決定された最適化されたしきい値設定ごとにしきい値の上限と下限を調整し、[ 適用]をクリックします。
- 培養細胞の場合、MAP2チャネルとフリーハンド関心領域(ROI)ツールを使用して、樹状突起およびソーマを含む各ニューロンのROIを描画します。スライスされた組織の場合、関心領域(例えば、海馬内のCA1の層径)をカプセル化するスライス画像内にフリーハンドROIを描画する。
- ROI の領域を取得します。ImageJ で、[ 分析] メニューの下にある領域>測定します。
- 手順 9.7 ~ 9.9 に従って、ImageJ の粒子分析などの自動検出ツールを使用して、各 ROI 内のプンクタの数を検出します。
- [粒子の解析]メニューの[粒子解析]オプション>[解析]を見つけます。まず、穿刺サイズの直径(通常は0.1〜3μm2)を定義します。
- 次に、「表示」ドロップダウンメニューから「オーバーレイマスク」オプションを選択し、「結果の表示」オプションをチェックします。次に、[OK]をクリックします。
- 選択した直径範囲で穿刺が検出されない場合は、この解析ですべての穿刺が検出されるまで範囲を調整します。すべての画像に同じパーティクル解析設定を使用してください。
- 手順 9.11 ~ 9.13 に従って、プンクタの数を個々の関心領域の面積で割ります。
- 各イメージの結果をコピーして、ImageJ の [結果] ポップアップからスプレッドシートに貼り付けます。
- まず、データがどのファイルとサンプルから取得されたかを特定します。次に、ROIの面積をプンクタの数で割る。
- 次に、[結果] ポップアップからデータをクリアし、手順 9.2 ~ 9.12 を繰り返します。
- 対照サンプルへの結果を正規化する: 対照サンプルの結果(プンクタ/ROI領域の数)を平均化します。次いで、得られた全サンプルの結果を対照の平均で除算し、正規化された結果を得る。対照サンプルの新しい平均は1に等しくなければなりません。
結果
Heaney et al.6から改変されたデータは、シナプス形成の増加が予想される実験を実証するために提示されている(メカニズムの詳細およびより詳細な議論については、6を参照されたい)。以前に、迅速な抗うつ薬が有効であるためには、阻害性代謝受容体GABAB(γ-アミノ酪酸サブタイプB)の活性化が必要であることが実証された13。さらに、以前?...
ディスカッション
DetectSynは、近接ライゲーションアッセイを使用して互いに40nm以内のタンパク質を検出する迅速なアッセイであり、シナプス形成の検出を可能にする。この技術は、シナプス形成の代理測定としてのみ役立つ現在の蛍光アッセイを改善する。DetectSynは、互いの40nm以内、すなわちシナプス間隙内に局在するシナプスタンパク質の定量可能な変化を検出する。さらに、DetectSynは、シナプスを測定?...
開示事項
著者らは利益相反を報告していない。
謝辞
この研究は、米国国立衛生研究所 NINDS R01 NS105005 (KRG) および NS105005-03S1 (KRG)、国防総省 USAMRMC W81XWH-14-1-0061 (KRG)、NIAAA R01AA016852、NIAAA T32AA007565 (CFH)、および FRAXA Research (CFH) および Alzheimer's Association, AARG-NTF-21-852843 (KRG), AARF-19-614794-RAPID (KRG) からの助成金によって支援されました。
資料
| Name | Company | Catalog Number | Comments |
| 10x PBS | Fisher Scientific | BP39920 | PBS made in house works, as well. |
| 24 well plates | Fisher Scientific | FB012929 | For tissue slices, pre-sterilized plates may be unnecessary. |
| 50 mL conical tubes | Fisher Scientific | 14-432-22 | |
| Aluminium foil | Fisher Scientific | 15-078-290 | |
| Chicken anti-MAP2 antibody | Abcam | ab5392 | |
| Clear nail polish | Fisher Scientific | NC1849418 | Other clear nail polish works, as well. |
| Cold block | Fisher Scientific | 13131012 | |
| Computer workstation | HP | ||
| Confocal or fluorescent microscope | Nikon | A1R HD25 | |
| Donkey anti-chicken FITC | Fisher Scientific | SA1-72000 | |
| Duolink donkey anti-Mouse PLUS | Sigma | DUO92001 | |
| Duolink donkey anti-Rabbit MINUS | Sigma | DUO92005 | |
| Duolink In Situ Detection Reagents Far Red | Sigma | DUO92013 | Contains ligation stock, amplification stock, ligase, and polymerase. |
| Duolink In Situ Mounting Medium with DAPI | Sigma | DUO82040 | |
| Duolink In Situ Wash Buffers, Fluorescence | Sigma | DUO82049 | Contains Wash Buffer A and Wash Buffer B; dilute Wash Buffer B to 1% in diH20 for 1% Wash Buffer B. |
| Fine-tipped paintbrush | Fisher Scientific | NC9691026 | Sable hair, size 00 or 000, can also find at craft stores |
| Fisherbrand Cover Glasses: Rectangles | Fisher Scientific | 12545MP | Cover glass is unnecessary for cultured neurons already on glass coverslips. |
| Fisherbrand Superfrost Plus Microscope Slides | Fisher Scientific | 1255015 | For cultured neurons already on glass coverslips, Superfrost slides may be unnecessary. |
| Freezer, -20°C | VWR | 76449-108 | |
| Glass coverslips | Fisher Scientific | 125480 | |
| Glycine | Fisher Scientific | BP381-1 | |
| Image processing software | e.g. NIS Elements, ImageJ | ||
| Incubator | Fisher Scientific | 15-015-2633 | |
| Large petri dish, 100mm | Fisher Scientific | FB0875712 | |
| Molecular grade water | Fisher Scientific | BP24701 | |
| Mouse anti-Synapsin1 antibody | Synaptic Systems | 106-011 | |
| Normal donkey serum | Jackson ImmunoResearch | 017-000-121 | |
| Orbital shaker | Fisher Scientific | 02-106-1013 | |
| Parafilm | Fisher Scientific | 13-374-10 | |
| Pipette tips | Fisher Scientific | 02-707-025 | |
| Pipettes | Fisher Scientific | 14-388-100 | Working volumes range from 3 µL to 500 µL |
| Plastic pasteur pipette | Fisher Scientific | 02-708-006 | |
| Precision tweezers/foreceps | Fisher Scientific | 12-000-122 | |
| Rabbit anti-PSD95 antibody | Abcam | ab18258 | Other antibody pairs may work, as well, with optimization. |
| Refrigerator | VWR | 76470-402 | |
| Small petri dish, 60 mm | Fisher Scientific | FB0875713A | |
| Timer | Fisher Scientific | 14-649-17 | |
| Tween 20 | Fisher Scientific | BP337-100 |
参考文献
- Südhof, T. C. Towards an understanding of synapse formation. Neuron. 100 (2), 276-293 (2018).
- Bliss, T. V., Collingridge, G. L. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 361 (6407), 31-39 (1993).
- Heaney, C. F., Raab-Graham, K. F. Dysregulated protein synthesis in major depressive disorder. The Oxford Handbook of Neuronal Protein Synthesis. , 510-532 (2018).
- Masliah, E., Crews, L., Hansen, L. Synaptic remodeling during aging and in Alzheimer's disease. Journal of Alzheimer's Disease. 9, 91-99 (2006).
- van Spronsen, M., Hoogenraad, C. C. Synapse pathology in psychiatric and neurologic disease. Current Neurology and Neuroscience Reports. 10 (3), 207-214 (2010).
- Heaney, C. F., Namjoshi, S. V., Uneri, A., Bach, E. C., Weiner, J. L., Raab-Graham, K. F. Role of FMRP in rapid antidepressant effects and synapse regulation. Molecular Psychiatry. 26 (6), 2350-2362 (2021).
- Pchitskaya, E., Bezprozvanny, I. Dendritic spines shape analysis-Classification or clusterization? Perspective. Frontiers in Synaptic Neuroscience. 12, 31 (2020).
- Alvarez, V. A., Sabatini, B. L. Anatomical and physiological plasticity of dendritic spines. Annual Review of Neuroscience. 30 (1), 79-97 (2007).
- Berry, K. P., Nedivi, E. Spine Dynamics: Are they all the same. Neuron. 96 (1), 43-55 (2017).
- Micheva, K. D., Smith, S. J. Array tomography: A new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron. 55 (1), 25-36 (2007).
- Kaech, S., Banker, G. Culturing hippocampal neurons. Nature Protocols. 1 (5), 2406-2415 (2006).
- Gage, G. J., Kipke, D. R., Shain, W. Whole animal perfusion fixation for rodents. Journal of Visualized Experiments: JoVE. (65), e3564 (2012).
- Workman, E. R., Niere, F., Raab-Graham, K. F. mTORC1-dependent protein synthesis underlying rapid antidepressant effect requires GABABR signaling. Neuropharmacology. 73, 192-203 (2013).
- Li, N., et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 329 (5994), 959-964 (2010).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved