
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
DetectSyn: Eine schnelle, unvoreingenommene fluoreszierende Methode zur Erkennung von Änderungen der Synapsendichte
In diesem Artikel
Zusammenfassung
DetectSyn ist ein unvoreingenommener, schneller fluoreszierender Assay, der Veränderungen der relativen Synapsenzahl (prä- und postsynaptisches Engagement) über Behandlungen oder Krankheitszustände hinweg misst. Diese Technik verwendet eine Proximity-Ligationstechnik, die sowohl in kultivierten Neuronen als auch in festem Gewebe eingesetzt werden kann.
Zusammenfassung
Synapsen sind der Ort der Kommunikation zwischen Neuronen. Die Stärke des neuronalen Schaltkreises hängt mit der synaptischen Dichte zusammen, und der Abbau von Synapsen ist charakteristisch für Krankheitszustände wie schwere depressive Störungen (MDD) und Alzheimer-Krankheit. Traditionelle Techniken zur Untersuchung der Synapsenzahlen umfassen die genetische Expression von fluoreszierenden Markern (z. B. grün fluoreszierendes Protein (GFP)), Farbstoffe, die ein Neuron füllen (z. B. Carbocyaninfarbstoff, DiI), und der immunfluoreszierende Nachweis von Wirbelsäulenmarkern (z. B. postsynaptische Dichte 95 (PSD95)). Ein wichtiger Vorbehalt gegenüber diesen Proxy-Techniken ist, dass sie nur postsynaptische Veränderungen identifizieren. Eine Synapse ist jedoch eine Verbindung zwischen einem präsynaptischen Terminal und einer postsynaptischen Wirbelsäule. Der Goldstandard zur Messung der Synapsenbildung/-elimination erfordert zeitaufwändige Elektronenmikroskopie- oder Array-Tomographie-Techniken. Diese Techniken erfordern spezielle Schulungen und teure Ausrüstung. Darüber hinaus kann nur eine begrenzte Anzahl von Neuronen beurteilt werden und wird verwendet, um Veränderungen in einer ganzen Gehirnregion darzustellen. DetectSyn ist eine schnelle fluoreszierende Technik, die Veränderungen der Synapsenbildung oder -eliminierung aufgrund eines Krankheitszustands oder einer Arzneimittelaktivität identifiziert. DetectSyn verwendet einen schnellen Proximity-Ligationsassay, um nebeneinander liegende prä- und postsynaptische Proteine und die Standard-Fluoreszenzmikroskopie nachzuweisen, eine Technik, die den meisten Labors leicht zur Verfügung steht. Die fluoreszierende Detektion der resultierenden Puncta ermöglicht eine schnelle und unvoreingenommene Analyse von Experimenten. DetectSyn liefert repräsentativere Ergebnisse als die Elektronenmikroskopie, da größere Bereiche analysiert werden können als eine begrenzte Anzahl fluoreszierender Neuronen. Darüber hinaus funktioniert DetectSyn für in vitro kultivierte Neuronen und festsitzende Gewebeschnitte. Schließlich wird eine Methode bereitgestellt, um die von dieser Technik gesammelten Daten zu analysieren. Insgesamt bietet DetectSyn ein Verfahren zur Erkennung relativer Veränderungen der Synapsendichte über Behandlungen oder Krankheitszustände hinweg und ist zugänglicher als herkömmliche Techniken.
Einleitung
Synapsen sind die grundlegende Einheit der Kommunikation zwischen Neuronen1. Viele Synapsen zwischen Neuronen innerhalb derselben Regionen führen zu Schaltkreisen, die das Verhalten vermitteln2. Synapsen bestehen aus einem präsynaptischen Terminal von einem Neuron, das Neurotransmitter oder Neuropeptide freisetzt, die Informationen an postsynaptische Rezeptoren eines anderen Neurons weiterleiten. Die Summe präsynaptischer Signale bestimmt, ob das postsynaptische Neuron ein Aktionspotential auslöst und die Nachricht an andere Neuronen weitergibt.
Synaptopathologie, der Abbau von Synapsen, entsteht bei Krankheiten und Störungen, die durch ein vermindertes neuronales Volumen gekennzeichnet sind, wie Alzheimer-Krankheit und schwere depressive Störung, was zu Schaltkreisen führt, die nicht mehr optimal funktionieren 3,4,5. Die Wiederherstellung der Synapsendichte liegt wahrscheinlich der Wirksamkeit potenzieller Behandlungen für diese Erkrankungen zugrunde. Zum Beispiel wurde kürzlich gezeigt, dass zunehmende Synapsen der Verhaltenswirksamkeit von schnellen Antidepressivazugrunde liegen 6. Um mögliche synaptopathologische Behandlungen schnell zu screenen, benötigen Forscher Techniken, die Veränderungen der Synapsenzahlen schnell identifizieren.
Aktuelle Methoden sind entweder zeitaufwendig und teuer (Elektronenmikroskopie, Array-Tomographie) oder sie untersuchen nur postsynaptische Veränderungen, ohne präsynaptische Interaktion (Wirbelsäulenanalysen, Immunfluoreszenz/-kolokalisation) einzubeziehen. Farbstoffe wie DiI oder fluoreszierende Proteine wie GFP helfen, Neuronen sichtbar zu machen und postsynaptische Stacheln zu charakterisieren. Die Wirbelsäulenanalyse verwendet jedoch von Forschern definierte Verhältnisse, um die Morphologie zu bestimmen, was die Reproduzierbarkeit verringernkann 7. Darüber hinaus wird immer noch aufgedeckt, wie sich die verschiedenen Wirbelsäulenklassen auf funktionelle Synapsen beziehen8. Die Wirbelsäulenbildung kann vorübergehend sein und die postsynaptische Plastizität widerspiegeln, aber diese Stacheln könnten eliminiert werden, bevor sie sich mit einem präsynaptischen Neuron9 zu einer Synapse stabilisieren.
Die Kolokalisation bietet einen besseren Proxy für Synapsen als die Wirbelsäulenanalyse, da man eine Immunstrukturierung für präsynaptische und postsynaptische Proteine durchführen kann. Synaptische Proteine können jedoch niedrige Kolokalisationswerte ergeben, da die Proteine nebeneinander stehen und sich möglicherweise nicht konsistent überlappen. Da die Proteine nicht vollständig überlagert sind, können Kolokalisierungstechniken aufgrund dieser fehlenden Informationen Änderungen der Synapsenbildung möglicherweise nicht genau messen. Obwohl sowohl die Elektronenmikroskopie (EM) als auch die Array-Tomographie hochauflösende Bilder von Synapsen liefern, sind sie zeitaufwendig. EM erfordert außerdem spezielle Ausrüstung, und die Forscher sind auf kleine Gewebevolumina für ein bestimmtes Experiment beschränkt. Während die Array-Tomographie elegant die Möglichkeit bietet, auf ultradünne Abschnitte nach vielen Proteinen zu suchen und mit EM10 kombiniert werden kann, kann diese Technik zu arbeitsintensiv sein und den Rahmen von Experimenten sprengen, die schnell nach Veränderungen der Synapsenbildung suchen müssen.
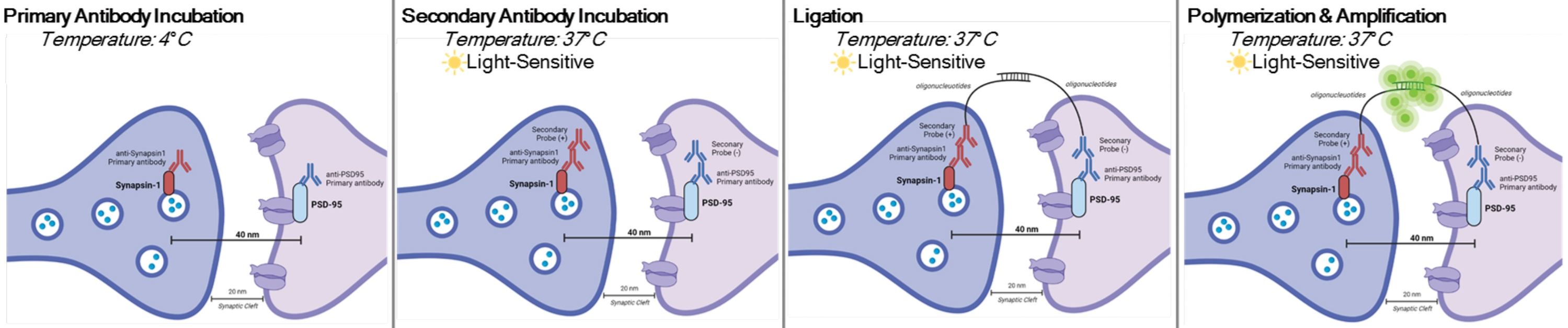
DetectSyn ist eine spezifische Anwendung des Duolink Proximity Ligation Assay. Der PLA-Assay ermöglicht den allgemeinen Nachweis von Protein-Protein-Interaktionen. DetectSyn überbrückt postsynaptische Proxy-Messungen, indem es ein fluoreszierendes Signal verstärkt, das von markierten prä- und postsynaptischen Proteinen innerhalb von 40 nm voneinander emittiert wird. Wenn sich die synaptischen Proteine innerhalb von 40 nm befinden, wie in einem synaptischen Spalt, dann hybridisieren die sekundären Antikörper, die DNA-Sonden enthalten, zu zirkulärer DNA. Diese hybridisierte zirkuläre DNA exprimiert eine fluoreszierende Sonde, die dann mit Standard-Fluoreszenzmikroskopie-Techniken amplifiziert und nachgewiesen wird (siehe Abbildung 1). Entscheidend ist, dass diese Technik im Gegensatz zu EM und Array-Tomographie keine spezielle Ausrüstung erfordert und etwa die gleiche Zeit in Anspruch nimmt wie die Standard-Immunhistochemie. Die Zugänglichkeit dieser Technik ermöglicht es somit Forschern außerhalb forschungsintensiver Institutionen, an der synaptopathologischen Forschung teilzunehmen. Darüber hinaus kann diese Technik Veränderungen der synaptischen Dichte in mehreren Gehirnregionen innerhalb eines einzigen Experiments untersuchen und bietet eine ganzheitlichere Darstellung synaptischer Veränderungen aufgrund von Krankheit oder Behandlung.
Protokoll
Die Isolierung von Zellen und Gewebe von Tieren erfolgte in Übereinstimmung mit dem Leitfaden der National Institutes of Health für die Pflege und Verwendung von Labortieren und wurde vom Wake Forest Institutional Animal Care and Use Committee genehmigt.
HINWEIS: Dieses Protokoll wird für Proben verwendet, die bereits gemäß bestimmten experimentellen Paradigmen und Anforderungen behandelt und fixiert wurden. Zu Demonstrationszwecken wird die Synapsenbildung durch eine schnelle antidepressive Behandlung verwendet, um diese Synapsennachweistechnikhervorzuheben 6. Neuronen, die zuvor auf Deckgläsern kultiviert, behandelt, in 4% Paraformaldehyd (PFA) fixiert und in 1x phosphatgepufferter Kochsalzlösung (PBS) gelagert wurden, werden verwendet, um die In-vitro-Verfahren hervorzuheben. Zuvor geschnittenes Hippocampusgewebe (25 μm dick) von behandelten Mäusen, transkardial perfundiert mit eiskaltem PBS und 4% PFA und dann in Kryoprotektor gelagert, wird verwendet, um die Schneideverfahren hervorzuheben. Bitte beachten Sie11,12 für weitere Informationen darüber, wie man Neuronen oder transkardial perfusionierte Nagetiere. Eine grafische Darstellung dieses Verfahrens finden Sie in Abbildung 1.

Abbildung 1: Grafische Darstellung des DetectSyn-Assays. Nach der Permeabilisierung der Zellmembranen binden primäre Antikörper für Synapsin1 und PSD95 an diese synaptischen Proteine. Secondaries mit Oligonukleotid-Tags binden dann an die primären Antikörper. Wenn Synapsin1 und PSD95 wie bei einer Synapse innerhalb von 40 nm liegen, interagieren die Oligonukleotide und ein fluoreszierender Tag wird verstärkt. Dieses fluoreszierende Signal kann dann mittels Standardmikroskopie abgebildet und analysiert werden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
1. Proben spülen
- Spülen Sie die Proben mit 500 μL 1x PBS + 0,75% Glycin für 5 min 3 mal mit sanftem Rühren auf einem Orbitalschüttler, um Reste von PFA oder Kryoprotektivum zu entfernen.
2. Proben blockieren und durchpermeabilisieren
- Bereiten Sie Blockierungs- und Permeabilisierungslösung (10% normales Eselserum, 0,25% Tween 20) in 1x PBS vor. Bereiten Sie sich ausreichend für Blockierung, primäre und sekundäre Inkubationen vor.
- Zu Proben (z. B. Deckgläser oder frei schwebende Scheiben) in 24 Vertiefungsplatten sind 500 μL Blockier- und Permeabilisierungslösung hinzuzufügen. Stellen Sie sicher, dass jede Vertiefung eine andere Probe enthält und entsprechend gekennzeichnet ist, um zu verhindern, dass Proben gewechselt werden.
- Inkubieren Sie die Proben bei Raumtemperatur (RT) für 60 min für kultivierte Zellen oder 2 h für geschnittenes Gewebe. Verwenden Sie einen Orbitalschüttler für sanftes Rühren.
3. Inkubieren von Proben in primären Antikörpern
- Primäre Antikörper im Blockpuffer vorbereiten:
- Prepare Postsynaptische Dichte 95 (PSD95; 1:500, Kaninchen polyklonal), Synapsin1 (1:500, Maus monoklonal), MAP2 (1:400, Huhn polyklonal)
- Bereiten Sie ein Negativkontrollaliquot vor, das eines der synaptischen Paare auslässt (z. B. ohne PSD95)
- Entfernen Sie die blockierende Lösung vorsichtig mit einer Pasteur-Pipette aus Kunststoff. Versuchen Sie, so viel wie möglich zu entfernen, ohne die Zellen zu stören oder das Gewebe zu zerreißen.
- Für kultivierte Zellen:
- Eine große Petrischale aus Kunststoff mit Parafilm auslegen. Übertragen Sie die Deckgläser vorsichtig mit einer Pinzette auf den Parafilm.
- Geben Sie vorsichtig 60 μL der primären Antikörperlösung auf die Oberseite der Deckgläser. Achten Sie darauf, die primäre Antikörperlösung nicht über die Seite des Deckglases zu verschütten.
- Um Feuchtigkeit zu spenden und zu verhindern, dass Proben während der Inkubationszeit austrocknen, fügen Sie einer kleineren Petrischale hochreines Wasser hinzu und ordnen Sie die kleine Petrischale sorgfältig um die Deckblätter an.
- Decken Sie die große Petrischale ab und inkubieren Sie kultivierte Zellen für 1 h bei RT.
- Für geschnittenes Gewebe:
- Geben Sie vorsichtig 250 μL der primären Antikörperlösung zu frei schwebenden Scheiben in einer 24-Well-Platte.
- Bedecken Sie die Platte und inkubieren Sie das Gewebe über Nacht bei 4 °C mit sanfter Bewegung auf einem Orbitalschüttler.
4. Proben waschen und dann in sekundären Antikörpern inkubieren
- Sekundäre Antikörper im Sperrpuffer vorbereiten:
- Bereiten Sie Esel-Anti-Maus (1:5), Esel-Anti-Kaninchen (1:5), Esel-Anti-Huhn (1:400) vor.
- In diesem Schritt können zusätzliche technische Kontrollen erhalten werden, indem ein sekundäres Aliquot hergestellt wird, das entweder das Anti-Maus- oder das Anti-Kaninchen-Sekundärsystem auslässt.
- Für kultivierte Zellen:
- Klopfen Sie mit einer Pinzette vorsichtig die Primärlösung von Deckgläsern auf ein Papiertuch
- Übertragen Sie die Deckgläser vorsichtig mit einer Pinzette zurück auf ihre ursprüngliche 24-Well-Platte, die mit 500 μL 1x PBS gefüllt ist.
- Für geschnittenes Gewebe:
- Entfernen Sie vorsichtig die primäre Antikörperlösung mit einer Pasteur-Pipette aus Kunststoff. Versuchen Sie, so viel wie möglich zu entfernen, ohne das Gewebe zu zerreißen.
- Fügen Sie 500 μL von 1x PBS hinzu
- Waschen Sie die Proben für 10 min 3 mal in 1x PBS mit sanftem Rühren auf einem Orbitalschüttler. Bringen Sie während dieser Zeit alle Waschpuffer zu RT.
- Wechseln Sie in dieser Zeit den Parafilm in der großen Petrischale
- Für kultivierte Zellen:
- Übertragen Sie die Deckgläser vorsichtig mit einer Pinzette zurück auf die parafilmierte große Petrischale
- Fügen Sie vorsichtig 40 μL der sekundären Antikörperlösung auf die Oberseite der Deckgläser auf. Achten Sie darauf, die sekundäre Antikörperlösung nicht über die Seite des Deckglases zu verschütten.
- Bei Bedarf mehr Reinstwasser in eine kleinere Petrischale geben und die kleine Petrischale sorgfältig um Deckgläser herum anordnen.
- Decken Sie die große Petrischale ab
- Für geschnittenes Gewebe:
- Fügen Sie vorsichtig 250 μL der sekundären Antikörperlösung zu frei schwebenden Scheiben in einer 24-Well-Platte hinzu.
- Decken Sie die Platte ab
HINWEIS: Schützen Sie die Proben von nun an vor Licht, indem Sie die Oberseiten der Platten mit Folie umwickeln.
- Inkubieren Sie die Proben bei 37 °C für 1 h
5. Ligation
- Mischen Sie den Ligationsstock 1:5 in molekularem Wasser.
- Übertragen Sie wie in Abschnitt 4 vorsichtig die Deckgläser und entfernen Sie die sekundäre Mischung aus dem geschnittenen Gewebe.
- Waschen Sie die Proben in 500 μL Waschpuffer A
- Für kultivierte Zellen 2 mal für 5 min waschen. Für geschnittenes Tuch 2 mal für 10 min waschen. Verwenden Sie für beide ein sanftes Rühren auf einem Orbitalschüttler.
- Wechseln Sie in dieser Zeit den Parafilm in der großen Petrischale
- Während Sie die Ligase auf einem kalten Block halten, verdünnen Sie die Ligase 1:40 im Ligationsstock aus Schritt 5.1. Führen Sie diese Verdünnung unmittelbar vor dem Hinzufügen der Ligase zu den Proben durch.
- Wie in Abschnitt 4 ist so viel Waschpuffer A wie möglich aus den Proben zu entfernen, bevor die Ligase hinzugefügt wird.
- Für kultivierte Zellen: Coverslips zurück in die parafilmierte Petrischale geben. 40 μL der Ligationsmischung in Deckgläser geben, kleine mit Wasser gefüllte Petrischalen um die Deckgläser herum anordnen und die große Petrischale abdecken.
- Für geschnittenes Gewebe: 250 μL der Ligationsmischung aus Schritt 5.4 in jede Vertiefung geben und die Platte abdecken.
- Die Proben 30 min bei 37 °C inkubieren.
6. Verstärkung
- Mischen Sie den Verstärkungsstock 1:5 in molekularem Wasser.
- Übertragen Sie wie in Abschnitt 4 vorsichtig die Deckgläser und entfernen Sie die Ligaturmischung aus dem geschnittenen Gewebe.
- Waschproben in 500 μL Waschpuffer A
- Für kultivierte Zellen 2 mal für 2 min waschen. Für geschnittenes Tuch 2 mal für 10 min waschen. Verwenden Sie für beide ein sanftes Rühren auf einem Orbitalschüttler.
- Wechseln Sie in dieser Zeit den Parafilm in der großen Petrischale
- Führen Sie diese Verdünnung unmittelbar vor dem Hinzufügen der Polymerase zu Proben durch. Während die Polymerase auf einem kalten Block gehalten wird, verdünnen Sie die Polymerase
- Bei kultivierten Zellen wird die Polymerase 1:80 im Amplifikationsmaterial aus Schritt 6.1 verdünnt.
- Für geschnittenes Gewebe wird die Polymerase 1:40 im Amplifikationsmaterial aus Schritt 6.1 verdünnt.
- Entfernen Sie wie in Schritt 4 so viel Waschpuffer A wie möglich aus den Proben, bevor Sie die Polymerase hinzufügen.
- Für kultivierte Zellen: Übertragen Sie die Deckgläser zurück in die parafilmierte Petrischale. 40 μL der Verstärkungsmischung aus Schritt 6.4.1 in Deckgläser geben, kleine mit Wasser gefüllte Petrischalen um die Deckgläser herum anordnen und die große Petrischale abdecken. Die Proben 100 min bei 37 °C inkubieren.
- Für geschnittenes Gewebe: 250 μL der Verstärkungsmischung aus Schritt 6.4.2 in jede Vertiefung geben und die Platte abdecken. Die Proben für 2 h bei 37 °C inkubieren.
HINWEIS: Bereiten Sie während dieser Zeit Folien vor und beschriften Sie sie.
7. Montage
- Übertragen Sie wie in Schritt 4 vorsichtig die Deckgläser und entfernen Sie die Verstärkungsmischung aus dem geschnittenen Gewebe.
- Waschen Sie die Proben in 500 μL Waschpuffer B 2 mal für 10 min mit sanftem Rühren auf einem Orbitalschüttler.
- Waschen Sie die Proben in 500 μL 1% Waschpuffer B für 1 min mit sanftem Rühren auf einem Orbitalschüttler.
- Für kultivierte Zellen:
- Legen Sie 3 μL Montagemedien auf ein Dia
- Klopfen Sie überschüssigen Waschpuffer vom Deckglas ab und legen Sie dann das Deckglas (mit Zellen nach unten) in das Montagemedium. Versiegeln Sie die Seiten mit einer kleinen Menge klarem Nagellack, um das Deckglas an Ort und Stelle zu versiegeln.
- Für geschnittenes Gewebe:
- Legen Sie vorsichtig eine Gewebescheibe auf den vorbereiteten Objektträger und ordnen Sie sie so an, dass die Scheibe flach liegt. Tropfen Sie zwischen 5-10 μL Montagemedium (Menge hängt von der Größe der Scheibe ab) auf die Gewebescheibe
- Legen Sie vorsichtig ein Glasdeckglas über die Gewebescheibe und verschließen Sie es mit einer kleinen Menge klarer Nagellacke entlang der Kante, um das Deckglas an Ort und Stelle zu versiegeln.
- Warten Sie mindestens 15 Minuten, bevor Sie unter dem Mikroskop analysieren, oder lagern Sie es bei -20 ° C.
8. Erhalten Sie digitale Bilder mit einem konfokalen Mikroskop
- Optimieren Sie die Erfassungseinstellungen (z. B. Laserleistung, Verstärkung, Offset) über Proben aus allen Behandlungen hinweg. Stellen Sie sicher, dass die Optimierung die Verringerung des Hintergrundrauschens und die Verbesserung des Signals umfasst, ohne die Intensität der Fluoreszenzsignale zu übersättigen. Sobald die Einstellungen festgelegt wurden, wenden Sie die gleichen Aufnahmeeinstellungen auf alle erhaltenen Bilder an.
HINWEIS: Die folgenden Erfassungsdetails können mit einem Nikon A1 Konfokalmikroskop und der Nikon NIS AR Elements Software verwendet werden. - Stellen Sie den Objektträger mit der Probe auf die Bühne und finden Sie die Brennebene für die Probe mit DAPI durch das Okular.
- Schalten Sie den Eyeport aus, indem Sie auf Eye port klicken und eine optische Konfigurationstaste auswählen, um die Einstellungen anzupassen.
- Passen Sie die Verstärkung, den Offset und die Laserleistung für jeden fluoreszierenden Kanal an, um Hintergrundgeräusche zu verringern und das Fluoreszenzsignal zu verbessern. Stellen Sie sicher, dass das Fluoreszenzsignal nicht übersättigt wird, wie in den Schritten 8.5-8.6 erwähnt.
- Überwachen Sie die Übersättigung mit einer Pseudofarbe für das fluoreszierende Signal. Klicken Sie unten im Livebild mit der rechten Maustaste auf die Registerkarte, die mit dem aktuell verwendeten Fluoreszenzkanal beschriftet ist.
- Als nächstes wählen Sie Kanalfärbung und wählen Sie eine Pseudofarbe wie Rainbow Dark, um die Fluoreszenzintensität in einer Heatmap-ähnlichen Pseudofarbe zu visualisieren. In Rainbow Dark zeigen kühlere Farben eine geringere Fluoreszenzintensität und heißere Farben eine höhere Fluoreszenzintensität an.
- Sobald alle Leuchtstoffkanäle optimiert sind, klicken Sie mit der rechten Maustaste auf die zuvor gewählte optische Konfigurationsschaltfläche und wählen Sie für diese Schaltfläche Aktuelle Kameraeinstellung zuweisen .
- Stellen Sie sicher, dass die gewählten Einstellungen für eine Zufallsstichprobe aus jeder Behandlungsgruppe ausreichen. Wenn die ausgewählten Einstellungen eine dieser Stichproben übersättigen, wiederholen Sie Schritt 8.4, um die Übersättigung zu beseitigen.
- Führen Sie für kultivierte Neuronen die Schritte 8.10-8.16 aus.
- Suchen Sie mit dem Augenhafen nach einem Neuron mit Dendriten, die sich nur minimal mit anderen Dendriten überlappen.
- Schalten Sie den Augenport aus und verwenden Sie den DAPI-Kanal, um den Zellkörper des ausgewählten Neurons zu visualisieren. Doppelklicken Sie auf die Mitte des Somas, um das Neuron in der Mitte des Sichtfeldes zu zentrieren.
- Finden Sie mit dem MAP2-Kanal die beste Fokusebene für das MAP2-Signal mit Live-Scanning.
- Klicken Sie auf der Registerkarte ND-Erfassung auf In Datei speichern und wählen Sie unter Durchsuchen eine Datei aus, in der das Bild gespeichert werden soll. Geben Sie dann den Dateinamen ein.
- Wählen Sie auf der Registerkarte Z die Option Symmetrischer Modus definiert durch Bereich . Setzen Sie den Fokus auf die beste MAP2-Ebene und klicken Sie auf die Schaltfläche Relativ, um diese Fokusebene als Mitte des Z-Stacks festzulegen.
- Stellen Sie den Bereich auf 5 μm mit 1 μm Schritten ein und stellen Sie sicher, dass Close Active Shutter During Z Movement aktiviert ist. Wählen Sie auf der Registerkarte Wellenlänge den Namen der optischen Taste mit den zuvor konfigurierten Erfassungseinstellungen unter Optical Conf aus. Klicken Sie dann auf Jetzt ausführen.
- Wiederholen Sie die Schritte 8.1.10-8.1.15 für etwa 10 Neuronen pro Deckglas/Behandlung.
- Führen Sie für geschnittenes Gewebe die Schritte 8.18-8.22 aus.
- Suchen Sie mit dem Eyeport nach der Region von Interesse. Suchen Sie beispielsweise CA1 des Hippocampus.
- Schalten Sie den Eye-Port aus und verwenden Sie den MAP2-Kanal, um die beste Fokusebene für das MAP2-Signal mit Live-Scan zu finden.
- Wählen Sie im Menü " Erfassen " die Option "Großes Bild scannen". Wählen Sie als Nächstes den Namen der optischen Taste mit den zuvor konfigurierten Erfassungseinstellungen unter dem Aufnahmefenster des sich öffnenden Bedienfelds aus. Stellen Sie außerdem sicher, dass Sie in diesem Bereich das richtige Ziel auswählen.
- Verwenden Sie unter dem Bedienfeld "Bereich" und dem Okular die Pfeiltasten, um die Grenzen des gewünschten Bereichs festzulegen. Klicken Sie anschließend auf Großes Bild in Datei speichern und erstellen Sie einen Dateinamen für den Speicherpfad für das Bild.
- Stellen Sie im Setup-Bedienfeld sicher, dass Multichannel Capture aktiviert ist, und wählen Sie dann den Namen der optischen Taste mit den zuvor konfigurierten Erfassungseinstellungen unter Optical Conf.
HINWEIS: Ein Z-Stack für ein großes Bild ist möglich, erhöht aber die Scanzeit.
9. Analyse
- Ähnlich wie bei den Erfassungseinstellungen können Sie Proben aus allen Behandlungen verwenden, um die Schwellenwerteinstellungen zu optimieren. Stellen Sie sicher, dass sich die Schwellenwertoptimierung auf die Verringerung des Hintergrundrauschens und die Verbesserung des Signals konzentriert, ohne die Intensität der Fluoreszenzsignale zu übersättigen. Sobald diese Einstellungen festgelegt wurden, wenden Sie dieselben Schwellenwerteinstellungen auf alle für die Analyse verwendeten Bilder an, wie in den Schritten 9.2-9.3 beschrieben.
- In ImageJ befindet sich die Schwellenwertoption unter dem Menü Image > Adjust > Threshold. Wählen Sie die Option "Dunkler Hintergrund", wenn das Bild einen dunklen Hintergrund hat.
- Passen Sie als Nächstes die oberen und unteren Grenzen des Schwellenwerts gemäß den zuvor festgelegten optimierten Schwellenwerteinstellungen an und klicken Sie dann auf Übernehmen.
- Verwenden Sie für kultivierte Zellen den MAP2-Kanal und ein ROI-Tool (Freehand Region of Interest), um einen ROI für jedes Neuron zu zeichnen, einschließlich Dendriten und Soma. Zeichnen Sie für geschnittenes Gewebe einen Freihand-ROI innerhalb des Slice-Bildes, der den interessierenden Bereich (z. B. Stratum radiatum des CA1 innerhalb des Hippocampus) einkapselt.
- Erhalten Sie den Bereich des ROI. Messen Sie in ImageJ den Bereich unter dem Menü Analyze > Measure.
- Erkennen Sie die Anzahl der Puncta innerhalb jedes ROI mit einem automatischen Erkennungstool wie Particle Analysis in ImageJ gemäß den Schritten 9.7-9.9.
- Suchen Sie die Option Partikelanalyse im Menü Analysieren > Partikel analysieren. Definieren Sie zunächst den Durchmesser der Puncta-Größe, typischerweise 0,1-3 μm2.
- Wählen Sie als Nächstes die Option Überlagern von Masken aus dem Dropdown-Menü Anzeigen und aktivieren Sie die Option Ergebnisse anzeigen. Klicken Sie dann auf OK.
- Wenn Puncta mit dem gewählten Durchmesserbereich nicht erkannt werden, passen Sie den Bereich an, bis alle Puncta mit dieser Analyse erkannt wurden. Stellen Sie sicher, dass Sie für alle Bilder dieselben Einstellungen für die Partikelanalyse verwenden.
- Teilen Sie die Anzahl der Puncta durch den Bereich einer einzelnen Region von Interesse, indem Sie die Schritte 9.11-9.13 ausführen.
- Kopieren Sie die Ergebnisse für jedes Bild aus dem Popup-Fenster Ergebnisse aus ImageJ und fügen Sie sie in eine Tabelle ein.
- Identifizieren Sie zunächst, aus welcher Datei und Probe die Daten gewonnen wurden. Teilen Sie dann den Bereich des ROI durch die Anzahl der Puncta.
- Löschen Sie dann die Daten aus dem Popup-Fenster Ergebnisse, und wiederholen Sie die Schritte 9.2 bis 9.12.
- Normalisieren Sie die Ergebnisse, um die Proben zu kontrollieren: Mittelwert der Ergebnisse (Anzahl der Puncta/ROI-Bereiche) für die Kontrollproben. Teilen Sie dann die erhaltenen Ergebnisse aller Proben durch den Durchschnitt der Kontrolle, um die normalisierten Ergebnisse zu erhalten. Der neue Mittelwert der Kontrollstichproben sollte gleich 1 sein.
Ergebnisse
Daten, die von Heaney et al.6 modifiziert wurden, werden vorgestellt, um ein Experiment zu demonstrieren, bei dem eine erhöhte Synapsenbildung erwartet wird (siehe6 für weitere Informationen und eine eingehendere Diskussion des Mechanismus). Zuvor konnte gezeigt werden, dass schnelle Antidepressiva die Aktivierung des inhibitorischen metabotropen Rezeptors GABAB (Gamma-Aminobuttersäure-Subtyp B) erfordern, um wirksam zu sein13. Darüber hinaus deu...
Diskussion
DetectSyn ist ein Schnellassay, der einen Proximity-Ligationsassay verwendet, um Proteine innerhalb von 40 nm voneinander nachzuweisen, was den Nachweis der Synapsenbildung ermöglicht. Diese Technik verbessert aktuelle fluoreszierende Assays, die nur als Proxy-Messungen für die Synapsenbildung dienen. DetectSyn erkennt quantifizierbare Veränderungen in synaptischen Proteinen, die innerhalb von 40 nm, d.h. innerhalb des synaptischen Spalts, voneinander lokalisiert sind. Darüber hinaus ist DetectSyn kostengünstiger un...
Offenlegungen
Die Autoren berichten von keinem Interessenkonflikt.
Danksagungen
Diese Arbeit wurde von den National Institutes of Health NINDS R01 NS105005 (KRG) und NS105005-03S1 (KRG), dem Verteidigungsministerium USAMRMC W81XWH-14-1-0061 (KRG), NIAAA R01AA016852, NIAAA T32AA007565 (CFH) und einem Zuschuss von FRAXA Research (CFH) und der Alzheimer's Association, AARG-NTF-21-852843 (KRG), AARF-19-614794-RAPID (KRG), unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| 10x PBS | Fisher Scientific | BP39920 | PBS made in house works, as well. |
| 24 well plates | Fisher Scientific | FB012929 | For tissue slices, pre-sterilized plates may be unnecessary. |
| 50 mL conical tubes | Fisher Scientific | 14-432-22 | |
| Aluminium foil | Fisher Scientific | 15-078-290 | |
| Chicken anti-MAP2 antibody | Abcam | ab5392 | |
| Clear nail polish | Fisher Scientific | NC1849418 | Other clear nail polish works, as well. |
| Cold block | Fisher Scientific | 13131012 | |
| Computer workstation | HP | ||
| Confocal or fluorescent microscope | Nikon | A1R HD25 | |
| Donkey anti-chicken FITC | Fisher Scientific | SA1-72000 | |
| Duolink donkey anti-Mouse PLUS | Sigma | DUO92001 | |
| Duolink donkey anti-Rabbit MINUS | Sigma | DUO92005 | |
| Duolink In Situ Detection Reagents Far Red | Sigma | DUO92013 | Contains ligation stock, amplification stock, ligase, and polymerase. |
| Duolink In Situ Mounting Medium with DAPI | Sigma | DUO82040 | |
| Duolink In Situ Wash Buffers, Fluorescence | Sigma | DUO82049 | Contains Wash Buffer A and Wash Buffer B; dilute Wash Buffer B to 1% in diH20 for 1% Wash Buffer B. |
| Fine-tipped paintbrush | Fisher Scientific | NC9691026 | Sable hair, size 00 or 000, can also find at craft stores |
| Fisherbrand Cover Glasses: Rectangles | Fisher Scientific | 12545MP | Cover glass is unnecessary for cultured neurons already on glass coverslips. |
| Fisherbrand Superfrost Plus Microscope Slides | Fisher Scientific | 1255015 | For cultured neurons already on glass coverslips, Superfrost slides may be unnecessary. |
| Freezer, -20°C | VWR | 76449-108 | |
| Glass coverslips | Fisher Scientific | 125480 | |
| Glycine | Fisher Scientific | BP381-1 | |
| Image processing software | e.g. NIS Elements, ImageJ | ||
| Incubator | Fisher Scientific | 15-015-2633 | |
| Large petri dish, 100mm | Fisher Scientific | FB0875712 | |
| Molecular grade water | Fisher Scientific | BP24701 | |
| Mouse anti-Synapsin1 antibody | Synaptic Systems | 106-011 | |
| Normal donkey serum | Jackson ImmunoResearch | 017-000-121 | |
| Orbital shaker | Fisher Scientific | 02-106-1013 | |
| Parafilm | Fisher Scientific | 13-374-10 | |
| Pipette tips | Fisher Scientific | 02-707-025 | |
| Pipettes | Fisher Scientific | 14-388-100 | Working volumes range from 3 µL to 500 µL |
| Plastic pasteur pipette | Fisher Scientific | 02-708-006 | |
| Precision tweezers/foreceps | Fisher Scientific | 12-000-122 | |
| Rabbit anti-PSD95 antibody | Abcam | ab18258 | Other antibody pairs may work, as well, with optimization. |
| Refrigerator | VWR | 76470-402 | |
| Small petri dish, 60 mm | Fisher Scientific | FB0875713A | |
| Timer | Fisher Scientific | 14-649-17 | |
| Tween 20 | Fisher Scientific | BP337-100 |
Referenzen
- Südhof, T. C. Towards an understanding of synapse formation. Neuron. 100 (2), 276-293 (2018).
- Bliss, T. V., Collingridge, G. L. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 361 (6407), 31-39 (1993).
- Heaney, C. F., Raab-Graham, K. F. Dysregulated protein synthesis in major depressive disorder. The Oxford Handbook of Neuronal Protein Synthesis. , 510-532 (2018).
- Masliah, E., Crews, L., Hansen, L. Synaptic remodeling during aging and in Alzheimer's disease. Journal of Alzheimer's Disease. 9, 91-99 (2006).
- van Spronsen, M., Hoogenraad, C. C. Synapse pathology in psychiatric and neurologic disease. Current Neurology and Neuroscience Reports. 10 (3), 207-214 (2010).
- Heaney, C. F., Namjoshi, S. V., Uneri, A., Bach, E. C., Weiner, J. L., Raab-Graham, K. F. Role of FMRP in rapid antidepressant effects and synapse regulation. Molecular Psychiatry. 26 (6), 2350-2362 (2021).
- Pchitskaya, E., Bezprozvanny, I. Dendritic spines shape analysis-Classification or clusterization? Perspective. Frontiers in Synaptic Neuroscience. 12, 31 (2020).
- Alvarez, V. A., Sabatini, B. L. Anatomical and physiological plasticity of dendritic spines. Annual Review of Neuroscience. 30 (1), 79-97 (2007).
- Berry, K. P., Nedivi, E. Spine Dynamics: Are they all the same. Neuron. 96 (1), 43-55 (2017).
- Micheva, K. D., Smith, S. J. Array tomography: A new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron. 55 (1), 25-36 (2007).
- Kaech, S., Banker, G. Culturing hippocampal neurons. Nature Protocols. 1 (5), 2406-2415 (2006).
- Gage, G. J., Kipke, D. R., Shain, W. Whole animal perfusion fixation for rodents. Journal of Visualized Experiments: JoVE. (65), e3564 (2012).
- Workman, E. R., Niere, F., Raab-Graham, K. F. mTORC1-dependent protein synthesis underlying rapid antidepressant effect requires GABABR signaling. Neuropharmacology. 73, 192-203 (2013).
- Li, N., et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 329 (5994), 959-964 (2010).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten