
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
DetectSyn: un metodo fluorescente rapido e imparziale per rilevare i cambiamenti nella densità delle sinapsi
In questo articolo
Riepilogo
DetectSyn è un saggio fluorescente rapido e imparziale che misura i cambiamenti nel numero relativo di sinapsi (impegno pre e postsinaptico) tra trattamenti o stati patologici. Questa tecnica utilizza una tecnica di legatura di prossimità che può essere utilizzata sia nei neuroni in coltura che nei tessuti fissi.
Abstract
Le sinapsi sono il sito di comunicazione tra i neuroni. La forza del circuito neuronale è correlata alla densità sinaptica e la rottura delle sinapsi è caratteristica di stati patologici come il disturbo depressivo maggiore (MDD) e il morbo di Alzheimer. Le tecniche tradizionali per studiare il numero di sinapsi includono l'espressione genetica di marcatori fluorescenti (ad esempio, proteina fluorescente verde (GFP)), coloranti che riempiono un neurone (ad esempio, colorante carbocianine, DiI) e rilevamento immunofluorescente di marcatori spinali (ad esempio, densità postsinaptica 95 (PSD95)). Un avvertimento importante a queste tecniche proxy è che identificano solo i cambiamenti postsinaptici. Tuttavia, una sinapsi è una connessione tra un terminale presinaptico e una colonna vertebrale postsinaptica. Il gold standard per misurare la formazione / eliminazione delle sinapsi richiede tecniche di microscopia elettronica o tomografia array che richiedono tempo. Queste tecniche richiedono una formazione specializzata e attrezzature costose. Inoltre, solo un numero limitato di neuroni può essere valutato e viene utilizzato per rappresentare i cambiamenti in un'intera regione del cervello. DetectSyn è una tecnica fluorescente rapida che identifica i cambiamenti nella formazione o nell'eliminazione delle sinapsi a causa di uno stato di malattia o di un'attività farmacologica. DetectSyn utilizza un test di legatura di prossimità rapida per rilevare proteine pre e postsinaptiche giustapposte e microscopia fluorescente standard, una tecnica prontamente disponibile per la maggior parte dei laboratori. Il rilevamento fluorescente del puncta risultante consente un'analisi rapida e imparziale degli esperimenti. DetectSyn fornisce risultati più rappresentativi rispetto alla microscopia elettronica perché è possibile analizzare aree più grandi rispetto a un numero limitato di neuroni fluorescenti. Inoltre, DetectSyn funziona per neuroni in coltura in vitro e fette di tessuto fisse. Infine, viene fornito un metodo per analizzare i dati raccolti da questa tecnica. Nel complesso, DetectSyn offre una procedura per rilevare i cambiamenti relativi nella densità delle sinapsi tra trattamenti o stati patologici ed è più accessibile rispetto alle tecniche tradizionali.
Introduzione
Le sinapsi sono l'unità fondamentale della comunicazione tra i neuroni1. Molte sinapsi tra neuroni all'interno delle stesse regioni danno origine a circuiti che mediano il comportamento2. Le sinapsi sono costituite da un terminale presinaptico di un neurone che rilascia neurotrasmettitori o neuropeptidi che trasmettono informazioni ai recettori postsinaptici di un altro neurone. La somma dei segnali presinaptici determina se il neurone postsinaptico attiverà un potenziale d'azione e propagherà il messaggio ad altri neuroni.
La sinaptopatologia, la rottura delle sinapsi, si manifesta in malattie e disturbi caratterizzati da diminuzione del volume neurale, come il morbo di Alzheimer e il disturbo depressivo maggiore, con conseguente circuiti che non funzionano più in modo ottimale 3,4,5. Il ripristino della densità delle sinapsi probabilmente è alla base dell'efficacia dei potenziali trattamenti per questi disturbi. Ad esempio, è stato recentemente dimostrato che l'aumento delle sinapsi è alla base dell'efficacia comportamentale degli antidepressivi rapidi6. Per esaminare rapidamente possibili trattamenti sinaptopatologici, i ricercatori richiedono tecniche che identifichino rapidamente i cambiamenti nei numeri delle sinapsi.
Le metodologie attuali sono lunghe e costose (microscopia elettronica, tomografia ad array), oppure esaminano solo i cambiamenti postsinaptici senza incorporare l'impegno presinaptico (analisi della colonna vertebrale, immunofluorescenza / colocalizzazione). Coloranti come DiI o proteine fluorescenti come GFP aiutano a visualizzare i neuroni e caratterizzano le spine postsinaptiche. Tuttavia, l'analisi della colonna vertebrale utilizza rapporti definiti dal ricercatore per determinare la morfologia, che può ridurre la riproducibilità7. Inoltre, il modo in cui le diverse classi della colonna vertebrale si riferiscono alle sinapsi funzionali è ancora in fase di scoperta8. La formazione della colonna vertebrale può essere transitoria e può riflettere la plasticità postsinaptica, ma queste spine potrebbero essere eliminate prima di stabilizzarsi in una sinapsi con un neurone presinaptico9.
La colocalizzazione fornisce un proxy migliore per le sinapsi rispetto all'analisi della colonna vertebrale perché si può immunostain per le proteine presinaptiche e postsinaptiche. Tuttavia, le proteine sinaptiche possono produrre bassi valori di colocalizzazione perché le proteine sono giustapposte e potrebbero non sovrapporsi in modo coerente. Pertanto, poiché le proteine non sono completamente sovrapposte, le tecniche di colocalizzazione potrebbero non misurare con precisione i cambiamenti nella formazione delle sinapsi a causa di queste informazioni mancanti. Infine, sebbene sia la microscopia elettronica (EM) che la tomografia array forniscano immagini ad alta risoluzione delle sinapsi, richiedono molto tempo. EM richiede inoltre attrezzature specializzate e i ricercatori sono limitati a piccoli volumi di tessuto per ogni dato esperimento. Mentre la tomografia ad array fornisce elegantemente la possibilità di selezionare molte proteine su sezioni ultrasottili e può essere combinata con EM10, questa tecnica può essere troppo laboriosa e al di là dello scopo degli esperimenti che devono eseguire rapidamente la scansione per i cambiamenti nella formazione delle sinapsi.
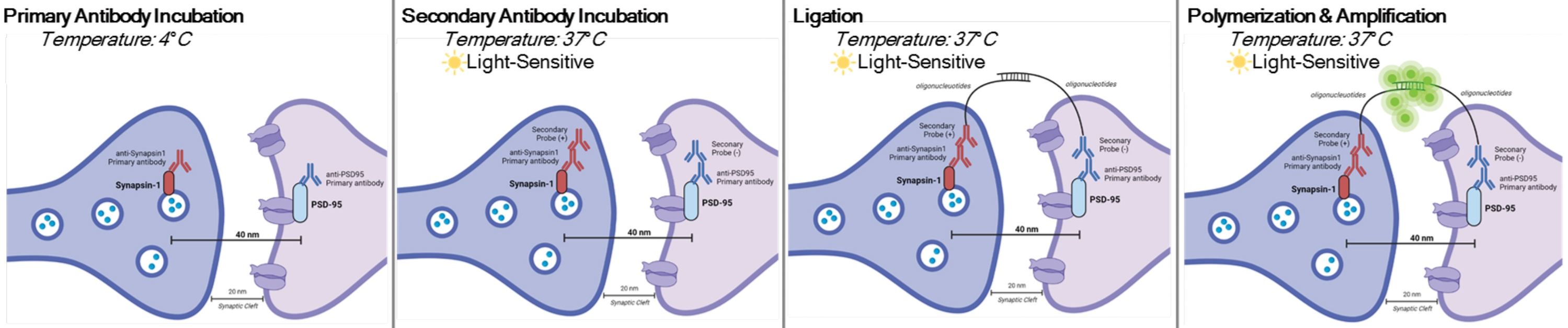
DetectSyn è un'applicazione specifica del Duolink Proximity Ligation Assay. Il test PLA consente la rilevazione generale delle interazioni proteina-proteina. DetectSyn collega le misure postsinaptiche proxy amplificando un segnale fluorescente emesso da proteine pre- e postsinaptiche taggate entro 40 nm l'una dall'altra. Se le proteine sinaptiche sono entro 40 nm, come all'interno di una fessura sinaptica, allora gli anticorpi secondari, che contengono sonde di DNA, si ibridano in DNA circolare. Questo DNA circolare ibridato esprime una sonda fluorescente, che viene poi amplificata e rilevata con tecniche standard di microscopia fluorescente (vedi Figura 1). Fondamentalmente, a differenza della tomografia EM e array, questa tecnica non richiede attrezzature specializzate e richiede circa la stessa quantità di tempo dell'immunoistochimica standard. L'accessibilità di questa tecnica, quindi, consente ai ricercatori al di fuori delle istituzioni ad alta intensità di ricerca di partecipare alla ricerca sinaptopatologica. Inoltre, questa tecnica può esaminare i cambiamenti della densità sinaptica in più regioni del cervello all'interno di un singolo esperimento, offrendo una rappresentazione più olistica dei cambiamenti sinaptici dovuti a malattie o trattamenti.
Protocollo
L'isolamento di cellule e tessuti dagli animali è stato conforme alla Guida del National Institutes of Health per la cura e l'uso degli animali da laboratorio e approvato dal Comitato istituzionale per la cura e l'uso degli animali di Wake Forest
NOTA: Questo protocollo viene utilizzato su campioni già trattati e fissati secondo specifici paradigmi e requisiti sperimentali. A scopo dimostrativo, la formazione di sinapsi dovuta al rapido trattamento antidepressivo viene utilizzata per evidenziare questa tecnica di rilevamento delle sinapsi6. I neuroni precedentemente coltivati su coverslip, trattati, fissati in paraformaldeide al 4% (PFA) e conservati in 1x soluzione salina tamponata con fosfato (PBS) saranno utilizzati per evidenziare le procedure in vitro. Il tessuto ippocampale precedentemente affettato (spesso 25 μm) da topi trattati, perfuso transcardialmente con PBS ghiacciato e PFA al 4% e quindi conservato in crioprotettore sarà utilizzato per evidenziare le procedure della fetta. Si prega di vedere 11,12 per ulteriori informazioni su come coltivare neuroni o perfondere transcardialmente i roditori. Vedere la Figura 1 per una rappresentazione grafica di questa procedura.

Figura 1: Rappresentazione grafica del test DetectSyn. Dopo aver permeabilizzato le membrane cellulari, gli anticorpi primari per Synapsin1 e PSD95 si legano a queste proteine sinaptiche. I secondari con tag oligonucleotidici si legano quindi agli anticorpi primari. Se Synapsin1 e PSD95 sono entro 40 nm, come in una sinapsi, allora gli oligonucleotidi interagiscono e un tag fluorescente viene amplificato. Questo segnale fluorescente può quindi essere ripreso tramite microscopia standard e analizzato. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
1. Risciacquare i campioni
- Risciacquare i campioni con 500 μL di 1x PBS + 0,75% glicina per 5 min 3 volte con agitazione delicata su uno shaker orbitale per rimuovere PFA residuo o crioprotettore.
2. Blocca e permeabilizza i campioni
- Preparare la soluzione di blocco e permeabilizzazione (10% di siero d'asino normale, 0,25% Tween 20) in 1x PBS. Preparare abbastanza da usare per il blocco, le incubazioni primarie e secondarie.
- Ai campioni (ad esempio, coverslips o fette fluttuanti) in 24 piastre di pozzetto, aggiungere 500 μL di soluzione di blocco e permeabilizzazione. Assicurarsi che ogni pozzetto contenga un campione diverso e sia opportunamente etichettato per evitare che i campioni vengano scambiati.
- Incubare i campioni a temperatura ambiente (RT) per 60 minuti per le cellule in coltura o 2 ore per il tessuto affettato. Utilizzare uno shaker orbitale per un'agitazione delicata.
3. Incubare campioni in anticorpi primari
- Preparare gli anticorpi primari nel tampone di blocco:
- Preparare densità postsinaptica 95 (PSD95; 1:500, policlonale di coniglio), Synapsin1 (1:500, monoclonale di topo), MAP2 (1:400, policlonale di pollo)
- Preparare un'aliquota di controllo negativa che ometta una delle coppie sinaptiche (ad esempio, senza PSD95)
- Rimuovere con attenzione la soluzione di blocco con una pipetta Pasteur di plastica. Cerca di rimuovere il più possibile senza disturbare le cellule o strappare il tessuto.
- Per le cellule coltivate:
- Foderare una grande capsula di Petri di plastica con parafilm. Trasferire con cura le coperture sul parafilm usando una pinza.
- Aggiungere con attenzione 60 μL della soluzione anticorpale primaria nella parte superiore dei coverslip. Assicurarsi di non versare la soluzione anticorpale primaria sul lato del coverslip.
- Per fornire umidità ed evitare che i campioni si secchino durante il periodo di incubazione, aggiungere acqua ultrapura a una capsula di Petri più piccola e disporre con cura la piccola capsula di Petri attorno alle coperture.
- Coprire la grande capsula di Petri e incubare le cellule coltivate per 1 ora a RT.
- Per il tessuto affettato:
- Aggiungere con attenzione 250 μL della soluzione anticorpale primaria alle fette fluttuanti in una piastra a 24 pozzetti.
- Coprire la piastra e incubare il tessuto durante la notte a 4 °C con una delicata agitazione su uno shaker orbitale.
4. Lavare i campioni, quindi incubare in anticorpi secondari
- Preparare gli anticorpi secondari nel tampone bloccante:
- Prepara Asino anti-mouse (1:5), asino anti-coniglio (1:5), asino anti-pollo (1:400).
- In questa fase, è possibile ottenere ulteriori controlli tecnici preparando un'aliquota secondaria che omette il secondario anti-topo o anti-coniglio.
- Per le cellule coltivate:
- Usando una pinza, toccare con attenzione la soluzione primaria dalle coperture su un tovagliolo di carta
- Utilizzando una pinza, trasferire con cura le coperture alla loro piastra originale a 24 pozzetti riempita con 500 μL di 1x PBS.
- Per il tessuto affettato:
- Rimuovere con attenzione la soluzione anticorpale primaria con una pipetta pasteur di plastica. Cerca di rimuovere il più possibile senza strappare il tessuto.
- Aggiungere 500 μL di 1x PBS
- Lavare i campioni per 10 min 3 volte in 1x PBS con agitazione delicata su uno shaker orbitale. Durante questo periodo, portare tutti i tamponi di lavaggio a RT.
- Durante questo periodo, cambia il parafilm nella grande capsula di Petri
- Per le cellule coltivate:
- Usando la pinza, trasferire con cura le coperture sulla grande capsula di Petri parafilmata
- Aggiungere con attenzione 40 μL della soluzione anticorpale secondaria nella parte superiore dei coverslip. Assicurarsi di non versare la soluzione anticorpale secondaria sul lato del coperchio.
- Se necessario, aggiungere più acqua ultrapura a una capsula di Petri più piccola e disporre con cura la piccola capsula di Petri attorno alle coperture.
- Coprire la grande capsula di Petri
- Per il tessuto affettato:
- Aggiungere con attenzione 250 μL della soluzione anticorpale secondaria alle fette fluttuanti in una piastra a 24 pozzetti.
- Coprire il piatto
NOTA: Da qui in poi, proteggere i campioni dalla luce avvolgendo le parti superiori delle piastre con un foglio.
- Incubare i campioni a 37 °C per 1 ora
5. Legatura
- Mescolare il calcio di legatura 1:5 in acqua di grado molecolare.
- Come nel paragrafo 4, trasferire con cura i coverslip e rimuovere la miscela secondaria dal tessuto affettato.
- Lavare i campioni in 500 μL di tampone di lavaggio A
- Per le cellule coltivate, lavare 2 volte per 5 minuti. Per il tessuto affettato, lavare 2 volte per 10 minuti. Utilizzare un'agitazione delicata su uno shaker orbitale per entrambi.
- Durante questo periodo, cambia il parafilm nella grande capsula di Petri
- Mantenendo la ligasi su un blocco freddo, diluire la ligasi 1:40 nel calcio di legatura dal passaggio 5.1. Eseguire questa diluizione immediatamente prima di aggiungere la ligasi ai campioni.
- Come nel paragrafo 4, rimuovere il più possibile il tampone di lavaggio A dai campioni prima di aggiungere la ligasi.
- Per le cellule in coltura: trasferire le coperture alla capsula di Petri parafilmata. Aggiungere 40 μL della miscela di legatura alle coperture, disporre piccole piastre di Petri piene d'acqua attorno alle coperture e coprire la grande capsula di Petri.
- Per il tessuto affettato: aggiungere 250 μL della miscela di legatura dal passaggio 5.4 a ciascun pozzetto e coprire la piastra.
- Incubare i campioni per 30 minuti a 37 °C.
6. Amplificazione
- Mescolare lo stock di amplificazione 1:5 in acqua di grado molecolare.
- Come nella sezione 4, trasferire con attenzione le coperture e rimuovere la miscela di legatura dal tessuto affettato.
- Lavare i campioni in 500 μL di tampone di lavaggio A
- Per le cellule coltivate, lavare 2 volte per 2 minuti. Per il tessuto affettato, lavare 2 volte per 10 minuti. Utilizzare un'agitazione delicata su uno shaker orbitale per entrambi.
- Durante questo periodo, cambia il parafilm nella grande capsula di Petri
- Eseguire questa diluizione immediatamente prima di aggiungere la polimerasi ai campioni. Mantenendo la polimerasi su un blocco freddo, diluire la polimerasi
- Per le cellule in coltura, diluire la polimerasi 1:80 nello stock di amplificazione dal punto 6.1.
- Per il tessuto affettato, diluire la polimerasi 1:40 nel materiale di amplificazione dal punto 6.1.
- Come nel passaggio 4, rimuovere il più possibile il tampone di lavaggio A dai campioni prima di aggiungere la polimerasi.
- Per le cellule in coltura: trasferire i coverslip nella capsula di Petri parafilmata. Aggiungere 40 μL della miscela di amplificazione dal passaggio 6.4.1 alle coperture, disporre piccole piastre di Petri piene d'acqua attorno alle coperture e coprire la grande capsula di Petri. Incubare i campioni per 100 minuti a 37 °C.
- Per il tessuto affettato: aggiungere 250 μL della miscela di amplificazione dal punto 6.4.2 a ciascun pozzetto e coprire la piastra. Incubare i campioni per 2 ore a 37 °C.
NOTA: durante questo periodo, preparare ed etichettare le diapositive.
7. Montaggio
- Come nel passaggio 4, trasferire con cura i coverslip e rimuovere la miscela di amplificazione dal tessuto affettato.
- Lavare i campioni in 500 μL di tampone di lavaggio B 2 volte per 10 minuti con una leggera agitazione su uno shaker orbitale.
- Lavare i campioni in 500 μL di tampone di lavaggio B all'1% per 1 minuto con agitazione delicata su uno shaker orbitale.
- Per le cellule coltivate:
- Caduta di 3 μL di supporto di montaggio su una slitta
- Toccare il tampone di lavaggio in eccesso dal coperchio e quindi posizionare il coperchio (con le celle rivolte verso il basso) nel supporto di montaggio. Sigillare i lati con una piccola quantità di smalto trasparente per sigillare il coperchio in posizione.
- Per il tessuto affettato:
- Trasferire con cura una fetta di tessuto sul vetrino preparato e disporla, in modo che la fetta sia piatta. Caduta tra 5-10 μL di supporto di montaggio (la quantità dipenderà dalle dimensioni della fetta) sulla fetta di tessuto
- Posizionare con cura una copertura di vetro sulla fetta di tessuto e sigillare con una piccola quantità di smalto trasparente lungo il bordo per sigillare la copertina in posizione.
- Attendere almeno 15 minuti prima di analizzare al microscopio o conservare a -20 °C.
8. Ottenere immagini digitali con un microscopio confocale
- Ottimizza le impostazioni di acquisizione (ad esempio, potenza laser, guadagno, offset) su campioni di tutti i trattamenti. Assicurarsi che l'ottimizzazione includa la riduzione del rumore di fondo e il miglioramento del segnale senza sovrasaturare l'intensità dei segnali fluorescenti. Una volta determinate le impostazioni, applicare le stesse impostazioni di acquisizione a tutte le immagini ottenute.
NOTA: i seguenti dettagli di acquisizione possono essere utilizzati con un microscopio confocale Nikon A1 e il software Nikon NIS AR Elements. - Impostare la diapositiva con il campione sul palco e trovare il piano focale per il campione utilizzando DAPI attraverso l'oculare.
- Spegnere la porta dell'occhio facendo clic su Porta occhio e scegliere un pulsante di configurazione ottica per regolare le impostazioni.
- Regola il guadagno, l'offset e la potenza laser per ciascun canale fluorescente per ridurre il rumore di fondo e migliorare il segnale fluorescente. Assicurarsi che il segnale fluorescente non diventi troppo saturo come indicato nei passaggi 8.5-8.6.
- Monitorare la sovrasaturazione utilizzando uno pseudocolore per il segnale fluorescente. Nella parte inferiore dell'immagine live, fai clic con il pulsante destro del mouse sulla scheda etichettata con il canale fluorescente attualmente in uso.
- Quindi, scegli Colorazione canale e scegli uno pseudocolore come Rainbow Dark per visualizzare l'intensità della fluorescenza in uno pseudocolore simile a una mappa di calore. In Rainbow Dark, i colori più freddi indicano un'intensità meno fluorescente e i colori più caldi indicano un'intensità più fluorescente.
- Una volta ottimizzati tutti i canali fluorescenti, fare clic con il pulsante destro del mouse sul pulsante di configurazione ottica precedentemente scelto e scegliere Assegna impostazione corrente fotocamera per questo pulsante.
- Verificare che le impostazioni scelte siano sufficienti per un campione casuale di ciascun gruppo di trattamento. Se le impostazioni scelte sovrasaturano uno di questi campioni, ripetere il passaggio 8.4 per eliminare la sovrasaturazione.
- Per i neuroni in coltura, seguire i passaggi 8.10-8.16.
- Usando la porta dell'occhio, cerca un neurone con dendriti che hanno una sovrapposizione minima con altri dendriti.
- Spegnere la porta dell'occhio e utilizzare il canale DAPI per visualizzare il corpo cellulare del neurone scelto. Fare doppio clic sul centro del soma per centrare il neurone al centro del campo visivo.
- Utilizzando il canale MAP2, trova il miglior piano di messa a fuoco per il segnale MAP2 con la scansione dal vivo.
- Nella scheda Acquisizione ND , fare clic su Salva su file e scegliere un file in cui salvare l'immagine in Sfoglia. Quindi, inserisci il nome del file.
- Nella scheda Z selezionare l'opzione Modalità simmetrica definita da Intervallo . Impostare lo stato attivo sul piano MAP2 migliore e fare clic sul pulsante Relativo per impostare questo piano focale come centro dello z-stack.
- Impostare l'intervallo su 5 μm con passi di 1 μm e assicurarsi di selezionare Chiudi otturatore attivo durante il movimento Z. Nella scheda Lunghezza d'onda selezionare il nome del pulsante ottico con le impostazioni di acquisizione configurate in precedenza in Conf ottico. Quindi, fai clic su Esegui ora.
- Ripetere i passaggi 8.1.10-8.1.15 per circa 10 neuroni per coverslip/trattamento.
- Per il tessuto affettato, seguire i passaggi 8.18-8.22.
- Usando la porta dell'occhio, cerca la regione di interesse. Ad esempio, individuare CA1 dell'ippocampo.
- Spegnere la porta oculare e utilizzare il canale MAP2 per trovare il miglior piano di messa a fuoco per il segnale MAP2 con la scansione dal vivo.
- Nel menu Acquisisci , scegliere Scansione immagine di grandi dimensioni. Quindi, selezionare il nome del pulsante ottico con le impostazioni di acquisizione configurate in precedenza nel pannello Acquisizione dal pannello che si apre. Inoltre, assicurati di selezionare l'obiettivo corretto in questo pannello.
- Sotto il pannello Area e l'oculare, usate i tasti freccia per impostare i confini della regione di interesse. Quindi, fai clic su Salva immagine di grandi dimensioni su file e crea un nome file del percorso di salvataggio per l'immagine.
- Nel pannello Configurazione, assicurarsi che Acquisizione multicanale sia selezionata, quindi scegliere il nome del pulsante ottico con le impostazioni di acquisizione configurate in precedenza in Conf ottico.
NOTA: è possibile uno z-stack per un'immagine di grandi dimensioni, ma aumenterà il tempo di scansione.
9. Analisi
- Analogamente alle impostazioni di acquisizione, utilizzare i campioni di tutti i trattamenti per ottimizzare le impostazioni di soglia. Assicurarsi che l'ottimizzazione della soglia si concentri sulla riduzione del rumore di fondo e sul miglioramento del segnale senza sovrasaturare l'intensità dei segnali fluorescenti. Una volta determinate queste impostazioni, applicare le stesse impostazioni di soglia a tutte le immagini utilizzate per l'analisi come descritto nei passaggi 9.2-9.3.
- In ImageJ, l'opzione di soglia si trova sotto il menu Immagine > Regola > soglia. Scegli l'opzione Sfondo scuro se l'immagine ha uno sfondo scuro.
- Quindi, regola i limiti superiore e inferiore della soglia per le impostazioni di soglia ottimizzate precedentemente determinate, quindi fai clic su Applica.
- Per le cellule coltivate, utilizzare il canale MAP2 e uno strumento di regione di interesse a mano libera (ROI) per disegnare un ROI per ciascun neurone, inclusi dendriti e soma. Per il tessuto affettato, disegna un ROI a mano libera all'interno dell'immagine della fetta che incapsula l'area di interesse (ad esempio, strato radiato del CA1 all'interno dell'ippocampo).
- Ottenere l'area del ROI. In ImageJ, misurare l'area nel menu Analizza > Misura.
- Rileva il numero di puncta all'interno di ciascun ROI utilizzando uno strumento di rilevamento automatico come Analisi delle particelle in ImageJ seguendo i passaggi 9.7-9.9.
- Trova l'opzione Analisi particelle nel menu Analizza > analizza particelle. Innanzitutto, definisci il diametro della dimensione del punto, in genere 0,1-3 μm2.
- Quindi, scegli l'opzione Maschere sovrapposte dal menu a discesa Mostra e seleziona l'opzione Visualizza risultati. Quindi, fare clic su OK.
- Se i puncta non vengono rilevati con l'intervallo di diametro scelto, regolare l'intervallo fino a quando tutti i puncta non vengono rilevati con questa analisi. Assicurati di utilizzare le stesse impostazioni di analisi delle particelle per tutte le immagini.
- Dividere il numero di puncta per l'area di una singola regione di interesse seguendo i passaggi 9.11-9.13.
- Copia e incolla i risultati per ogni immagine dal popup Risultati da ImageJ in un foglio di calcolo.
- Innanzitutto, identificare da quale file e campione sono stati ottenuti i dati. Quindi, dividi l'area del ROI per il numero di puncta.
- Quindi, cancella i dati dal popup Risultati e ripeti i passaggi 9.2-9.12.
- Normalizzare i risultati per controllare i campioni: media dei risultati (numero di aree puncta/ROI) per i campioni di controllo. Quindi, dividere i risultati ottenuti di tutti i campioni per la media del controllo per ottenere i risultati normalizzati. La nuova media dei campioni di controllo dovrebbe essere pari a 1.
Risultati
I dati modificati da Heaney et al.6 sono presentati per dimostrare un esperimento in cui è previsto un aumento della formazione di sinapsi (vedere6 per ulteriori informazioni e una discussione più approfondita del meccanismo). In precedenza, è stato dimostrato che gli antidepressivi rapidi richiedono l'attivazione del recettore metabotropico inibitorio, GABAB (sottotipo di acido gamma-aminobutirrico B), per essere efficace13. Inoltre, i dati prece...
Discussione
DetectSyn è un test rapido che utilizza un test di legatura di prossimità per rilevare proteine entro 40 nm l'una dall'altra, che consente il rilevamento della formazione di sinapsi. Questa tecnica migliora gli attuali saggi fluorescenti, che servono solo come misure proxy per la formazione delle sinapsi. DetectSyn rileva cambiamenti quantificabili nelle proteine sinaptiche localizzate entro 40 nm, cioè all'interno della fessura sinaptica, l'una dell'altra. Inoltre, DetectSyn è più conveniente e richiede meno tempo ...
Divulgazioni
Gli autori non segnalano alcun conflitto di interessi.
Riconoscimenti
Questo lavoro è stato supportato da National Institutes of Health NINDS R01 NS105005 (KRG) e NS105005-03S1 (KRG), Department of Defense USAMRMC W81XWH-14-1-0061 (KRG), NIAAA R01AA016852, NIAAA T32AA007565 (CFH), e una sovvenzione da FRAXA Research (CFH) e Alzheimer's Association, AARG-NTF-21-852843 (KRG), AARF-19-614794-RAPID (KRG).
Materiali
| Name | Company | Catalog Number | Comments |
| 10x PBS | Fisher Scientific | BP39920 | PBS made in house works, as well. |
| 24 well plates | Fisher Scientific | FB012929 | For tissue slices, pre-sterilized plates may be unnecessary. |
| 50 mL conical tubes | Fisher Scientific | 14-432-22 | |
| Aluminium foil | Fisher Scientific | 15-078-290 | |
| Chicken anti-MAP2 antibody | Abcam | ab5392 | |
| Clear nail polish | Fisher Scientific | NC1849418 | Other clear nail polish works, as well. |
| Cold block | Fisher Scientific | 13131012 | |
| Computer workstation | HP | ||
| Confocal or fluorescent microscope | Nikon | A1R HD25 | |
| Donkey anti-chicken FITC | Fisher Scientific | SA1-72000 | |
| Duolink donkey anti-Mouse PLUS | Sigma | DUO92001 | |
| Duolink donkey anti-Rabbit MINUS | Sigma | DUO92005 | |
| Duolink In Situ Detection Reagents Far Red | Sigma | DUO92013 | Contains ligation stock, amplification stock, ligase, and polymerase. |
| Duolink In Situ Mounting Medium with DAPI | Sigma | DUO82040 | |
| Duolink In Situ Wash Buffers, Fluorescence | Sigma | DUO82049 | Contains Wash Buffer A and Wash Buffer B; dilute Wash Buffer B to 1% in diH20 for 1% Wash Buffer B. |
| Fine-tipped paintbrush | Fisher Scientific | NC9691026 | Sable hair, size 00 or 000, can also find at craft stores |
| Fisherbrand Cover Glasses: Rectangles | Fisher Scientific | 12545MP | Cover glass is unnecessary for cultured neurons already on glass coverslips. |
| Fisherbrand Superfrost Plus Microscope Slides | Fisher Scientific | 1255015 | For cultured neurons already on glass coverslips, Superfrost slides may be unnecessary. |
| Freezer, -20°C | VWR | 76449-108 | |
| Glass coverslips | Fisher Scientific | 125480 | |
| Glycine | Fisher Scientific | BP381-1 | |
| Image processing software | e.g. NIS Elements, ImageJ | ||
| Incubator | Fisher Scientific | 15-015-2633 | |
| Large petri dish, 100mm | Fisher Scientific | FB0875712 | |
| Molecular grade water | Fisher Scientific | BP24701 | |
| Mouse anti-Synapsin1 antibody | Synaptic Systems | 106-011 | |
| Normal donkey serum | Jackson ImmunoResearch | 017-000-121 | |
| Orbital shaker | Fisher Scientific | 02-106-1013 | |
| Parafilm | Fisher Scientific | 13-374-10 | |
| Pipette tips | Fisher Scientific | 02-707-025 | |
| Pipettes | Fisher Scientific | 14-388-100 | Working volumes range from 3 µL to 500 µL |
| Plastic pasteur pipette | Fisher Scientific | 02-708-006 | |
| Precision tweezers/foreceps | Fisher Scientific | 12-000-122 | |
| Rabbit anti-PSD95 antibody | Abcam | ab18258 | Other antibody pairs may work, as well, with optimization. |
| Refrigerator | VWR | 76470-402 | |
| Small petri dish, 60 mm | Fisher Scientific | FB0875713A | |
| Timer | Fisher Scientific | 14-649-17 | |
| Tween 20 | Fisher Scientific | BP337-100 |
Riferimenti
- Südhof, T. C. Towards an understanding of synapse formation. Neuron. 100 (2), 276-293 (2018).
- Bliss, T. V., Collingridge, G. L. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 361 (6407), 31-39 (1993).
- Heaney, C. F., Raab-Graham, K. F. Dysregulated protein synthesis in major depressive disorder. The Oxford Handbook of Neuronal Protein Synthesis. , 510-532 (2018).
- Masliah, E., Crews, L., Hansen, L. Synaptic remodeling during aging and in Alzheimer's disease. Journal of Alzheimer's Disease. 9, 91-99 (2006).
- van Spronsen, M., Hoogenraad, C. C. Synapse pathology in psychiatric and neurologic disease. Current Neurology and Neuroscience Reports. 10 (3), 207-214 (2010).
- Heaney, C. F., Namjoshi, S. V., Uneri, A., Bach, E. C., Weiner, J. L., Raab-Graham, K. F. Role of FMRP in rapid antidepressant effects and synapse regulation. Molecular Psychiatry. 26 (6), 2350-2362 (2021).
- Pchitskaya, E., Bezprozvanny, I. Dendritic spines shape analysis-Classification or clusterization? Perspective. Frontiers in Synaptic Neuroscience. 12, 31 (2020).
- Alvarez, V. A., Sabatini, B. L. Anatomical and physiological plasticity of dendritic spines. Annual Review of Neuroscience. 30 (1), 79-97 (2007).
- Berry, K. P., Nedivi, E. Spine Dynamics: Are they all the same. Neuron. 96 (1), 43-55 (2017).
- Micheva, K. D., Smith, S. J. Array tomography: A new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron. 55 (1), 25-36 (2007).
- Kaech, S., Banker, G. Culturing hippocampal neurons. Nature Protocols. 1 (5), 2406-2415 (2006).
- Gage, G. J., Kipke, D. R., Shain, W. Whole animal perfusion fixation for rodents. Journal of Visualized Experiments: JoVE. (65), e3564 (2012).
- Workman, E. R., Niere, F., Raab-Graham, K. F. mTORC1-dependent protein synthesis underlying rapid antidepressant effect requires GABABR signaling. Neuropharmacology. 73, 192-203 (2013).
- Li, N., et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 329 (5994), 959-964 (2010).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati