
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
DetectSyn: Une méthode fluorescente rapide et impartiale pour détecter les changements dans la densité des synapses
Dans cet article
Résumé
DetectSyn est un test fluorescent rapide et impartial qui mesure les changements dans le nombre relatif de synapses (engagement pré- et postsynaptique) entre les traitements ou les états pathologiques. Cette technique utilise une technique de ligature de proximité qui peut être utilisée à la fois dans les neurones cultivés et les tissus fixes.
Résumé
Les synapses sont le site de communication entre les neurones. La force du circuit neuronal est liée à la densité synaptique, et la dégradation des synapses est caractéristique des états pathologiques comme le trouble dépressif majeur (TDM) et la maladie d’Alzheimer. Les techniques traditionnelles pour étudier le nombre de synapses comprennent l’expression génétique de marqueurs fluorescents (p. ex., protéine fluorescente verte (GFP)), les colorants qui remplissent un neurone (p. ex., colorant à la carbocyanine, DiI) et la détection immunofluorescente des marqueurs de la colonne vertébrale (p. ex., densité postsynaptique 95 (DSP95)). Une mise en garde majeure à ces techniques de proxy est qu’elles n’identifient que les changements postsynaptiques. Pourtant, une synapse est une connexion entre une borne présynaptique et une colonne vertébrale postsynaptique. L’étalon-or pour mesurer la formation / élimination des synapses nécessite des techniques de microscopie électronique ou de tomographie par réseau qui prennent beaucoup de temps. Ces techniques nécessitent une formation spécialisée et un équipement coûteux. De plus, seul un nombre limité de neurones peut être évalué et est utilisé pour représenter les changements dans toute une région du cerveau. DetectSyn est une technique fluorescente rapide qui identifie les changements dans la formation ou l’élimination des synapses en raison d’un état pathologique ou d’une activité médicamenteuse. DetectSyn utilise un test de ligature de proximité rapide pour détecter les protéines pré- et postsynaptiques juxtaposées et la microscopie fluorescente standard, une technique facilement disponible pour la plupart des laboratoires. La détection fluorescente de la ponctuation résultante permet une analyse rapide et impartiale des expériences. DetectSyn fournit des résultats plus représentatifs que la microscopie électronique car de plus grandes zones peuvent être analysées qu’un nombre limité de neurones fluorescents. De plus, DetectSyn fonctionne pour les neurones cultivés in vitro et les tranches de tissus fixes. Enfin, une méthode est fournie pour analyser les données collectées à partir de cette technique. Dans l’ensemble, DetectSyn offre une procédure pour détecter les changements relatifs dans la densité des synapses à travers les traitements ou les états pathologiques et est plus accessible que les techniques traditionnelles.
Introduction
Les synapses sont l’unité fondamentale de communication entre les neurones1. De nombreuses synapses entre neurones au sein d’une même région donnent naissance à des circuits qui médient le comportement2. Les synapses sont constituées d’un terminal présynaptique d’un neurone qui libère des neurotransmetteurs ou des neuropeptides qui relaient l’information aux récepteurs postsynaptiques d’un autre neurone. La somme des signaux présynaptiques détermine si le neurone postsynaptique déclenchera un potentiel d’action et propagera le message à d’autres neurones.
La synaptopathologie, la décomposition des synapses, survient dans les maladies et les troubles marqués par une diminution du volume neuronal, comme la maladie d’Alzheimer et le trouble dépressif majeur, entraînant des circuits qui ne fonctionnent plus de manière optimale 3,4,5. La restauration de la densité synaptique sous-tend probablement l’efficacité des traitements potentiels pour ces troubles. Par exemple, il a été récemment démontré que l’augmentation des synapses sous-tend l’efficacité comportementale des antidépresseurs rapides6. Pour dépister rapidement les traitements synaptopathologiques possibles, les chercheurs ont besoin de techniques qui identifient rapidement les changements dans le nombre de synapses.
Les méthodologies actuelles sont soit longues et coûteuses (microscopie électronique, tomographie en réseau), soit elles n’examinent que les changements postsynaptiques sans intégrer l’engagement présynaptique (analyses de la colonne vertébrale, immunofluorescence/colocalisation). Les colorants comme DiI ou les protéines fluorescentes comme GFP aident à visualiser les neurones et à caractériser les épines postsynaptiques. Cependant, l’analyse de la colonne vertébrale utilise des ratios définis par les chercheurs pour déterminer la morphologie, ce qui peut diminuer la reproductibilité7. De plus, la façon dont les différentes classes de colonne vertébrale se rapportent aux synapses fonctionnelles est encore en cours dedécouverte 8. La formation de la colonne vertébrale peut être transitoire et peut refléter une plasticité postsynaptique, mais ces épines pourraient être éliminées avant de se stabiliser en une synapse avec un neurone présynaptique9.
La colocalisation fournit un meilleur indicateur pour les synapses que l’analyse de la colonne vertébrale, car on peut s’immunocolorer pour les protéines présynaptiques et postsynaptiques. Cependant, les protéines synaptiques peuvent donner de faibles valeurs de colocalisation parce que les protéines sont juxtaposées et peuvent ne pas se chevaucher de manière cohérente. Ainsi, comme les protéines ne sont pas entièrement superposées, les techniques de colocalisation peuvent ne pas mesurer avec précision les changements dans la formation des synapses en raison de ces informations manquantes. Enfin, bien que la microscopie électronique (EM) et la tomographie en réseau fournissent des images à haute résolution des synapses, elles prennent beaucoup de temps. L’EM nécessite en outre un équipement spécialisé, et les chercheurs sont limités à de petits volumes de tissus pour une expérience donnée. Alors que la tomographie en réseau offre élégamment la possibilité de dépister de nombreuses protéines sur des sections ultraminces et peut être combinée avec EM10, cette technique peut être trop laborieuse et dépasser la portée des expériences qui doivent rechercher rapidement des changements dans la formation de synapses.
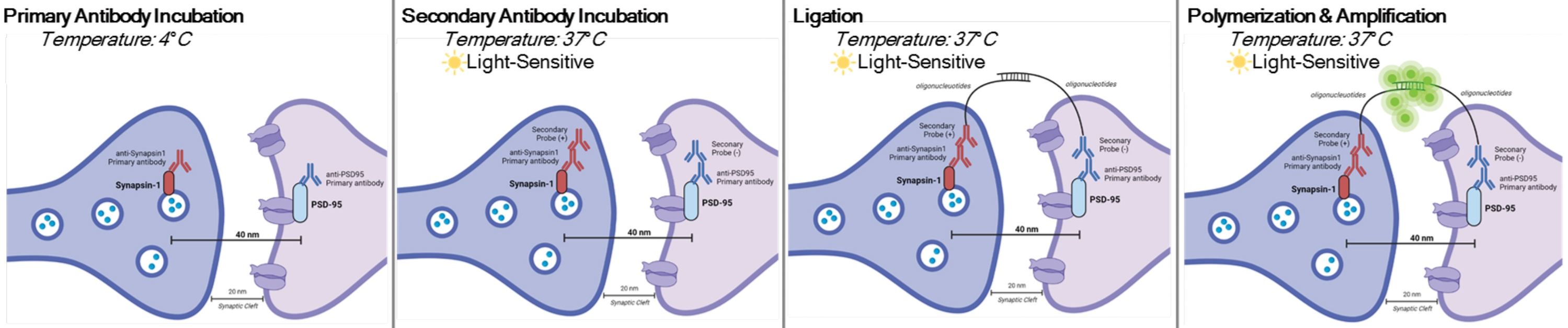
DetectSyn est une application spécifique du test de ligature de proximité Duolink. Le test PLA permet la détection générale des interactions protéine-protéine. DetectSyn relie les mesures postsynaptiques proxy en amplifiant un signal fluorescent émis par des protéines pré- et postsynaptiques marquées à moins de 40 nm les unes des autres. Si les protéines synaptiques sont à moins de 40 nm, comme dans une fente synaptique, alors les anticorps secondaires, qui contiennent des sondes d’ADN, s’hybrideront en ADN circulaire. Cet ADN circulaire hybridé exprime une sonde fluorescente, qui est ensuite amplifiée et détectée avec des techniques de microscopie fluorescente standard (voir figure 1). Fondamentalement, contrairement à la TOMOGRAPHIE PAR ET À LA TOMOGRAPHIE PAR RÉSEAU, cette technique ne nécessite pas d’équipement spécialisé et prend à peu près le même temps que l’immunohistochimie standard. L’accessibilité de cette technique permet donc aux chercheurs en dehors des institutions à forte intensité de recherche de participer à la recherche en synaptopathologie. En outre, cette technique peut examiner les changements de densité synaptique dans plusieurs régions du cerveau au sein d’une seule expérience, offrant une représentation plus holistique des changements synaptiques dus à la maladie ou au traitement.
Protocole
L’isolement des cellules et des tissus des animaux était conforme au Guide pour le soin et l’utilisation des animaux de laboratoire des National Institutes of Health et approuvé par le Wake Forest Institutional Animal Care and Use Committee
REMARQUE: Ce protocole est utilisé sur des échantillons déjà traités et fixés selon des paradigmes et des exigences expérimentaux spécifiques. À des fins de démonstration, la formation de synapses due à un traitement antidépresseur rapide est utilisée pour mettre en évidence cette technique de détection des synapses6. Des neurones précédemment cultivés sur des couvercles, traités, fixés dans du paraformaldéhyde (PFA) à 4 % et stockés dans 1x solution saline tamponnée au phosphate (PBS) seront utilisés pour mettre en évidence les procédures in vitro. Du tissu hippocampique préalablement tranché (25 μm d’épaisseur) provenant de souris traitées, perfusés transcardiquement avec du PBS glacé et du PFA à 4%, puis stockés dans un cryoprotecteur seront utilisés pour mettre en évidence les procédures de tranche. Veuillez consulter11,12 pour plus d’informations sur la façon de cultiver des neurones ou de perfuser des rongeurs de manière transcardique. Reportez-vous à la figure 1 pour une représentation graphique de cette procédure.

Figure 1 : Représentation graphique du test DetectSyn. Après perméabilisation des membranes cellulaires, les anticorps primaires de Synapsin1 et de PSD95 se lient à ces protéines synaptiques. Les secondaires avec des marqueurs oligonucléotidiques se lient ensuite aux anticorps primaires. Si Synapsin1 et PSD95 sont à moins de 40 nm, comme à une synapse, alors les oligonucléotides interagissent et une étiquette fluorescente est amplifiée. Ce signal fluorescent peut ensuite être imagé par microscopie standard et analysé. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
1. Rincer les échantillons
- Rincer les échantillons avec 500 μL de 1x PBS + 0,75% de glycine pendant 5 min 3 fois avec une agitation douce sur un agitateur orbital pour éliminer le PFA résiduel ou le cryoprotecteur.
2. Bloquer et perméabiliser les échantillons
- Préparer une solution de blocage et de perméabilisation (10% de sérum d’âne normal, 0,25% de Tween 20) dans 1x PBS. Préparez-vous suffisamment pour les incubations bloquantes, primaires et secondaires.
- Pour échantillonner (p. ex. des couvercles ou des tranches flottantes) dans 24 plaques de puits, ajouter 500 μL de solution de blocage et de perméabilisation. Assurez-vous que chaque puits contient un échantillon différent et qu’il est étiqueté de manière appropriée pour éviter que les échantillons ne soient changés.
- Incuber les échantillons à température ambiante (RT) pendant 60 min pour les cellules cultivées ou 2 h pour les tissus tranchés. Utilisez un agitateur orbital pour une agitation douce.
3. Incuber des échantillons dans des anticorps primaires
- Préparer des anticorps primaires dans un tampon bloquant :
- Préparer la densité postsynaptique 95 (PSD95; 1:500, lapin polyclonal), Synapsin1 (1:500, souris monoclonale), MAP2 (1:400, poulet polyclonal)
- Préparer une aliquote témoin négative qui omet l’une des paires synaptiques (p. ex., sans DSP95)
- Retirez soigneusement la solution bloquante à l’aide d’une pipette Pasteur en plastique. Essayez d’en enlever autant que possible sans perturber les cellules ou déchirer les tissus.
- Pour les cellules cultivées :
- Tapisser une grande boîte de Pétri en plastique avec un parafilm. Transférez soigneusement les couvercles sur le parafilm à l’aide de pinces.
- Ajouter soigneusement 60 μL de la solution d’anticorps primaires sur le dessus des couvercles. Assurez-vous de ne pas renverser la solution d’anticorps primaire sur le côté du couvercle.
- Pour fournir de l’humidité et empêcher les échantillons de se dessécher pendant la période d’incubation, ajoutez de l’eau ultrapure à une boîte de Petri plus petite et disposez soigneusement la petite boîte de Petri autour des couvercles.
- Couvrir la grande boîte de Pétri et incuber les cellules cultivées pendant 1 h à TA.
- Pour les tissus tranchés :
- Ajouter soigneusement 250 μL de la solution d’anticorps primaires aux tranches flottantes dans une plaque de 24 puits.
- Couvrir la plaque et incuber le tissu pendant la nuit à 4 °C avec une légère agitation sur un agitateur orbital.
4. Laver les échantillons, puis incuber dans des anticorps secondaires
- Préparer des anticorps secondaires dans le tampon bloquant :
- Préparez l’âne anti-souris (1:5), l’âne anti-lapin (1:5), l’âne anti-poulet (1:400).
- À cette étape, des contrôles techniques supplémentaires peuvent être obtenus en préparant une aliquote secondaire qui omet soit l’anti-souris, soit l’anti-lapin secondaire.
- Pour les cellules cultivées :
- À l’aide de pinces, tapotez soigneusement la solution primaire des couvercles sur une serviette en papier
- À l’aide de pinces, transférez soigneusement les couvercles dans leur plaque d’origine de 24 puits remplie de 500 μL de 1x PBS.
- Pour les tissus tranchés :
- Retirez soigneusement la solution d’anticorps primaires à l’aide d’une pipette Pasteur en plastique. Essayez d’en enlever autant que possible sans déchirer le tissu.
- Ajouter 500 μL de 1x PBS
- Lavez les échantillons pendant 10 min 3 fois dans 1x PBS avec une agitation douce sur un agitateur orbital. Pendant ce temps, apportez tous les tampons de lavage à RT.
- Pendant ce temps, changez le parafilm dans la grande boîte de Pétri
- Pour les cellules cultivées :
- À l’aide d’une pince, transférez soigneusement les couvercles dans la grande boîte de Petri parafilmée.
- Ajouter soigneusement 40 μL de la solution d’anticorps secondaires sur le dessus des couvercles. Assurez-vous de ne pas renverser la solution d’anticorps secondaire sur le côté du couvercle.
- Si nécessaire, ajoutez plus d’eau ultrapure à une boîte de Pétri plus petite et disposez soigneusement la petite boîte de Pétri autour des couvercles.
- Couvrir la grande boîte de Pétri
- Pour les tissus tranchés :
- Ajouter soigneusement 250 μL de la solution d’anticorps secondaires aux tranches flottantes dans une plaque de 24 puits.
- Couvrir la plaque
REMARQUE: À partir de là, protégez les échantillons de la lumière en enveloppant le dessus des plaques avec du papier d’aluminium.
- Incuber les échantillons à 37 °C pendant 1 h
5. Ligature
- Mélanger le stock de ligature 1:5 dans de l’eau de qualité moléculaire.
- Comme dans la section 4, transférer soigneusement les couvercles et retirer le mélange secondaire du tissu tranché.
- Laver les échantillons dans 500 μL de tampon de lavage A
- Pour les cellules cultivées, laver 2 fois pendant 5 min. Pour les mouchoirs tranchés, laver 2 fois pendant 10 min. Utilisez une agitation douce sur un agitateur orbital pour les deux.
- Pendant ce temps, changez le parafilm dans la grande boîte de Pétri
- Tout en gardant la ligase sur un bloc froid, diluez la ligase 1:40 dans le stock de ligature à partir de l’étape 5.1. Effectuez cette dilution immédiatement avant d’ajouter la ligase aux échantillons.
- Comme dans la section 4, retirez autant que possible le tampon de lavage A des échantillons avant d’ajouter la ligase.
- Pour les cellules cultivées : Transférer les couvercles vers la boîte de Petri parafilmée. Ajouter 40 μL du mélange de ligature aux couvercles, disposer de petites boîtes de Petri remplies d’eau autour des couvercles et couvrir la grande boîte de Pétri.
- Pour les tissus tranchés : Ajouter 250 μL du mélange de ligature de l’étape 5.4 à chaque puits et couvrir la plaque.
- Incuber les échantillons pendant 30 min à 37 °C.
6. Amplification
- Mélanger la masse d’amplification 1:5 dans de l’eau de qualité moléculaire.
- Comme dans la section 4, transférez soigneusement les couvercles et retirez le mélange de ligature du tissu tranché.
- Laver les échantillons dans 500 μL de tampon de lavage A
- Pour les cellules cultivées, laver 2 fois pendant 2 min. Pour les mouchoirs tranchés, laver 2 fois pendant 10 min. Utilisez une agitation douce sur un agitateur orbital pour les deux.
- Pendant ce temps, changez le parafilm dans la grande boîte de Pétri
- Effectuez cette dilution immédiatement avant d’ajouter la polymérase aux échantillons. Tout en gardant la polymérase sur un bloc froid, diluer la polymérase
- Pour les cellules cultivées, diluer la polymérase 1:80 dans la souche d’amplification à partir de l’étape 6.1.
- Pour les tissus tranchés, diluer la polymérase 1:40 dans la crosse d’amplification à partir de l’étape 6.1.
- Comme à l’étape 4, retirez autant que possible le tampon de lavage A des échantillons avant d’ajouter la polymérase.
- Pour les cellules cultivées : Transférez les couvercles dans la boîte de Petri parafilmée. Ajouter 40 μL du mélange d’amplification de l’étape 6.4.1 aux couvercles, disposer de petites boîtes de Petri remplies d’eau autour des couvercles et recouvrir la grande boîte de Pétri. Incuber les échantillons pendant 100 min à 37 °C.
- Pour les tissus tranchés : Ajouter 250 μL du mélange d’amplification de l’étape 6.4.2 à chaque puits et couvrir la plaque. Incuber les échantillons pendant 2 h à 37 °C.
REMARQUE: Pendant ce temps, préparez et étiquetez les diapositives.
7. Montage
- Comme à l’étape 4, transférez soigneusement les couvercles et retirez le mélange d’amplification du tissu tranché.
- Laver les échantillons dans 500 μL de tampon de lavage B 2 fois pendant 10 min avec une agitation douce sur un agitateur orbital.
- Laver les échantillons dans 500 μL de tampon de lavage B à 1% pendant 1 min avec une agitation douce sur un agitateur orbital.
- Pour les cellules cultivées :
- Déposer 3 μL de support de montage sur une glissière
- Retirez l’excès de tampon de lavage du couvercle, puis placez le couvercle (avec les cellules vers le bas) dans le support de montage. Scellez les côtés avec une petite quantité de vernis à ongles transparent pour sceller le couvercle en place.
- Pour les tissus tranchés :
- Transférez soigneusement une tranche de tissu sur la lame préparée et disposez-la, de sorte que la tranche soit à plat. Déposer entre 5 et 10 μL de support de montage (la quantité dépendra de la taille de la tranche) sur la tranche de tissu
- Placez soigneusement un couvercle en verre sur une tranche de tissu et scellez avec une petite quantité de vernis à ongles transparent le long du bord pour sceller le couvercle en place.
- Attendez au moins 15 minutes avant d’analyser au microscope, ou conservez à -20 °C.
8. Obtenez des images numériques avec un microscope confocal
- Optimisez les paramètres d’acquisition (par exemple, puissance laser, gain, décalage) sur les échantillons de tous les traitements. Assurez-vous que l’optimisation comprend la diminution du bruit de fond et l’amélioration du signal sans sursaturer l’intensité des signaux fluorescents. Une fois les paramètres déterminés, appliquez les mêmes paramètres d’acquisition à toutes les images obtenues.
REMARQUE: Les détails d’acquisition suivants peuvent être utilisés avec un microscope confocal Nikon A1 et le logiciel Nikon NIS AR Elements. - Placez la diapositive avec l’échantillon sur la scène et trouvez le plan focal de l’échantillon à l’aide de DAPI à travers l’oculaire.
- Éteignez le port oculaire en cliquant sur Port œil et choisissez un bouton de configuration optique pour ajuster les paramètres.
- Ajustez le gain, le décalage et la puissance laser pour chaque canal fluorescent afin de réduire le bruit de fond et d’améliorer le signal fluorescent. Assurez-vous que le signal fluorescent ne devient pas sursaturé comme mentionné aux étapes 8.5-8.6.
- Surveillez la sursaturation à l’aide d’une pseudocolore pour le signal fluorescent. Au bas de l’image en direct, cliquez avec le bouton droit sur l’onglet étiqueté avec le canal fluorescent actuellement utilisé.
- Ensuite, choisissez Channel Coloring et choisissez une pseudocolore comme Rainbow Dark pour visualiser l’intensité de fluorescence dans une pseudocolore de type carte thermique. Dans Rainbow Dark, les couleurs plus froides indiquent moins d’intensité fluorescente et les couleurs plus chaudes indiquent plus d’intensité fluorescente.
- Une fois tous les canaux fluorescents optimisés, cliquez avec le bouton droit de la souris sur le bouton de configuration optique précédemment choisi et choisissez Assign Current Camera Setting pour ce bouton.
- Vérifiez que les paramètres choisis sont suffisants pour un échantillon aléatoire de chaque groupe de traitement. Si les paramètres choisis sursaturent l’un de ces échantillons, répétez l’étape 8.4 pour éliminer la sursaturation.
- Pour les neurones cultivés, suivez les étapes 8.10-8.16.
- À l’aide du port oculaire, recherchez un neurone avec des dendrites qui ont un chevauchement minimal avec d’autres dendrites.
- Éteignez le port oculaire et utilisez le canal DAPI pour visualiser le corps cellulaire du neurone choisi. Double-cliquez sur le centre du soma pour centrer le neurone au milieu du champ de vision.
- À l’aide du canal MAP2, trouvez le meilleur plan de mise au point pour le signal MAP2 avec balayage en direct.
- Sous l’onglet Acquisition ND , cliquez sur Enregistrer dans un fichier et choisissez un fichier dans lequel enregistrer l’image sous Parcourir. Ensuite, entrez le nom du fichier.
- Sous l’onglet Z , sélectionnez l’option Mode symétrique défini par plage . Réglez le focus sur le meilleur plan MAP2 et cliquez sur le bouton Relatif pour définir ce plan focal comme le milieu de la pile z.
- Réglez la plage sur 5 μm avec des pas de 1 μm et assurez-vous de cocher Fermer l’obturateur actif pendant le mouvement Z. Sous l’onglet Longueur d’onde , sélectionnez le nom du bouton optique avec les paramètres d’acquisition précédemment configurés sous Conf optique. Ensuite, cliquez sur Exécuter maintenant.
- Répétez les étapes 8.1.10-8.1.15 pour environ 10 neurones par coverslip/traitement.
- Pour les tissus tranchés, suivez les étapes 8.18 à 8.22.
- À l’aide du port oculaire, recherchez la région d’intérêt. Par exemple, localisez CA1 de l’hippocampe.
- Éteignez le port oculaire et utilisez le canal MAP2 pour trouver le meilleur plan de mise au point pour le signal MAP2 avec balayage en direct.
- Dans le menu Acquérir , choisissez Numériser une grande image. Ensuite, sélectionnez le nom du bouton optique avec les paramètres d’acquisition précédemment configurés sous le panneau Capture du panneau qui s’ouvre. Assurez-vous également de sélectionner le bon objectif dans ce panneau.
- Sous le panneau Zone et l’oculaire, utilisez les touches fléchées pour définir les limites de la région d’intérêt. Ensuite, cliquez sur Enregistrer une grande image dans un fichier et créez un nom de fichier de chemin d’enregistrement pour l’image.
- Sous le panneau Configuration , assurez-vous que l’option Capture multicanal est cochée, puis choisissez le nom du bouton optique avec les paramètres d’acquisition précédemment configurés sous Conf optique.
REMARQUE: Une pile z pour une grande image est possible, mais augmentera le temps de numérisation.
9. Analyse
- Semblable aux paramètres d’acquisition, utilisez des échantillons de tous les traitements pour optimiser les paramètres de seuil. Assurez-vous que l’optimisation du seuil se concentre sur la diminution du bruit de fond et l’amélioration du signal sans sursaturer l’intensité des signaux fluorescents. Une fois ces paramètres déterminés, appliquez les mêmes paramètres de seuil à toutes les images utilisées pour l’analyse, comme décrit aux étapes 9.2 à 9.3.
- Dans ImageJ, l’option de seuil se trouve sous le menu Image > Ajuster > Seuil. Choisissez l’option Arrière-plan sombre si l’image a un arrière-plan sombre.
- Ensuite, ajustez les limites supérieure et inférieure du seuil selon les paramètres de seuil optimisés précédemment déterminés, puis cliquez sur Appliquer.
- Pour les cellules cultivées, utilisez le canal MAP2 et un outil de région d’intérêt (ROI) à main levée pour dessiner un retour sur investissement pour chaque neurone, y compris les dendrites et le soma. Pour les tissus tranchés, dessinez un retour sur investissement à main levée dans l’image de la tranche qui encapsule la zone d’intérêt (par exemple, la strate rayonnée du CA1 dans l’hippocampe).
- Obtenez la zone du roi. Dans ImageJ, mesurez la zone sous le menu Analyser > Mesure.
- Détectez le nombre de ponctuations dans chaque roi à l’aide d’un outil de détection automatique tel que l’analyse des particules dans ImageJ en suivant les étapes 9.7-9.9.
- Recherchez l’option Analyse des particules dans le menu Analyser > Analyser les particules. Tout d’abord, définissez le diamètre de la taille de puncta, généralement de 0,1 à 3 μm2.
- Ensuite, choisissez l’option Masques de superposition dans le menu déroulant Afficher et cochez l’option Afficher les résultats. Ensuite, cliquez sur OK.
- Si les puncta ne sont pas détectés avec la plage de diamètre choisie, ajustez la plage jusqu’à ce que tous les puncta soient détectés avec cette analyse. Assurez-vous d’utiliser les mêmes paramètres d’analyse des particules pour toutes les images.
- Divisez le nombre de puncta par la zone d’une région d’intérêt individuelle en suivant les étapes 9.11 à 9.13.
- Copiez et collez les résultats de chaque image à partir de la fenêtre contextuelle Résultats d’ImageJ dans une feuille de calcul.
- Tout d’abord, identifiez le fichier et l’échantillon à partir desquels les données ont été obtenues. Ensuite, divisez la zone du ROI par le nombre de puncta.
- Ensuite, effacez les données de la fenêtre contextuelle Résultats et répétez les étapes 9.2 à 9.12.
- Normaliser les résultats pour contrôler les échantillons : Faire la moyenne des résultats (nombre de zones de ponctuation/retour sur investissement) pour les échantillons témoins. Ensuite, divisez les résultats obtenus de tous les échantillons par la moyenne du témoin pour obtenir les résultats normalisés. La nouvelle moyenne des échantillons témoins devrait être égale à 1.
Résultats
Les données modifiées à partir de Heaney et al.6 sont présentées pour démontrer une expérience où l’on s’attend à une formation accrue de synapses (voir6 pour plus d’informations et une discussion plus approfondie du mécanisme). Auparavant, il a été démontré que les antidépresseurs rapides nécessitent l’activation du récepteur métabotropique inhibiteur, gabab (acide gamma-aminobutyrique sous-type B), pour êtreefficaces 13. ...
Discussion
DetectSyn est un test rapide qui utilise un test de ligature de proximité pour détecter les protéines à moins de 40 nm les unes des autres, ce qui permet de détecter la formation de synapses. Cette technique améliore les tests fluorescents actuels, qui ne servent que de mesures indirectes pour la formation de synapses. DetectSyn détecte des changements quantifiables dans les protéines synaptiques localisées à moins de 40 nm, c’est-à-dire dans la fente synaptique, les unes des autres. En outre, DetectSyn est ...
Déclarations de divulgation
Les auteurs ne signalent aucun conflit d’intérêts.
Remerciements
Ce travail a été soutenu par les National Institutes of Health NINDS R01 NS105005 (KRG) et NS105005-03S1 (KRG), le ministère de la Défense USAMRMC W81XWH-14-1-0061 (KRG), NIAAA R01AA016852, NIAAA T32AA007565 (CFH), et une subvention de FRAXA Research (CFH) et de l’Alzheimer’s Association, AARG-NTF-21-852843 (KRG), AARF-19-614794-RAPID (KRG).
matériels
| Name | Company | Catalog Number | Comments |
| 10x PBS | Fisher Scientific | BP39920 | PBS made in house works, as well. |
| 24 well plates | Fisher Scientific | FB012929 | For tissue slices, pre-sterilized plates may be unnecessary. |
| 50 mL conical tubes | Fisher Scientific | 14-432-22 | |
| Aluminium foil | Fisher Scientific | 15-078-290 | |
| Chicken anti-MAP2 antibody | Abcam | ab5392 | |
| Clear nail polish | Fisher Scientific | NC1849418 | Other clear nail polish works, as well. |
| Cold block | Fisher Scientific | 13131012 | |
| Computer workstation | HP | ||
| Confocal or fluorescent microscope | Nikon | A1R HD25 | |
| Donkey anti-chicken FITC | Fisher Scientific | SA1-72000 | |
| Duolink donkey anti-Mouse PLUS | Sigma | DUO92001 | |
| Duolink donkey anti-Rabbit MINUS | Sigma | DUO92005 | |
| Duolink In Situ Detection Reagents Far Red | Sigma | DUO92013 | Contains ligation stock, amplification stock, ligase, and polymerase. |
| Duolink In Situ Mounting Medium with DAPI | Sigma | DUO82040 | |
| Duolink In Situ Wash Buffers, Fluorescence | Sigma | DUO82049 | Contains Wash Buffer A and Wash Buffer B; dilute Wash Buffer B to 1% in diH20 for 1% Wash Buffer B. |
| Fine-tipped paintbrush | Fisher Scientific | NC9691026 | Sable hair, size 00 or 000, can also find at craft stores |
| Fisherbrand Cover Glasses: Rectangles | Fisher Scientific | 12545MP | Cover glass is unnecessary for cultured neurons already on glass coverslips. |
| Fisherbrand Superfrost Plus Microscope Slides | Fisher Scientific | 1255015 | For cultured neurons already on glass coverslips, Superfrost slides may be unnecessary. |
| Freezer, -20°C | VWR | 76449-108 | |
| Glass coverslips | Fisher Scientific | 125480 | |
| Glycine | Fisher Scientific | BP381-1 | |
| Image processing software | e.g. NIS Elements, ImageJ | ||
| Incubator | Fisher Scientific | 15-015-2633 | |
| Large petri dish, 100mm | Fisher Scientific | FB0875712 | |
| Molecular grade water | Fisher Scientific | BP24701 | |
| Mouse anti-Synapsin1 antibody | Synaptic Systems | 106-011 | |
| Normal donkey serum | Jackson ImmunoResearch | 017-000-121 | |
| Orbital shaker | Fisher Scientific | 02-106-1013 | |
| Parafilm | Fisher Scientific | 13-374-10 | |
| Pipette tips | Fisher Scientific | 02-707-025 | |
| Pipettes | Fisher Scientific | 14-388-100 | Working volumes range from 3 µL to 500 µL |
| Plastic pasteur pipette | Fisher Scientific | 02-708-006 | |
| Precision tweezers/foreceps | Fisher Scientific | 12-000-122 | |
| Rabbit anti-PSD95 antibody | Abcam | ab18258 | Other antibody pairs may work, as well, with optimization. |
| Refrigerator | VWR | 76470-402 | |
| Small petri dish, 60 mm | Fisher Scientific | FB0875713A | |
| Timer | Fisher Scientific | 14-649-17 | |
| Tween 20 | Fisher Scientific | BP337-100 |
Références
- Südhof, T. C. Towards an understanding of synapse formation. Neuron. 100 (2), 276-293 (2018).
- Bliss, T. V., Collingridge, G. L. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 361 (6407), 31-39 (1993).
- Heaney, C. F., Raab-Graham, K. F. Dysregulated protein synthesis in major depressive disorder. The Oxford Handbook of Neuronal Protein Synthesis. , 510-532 (2018).
- Masliah, E., Crews, L., Hansen, L. Synaptic remodeling during aging and in Alzheimer's disease. Journal of Alzheimer's Disease. 9, 91-99 (2006).
- van Spronsen, M., Hoogenraad, C. C. Synapse pathology in psychiatric and neurologic disease. Current Neurology and Neuroscience Reports. 10 (3), 207-214 (2010).
- Heaney, C. F., Namjoshi, S. V., Uneri, A., Bach, E. C., Weiner, J. L., Raab-Graham, K. F. Role of FMRP in rapid antidepressant effects and synapse regulation. Molecular Psychiatry. 26 (6), 2350-2362 (2021).
- Pchitskaya, E., Bezprozvanny, I. Dendritic spines shape analysis-Classification or clusterization? Perspective. Frontiers in Synaptic Neuroscience. 12, 31 (2020).
- Alvarez, V. A., Sabatini, B. L. Anatomical and physiological plasticity of dendritic spines. Annual Review of Neuroscience. 30 (1), 79-97 (2007).
- Berry, K. P., Nedivi, E. Spine Dynamics: Are they all the same. Neuron. 96 (1), 43-55 (2017).
- Micheva, K. D., Smith, S. J. Array tomography: A new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron. 55 (1), 25-36 (2007).
- Kaech, S., Banker, G. Culturing hippocampal neurons. Nature Protocols. 1 (5), 2406-2415 (2006).
- Gage, G. J., Kipke, D. R., Shain, W. Whole animal perfusion fixation for rodents. Journal of Visualized Experiments: JoVE. (65), e3564 (2012).
- Workman, E. R., Niere, F., Raab-Graham, K. F. mTORC1-dependent protein synthesis underlying rapid antidepressant effect requires GABABR signaling. Neuropharmacology. 73, 192-203 (2013).
- Li, N., et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 329 (5994), 959-964 (2010).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.